



Introduction
Fraser syndrome (FS) (MIM#219000) is a rare genetic disorder that often presents with ocular, renal, genital and limb's congenital anomalies [1]. The diagnosis of FS is based on major criteria (syndactyly, cryptophthalmos spectrum, urinary tract abnormalities, ambiguous genitalia, laryngeal and tracheal anomalies, positive family history) and minor criteria (anorectal defects, dysplastic ears, nasal anomalies, skull ossification defects, umbilical abnormalities) [2, 3]. In human, homozygous or heterozygous mutations in FS protein 1 (FRAS1), FRAS1-related extracellular matrix protein 1 (FREM1), FREM2, and glutamate receptor-interacting protein 1 (GRIPI) genes result in classic FS phenotype [4]. Functionally, FRAS1, FREM1 and FREM2 genes encode members of extracellular matrix proteins [5]. These proteins are secreted by mesenchymal cells during diaphragmatic development and form a ternary complex at the basement membrane, which plays a crucial role in forming human and rodent diaphragms [6]. GRIP1 encodes a multi-PDZ domain-containing protein involved in the basolateral trafficking and export of FRAS1 and FREM2 in epithelial cells [7]. Defects of any of these proteins destabilize the extracellular matrix, causing dermal-epidermal detachment [8].
Clinically, the malformations of FS are challenging to detect prenatally [9]. In order to examine genetic disease or chromosome abnormalities in embryos before implantation to the uterus, as one of the most effective technologies, next-generation sequencing (NGS) in preimplantation genetic diagnosis (PGD) is developed [10]. It allows achieving normal pregnancy by transferring embryos without the target gene mutation. However, embryologists still face severe challenges in detecting mutations in human embryos due to a small number of embryos producing the issues of high amplification bias, low accuracy and reproducibility in NGS–based preimplantation genetic diagnosis [11]. Another challenge is from allele drop out (ADO) arising from non-amplification of one allele, which produces the false-negative result and typically results in misdiagnosis [12]. In recent years, as an economical, user-friendly, and accurate method, next-generation sequencing (NGS)-based single nucleotide polymorphism (SNP) haplotyping has been applied to detect the mutation in human preimplantation embryos [13, 14].
In this study, we first report two novel causative mutations in the FREM2 gene in an FS family. Furthermore, NGS-based SNP haplotyping was used to help in selecting the ‘right’ embryos for in vitro fertilization treatment. Finally, the woman successfully conceived on this embryo transfer.
Results
Two novel mutations in FREM2
A Chinese family with FS was recruited in this study (Figure 1). Two novel FREM2 gene mutations were found: one frameshift mutation c.7542_7543insG was carried by the female (II:2), and another nonsense mutation c.2689C>T was carried by the male (II:1) (Figure 2A). These two mutations were not detected in 80 unrelated healthy individuals. The couple was introduced to our center for PGD testing. These two mutations in FREM2 (c.7542_7543insG, p.V2516GfsX10; c.2689C>T, p.Gln897Ter) (Figure 2B) were submitted to the ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar), and the received the IDs SUB5692939 and SUB5690409, respectively. Clustal analysis indicated that these two amino acids are highly conserved in FREM2 proteins across vertebrate species, including the chimpanzee, Macaque, Cat, Musculus, and Chicken (Figure 3).
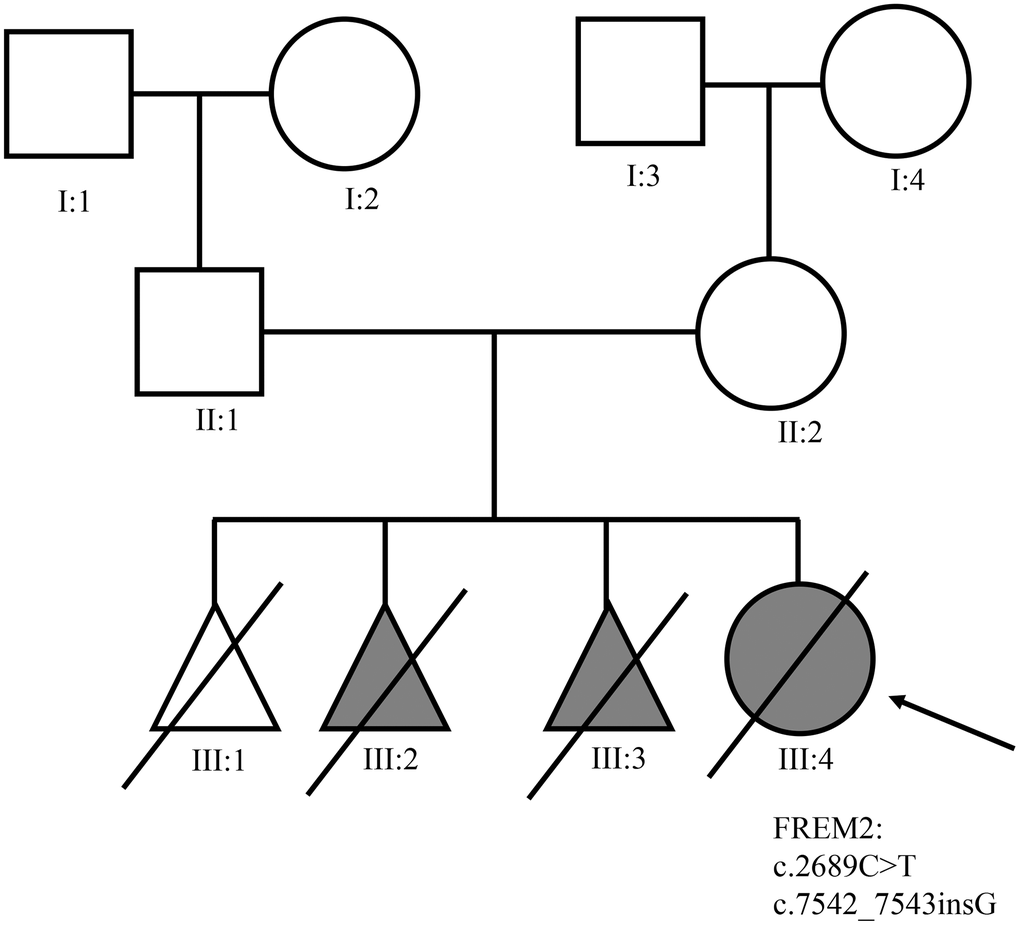
Figure 1. The pedigree of the family with Fraser syndrome. Roman numerals indicate generations, and individuals within a generation are numbered from left to right. The proband (III: 4) is denoted with an arrow.
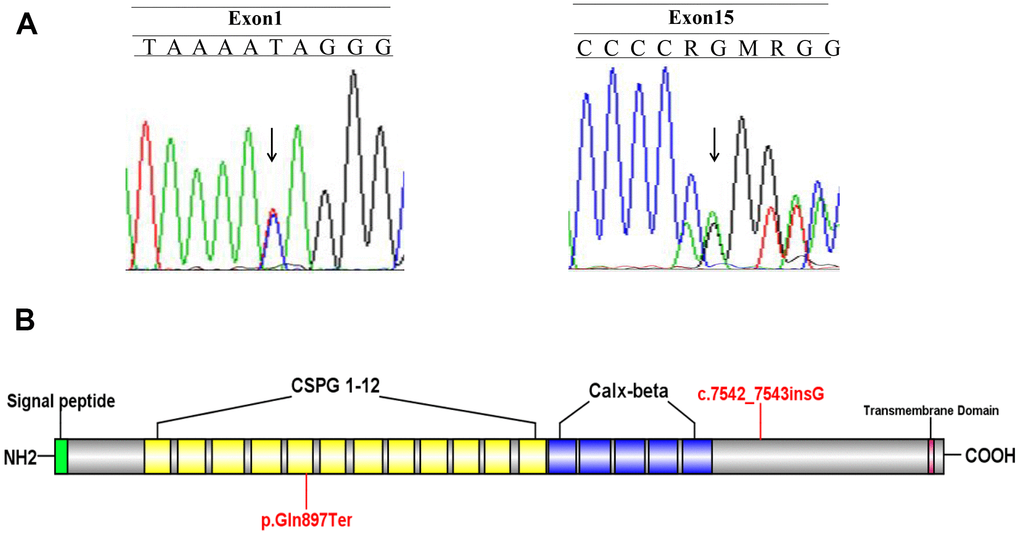
Figure 2. Identify the disease-causing mutation in the Fraser syndrome family. (A) Two mutations c.7542_7543insG and c.2689C>T were identified. (B) A schematic of the FREM2 protein and location of the mutations.

Figure 3. Conservation analysis of affected amino acids among six primate species. Evolutionary conservation of the mutations within FREM2 across species is analyzed. The positions of 2 mutations p.Gln897Term (A) and p.V2516GfsX10 (B) are indicated in red boxes.
Sanger sequencing revealed that the mutation c.2689C>T (p.Gln897Ter) in the male (II:1) was inherited from his father I:1. The mutation c.7542_7543insG (p.V2516GfsX10) in female II:2 was inherited from her mother I:4 (Table 1). These two mutations were identified at the level of PVS1 according to the guidelines for assigning disease causality. Mutation Taster and PROVEAN software predicted both mutations appeared to produce truncated proteins, which seriously affected the function of FREM2.
Table 1. Sequencing results of the family members.
| c.2689C>T, p.Gln897Ter | c.7542_7543insG, p.V2516GfsX10 | |
| Female | Wild-type | Heterozygous mutation |
| Male | Heterozygous mutation | Wild-type |
| Female’ father | Wild-type | Wild-type |
| Female’ mother | Wild-type | Heterozygous mutation |
| Male’ father | Heterozygous mutation | Wild-type |
| Male’ mother | Wild-type | Wild-type |
| Fetus | Heterozygous mutation | Wild-type |
Biopsied trophectoderm cells
In order to help these couples with the issues of FREM2 mutations in having a healthy baby, seventeen matured oocytes from the patients at MII stage were collected after ovarian stimulation and then fertilized by intracytoplasmic sperm injection. Nine blastocysts were utilized for trophectoderm biopsy when the trophectoderm cells herniated out of the zona pellucid on day 5 or 6 after fertilization.
Pedigree haplotype construction and linkage analyses
Analysis of the detected SNP loci suggested that 68 SNP loci, including 20 loci from the female (II:2) and 48 loci from the male (II:1), could provide information for linkage analyses. Among these 68 loci, 5 were on the FREM2 gene, including 4 in the female (II:2) and 1 in the male (II:1). The pedigree haplotype and linkage analysis were used to determine whether the embryo had carried the mutations. For example, on SNP rs38089800, the male had homozygous C/C, and the female had heterozygous T/C, suggesting that the male transmitted “C” and the female transmitted “T” to E1. Meanwhile, the linkage analysis showed that E5 and E15 were wild-types, E4, E7, E8 and E14 were heterozygous carriers, and E1, E2 and E17 were homozygous carriers (Table 2).
Table 2. Twenty-two SNP markers were respectively selected for identifying disease-associated allele in the embryos.
| ID | Chr | POS | REF | ALT | MF | MM | M | FF | FM | F | E1 | E2 | E4 | E5 | E7 | E8 | E14 | E15 | E17 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_8 | Chr13 | 38089800 | C | T | C/T | C/C | C/C | C/C | T/T | T/C | C/T | C/T | C/T | C/C | C/C | C/C | C/T | C/T | C/T | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_9 | Chr13 | 38174781 | T | C | T/C | T/T | T/T | T/T | C/C | C/T | T/C | T//C | T/T | T/T | T/T | T/T | T/T | ?/? | T/C | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_14 | Chr13 | 38361871 | A | G | G/G | G/G | G/G | G/A | G/A | G/A | G/G | G/G | G/G | G/A | G/A | G/A | G/G | G/A | G/G | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_F_2 | Chr13 | 38382809 | A | G | A/A | G/G | A/G | G/A | A/A | A/A | G/G | G/G | G/A | G/A | A/A | G/A | G/A | G/A | A/A | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_15 | Chr13 | 38382875 | C | T | C/C | T/T | C/T | T/C | C/C | C/C | C/C | C/C | T/C | T/C | C/C | T/C | T/C | T/C | C/C | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_F_3 | Chr13 | 38382982 | G | C | G/G | C/C | G/C | C/G | G/G | G/G | G/G | G/G | C/G | C/G | G/G | C/G | C/G | C/G | G/G | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_16 | Chr13 | 38406297 | G | A | G/G | A/A | G/A | A/G | G/G | G/G | G/G | G/G | A/G | A/G | G/G | A/G | A/G | A/G | G/G | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_F_4 | Chr13 | 38406309 | C | A | C/C | A/A | C/A | A/C | C/C | C/C | C/C | C/C | A/C | A/C | C/C | A/C | A/C | A/C | C/C | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_F_6 | Chr13 | 38501932 | G | T | G/G | T/T | G/T | G/G | G/G | G/G | G/G | G/G | T/G | T/G | G/G | T/G | T/G | T/G | G/G | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_22 | Chr13 | 38552205 | G | A | G/G | A/A | G/A | G/G | G/G | G/G | G/G | G/G | A/G | A/G | G/G | A/G | A/G | A/G | G/G | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_M_1 | Chr13 | 38625874 | G | A | G/G | G/G | G/G | G/A | G/G | G/A | G/G | G/G | G/G | G/A | G/A | G/A | G/G | ?/? | G/G | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_F_14 | Chr13 | 39264690 | T | C | T/T | T/C | T/C | T/T | T/T | T/T | ?? | ?? | C/T | C/T | ?/? | ?/? | C/T | ?/? | ?/? | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_M_7 | Chr13 | 39430443 | G | A | G/G | G/G | G/G | A/A | G/G | G/A | G/G | G/G | G/G | G/A | G/A | G/G | G/G | G/A | G/G | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_M_8 | Chr13 | 39431945 | T | C | T/T | T/T | T/T | C/C | T/T | T/C | T/T | ?? | T/T | ?? | ?/? | T/T | T/T | ?/? | ?/? | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_M_10 | Chr13 | 39446869 | G | A | G/G | G/G | G/G | A/A | G/G | G/A | G/G | ?? | G/G | G/A | G/A | G/G | G/G | G/A | ?/? | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_M_11 | Chr13 | 39452934 | C | T | C/T | T/C | C/C | C/C | T/T | T/C | ?? | C/T | C/T | C/C | C/C | ?/? | C/T | C/C | C/T | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_62 | Chr13 | 39477115 | G | C | C/G | C/C | C/C | C/C | G/G | G/C | C/G | C/G | C/G | C/C | C/C | C/G | C/G | C/C | C/G | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_80 | Chr13 | 39792426 | C | T | C/C | T/C | C/C | T/T | C/C | C/T | C/C | C/C | C/C | T/C | C/T | C/C | C/C | C/T | C/C | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_82 | Chr13 | 39821473 | G | A | G/G | A/G | G/G | G/A | G/G | G/A | G/G | G/G | G/G | A/G | G/A | G/G | G/G | G/A | G/G | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_90 | Chr13 | 39949981 | T | C | T/T | C/C | T/C | T/T | T/T | T/T | T/T | T/T | C/T | T/C | T/T | C/T | C/T | C/T | T/T | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_M_12 | Chr13 | 39991091 | T | A | T/T | A/T | T/T | T/T | A/T | A/T | T/A | T/A | T/A | T/T | T/T | T/A | T/A | T/T | T/A | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FREM2_111 | Chr13 | 40464364 | G | A | G/A | A/A | G/A | A/A | A/A | A/A | G/A | G/A | A/A | A/A | G/A | A/A | A/A | A/A | G/A | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| E5 and E15 were wild-type, E4, E7, E8 and E14 were heterozygous carriers, and E1, E2 and E17 were carried two pathogenic mutations. ID, reference SNP cluster ID; Chr, chromosome number; POS, genomic position; REF, reference allele of the SNPs; ALT, alternative allele of the SNPs; FF, female’s father; FM, female’s mother; MF, male’s father; MM, male’s mother; E, embryo. The red base indicates associated mutation, while the black base indicates that the SNP links with wild type. The green background indicates uncertain recombination. “?”means that the alleles are not covered. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Clinical outcomes
Wild-type blastocysts E5 and E15 were selected for the first two transplantations. Unfortunately, no clinical pregnancy was achieved. We then transferred the heterozygous blastocyst E4 with a good grade, which still did not generate a clinical pregnancy. The first three transplantations were all carried out under the artificial cycle scheme. Due to repeated implantation failure, the woman was recommended to perform hysteroscopy. It showed that the uterine cavity had adhesion on the right side. The shape of the uterine cavity returned to normal after operation. Endometrial tissue pathology suggested that the proliferative endometrium was associated with polyp formation.
We then adopted the down-regulation scheme combined with the artificial cycle scheme to transplant E7 blastocyst. The woman finally got a clinical pregnancy. Sequencing of the fetal amniotic cell’s DNA revealed that the fetus was a heterozygous carrier with one FREM2 gene mutation (c.2689C>T, p.Gln897Ter). The mutation was inherited from its father (II1). The fetus’ karyotype was normal. The linkage analysis, D13S218, D13S894 and D13s1253 STR markers confirmed the above findings (Figure 4). Thus, continued pregnancy was recommended. During pregnancy, all ultrasonographic examinations did not show any morphological fetal abnormality. The baby was born on March 17, 2019 and showed normal development and growth after a follow-up of 12 months.
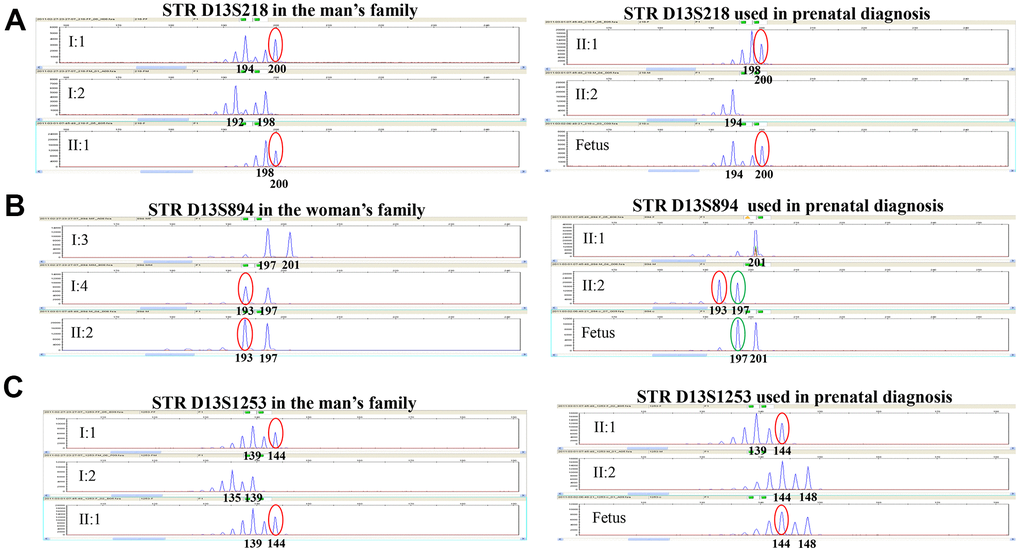
Figure 4. Genotyping results of STR D13S218, STR D13S894, and STR D13S1253 in this family. STR D13S218, D13S894 and D13S1253 showed that the fetus was a heterozygous carrier with one FREM2 gene mutation (c.2689C>T, p.Gln897Ter) inherited from the father, which is consistent with the PGD result. (A) Genotyping for STR D13S218 in the man’s family and used in prenatal diagnosis indicates that the chromosome carrying pathogenic mutation originated from his father. The fetus inherited pathogenic chromosome from his father. (B) Genotyping for STR D13S894 in the woman’s family and used in prenatal diagnosis indicates that the chromosome carrying pathogenic mutation originated from her mother. The fetus inherited the normal chromosome from his mother. (C) Genotyping for STR D13S1253 in the man’s family and used in prenatal diagnosis indicates that the chromosome carrying pathogenic mutation originated from his father. The fetus inherited the pathogenic chromosome from his father.
Discussion
FS is a rare genetic malformation with an autosomal recessive inheritance pattern in which the life expectancy is <1 year [15]. Genetics factors are well recognized in the cause of FS, but the underlying causes of FS remain unclear [16]. In the present study, the woman (II: 2) terminated pregnancies for fetal abnormality. PCR-based sequencing revealed that the woman carried a novel mutation c.7542_7543insG, and her husband (II: 1) carried a novel mutation c.2689C>T. Further analysis revealed that the woman’s mutation inherited from her mother (I: 4) and her husband’s mutation inherited from his father (I: 1). Since the postmortem examination of the proband (III: 4) conformed to the clinical diagnostic criteria of FS, we analyzed the pathogenicity of these two novel mutations in the FREM2 gene. The mutation p.Gln897Ter is located in the fifth chondroitin sulfate proteoglycan (CSPG) domain and can produce a truncated protein. The mutation p.V2516GfsX10 is closely located with the Calx-β domain and also produce a truncated protein. Many experiments demonstrated that a stop codon resulting in a truncated protein is considered to lose the protein biological function [17]. The possible pathogenic mechanism of FS patients with p.Gln897Ter mutation is that this mutation produces a truncated protein with a break CSPG domain and without Calx-β domain. Both of these two truncated proteins will lose the transmembrane domain of FREM2 protein. Therefore, we here predicted both nonsense mutations have strong evidence in favor of disease-causing potential.
To date, 45 disease-causing mutations, including 6 nonsense mutation in the FREM2 gene, have been reported in HGMD Professional (https://portal.biobase-international.com/hgmd/pro/all.php). However, only four mutations including a substitution c. 5914G>A in the second of 5 consecutive Calx-β domains, a splicing mutation IVS14+1G >A resulting in a stop codon at residue 2549, a substitution mutation c.6499C>T, and a deletion mutation c.15delG in FREM2 gene have been reported to be associated with a risk of FS in OMIM database [18, 19]. In order to exclude the embryo containing no suspected mutations, PGD was introduced in the late 1980s and allows the detection of FREM2 gene mutations in embryos produced through in vitro fertilization [20]. Today, advanced molecular technologies with better resolution, such as array comparative genomic hybridization, quantitative PCR, and NGS, are on the verge of becoming the gold standard in embryo preimplantation screening [21, 22]. However, amplification failure and ADO are two common problems caused by NGS sequencing errors [23, 24]. In the present study, we describe the methods based on NGS-based SNP haplotype to distinguish the wild-type, heterozygous carriers, and homozygous carriers of FS embryos. Our results revealed that the D13S218, D13S894 and D13s1253 STR markers could be used to exclude the mutations c.7542_7543insG and c.2689C>T in FS patients. For this couple, the wild-type embryos and heterozygous carriers could be used to transfer, but the risk should be informed. The end of this story is that they accept to undergo embryo transfer and obtained one healthy baby who is a heterozygous carrier.
In conclusion, in the present study, we report two novel mutations in FREM2 gene associated with the risk of FS. Next, we describe a method based on NGS-based SNP haplotype to exclude the embryos carried homozygous mutation. Lastly, the patients received an embryo with heterozygous mutation and obtained one healthy baby.
Materials and Methods
Ethics statement
This study followed the principles of the Declaration of Helsinki. Informed consent was obtained from all participants. The study was approved by the institutional ethics committee of The First Affiliated Hospital of Hainan Medical University.
Family history
II: 1 (male, 32 years old) and II: 2 (female, 31 years old) had no clinical symptoms of FS (Figure 1). The female had pregnancies four times, including three adverse pregnancies (III:1, III:2, and III:3). The last pregnancy was terminated at 27 weeks of gestation. Prenatal ultrasound examination revealed that the proband (III:4) had 4.0 cm thickness of fetal ascites, edema of fetal skin, and a large amount of fluid in fetal abdominal cavity and enlargement of lungs. Postmortem examination showed that the proband did not have a left eyeball. It had bilateral syndactyly (toe) and mal-developed right kidney, which conformed to the three major diagnostic criteria of FS. Also, the location of both ears was symmetrically low, and the umbilical cord was swollen, which were the two minor criteria for FS diagnosis.
Mutation screening
The DNA samples from the aborted proband (III:4) and the couple (II:1 and II:2) were screened by next-generation sequencing in Beijing Genomics Institution. We confirmed the mutations in FREM2 gene by Sanger sequencing. The PCR primers are shown in Table 3. The pathogenicity of all the mutations were evaluated according to the guidelines of the American College of Medical Genetics (ACMG), Mutation Taster and Provean software. All sequencing readouts were mapped to compare to the FREM2 reference genome on NCBI (NM_207361.6). Eighty human random control DNAs were set as healthy control.
Table 3. Primers used in this study.
| Mutations | Primers | Sequence(5’-3’) | Genomic location |
| c.2689C>T | FREM2_1F | GTCCTCAACACCGGCTTCA | chr13:39261173-39461268 |
| FREM2_1R | GAGTGCCAGTAGGATGGCTC | ||
| c.7542_7543insG | FREM2_15F | TCCACAGAGAAGTTGAAAGTACACA | |
| FREM2_15R | GCTTACCCAAGTCACCTACCA | ||
| D13S218 | D13S218F | GATTTGAAAATGAGCAGTCC | chr13:38458094-38458429 |
| D13S218R | GTCGGGCACTACGTTTATCT | ||
| D13S1288 | D13S1288F | TTCAGAGACCATCACGGC | chr13:38949133-38949542 |
| D13S1288R | CTGGAAAAATCAGTTGAATCCTAGC | ||
| D13S1253 | D13S1253F | CCTGCATTTGTGTACGTGT | chr13:39572161-39572531 |
| D13S1253R | CAGAGCCGTGGTAGTATATTTTT |
Whole-genome amplification
Biopsied trophectoderm cells were washed in PBS (with 0.1% HAS) and transferred to the PCR tube. The whole genome of the cell was amplified by the Pico PLEX single-cell WGA kit (NEB-WGA) according to the manufacturer’s protocol. The sequencing library was constructed by a personalized genome library-building Kit (zykw-c-003) produced by Peking Jabrehoo Med Tech Co. Ltd. Sequencing was performed with the Ion Torrent Personal Genome Machine (Life technology, USA). At the same time, WGA products were also re-amplified using primers surrounding the mutated site, as shown in Table 3.
Haplotype construction
Since the DNA sample of the proband (III:4) was missing, haplotype construction was generated from the couple (II:1, II:2) and their parents (I:1, I:2, I:3 and I:4). A total of 120 linked SNPs markers located in 5’ and 3’ regions of FREM2 were analyzed. The genomic DNA of this pedigree was sequenced by NGS. The SNP readouts at the adjacent positions allowed the pathogenic alleles to be identified. A similar strategy was used to determine whether each embryos’ pathogenic allele was present from the informative SNPs in embryos.
Embryo transfer
Blastocysts with normal or heterozygous mutations without chromosomal abnormality were selected for transplantation. Only 1 blastocyst was transplanted per resuscitation cycle. At 17+ weeks of gestation, 20 mL of the fetal amniotic fluid is extracted for DNA extraction, cell culture, and subsequent prenatal diagnosis.
Prenatal diagnosis
The FREM2 gene mutation was detected by Sanger sequencing. For prenatal diagnosis and linkage analysis, 4 STR markers which are located in the regions of the FREM2 gene were chosen, with each STR’s forward primer being modified with Fam fluorescence. These primer sequences are shown in Table 3. After PCR amplification, the products were examined by fluorescence electrophoresis by ABI-3500 (Applied Biosystems, Foster City, CA, USA). A detailed prenatal ultrasound examination was performed to eliminate the chance of fetal malformations after a successful pregnancy.
Author Contributions
Yao Zhou, Yanlin Ma, and Qi Li designed the study. Yao Zhou, Xiaohui Yang, Zheng Liu, and Yuxin Hu performed the analysis of mutations in the family. Yao Zhou, Xiaohui Yang, and Zheng Liu performed the whole genome amplification and prenatal diagnosis. Yao Zhou, Yu Zhang, Huaye Chen, and Yongfang Zhang diagnosed the patients and performed embryo transfer. Yao Zhou, Zheng Liu, Yanlin Ma, and Qi Li wrote the manuscript.
Acknowledgments
The authors want to thank the patients for their participation in the study.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This work was supported by Natural Science Foundation of Hainan Province (No.2019CXTD408 and ZDYF2020117), Major Science and Technology Program of Hainan Province (No. ZDKJ202003 and ZDKJ2017007) National Natural Science Foundation of China (No. 82072880, 81960283, 81460034), and project supported by Hainan Province Clinical Medical Center.
References
- 1. Mbonda A, Endomba FT, Kanmounye US, Nkeck JR, Tochie JN. Diagnosis of Fraser syndrome missed out until the age of six months old in a low-resource setting: a case report. BMC Pediatr. 2019; 19:292. https://doi.org/10.1186/s12887-019-1673-6 [PubMed]
- 2. Saleem AA, Siddiqui SN. Fraser Syndrome. J Coll Physicians Surg Pak. 2015 (Suppl 2); 25:S124–26. https://doi.org/10.2015/JCPSP.S124S126 [PubMed]
- 3. Boussion S, Lyonnet S, Van Der Zwaag B, Vogel MJ, Smol T, Mezel A, Manouvrier-Hanu S, Vincent-Delorme C, Vanlerberghe C. Fraser syndrome without cryptophthalmos: Two cases. Eur J Med Genet. 2020; 63:103839. https://doi.org/10.1016/j.ejmg.2020.103839 [PubMed]
- 4. Yu Q, Lin B, Xie S, Gao S, Li W, Liu Y, Wang H, Huang D, Xie Z. A homozygous mutation p.Arg2167Trp in FREM2 causes isolated cryptophthalmos. Hum Mol Genet. 2018; 27:2357–66. https://doi.org/10.1093/hmg/ddy144 [PubMed]
- 5. Nikolova YS, Iruku SP, Lin CW, Conley ED, Puralewski R, French B, Hariri AR, Sibille E. FRAS1-related extracellular matrix 3 (FREM3) single-nucleotide polymorphism effects on gene expression, amygdala reactivity and perceptual processing speed: An accelerated aging pathway of depression risk. Front Psychol. 2015; 6:1377. https://doi.org/10.3389/fpsyg.2015.01377 [PubMed]
- 6. Takahashi T, Friedmacher F, Zimmer J, Puri P. Mesenchymal expression of the FRAS1/FREM2 gene unit is decreased in the developing fetal diaphragm of nitrofen-induced congenital diaphragmatic hernia. Pediatr Surg Int. 2016; 32:135–40. https://doi.org/10.1007/s00383-015-3824-7 [PubMed]
- 7. Al-Hamed MH, Sayer JA, Alsahan N, Tulbah M, Kurdi W, Ambusaidi Q, Ali W, Imtiaz F. Novel loss of function variants in FRAS1 AND FREM2 underlie renal agenesis in consanguineous families. J Nephrol. 2021; 34:893–900. https://doi.org/10.1007/s40620-020-00795-0 [PubMed]
- 8. Hines EA, Verheyden JM, Lashua AJ, Larson SC, Branchfield K, Domyan ET, Gao J, Harvey JF, Herriges JC, Hu L, Mcculley DJ, Throckmorton K, Yokoyama S, et al. Syndactyly in a novel Fras1(rdf) mutant results from interruption of signals for interdigital apoptosis. Dev Dyn. 2016; 245:497–507. https://doi.org/10.1002/dvdy.24389 [PubMed]
- 9. Tessier A, Sarreau M, Pelluard F, André G, Blesson S, Bucourt M, Dechelotte P, Faivre L, Frébourg T, Goldenberg A, Goua V, Jeanne-Pasquier C, Guimiot F, et al. Fraser syndrome: features suggestive of prenatal diagnosis in a review of 38 cases. Prenat Diagn. 2016; 36:1270–75. https://doi.org/10.1002/pd.4971 [PubMed]
- 10. Sullivan-Pyke C, Dokras A. Preimplantation Genetic Screening and Preimplantation Genetic Diagnosis. Obstet Gynecol Clin North Am. 2018; 45:113–25. https://doi.org/10.1016/j.ogc.2017.10.009 [PubMed]
- 11. Ruan Q, Ruan W, Lin X, Wang Y, Zou F, Zhou L, Zhu Z, Yang C. Digital-WGS: Automated, highly efficient whole-genome sequencing of single cells by digital microfluidics. Sci Adv. 2020; 6:eabd6454. https://doi.org/10.1126/sciadv.abd6454 [PubMed]
- 12. Zucca S, Villaraggia M, Gagliardi S, Grieco GS, Valente M, Cereda C, Magni P. Analysis of amplicon-based NGS data from neurological disease gene panels: a new method for allele drop-out management. BMC Bioinformatics. 2016 (Suppl 12); 17:339. https://doi.org/10.1186/s12859-016-1189-0 [PubMed]
- 13. Ji X, Zhang Z, Shi J, He B. Clinical application of NGS-based SNP haplotyping for the preimplantation genetic diagnosis of primary open angle glaucoma. Syst Biol Reprod Med. 2019; 65:258–63. https://doi.org/10.1080/19396368.2019.1590479 [PubMed]
- 14. Chen L, Diao Z, Xu Z, Zhou J, Yan G, Sun H. The clinical application of NGS-based SNP haplotyping for PGD of Hb H disease. Syst Biol Reprod Med. 2017; 63:212–17. https://doi.org/10.1080/19396368.2017.1296501 [PubMed]
- 15. Chevreau J, Rieu C, Page C. An unusual prenatal laryngeal image. Eur Ann Otorhinolaryngol Head Neck Dis. 2017; 134:135–36. https://doi.org/10.1016/j.anorl.2016.11.012 [PubMed]
- 16. Capone VP, Morello W, Taroni F, Montini G. Genetics of Congenital Anomalies of the Kidney and Urinary Tract: The Current State of Play. Int J Mol Sci. 2017; 18:796. https://doi.org/10.3390/ijms18040796 [PubMed]
- 17. Bartha I, di Iulio J, Venter JC, Telenti A. Human gene essentiality. Nat Rev Genet. 2018; 19:51–62. https://doi.org/10.1038/nrg.2017.75 [PubMed]
- 18. Jadeja S, Smyth I, Pitera JE, Taylor MS, van Haelst M, Bentley E, McGregor L, Hopkins J, Chalepakis G, Philip N, Perez Aytes A, Watt FM, Darling SM, et al. Identification of a new gene mutated in Fraser syndrome and mouse myelencephalic blebs. Nat Genet. 2005; 37:520–25. https://doi.org/10.1038/ng1549 [PubMed]
- 19. Shafeghati Y, Kniepert A, Vakili G, Zenker M. Fraser syndrome due to homozygosity for a splice site mutation of FREM2. Am J Med Genet A. 2008; 146A:529–31. https://doi.org/10.1002/ajmg.a.32091 [PubMed]
- 20. Kang X, He W, Huang Y, Yu Q, Chen Y, Gao X, Sun X, Fan Y. Introducing precise genetic modifications into human 3PN embryos by CRISPR/Cas-mediated genome editing. J Assist Reprod Genet. 2016; 33:581–88. https://doi.org/10.1007/s10815-016-0710-8 [PubMed]
- 21. Friedenthal J, Maxwell SM, Munné S, Kramer Y, McCulloh DH, McCaffrey C, Grifo JA. Next generation sequencing for preimplantation genetic screening improves pregnancy outcomes compared with array comparative genomic hybridization in single thawed euploid embryo transfer cycles. Fertil Steril. 2018; 109:627–32. https://doi.org/10.1016/j.fertnstert.2017.12.017 [PubMed]
- 22. Zhang W, Zhang L, Liu Y, Li J, Xu X, Niu W, Xu J, Sun B, Guo Y. Higher chromosomal aberration frequency in products of conception from women older than 32 years old with diminished ovarian reserve undergoing IVF/ICSI. Aging (Albany NY). 2021; 13:10128–40. https://doi.org/10.18632/aging.202772 [PubMed]
- 23. Remec ZI, Trebusak Podkrajsek K, Repic Lampret B, Kovac J, Groselj U, Tesovnik T, Battelino T, Debeljak M. Next-Generation Sequencing in Newborn Screening: A Review of Current State. Front Genet. 2021; 12:662254. https://doi.org/10.3389/fgene.2021.662254 [PubMed]
- 24. He F, Zhou W, Cai R, Yan T, Xu X. Systematic assessment of the performance of whole-genome amplification for SNP/CNV detection and β-thalassemia genotyping. J Hum Genet. 2018; 63:407–16. https://doi.org/10.1038/s10038-018-0411-5 [PubMed]