



Introduction
Advancing age is associated with a multitude of physical and physiological deteriorations that leave the elderly susceptible to a wide variety of pathological conditions [1]. Consequently, there is a steep decline in the health-related quality of life for the elderly [2]. Amongst a wide variety of conditions, increased susceptibility to severe infections (such as COVID-19) and inflammatory conditions (such as sepsis) is one such age-related phenomenon [3–6]. Despite representing under 25% of the population, people older than 60 account for more than 75% of sepsis related deaths [7]. With respect to COVID-19, people over 60 are three times more likely to die from a severe infection than people under 60 [8]. Santesmasses et al., estimated that the risk of dying from COVID-19 doubles with every 6-8 years of increase in chronological age, highlighting the importance of age as a risk factor. In a retrospective study, Chen et al. showed that even with a similar number of comorbidities, patients older than 60 had a significantly higher probability of developing a serious version of COVID-19 compared to younger demographics [9]. The severity of disease progression in these population upon infection is partially attributed to the higher prevalence of severe cytokine storm in the elderly [10, 11]. Though there are many theories as to what makes the elderly susceptible to severe cytokine storm, there is no commonly accepted explanation to this phenomenon. [5, 12].
Cellular senescence is a phenomenon by virtue of which stressed or damaged cells undergo a permanent cell cycle arrest [13, 14]. In healthy individuals, senescent cells (SnCs) are cleared rapidly by the immune system [15]. This clearance mechanism has been shown to become impaired with advancing age, leading to the accumulation of SnCs [16, 17]. In turn, the accumulation of SnCs has been implicated in many age-related pathologies and diseases [18–20]. The detrimental effects of SnCs are partly a consequence of their expression of the senescence-associated secretory phenotype (SASP) [21]. The SASP includes an extensive list of factors such as inflammatory cytokines, chemokines, and matrix metalloproteases (MMPs) [22], which are detrimental to the normal functioning of neighboring cells [23–25]. Hence, we hypothesized that SnCs contribute to the increased severity of infectious diseases and infection-mediated cytokine storm in the elderly through the expression of the SASP. To test this hypothesis, we examined whether SnCs exhibit hyper-activation to LPS, IL1β and TNFα stimulation. Our results show that SnCs indeed have a greater proclivity to become hyper-activated in response to inflammatory insults, resulting in the increased production of a variety of inflammatory cytokines and chemokines when compared to their non-senescent counterparts, which we term senescence-associated hyper-activation. Senescence-associated hyper-activation may be attributable to a higher basal activation of the p38 mitogen activated protein kinase (p38) and NF-κB pathways [26]. These findings lay a foundation to elucidate the important role of SnCs in the age-related increased susceptibility to severe infections and inflammatory conditions.
Results
SnCs exhibit a senescence-associated hyper-activation phenotype in response to inflammatory stimulation
While it is well known that SnCs are pro-inflammatory in nature by virtue of expression of the SASP [22, 27], whether inflammatory stimulus could further significantly exacerbate their pro-inflammatory phenotype has not been studied yet. Endothelial cells being a common cell type spread throughout the body in the form of a lining layer of the blood vessels, we decided to use human umbilical vein endothelial cells (HUVEC) to examine this prospect. We also know that HUVEC are inherently responsive to various inflammatory stimuli [28]. To determine if an inflammatory stimulus could significantly exacerbate the pro-inflammatory phenotype of senescent HUVEC compared to their normal counterparts, we examined the transcriptional response of non-senescent (NC HUVEC) and ionizing radiation (IR)-induced senescent HUVEC (IR HUVEC) to lipopolysaccharide (LPS). Using the methods reported by us previously [29], senescence was induced in HUVEC by exposure to ionizing radiation or serial passaging. Induction of senescence in HUVEC by these methods were evidenced by the permanent cell cycle arrest measured by EdU staining (Supplementary Figure 1A), elevated expression of senescence-associated beta galactosidase (SA-β-Gal) activity (Supplementary Figure 1B), increased expression of CDKN2A and CDKN1A mRNA (Supplementary Figure 1C, 1D) and several SASP factors at basal conditions (Figure 1A–1F and Supplementary Figure 2).
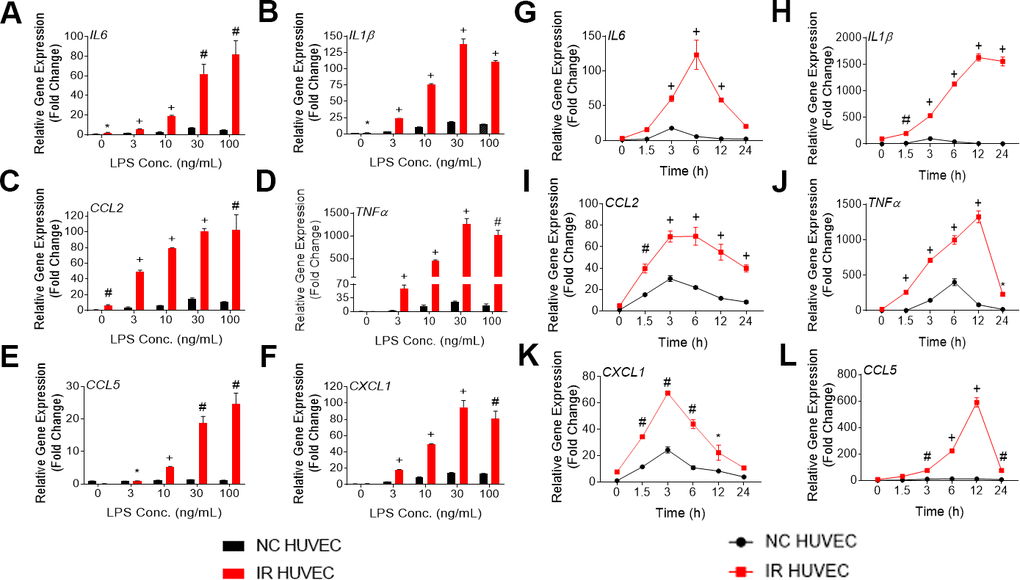
Figure 1. Lipopolysaccharide (LPS) induces a dose- and time-dependent induction of the senescence-associate secretory phenotype (SASP) gene expression in non-senescent HUVECs (NC HUVEC) and ionizing radiation (IR)-induced senescent HUVECs (IR HUVEC). (A–F) Dose response. Relative gene expression of IL6 (A), IL1β (B), CCL2 (C), TNFα (D), CCL5 (E), and CXCL1 (F) in NC HUVEC and IR HUVEC stimulated with 3-100 ng/ml LPS for 3 hours. (G–L), Time course. Relative gene expression of IL6 (G), IL1β (H), CCL2 (I), TNFα (J), CXCL1 (K), and CCL5 (L) in NC HUVEC and IR HUVEC as a function of time of stimulation with 30ng/mL LPS. Gene expression in unstimulated NC HUVEC was used as baseline and GAPDH was used as endogenous control. (n = 3; mean ± SEM; * p<0.05, # p <0.01, + p<0.001 vs. non-SnC).
Upon analyzing the transcriptional response of NC HUVEC and IR HUVEC to LPS stimulation, we observed that both types of cells showed a dose-dependent upregulation of mRNA expression for several cytokines and chemokines such as IL6, CCL2, CXCL1, CCL5, IL1β and TNFα (Figure 1A–1F). Moreover, at any given dose, IR HUVEC showed a significantly higher mRNA expression of these inflammatory mediators than their NC HUVEC counterparts (Figure 1A–1F).
Next, we examined the time-dependent dynamics of the mRNA expression of these inflammatory mediators upon LPS stimulation. Much like the dose-dependent response, both NC HUVEC and IR HUVEC exhibited a time-dependent response to LPS stimulation. Again, IR HUVEC expressed significantly higher levels of mRNA for the analyzed cytokines and chemokines, at most given time points, relative to NC HUVEC (Figure 1G–1L).
To verify if this exacerbated response of IR HUVEC was specific to LPS, we examined the response of NC and IR HUVEC to IL1β and TNFα, known inflammatory stimulants [28]. As observed with response to LPS, stimulation by IL1β and TNFα elicited a strong transcriptional activation of IL6, CCL2, CXCL1, CCL5, IL1β and TNFα mRNA expression in both NC HUVEC and IR HUVEC. However, stimulated IR HUVEC expressed significantly higher levels of mRNA for all tested cytokines and chemokines when compared to their NC counterparts (Figure 2A–2L). These results suggest that IR HUVEC are hyper-reactive to inflammatory stimulus, which we term senescence-associated hyper-activation.
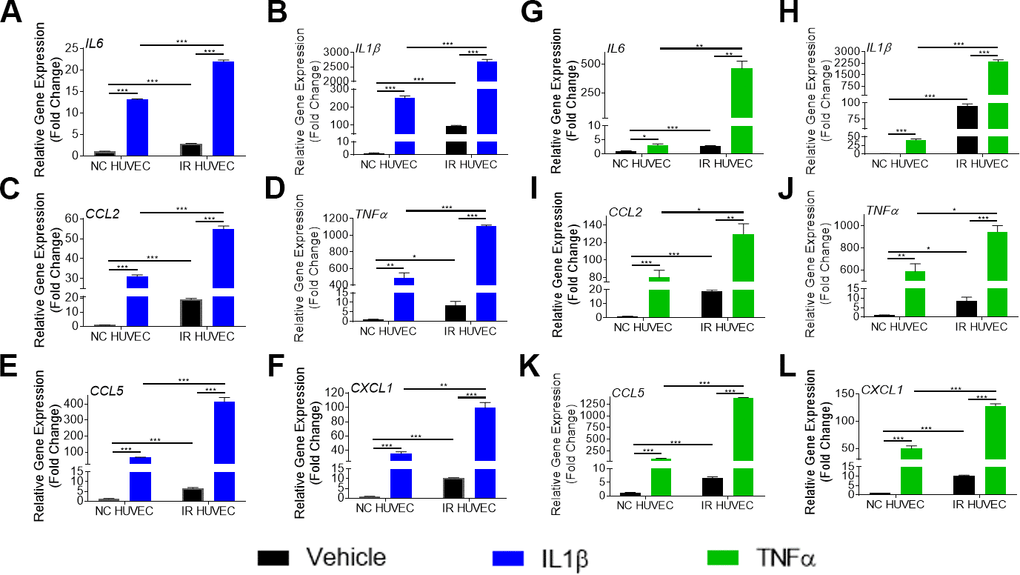
Figure 2. Comparison of the SASP gene expression in IL1β and TNFα-stimulated NC HUVEC and IR HUVEC. Relative fold change in gene expression of IL6 (A), IL1β (B), CCL2 (C), TNFα (D), CCL5 (E), and CXCL1 (F) in NC HUVEC and IR HUVEC 3 hours after stimulation with 3 ng/mL IL1β. Relative fold change in gene expression of IL6 (G), IL1β (H), CCL2 (I), TNFα (J), CCL5 (K), and CXCL1 (L) in NC HUVEC and IR HUVEC 3 hours after stimulation with 3 ng/mL TNFα. Gene expression in unstimulated NC HUVEC was used as baseline and GAPDH was used as endogenous control. (n = 3; mean ± SEM; * p<0.05, ** p<0.01, *** p<0.001).
To examine if this phenomenon was exclusive for IR HUVEC, we generated replicative senescent HUVEC (Rep-Sen HUVEC) and tested their response to LPS, IL1β and TNFα. Rep-Sen HUVEC, similar to IR HUVEC, showed exacerbated transcriptional activation of IL6, CXCL10 and CCL5 upon inflammatory stimulation (Supplementary Figure 2A–2I).
To explore whether senescence-associated hyper-activation is a general characteristic of SnCs, rather than being specific to endothelial cells, studies were extended to IR-induced senescent human adipose derived stem cells (ASCs) (Supplementary Figure 3A–3I), renal epithelial cells (RECs) (Supplementary Figure 4A–4I) and WI38 lung fibroblast (WI38) (Supplementary Figure 5A–5I). SnCs from all three cell types exhibited a higher basal level of IL6, CCL5 and CXCL10 mRNA expression as well as higher expression of these inflammatory mediators in response to LPS, IL1β and TNFα stimulation than non-SnCs with a few exceptions in which some of the cells were not very responsive to LPS stimulation. For example, non-senescent WI38 fibroblasts showed no significant change in expression upon LPS stimulation for any of the three genes analyzed, whereas senescent WI38 fibroblasts showed a significant upregulation of mRNA for CCL5, but not for IL6 and CXCL1 upon LPS stimulation.
Cumulatively, this data suggests that SnCs exhibit a senescence-associated hyper-activation phenotype upon being stimulated with a prominent inflammatory stimulant.
SnCs secrete high levels of inflammatory cytokines and chemokines
To investigate whether the increased mRNA levels for the multiple inflammatory mediators in SnCs translate into an elevated secretion of the corresponding factors, we analyzed the conditioned media from NC HUVEC and IR HUVEC with or without LPS stimulation. From the data presented in Figure 3 and Supplementary Table 1, it is evident that IR HUVEC secreted significantly higher levels of multiple cytokines and chemokines relative to NC HUVEC under basal condition. More importantly, when stimulated with LPS, IR HUVEC produced much greater levels of these factors than LPS-stimulated NC HUVEC (Figure 3 and Supplementary Table 1), confirming that SnCs indeed have a senescence-associated hyper-activation phenotype.
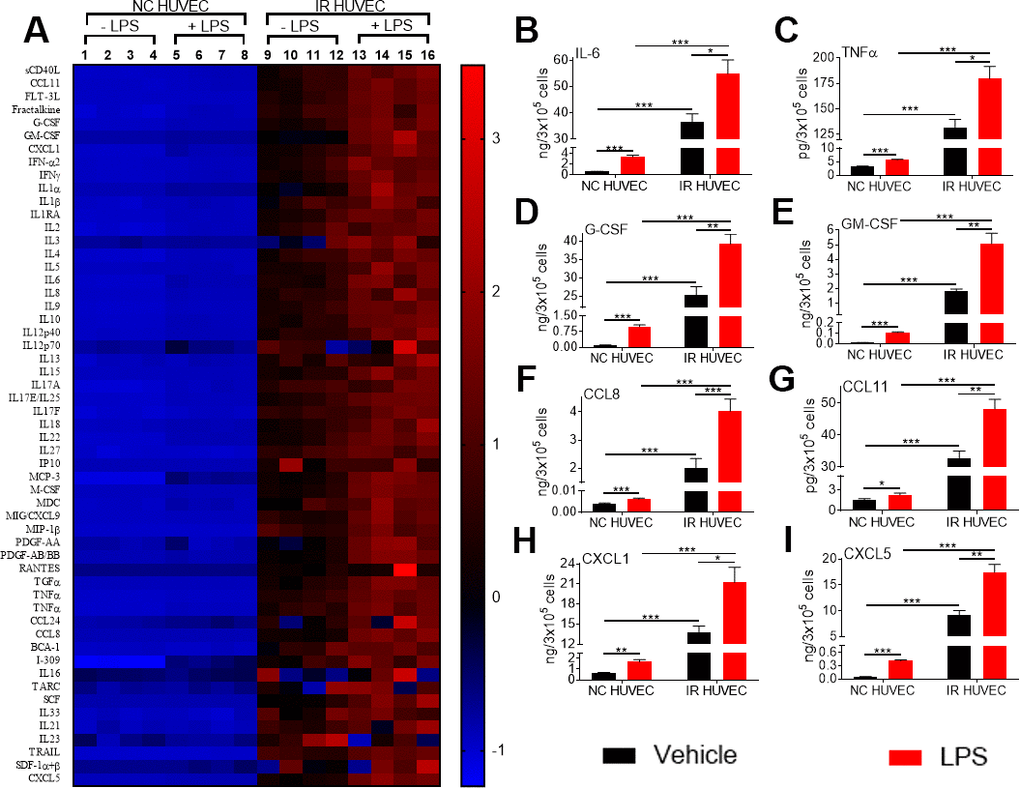
Figure 3. Comparison of the production of inflammatory cytokines and chemokines by NC HUVEC and IR HUVEC. (A) Heat-map representing the normalized concentrations of inflammatory cytokines and chemokines in the conditioned media of NC HUVEC and IR HUVEC stimulated with vehicle or LPS (30 ng/ml) for 24 hours. (B–I) Normalized concentration of IL6 (B), TNFα (C), G-CSF (D), GM-CSF (E), CCL8 (F), CCL11 (G), CXCL1 (H), and CXCL5 (I), produced by NC HUVEC and IR HUVEC stimulated with vehicle or LPS (30 ng/ml) for 24 hours. (n = 4; mean ± SEM; * p<0.05, ** p<0.01, *** p<0.001).
Senescence-associated hyper-activation is not associated with increased expression of the Toll-like receptor4 (TLR4)
To investigate whether senescence-associated hyper-activation is due to an increased expression of surface receptors for the inflammatory stimulants, we quantified the expression of TLR4 (a primary receptor for LPS [30]), IL1 receptor 1 (IL1R1), IL1R2 and TNF receptor 1 (TNFR1) in NC and IR HUVECs using western blotting (Figure 4). We found that both cells expressed similar levels of TLR4 (Figure 4B), IL1R1 (Figure 4C) and IL1R2 (Figure 4D), while IR HUVECs expressed a slightly higher level of TNFR1 than NC HUVECs (Figure 4E). This finding suggests that the senescence-associated hyper-activation in responses to LPS and IL1β stimulation is not due to an increased expression of surface receptors but possibly due to the changes in their downstream pathways. However, increased expression of TNFR1 may partially contribute to the senescence-associated hyper-activation to TNFα.
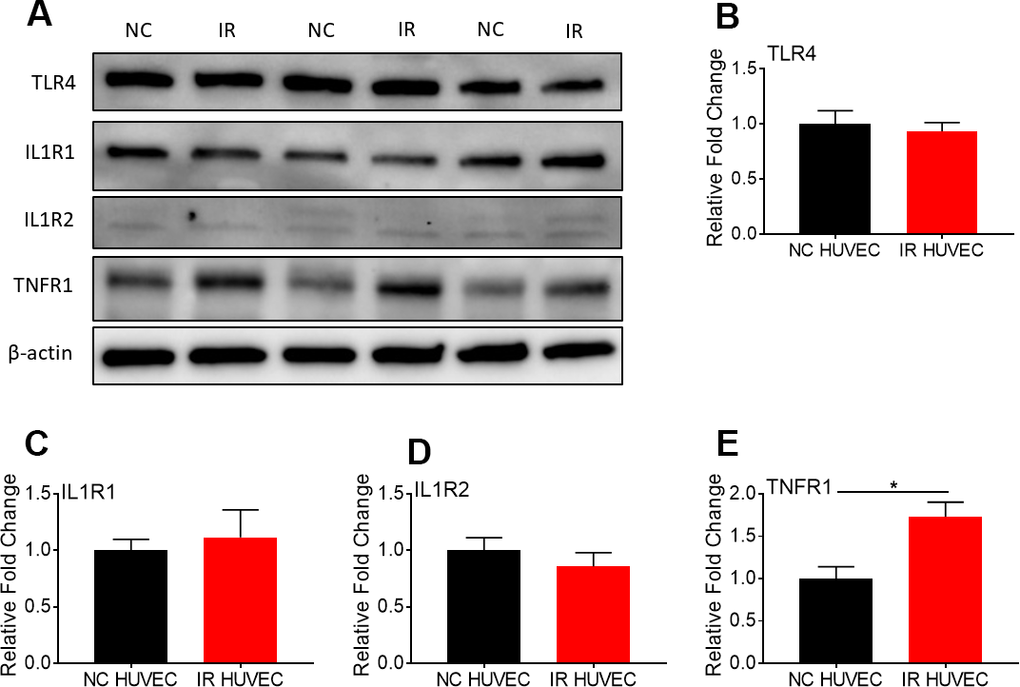
Figure 4. Comparison of the expression of surface receptors of inflammatory stimulants in NC HUVEC and IR HUVEC. Representative western-blot images (A) and densitometry based quantitative analysis of TLR4 (B) IL1R1 (C), IL1R2 (D), and TNFR1 (E) in NC HUVEC and IR HUVEC. β-actin was used as a loading control. (n = 3; mean ± SEM; * p<0.05).
Senescence-associated hyper-activation is mediated by the activation of p38
We hypothesized that senescence-associated hyper-activation is attributable to the hyper-activity of some of the intracellular signaling pathways responsible for the SASP phenotype. To test this hypothesis, we examined if the p38 pathway becomes hyper-activated in SnCs upon inflammatory stimulation. Our results not only corroborated previous demonstrations of SnCs having an elevated basal p38 activation [26], but also showed that IR HUVEC exhibited a significantly higher activation of p38 upon IPS stimulation compared to NC HUVEC (Figure 5A, 5B).
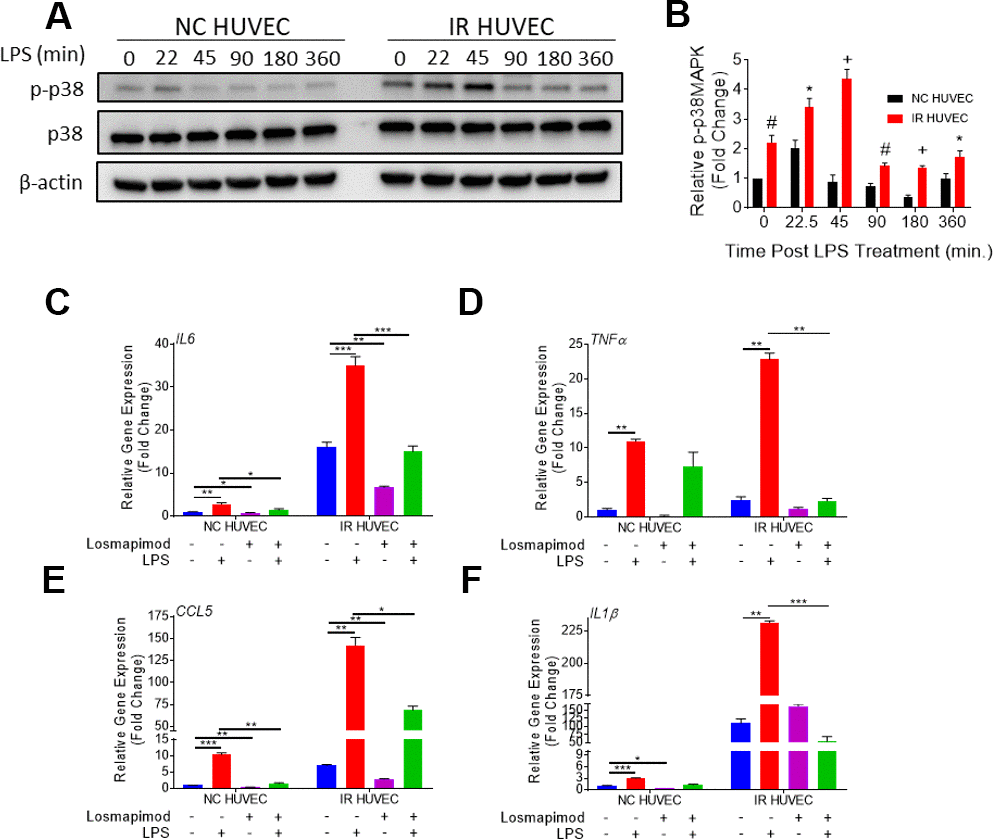
Figure 5. Regulation of senescence-associated hyper-activation via p38-MAPK (p38) pathway. (A, B) IR HUVEC exhibit higher activation of p38 than NC HUVEC. Representative western-blot images (A) and densitometry based quantitative analysis (B) of phosphorylated p38 (p-p38) and total p38 (p38) in NC HUVEC and IR HUVEC stimulated with LPS (30 ng/ml) for 0-6 hours. (n = 3; mean ± SEM; * p<0.05, # p <0.01, + p<0.001 vs. NC HUVEC). β-actin was used as a loading control. (C–F) p38 inhibition attenuates the expression of IL6 (C), TNFα (D), CCL5 (E), and IL1β (F) mRNA in IR-HUVEC. NC HUVEC and IR-HUVEC were exposed to LPS (30 ng/ml) or the p38 inhibitor losmapimod (1 μM) or their combination for 3 hours followed by mRNA analysis. Gene expression in unstimulated NC HUVEC was used as baseline and GAPDH was used as endogenous control (n = 3; mean ± SEM; * p<0.05, ** p<0.01, *** p<0.001).
To determine whether senescence-associated hyper-activation effect was dependent on the activation of p38, NC HUVEC and IR HUVEC were pre-treated with losmapimod, a specific p38 inhibitor [31], prior to LPS stimulation. Our analysis showed that the upregulation of IL6, TNFα, CCL5, and IL1β mRNA expressions in IR HUVEC with or with LIP stimulation were abrogated or significantly reduced by the losmapimod pretreatment, demonstrating that activation of p38 mediates not only the expression of SASP at the basal conditions but also that of senescence-associated hyper-activation in response to LPS stimulation (Figure 5C–5F).
Senescence-associated hyper-activation is also mediated by the activation of the NF-κB pathway
Based on the study by Freund et al., showing that p38-MAPK acts upstream of the NF-κB pathway to induce SASP, we hypothesized that NF-κB activation also mediates senescence-associated hyper-activation. To test this hypothesis, we analyzed NF-κB p65 levels in the cytoplasm and nucleus of NC HUVEC and IR HUVEC at baseline and upon stimulation with LPS, IL1β and TNFα, by immunocytochemistry (Supplementary Figure 6) and by immunoblotting (Figure 6A, 6B). Both results showed that IR HUVEC exhibited a significantly higher baseline level of nuclear NF-κB, as well as a significantly higher nuclear translocation of NF-κB upon LPS stimulation than NC HUVEC (Figure 6A, 6B and Supplementary Figure 6). Similarly, IR HUVEC showed a greater translocation of NF-κB p65 into the nucleus upon stimulation with IL1β and TNFα than NC HUVEC (Supplementary Figure 6). Collectively, these results suggest that IR HUVEC respond to inflammatory stimulation by hyper-activation of the NF-κB pathway.
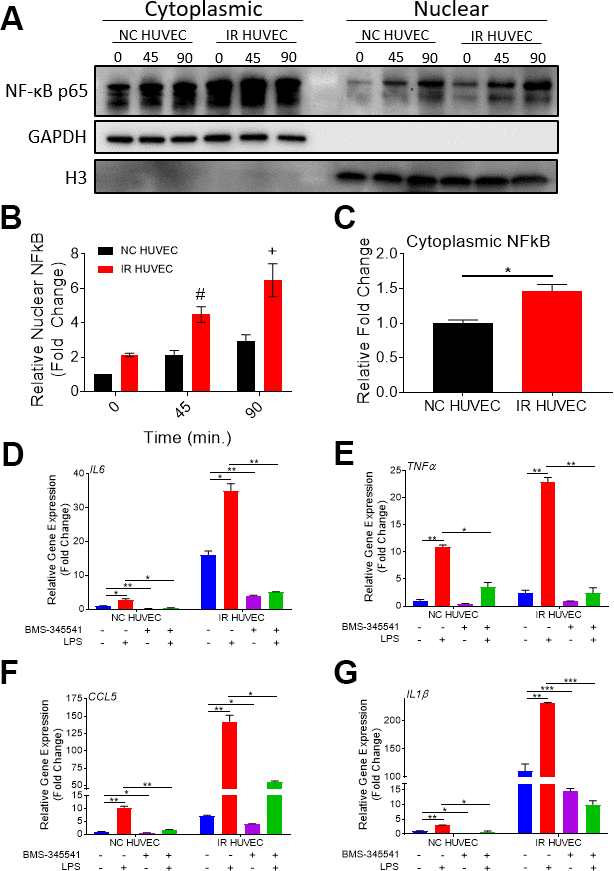
Figure 6. Regulation of senescence-associated hyper-activation via NF-κB pathway. (A–C) IR HUVEC exhibit higher baseline expression and activation of NF-κB when compared to NC HUVEC. Representative western-blot images (A) (the middle line is the molecular weight markers), densitometry based quantitative analysis of nuclear fraction (B) and cytoplasmic fraction (C) of NF-κB p65 in NC HUVEC and IR HUVEC stimulated with LPS (30 ng/ml) for 0-90 min (n = 3; mean ± SEM; * p<0.05, # p <0.01, + p<0.001 vs. NC HUVEC). Histone H3 and GAPDH were used as the loading control for nuclear and cytoplasmic proteins, respectively. (D–G) NF-κB inhibition attenuates the expression of IL6 (D), TNFα (E), CCL5 (F), and IL1β (G) mRNA in IR HUVEC. NC HUVEC and IR HUVEC were treated with LPS (30 ng/ml) or the NF-κB inhibitor BMS-345541 (10 μM) or their combination for three hours followed by mRNA analysis. Gene expression in unstimulated NC HUVEC was used as baseline and GAPDH was used as endogenous control. (n = 3; mean ± SEM; * p<0.05, ** p<0.01, *** p<0.001).
To validate the dependence of senescence-associated hyper-activation on NF-κB activity, NC HUVEC and IR HUVEC were treated with BMS-345541, a potent inhibitor of NF-κB activation [32], prior to LPS stimulation. Our analysis revealed that the upregulation of IL6, TNFα, CCL5 and IL1β mRNA expressions in IR HUVEC with or without LPS stimulation were abrogated or significantly reduced by the pretreatment with BMS-345541 (Figure 6C–6F), confirming that the NF-κB pathway plays an important role in the induction of both SASP and senescence-associated hyper-activation in IR HUVEC.
Discussion
An increased proclivity to severe infections and infection-induced adverse cytokine storm can be observed in the aging population [3, 5, 8, 11, 12]. Here we report the discovery of a novel phenomenon named senescence-associated hyper-activation in SnCs and demonstrated that common inflammatory stimuli such as LPS, IL1β and TNFα, which are relevant to many infections and inflammatory conditions, induce hyper-activation in SnCs. This senescence-associated hyper-activation makes SnCs produce a large amount of various inflammatory cytokines and chemokines including IL6, a cytokine believed to play a central role in the development of a cytokine storm [33, 34]. These findings are in agreement with the prior observation by Chambers et al., that skin biopsies from both young and old patients showed similar baseline expression of chemokines, such as CXCL1, CCL2 and CCL8 but skin biopsies from older population had a significantly higher expression of these chemokines under inflammatory conditions [6]. Additionally, Saito et al. also reported that old mice subjected to LPS injection or cecal ligation puncture (CLP) expressed significantly higher levels of IL6 and TNFα mRNA in lungs and hearts than the tissues from young mice [3].
Extrapolating from previously published studies, senescence-associated hyper-activation may be an underlying cause of the high susceptibility to infection-induced severe inflammation and cytokine storm in older populations. For example, IR HUVEC showed a significantly higher production of various inflammatory cytokines as IL6, G-CSF, and TNFα (Figure 3B–3I), known markers of a poor prognosis in severe infectious conditions including COVID-19 [35–37]. We speculate that senescence burden and senescence-associated hyper-activation may contribute to the severe cases of COVID-19 that disproportionately impact the elderly [5]. In addition, these findings could partly explain the findings by Saito et al., showing that while young and old mice had a similar baseline of serum cytokines, an inflammatory insult with LPS triggered the generation of significantly higher quantities of these cytokines in older mice compared to their younger counterparts [3]. Therefore, further studies are needed to determine if accumulation of SnCs is responsible for developing high susceptibility to severe cytokine storm seen in the elderly under various pathological conditions.
Our study also revealed that senescence-associated hyper-activation is not necessarily an outcome of an altered expression of surface receptors of the inflammatory stimulants but more probably attributable to the changes in the downstream pathways such as the p38 and NF-κB signaling pathways as reported previously [26]. As both these pathways have been shown to be crucial for the expression of the SASP [26, 38], it is likely that SnCs, by virtue of their SASP regulating networks, are primed to react more aggressively to inflammatory stimuli when compared to their normal counterparts despite not having an increased expression of receptors upstream of these pathways. The involvement of the p38 pathway in senescence-associated hyper-activation is in agreement with a report from Vukmanovic-Stejic et al., which shows that the enhanced cutaneous inflammation seen in the elderly injected with varicella zoster virus antigen, was attenuated by treatment with losmapimod, a highly specific p38 inhibitor [39]. Though the dysregulated cutaneous inflammation was initially not linked to cellular senescence, a recent study by Chambers et al., demonstrated the involvement of cellular senescence in this phenomenon, making it relevant to our study [6]. In addition, these results also provide additional evidence as to why p38 inhibition is a promising therapeutic option for the treatment of severe COVID-19 patients [40].
In conclusion, we discovered that SnCs exhibit hyper-activation upon an inflammatory insult, which we termed senescence-associated hyper-activation. Our results suggest that SnCs could contribute to the age-related predisposition of the body to develop stronger cytokine storm upon infections. This calls for a paradigm shifting study from considering SnCs as indirect participants in inflammatory pathologies to being recognized as central players in these processes. Discovering the senescence-associated hyper-activation phenomenon also highlights an opportunity and the urgent need for testing the possibilities that the newly developed senotherapeutics may have the potential to mitigate the incidence of life-threatening inflammatory conditions in the elderly and potentially lengthen their health-span.
Materials and Methods
Cell culture
Human umbilical vein endothelial cells (HUVEC, Cat. No. PCS-100-010), human WI38 fibroblasts (WI38, Cat. No. CCL-75) and human renal epithelial cells (REC, Cat. No. PCS-400-012) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Adipose stem/stromal cells (ASC) were previously isolated from adipose tissue (female donors) obtained during elective liposuction procedure (Traktuev et al., 2008). HUVEC were cultured in EBM-2 (Cat. No. CC-3156, Lonza, Basel, Switzerland) media supplemented with EGM-2 SingleQuots (Cat. No. CC-4176, Lonza, Basel, Switzerland). WI38 cells were cultured in DMEM medium (Cat. No. 12430054, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS, Cat. No. 89510-188, VWR, Radnor, PA, USA). REC were cultured in REBM medium (CC-3191, Lonza, Basel, Switzerland) supplemented with REGM SingleQuots (CC-4127, Lonza, Basel, Switzerland). ASC were cultured in EBM-2 (Cat. No. CC-3156, Lonza, Basel, Switzerland) media supplemented with EGM-2MV SingleQuots (Cat. No. CC-4147, Lonza, Basel, Switzerland). To prevent contamination all culture media were supplemented with 100 U/mL penicillin and 100 μg/mL streptomycin (Cat. No. 15140122, Thermo Fisher Scientific, Waltham, MA, USA). Cells were cultured in a humidified incubator at 37° C with 5% CO2.
Senescence induction
Cells of early passages (HUVEC < 10 passages; WI-38 < 25 passages; REC < 10 passages; and ASC < 4 passages) were considered to be non-senescent cells (NC). Two methods of senescence induction - replicative exhaustion and ionizing radiation, were used as previously described [29]. In short, to generate replicative senescent cells (SnCs), HUVEC were passaged until they stopped replicating further. To generate IR-induced SnCs, all cell types were exposed to 20 Gy of X-rays, followed by a culturing for 10 days. The induction of cellular senescence was validated by analyzing the expression of CDKN2A and CDKN1A mRNA, positive of SA-β-Gal, and lack of cell proliferation. SA-β-Gal was tested using SA-β-Gal staining kit (Cat. No. 9860, Cell Signaling Technologies, Danvers, MA, USA), and cell proliferation was assessed using EdU cell proliferation kit (Cat. No. C10337, Invitrogen, Carlsbad, CA, USA), following the manufacturers’ instructions.
Inflammatory stimulation
Various concentrations of LPS (Cat. No. L4391, Sigma-Aldrich, St. Louis, MO, USA), IL1β (Cat. No. 200-01B, Peprotech, Rocky Hill, NJ, USA), and TNFα (Cat. No. 300-01A, Peprotech, Rocky Hill, NJ, USA) were used to stimulate cells for different durations of time at the indicated concentrations and times presented in each figure legend.
Analysis of the production of various inflammatory cytokines and chemokines
NC HUVEC and IR HUVEC were cultured with medium alone or with 30 ng/mL LPS for 24 hours. The conditioned media from the cultures was then harvested and subjected to analysis using Human Cytokine Array/Chemokine Array 71-Plex Panel by the Eve Technologies Corporation (Calgary, AB, Canada). Values are listed in Supplementary Table 1 (Supplementary Table 1).
Quantitative polymerase chain reaction (qPCR)
RNA isolated from cells using RNeasy mini kit (Cat. No. 74106, Qiagen, Hilden, Germany) was converted into cDNA using a high capacity cDNA reverse transcription kit (Cat. No. 4368813, Applied Biosystems, Foster City, CA, USA). Gene expression was then quantified using gene specific primers (Supplementary Table 2) and fast SYBR green master-mix (Cat. No. 4385617, Applied Biosystems. Foster City, CA, USA) as per the manufacturer’s instructions. The expression of GAPDH was used for normalization. Level of gene expression in untreated NC cells was used as baseline and fold change in gene expression was defined based on ΔΔCT method.
Pathway inhibitory studies
NC HUVEC and IR HUVEC were pre-treated with 1μM of losmapimod (Cat. No. HY10402, Med Chem Express, New Jersey, USA) or BMS-345541 (Cat. No. HY-10519, Med Chem Express, New Jersey, USA) for 2 hours before the cells were stimulated with 30 ng/mL LPS for 3 hours before being harvested for RNA isolation for qPCR.
Whole protein isolation and sample preparation for immunoblot analysis
Control and treated cells were washed with 1X PBS before RIPA buffer (Cat. No. BP-115, Boston Bioproducts, Ashland, MA, USA) was added to the dish. The resultant lysate was then frozen at -80° C for later use. These protein lysates were then thawed and quantified using Pierce BCA protein assay kit (Cat. No. 23225, Thermo Fisher Scientific, Waltham, MA, USA) and normalized before adding 4X Laemmli loading buffer (Cat. No. BP-110R, Boston Bioproducts, Ashland, MA, USA). Samples were then heated to 95° C for 10 minutes before using for western blotting as we previously reported [29].
Cell fractionation and protein isolation
Cells were trypsinised and harvested as pellets after various treatments. They were lysed and then fractionated to obtain cytoplasmic and nuclear proteins for immunoblot analysis using the NE-PER nuclear and cytoplasmic extraction kit (Cat. No. 78833, Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s instructions.
Western blotting
Western blotting was done as previously described [29]. In short, 10 μL of each prepared protein sample was used to run western blot using 15-well 4-20% Mini-PROTEAN TGX Precast Protein Gels (Cat. No. 4561096, Bio-Rad Laboratories, Hercules, CA, USA). The samples were run at 200V for 45 minutes before transferring onto a PVDF membrane using the Trans-Blot turbo transfer system (Bio-Rad Laboratories, Hercules, CA, USA). The PVDF membranes were blocked with 5% milk for 1 hour at room temperature before incubating with primary antibody overnight at 4° C. The membranes were then washed and incubated with horse radish peroxidase (HRP)-conjugated secondary antibody for 1 hour at room temperature. Protein-antibody complexes were revealed with HRP substrate on ChemiDoc imaging system (Bio-Rad Laboratories, Hercules, CA, USA). Antibodies used are listed in Supplementary Table 3 (Supplementary Table 3).
Statistical analysis
Data are expressed as mean ± SEM, unless mentioned otherwise. When comparing more than two groups, data sets were analyzed using repeated measures analysis of variance (ANOVA) on Graphpad Prism (San Diego, CA, USA). Post hoc comparisons were performed between group means using Sidak multiple comparisons test. To compare two groups, Student's t test was used. p < 0.05 was considered significant.
Author Contributions
V.B. conceived the project, designed and performed the majority of the experiments, analyzed the data, and wrote the manuscript; S.M.S and Y.Y. assisted with some of the experiments; Y.H., D.O.T., and T.C.F. analyzed the data and revised the manuscript; V.B. and D.Z. conceived the project, designed the experiments, analyzed the data, and wrote and revised the manuscript.
Conflicts of Interest
The authors have no conflicts of interest.
Funding
This study was supported by US National Institutes of Health (NIH) grants R01 CA211963 (D.Z.), R01 AG037984 and R01 AG052258 (T.C.F), P30 AG028740, and the Evelyn F. McKnight Brain Research Foundation. The authors would like to thank Ms. Alexandra M. Fahnlander for editing the manuscript.
References
- 1. Franceschi C, Garagnani P, Morsiani C, Conte M, Santoro A, Grignolio A, Monti D, Capri M, Salvioli S. The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front Med (Lausanne). 2018; 5:61. https://doi.org/10.3389/fmed.2018.00061 [PubMed]
- 2. Olfson M, Wall M, Liu SM, Schoenbaum M, Blanco C. Declining Health-Related Quality of Life in the U.S. Am J Prev Med. 2018; 54:325–33. https://doi.org/10.1016/j.amepre.2017.11.012 [PubMed]
- 3. Saito H, Sherwood ER, Varma TK, Evers BM. Effects of aging on mortality, hypothermia, and cytokine induction in mice with endotoxemia or sepsis. Mech Ageing Dev. 2003; 124:1047–58. https://doi.org/10.1016/j.mad.2003.08.002 [PubMed]
- 4. Kale SS, Yende S. Effects of Aging on Inflammation and Hemostasis through the Continuum of Critical Illness. Aging Dis. 2011; 2:501–11. [PubMed]
- 5. Mueller AL, McNamara MS, Sinclair DA. Why does COVID-19 disproportionately affect older people? Aging (Albany NY). 2020; 12:9959–81. https://doi.org/10.18632/aging.103344 [PubMed]
- 6. Chambers ES, Vukmanovic-Stejic M, Shih BB, Trahair H, Subramanian P, Devine OP, Glanville J, Gilroy D, Rustin MHA, Freeman TC, Mabbott NA, Akbar AN. Recruitment of inflammatory monocytes by senescent fibroblasts inhibits antigen-specific tissue immunity during human aging. Nat Aging. 2021; 1:101–13. https://doi.org/10.1038/s43587-020-00010-6
- 7. Rhee C, Dantes R, Epstein L, Murphy DJ, Seymour CW, Iwashyna TJ, Kadri SS, Angus DC, Danner RL, Fiore AE, Jernigan JA, Martin GS, Septimus E, et al, and CDC Prevention Epicenter Program. Incidence and Trends of Sepsis in US Hospitals Using Clinical vs Claims Data, 2009-2014. JAMA. 2017; 318:1241–49. https://doi.org/10.1001/jama.2017.13836 [PubMed]
- 8. Santesmasses D, Castro JP, Zenin AA, Shindyapina AV, Gerashchenko MV, Zhang B, Kerepesi C, Yim SH, Fedichev PO, Gladyshev VN. COVID-19 is an emergent disease of aging. Aging Cell. 2020; 19:e13230. https://doi.org/10.1111/acel.13230 [PubMed]
- 9. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, Wang T, Zhang X, Chen H, Yu H, Zhang X, Zhang M, Wu S, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020; 130:2620–29. https://doi.org/10.1172/JCI137244 [PubMed]
- 10. Mauvais-Jarvis F. Aging, Male Sex, Obesity, and Metabolic Inflammation Create the Perfect Storm for COVID-19. Diabetes. 2020; 69:1857–63. https://doi.org/10.2337/dbi19-0023 [PubMed]
- 11. Song TZ, Zheng HY, Han JB, Jin L, Yang X, Liu FL, Luo RH, Tian RR, Cai HR, Feng XL, Liu C, Li MH, Zheng YT. Delayed severe cytokine storm and immune cell infiltration in SARS-CoV-2-infected aged Chinese rhesus macaques. Zool Res. 2020; 41:503–16. https://doi.org/10.24272/j.issn.2095-8137.2020.202 [PubMed]
- 12. Meftahi GH, Jangravi Z, Sahraei H, Bahari Z. The possible pathophysiology mechanism of cytokine storm in elderly adults with COVID-19 infection: the contribution of “inflame-aging”. Inflamm Res. 2020; 69:825–39. https://doi.org/10.1007/s00011-020-01372-8 [PubMed]
- 13. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965; 37:614–36. https://doi.org/10.1016/0014-4827(65)90211-9 [PubMed]
- 14. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018; 28:436–53. https://doi.org/10.1016/j.tcb.2018.02.001 [PubMed]
- 15. Prata LG, Ovsyannikova IG, Tchkonia T, Kirkland JL. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin Immunol. 2018; 40:101275. https://doi.org/10.1016/j.smim.2019.04.003 [PubMed]
- 16. Pereira BI, Devine OP, Vukmanovic-Stejic M, Chambers ES, Subramanian P, Patel N, Virasami A, Sebire NJ, Kinsler V, Valdovinos A, LeSaux CJ, Passos JF, Antoniou A, et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8+ T cell inhibition. Nat Commun. 2019; 10:2387. https://doi.org/10.1038/s41467-019-10335-5 [PubMed]
- 17. Muñoz DP, Yannone SM, Daemen A, Sun Y, Vakar-Lopez F, Kawahara M, Freund AM, Rodier F, Wu JD, Desprez PY, Raulet DH, Nelson PS, van’t Veer LJ, et al. Targetable mechanisms driving immunoevasion of persistent senescent cells link chemotherapy-resistant cancer to aging. JCI Insight. 2019; 5:e124716. https://doi.org/10.1172/jci.insight.124716 [PubMed]
- 18. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011; 479:232–36. https://doi.org/10.1038/nature10600 [PubMed]
- 19. Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015; 21:1424–35. https://doi.org/10.1038/nm.4000 [PubMed]
- 20. Olivieri F, Prattichizzo F, Grillari J, Balistreri CR. Cellular Senescence and Inflammaging in Age-Related Diseases. Mediators Inflamm. 2018; 2018:9076485. https://doi.org/10.1155/2018/9076485 [PubMed]
- 21. He S, Sharpless NE. Senescence in Health and Disease. Cell. 2017; 169:1000–11. https://doi.org/10.1016/j.cell.2017.05.015 [PubMed]
- 22. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010; 5:99–118. https://doi.org/10.1146/annurev-pathol-121808-102144 [PubMed]
- 23. Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, von Zglinicki T. A senescent cell bystander effect: senescence-induced senescence. Aging Cell. 2012; 11:345–49. https://doi.org/10.1111/j.1474-9726.2012.00795.x [PubMed]
- 24. Nelson G, Kucheryavenko O, Wordsworth J, von Zglinicki T. The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech Ageing Dev. 2018; 170:30–36. https://doi.org/10.1016/j.mad.2017.08.005 [PubMed]
- 25. da Silva PF, Ogrodnik M, Kucheryavenko O, Glibert J, Miwa S, Cameron K, Ishaq A, Saretzki G, Nagaraja-Grellscheid S, Nelson G, von Zglinicki T. The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell. 2019; 18:e12848. https://doi.org/10.1111/acel.12848 [PubMed]
- 26. Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011; 30:1536–48. https://doi.org/10.1038/emboj.2011.69 [PubMed]
- 27. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011; 192:547–56. https://doi.org/10.1083/jcb.201009094 [PubMed]
- 28. Makó V, Czúcz J, Weiszhár Z, Herczenik E, Matkó J, Prohászka Z, Cervenak L. Proinflammatory activation pattern of human umbilical vein endothelial cells induced by IL-1β, TNF-α, and LPS. Cytometry A. 2010; 77:962–70. https://doi.org/10.1002/cyto.a.20952 [PubMed]
- 29. He Y, Li W, Lv D, Zhang X, Zhang X, Ortiz YT, Budamagunta V, Campisi J, Zheng G, Zhou D. Inhibition of USP7 activity selectively eliminates senescent cells in part via restoration of p53 activity. Aging Cell. 2020; 19:e13117. https://doi.org/10.1111/acel.13117 [PubMed]
- 30. Park BS, Lee JO. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med. 2013; 45:e66. https://doi.org/10.1038/emm.2013.97 [PubMed]
- 31. Ino H, Takahashi N, Terao T, Igarashi H, Sarai N. Safety, tolerability, pharmacokinetics, and pharmacodynamics of losmapimod in healthy Japanese volunteers. Clin Pharmacol Drug Dev. 2015; 4:262–69. https://doi.org/10.1002/cpdd.190 [PubMed]
- 32. Burke JR, Pattoli MA, Gregor KR, Brassil PJ, MacMaster JF, McIntyre KW, Yang X, Iotzova VS, Clarke W, Strnad J, Qiu Y, Zusi FC. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J Biol Chem. 2003; 278:1450–56. https://doi.org/10.1074/jbc.M209677200 [PubMed]
- 33. McGonagle D, Sharif K, O’Regan A, Bridgewood C. The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun Rev. 2020; 19:102537. https://doi.org/10.1016/j.autrev.2020.102537 [PubMed]
- 34. Tanaka T, Narazaki M, Kishimoto T. Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy. 2016; 8:959–70. https://doi.org/10.2217/imt-2016-0020 [PubMed]
- 35. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020; 395:497–506. https://doi.org/10.1016/S0140-6736(20)30183-5 [PubMed]
- 36. Coomes EA, Haghbayan H. Interleukin-6 in Covid-19: A systematic review and meta-analysis. Rev Med Virol. 2020; 30:1–9. https://doi.org/10.1002/rmv.2141 [PubMed]
- 37. Han H, Ma Q, Li C, Liu R, Zhao L, Wang W, Zhang P, Liu X, Gao G, Liu F, Jiang Y, Cheng X, Zhu C, Xia Y. Profiling serum cytokines in COVID-19 patients reveals IL-6 and IL-10 are disease severity predictors. Emerg Microbes Infect. 2020; 9:1123–30. https://doi.org/10.1080/22221751.2020.1770129 [PubMed]
- 38. Salminen A, Kauppinen A, Kaarniranta K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal. 2012; 24:835–45. https://doi.org/10.1016/j.cellsig.2011.12.006 [PubMed]
- 39. Vukmanovic-Stejic M, Chambers ES, Suárez-Fariñas M, Sandhu D, Fuentes-Duculan J, Patel N, Agius E, Lacy KE, Turner CT, Larbi A, Birault V, Noursadeghi M, Mabbott NA, et al. Enhancement of cutaneous immunity during aging by blocking p38 mitogen-activated protein (MAP) kinase-induced inflammation. J Allergy Clin Immunol. 2018; 142:844–56. https://doi.org/10.1016/j.jaci.2017.10.032 [PubMed]
- 40. Grimes JM, Grimes KV. p38 MAPK inhibition: A promising therapeutic approach for COVID-19. J Mol Cell Cardiol. 2020; 144:63–65. https://doi.org/10.1016/j.yjmcc.2020.05.007 [PubMed]