




Introduction
Chronic low-grade inflammation is currently recognized as a core driver of cardiovascular disease and mortality [1–3]. Additionally, acute inflammatory episodes contribute to mortality risk through distinct but overlapping pathways, including sepsis, cytokine storm syndromes, and acute cardiovascular events [4]. Elevated circulating markers such as interleukin-6 (IL6), C-reactive protein (CRP) and growth differentiation factor-15 (GDF15) track with higher risks of cardiovascular disease, diabetes, cancer and neurodegeneration, yet their causal role remains uncertain because observational data are prone to confounding and reverse causation [5].
The IL6 axis plays a pivotal role in both acute and chronic inflammatory responses, operating through two distinct pathways: classical signalling via a membrane-bound IL6 receptor and trans-signalling mediated by a soluble IL6 receptor (IL6R), which enables IL6 activity in cells lacking the membrane receptor. While acute IL6 elevation drives immediate inflammatory responses essential for pathogen clearance, chronic IL6 activation promotes sustained inflammation contributing to cardiovascular disease development [6]. These parallel routes can exert opposing effects on cardiovascular homeostasis, so therapeutic strategies that inhibit IL6 or enhance IL6R function may have different implications for cardiovascular health and acute disease management [7]. Specifically, IL6R has been involved in the pathogenesis of major cardiovascular conditions including atrial fibrillation, coronary artery disease, and stroke through its modulation of inflammatory cascades in vascular tissues. In comparison, CRP and GDF15 are well-established systemic inflammation markers, reflecting both acute inflammatory states and chronic inflammatory processes, but whether they actively drive cardiovascular pathology or merely mirror it remains a matter of debate [5, 8].
Mendelian randomization (MR) exploits germline genetic variants as unconfounded proxies for modifiable exposures, offering a quasi-experimental route to infer causality across both acute and chronic inflammatory phenotypes [8, 9]. Although individual Mendelian randomization (MR) studies have linked single inflammatory biomarkers to specific diseases, a multi-marker MR assessment spanning multiple inflammatory pathways and focused on cardiovascular disease and all-cause mortality is still lacking.
Methods
Study design and overview
We conducted a comprehensive two-sample MR study following established guidelines and the STROBE-MR reporting standards [9]. Our analysis used publicly available summary statistics from large-scale genome-wide association studies [10, 11]. All analyses focused on individuals of European ancestry to minimize population stratification and ensure validity of genetic instruments.
Data harmonization and quality control
Prior to analysis, we implemented rigorous data harmonization procedures to ensure consistency across different GWAS datasets. Effect alleles were harmonized across exposure and outcome datasets to ensure consistent direction of effects. We excluded variants with minor allele frequency (MAF) <0.01, imputation quality score <0.8, or missing essential information (effect size, standard error, or sample size).
Palindromic single-nucleotide polymorphisms (SNPs) (A/T or G/C) were carefully handled using allele frequency information to infer strand alignment. When allele frequencies were ambiguous (close to 0.5), palindromic SNPs were excluded to prevent strand misalignment errors. We also excluded variants in the major histocompatibility complex (MHC) region (chromosome 6: 25–35 Mb) due to complex linkage disequilibrium patterns that could violate MR assumptions.
Selection of genetic instruments
Instrument selection criteria and pruning procedures
Genetic instruments were selected based on genome-wide significance for association with the respective biomarker levels. Because relatively few independent genetic variants are robustly associated with circulating inflammatory proteins, we selected instruments using a significance threshold of P < 5 × 10−5, combined with stringent LD pruning (r² < 0.001) and retention of variants with F-statistics >10 to avoid weak-instrument bias. This selection increased the precision of the causal estimates while preserving validity given our stringent linkage disequilibrium (LD) pruning (r2 < 0.001), strong instrument strength, and extensive pleiotropy checks. To ensure independence of instruments, we performed linkage LD pruning using PLINK v1.90 with the following parameters: window size of 10,000 kb, step size of 1,000 variants, and r2 threshold of 0.001. LD calculations were based on the 1000 Genomes Project Phase 3 European reference panel. The strength of genetic instruments was assessed using the F-statistic.
We also performed MR-Steiger filtering to ensure that the genetic instruments had stronger associations with the exposure than with the outcome, thereby supporting the assumed causal direction. Additionally, by reverse Mendelian randomization (MR) analysis we tested whether genetic liability to atrial fibrillation (AF), coronary artery disease (CAD), or ischemic stroke causally increases circulating IL-6 or IL-6R. All instruments passed this directionality test (P < 0.05 for all comparisons).
Outcome data sources
All-cause mortality data were derived from the FinnGen study, which includes 356,077 Finnish individuals followed through national health registries [12].
A total of 57,224 deaths were recorded over a median follow-up of 11.7 years, providing high statistical power for causal inference.
Causal effect on disease
Cardiovascular disease outcomes
Cardiovascular disease outcomes were obtained from large-scale genome-wide association studies and meta-analyses. Atrial fibrillation data were derived from the AFGen consortium including over 1 million individuals [13]. Coronary artery disease (CAD) data were obtained from the CARDIoGRAMplusC4D consortium meta-analysis including 185,000 CAD cases [14]. Stroke data were derived from the MEGASTROKE consortium including 521,612 individuals [15]. These datasets provided comprehensive coverage of major cardiovascular endpoints with substantial statistical power for detecting causal effects.
Similarly, other non-cardiovascular outcomes were tested. Among them there was a significant protective effect on cancer.
Statistical analysis
Primary analysis methods
We performed inverse-variance weighted (IVW) Mendelian randomization as our primary analytical method, which provides the most precise estimates when all genetic instruments are valid [16]. The IVW method combines individual Wald ratio estimates using inverse-variance weighting.
Effect estimates were expressed as odds ratios (OR) for clinical outcomes, with 95% confidence intervals (CI) calculated using the delta method.
Power calculations
Statistical power was calculated using the mRnd online calculator (https://shiny.cnsgenomics.com/mRnd/) based on the proportion of variance explained by genetic instruments (R2), sample sizes, and expected effect sizes. For IL6R, with R2 = 4.2% and outcome sample sizes ranging from 400,000 to over 1 million individuals for cardiovascular outcomes, we had >99% power to detect odds ratios of 0.95–0.97 for cardiovascular disease outcomes and mortality. For multi-variant instruments, power calculations incorporated the combined R2 values.
Sensitivity analyses and pleiotropy assessment
To assess the robustness of our findings and potential violations of MR assumptions, we conducted multiple sensitivity analyses using different methodological approaches.
MR-Egger Regression: This method provides estimates robust to horizontal pleiotropy by allowing for a non-zero intercept. A significant intercept (P < 0.05) indicates directional pleiotropy. MR-Egger estimates were calculated using the mr_egger_regression function with bootstrap confidence intervals (n = 1,000 iterations).
MR-PRESSO (Pleiotropy RESidual Sum and Outlier): This method identifies and corrects for horizontal pleiotropy by detecting outlying genetic variants [17]. The analysis includes three components: (1) global test for horizontal pleiotropy, (2) outlier detection and removal, and (3) distortion test comparing estimates before and after outlier removal. We used the mr_presso function with 5,000 iterations and significance threshold of P < 0.05.
Heterogeneity Assessment: We assessed heterogeneity using Cochran’s Q statistic. Cochran’s Q follows a chi-squared distribution with degrees of freedom equal to the number of instruments minus one.
Leave-One-Out Analysis: We systematically excluded each genetic instrument and recalculated MR estimates to identify variants driving the overall association. This analysis used the mr_leaveoneout function, with results visualized using forest plots.
Expression Quantitative Trait Loci (eQTL) Analysis: We investigated tissue-specific expression effects using data from the Genotype-Tissue Expression (GTEx) project version 8, which includes 54 tissue types from 838 individuals [18]. eQTL data were accessed through the GTEx Portal API and analyzed using the gtexr package. We focused on cis-eQTLs (within 1 Mb of the transcription start site) with false discovery rate (FDR) <0.05.
All statistical analyses were performed using R version 4.3.2 (R Foundation for Statistical Computing, Vienna, Austria). The primary MR analyses utilized the TwoSampleMR package and MendelianRandomization package. Linkage disequilibrium pruning was conducted using PLINK v1.90 (https://www.cog-genomics.org/plink/) with the 1000 Genomes Project Phase 3 European reference panel.
Data availability statement
Summary statistics used in this study are publicly available from the respective GWAS consortia and can be accessed through their designated repositories. GTEx data are available through the GTEx Portal (gtexportal.org) and dbGaP. Analysis code and supplementary data files are available upon reasonable request to the corresponding author.
Results
Characteristics and validation of genetic instruments
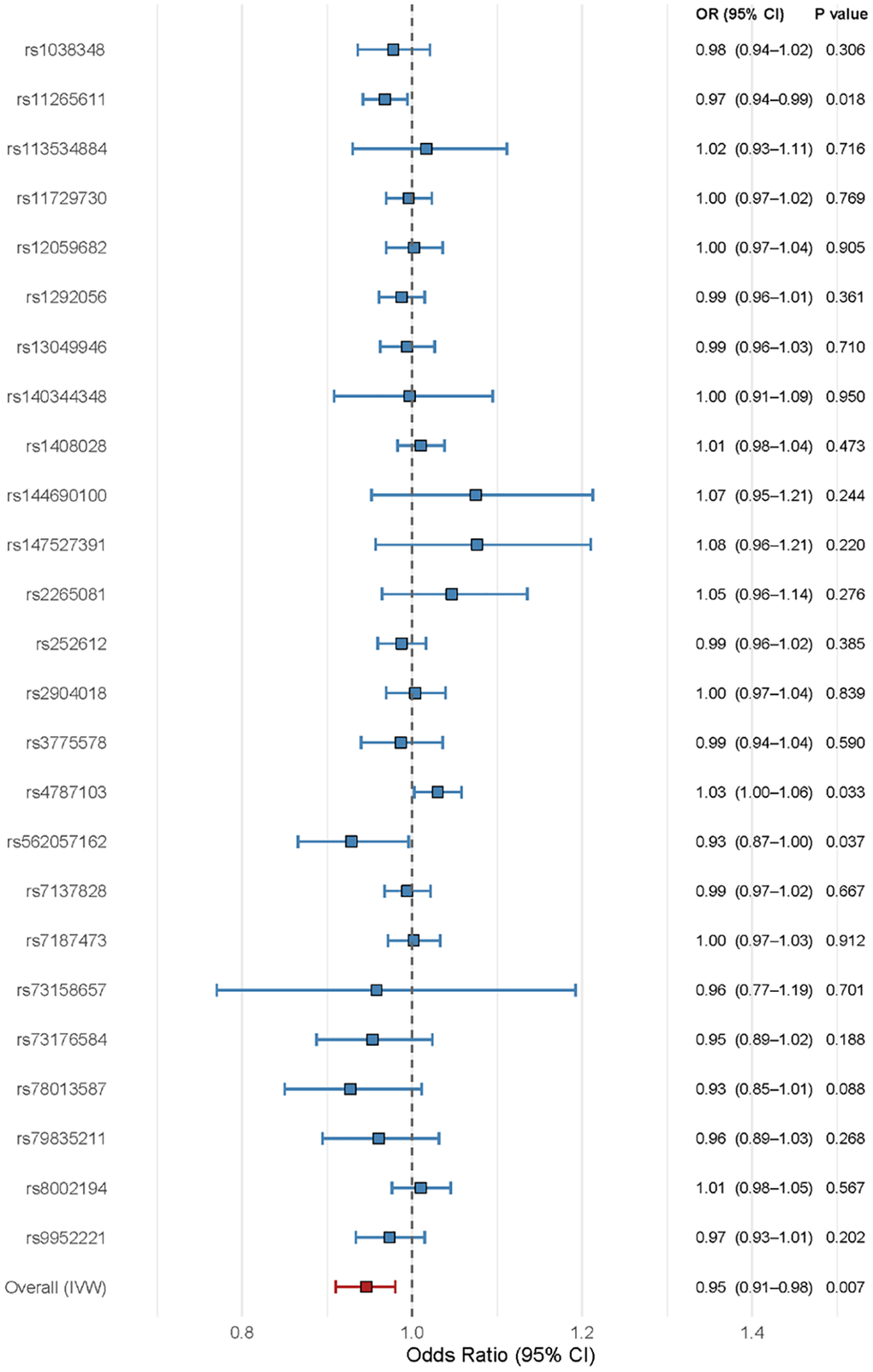
All genetic instruments demonstrated adequate strength for Mendelian randomization analysis, with F-statistics well above the threshold of 10. Main individual SNPs characteristics are reported in Supplementary Table 1. The IL6R instrument set comprised nine independent genetic variants with a mean combined F-statistic of 343.61. The multi-variant instruments for other biomarkers achieved combined F-statistics well above the threshold of 10.
Primary mendelian randomization results
IL6R protective effects
Genetically proxied higher IL6R levels were significantly associated with a lower risk of all-cause mortality (OR = 0.95; 95% CI: 0.91–0.98; P = 0.007; Figure 1), as well as with a reduced risk of major cardiovascular outcomes. These findings were derived from a Mendelian randomization analysis using genome-wide significant variants robustly associated with circulating IL6R levels.
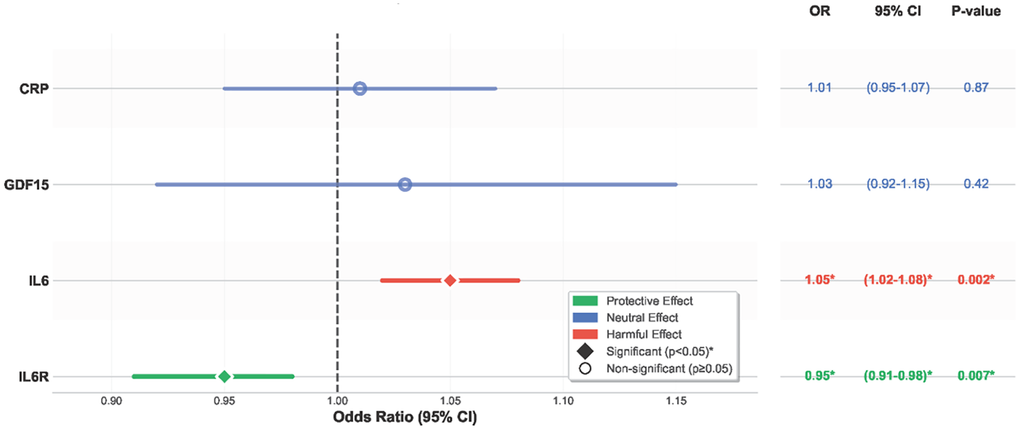
Figure 1. Causal effects of inflammatory biomarkers on all-cause mortality. Forest plot showing that higher IL6R is protective against mortality, whereas higher IL6 is harmful. CRP and GDF-15 are neutral. Abbreviations: IL6R: interleukin-6 receptor; IL6: interleukin-6; CRP: C-reactive protein; GDF-15: growth differentiation factor-15; OR: odds ratio; CI: confidence interval.
To further validate these results and to isolate potential cis-acting biological effects, we conducted a cis-Mendelian randomization analysis restricted to two independent variants located within the IL6R gene locus. This focused analysis confirmed the protective association with mortality (Supplementary Figure 1), reinforcing the causal relevance of IL6R in human survival and cardiovascular health.
Consistent protective effects were also observed for specific cardiovascular outcomes, including atrial fibrillation (OR = 0.96; 95% CI: 0.95–0.97; P < 0.001), coronary artery disease (OR = 0.97; 95% CI: 0.95–0.98; P < 0.001), and stroke (OR = 0.98; 95% CI: 0.97–1.0; P = 0.045) (Figure 2).
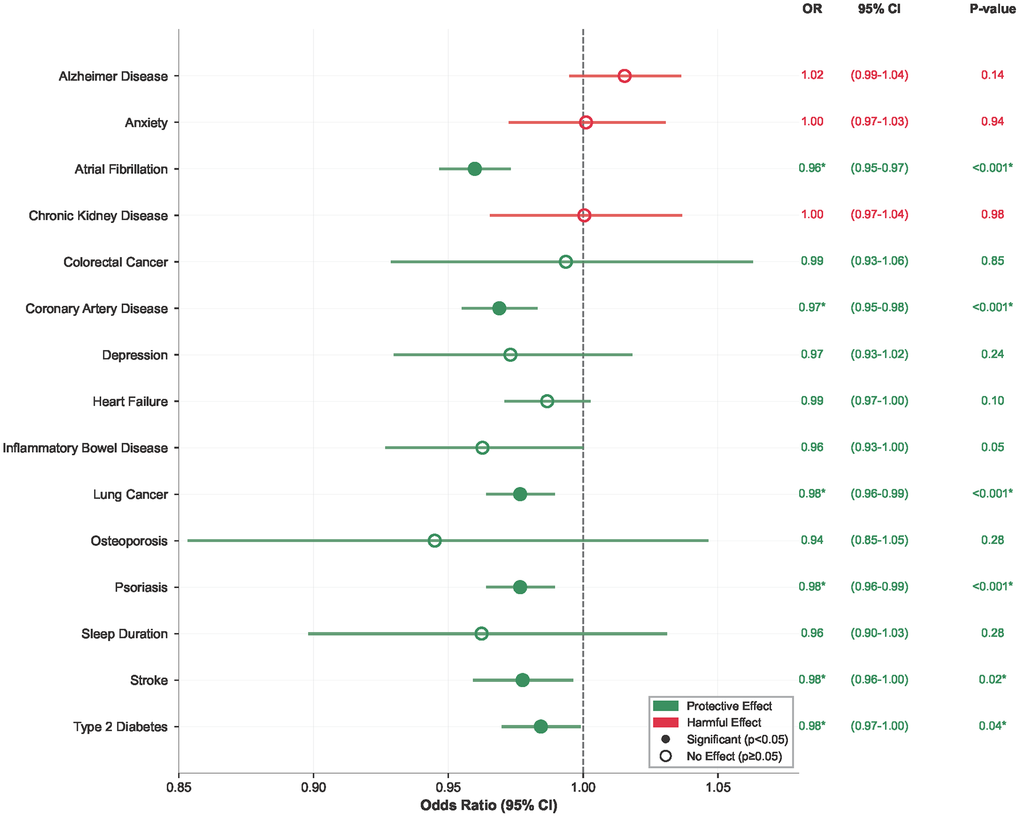
Figure 2. IL6R effects on cardiovascular disease outcomes. Forest plot showing protective effects of genetically predicted IL6R levels on major cardiovascular and non-cardiovascular outcomes. Abbreviations: OR: odds ratio; CI: confidence interval.
IL6 harmful effects
Genetically predicted higher IL6 levels showed significant harmful effects on mortality (OR 1.05, 95% CI 1.02-1.08, P = 0.002) (Figure 1). Sensitivity analyses confirmed these findings with no evidence of pleiotropy.
Reverse MR provided no evidence that genetic liability to CAD, AF, or ischemic stroke causally affects IL6 or IL6R: CAD and IL6 OR 1.03 (95% CI 0.95–1.12), P = 0.48; AF and IL6 OR 1.49 (95% CI 0.00–1097.74), P = 0.91; stroke and IL6 OR 1.03 (95% CI 0.97–1.10), P = 0.35; CAD and IL-6R OR 0.80 (95% CI 0.62–1.03), P = 0.10; AF and IL6R OR 0.49 (95% CI 0.00–66.81), P = 0.78; stroke and IL6R OR 0.96 (95% CI 0.89–1.04), P = 0.32. Sensitivity analyses were concordant and did not indicate directional pleiotropy.
CRP and GDF15
Neither CRP nor GDF15 demonstrated significant causal effects on mortality or cardiovascular disease outcomes. CRP showed neutral associations (mortality OR 1.01, P = 0.87, Figure 2), as did GDF15 (mortality OR 1.03, P = 0.42).
Individual SNP analysis confirmed the robustness of our findings across all nine IL6R genetic instruments (Figure 3). The consistent directional effects observed in both IL6 and IL6R analyses, with opposite biological directions, further validate the causal relationships identified in our primary analyses.
Expression quantitative trait loci analysis
eQTL analysis confirmed the biological relevance of our genetic instruments through their effects on target gene expression (Figure 4). Selected IL6R variants showed significant negative eQTL effects, indicating reduced IL6R gene expression, in line with their protective association with mortality.
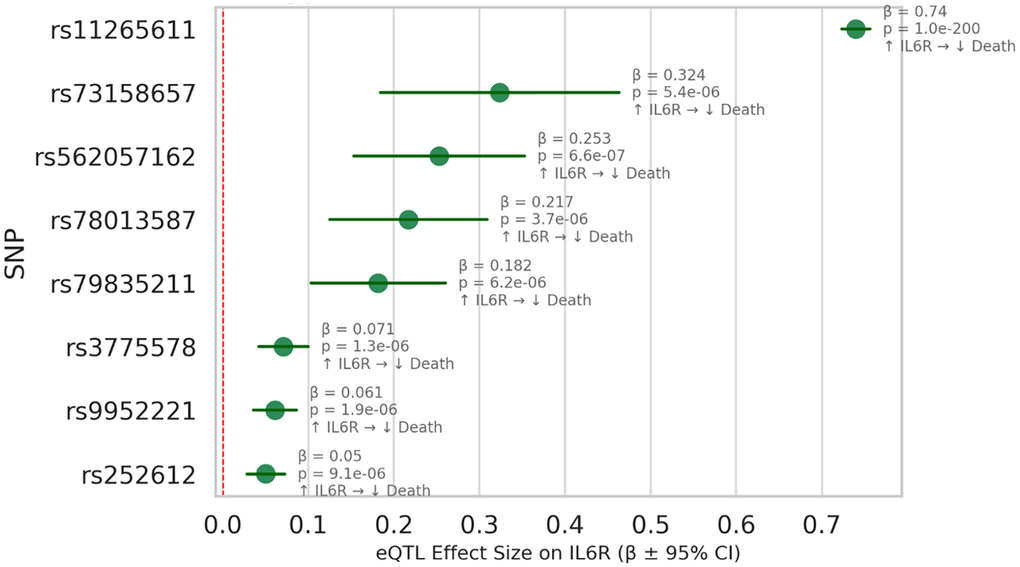
Figure 4. IL6R eQTL effects supporting protective association with mortality. The data are derived from GTEx v8 (Whole Blood) and large-scale GWAS consortia. This forest plot displays the cis-eQTL effects of genetic variants used as instrumental variables in our Mendelian randomization analysis of IL6R expression and all-cause mortality. Each row corresponds to a single nucleotide polymorphism (SNP), with effect sizes (β) representing the change in IL6R gene expression per allele, and horizontal bars indicating 95% confidence intervals. The red vertical line indicates no effect (β = 0). These results provide functional support for a protective role of higher IL6R expression in reducing mortality, and confirm the biological validity of the selected instruments. Abbreviations: eQTL: expression quantitative trait loci; SNP: single nucleotide polymorphism; remaining abbreviations as Figure 1.
Sensitivity analyses
Sensitivity analyses supported the robustness of these findings: Cochran’s Q-test showed no significant heterogeneity (IVW Q = 29.95, df = 25, P = 0.23), the MR-Egger intercept was close to zero and non-significant (−0.0034, SE = 0.0051, P = 0.51), leave-one-out analysis indicated that no single SNP drove the association (Supplementary Figure 2), and the funnel plot showed no evidence of asymmetry (Supplementary Figure 3). In addition, the scatter plot of SNP-specific effects demonstrated consistent alignment of variant estimates along the slope of the causal effect (Supplementary Figure 4), providing visual confirmation of the direction and magnitude of the protective association. Collectively, these results confirm a consistent and unbiased relationship between higher IL6R levels and reduced mortality risk.
Discussion
This large-scale Mendelian randomization study provides robust genetic evidence that inflammatory biomarkers causally influence long-term human survival (Figure 5).
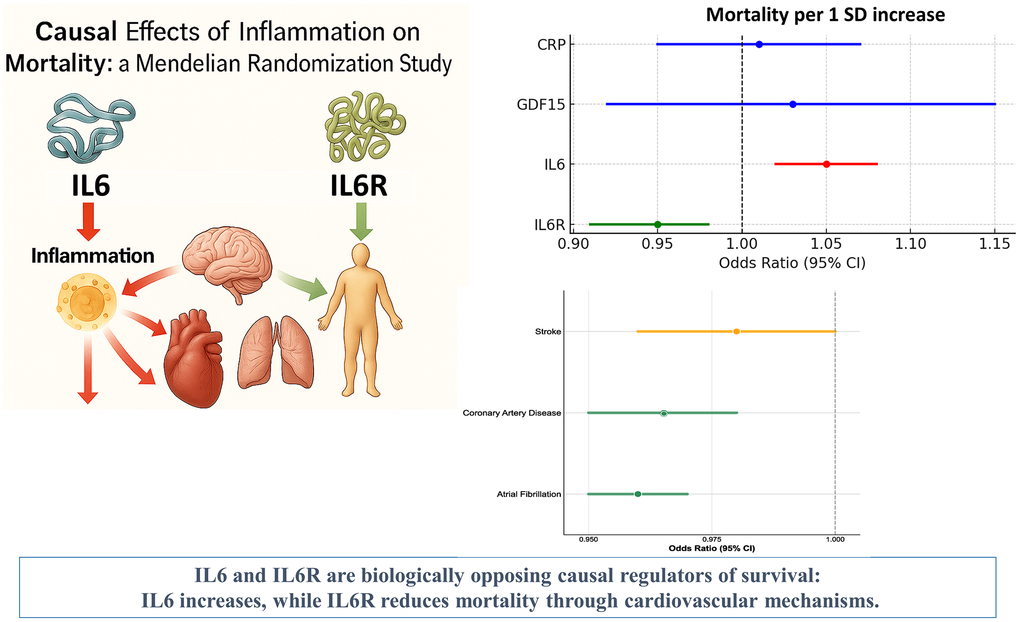
Figure 5. This illustration summarizes the Mendelian randomization findings on the causal impact of inflammatory biomarkers—IL6, IL6R, CRP, and GDF15—on all-cause mortality in over 750,000 genotyped individuals.
To our knowledge, this is the first MR analysis specifically focusing on the IL6/IL6R signaling pathway with long-term mortality as the primary outcome.
Using data from more than 750,000 individuals, we demonstrate that the two central components of this axis exert opposite effects: genetically higher soluble IL6 receptor (sIL6R) levels are protective, whereas elevated IL6 levels increase mortality risk [19].
A genetically predicted one–standard-deviation increase in sIL6R was associated with a ~5% reduction in all-cause mortality, while the same increment in IL6 corresponded to an increased mortality risk. The protective effect of sIL6R appears partly mediated by its favorable impact on major cardiovascular outcomes, including atrial fibrillation, coronary artery disease, and stroke. These findings are consistent with IL6R’s established role in modulating vascular inflammation and atherosclerosis. Beyond cardiovascular endpoints, we also observed a significant reduction in lung cancer, suggesting broader anti-inflammatory benefits.
The distinct directions of effect likely reflect different biological mechanisms. IL6R variants capture physiological modulation of receptor shedding, which may reduce IL6-mediated inflammation, whereas IL6 variants mirror sustained pro-inflammatory states that promote vascular injury and thrombosis [15, 17]. This targeted modulation of the IL6 pathway may offer a safer and more effective strategy for prevention than broad anti-inflammatory approaches. The cardiovascular and cancer protective effects of IL6R may offer a mechanistic explanation for its mortality benefits: by reducing atrial fibrillation, coronary artery disease, and stroke risk, IL6R directly addresses the leading causes of cardiovascular mortality. This cardiovascular-mediated protection offers a more targeted, potentially safer therapeutic avenue than broad anti-inflammatory approaches. In contrast, we found no evidence for causal effects of CRP or GDF-15 on mortality or cardiovascular disease, despite their strong epidemiological associations with cardiovascular outcomes [10, 20]. These findings underscore the importance of distinguishing prognostic correlates from true biological effectors, and suggests that these biomarkers may act as downstream indicators rather than upstream mediators of cardiovascular pathology.
Taken together, the results of the current mendelian randomization study strengthen the rationale for IL6R antagonism as a potential strategy to reduce cardiovascular disease and associated mortality.
The protective effects of sIL6R observed here are biologically plausible and mechanistically coherent with cardiovascular pathophysiology [19]. IL6 is a key driver of inflammation [4, 21]. The IL6R variants used as instruments increase the amount of receptor detectable in blood while proportionally reducing the receptor present on cell surfaces [22]. In vascular and myocardial tissues—where surface IL6R is naturally scarce—this shift, together with plasma regulators that bind IL-6–receptor complexes, dampens downstream IL6 activity and lowers inflammatory burden at the vessel wall and myocardium. In cardiovascular tissues, elevated IL-6R appears to exert anti-inflammatory effects by sequestering IL-6 and preventing its engagement with membrane-bound receptors, thereby reducing vascular inflammation, endothelial dysfunction, and thrombotic risk. The cardiovascular-specific protective effects we observed—particularly for atrial fibrillation and coronary artery disease—align with IL6R’s established role in modulating cardiac electrophysiology and coronary atherosclerosis. These cardiovascular benefits provide a clear mechanistic pathway through which IL6R reduces overall mortality, as cardiovascular disease remains the leading cause of death globally [23, 24].
Conversely, the harmful effects of IL6 are consistent with its established role as a pro-inflammatory cytokine with direct cardiovascular toxicity. IL6 promotes acute-phase responses, activates multiple inflammatory cascades, and contributes to insulin resistance, endothelial dysfunction, and thrombotic processes that directly contribute to cardiovascular disease development [6, 25]. Elevated IL6 has been linked to increased risk of myocardial infarction, stroke, and heart failure through its effects on coronary plaque instability, cardiac remodeling, and arrhythmogenesis, supporting its role as a causal mediator of cardiovascular morbidity and mortality [26].
Sensitivity analyses across multiple MR approaches yielded consistent findings for IL6R, with significant protective effects in all tested methods and minimal heterogeneity, reinforcing the robustness of the results. These genetic data align with clinical trial evidence: IL6R antagonists such as tocilizumab have demonstrated survival benefits in several inflammatory conditions, including COVID-19, without major long-term safety concerns in approved indications [27, 28].
Our findings suggest that precision targeting of IL6 signaling—particularly through selective modulation of trans-signaling—may represent a promising and safe strategy for preventing cardiovascular disease and reducing mortality risk. The cardiovascular-specific benefits we identified provide a strong therapeutic rationale for IL6R-targeted interventions in high-risk populations. However, translating these findings into preventive clinical applications will require careful risk–benefit evaluation and rigorous long-term safety assessments in dedicated cardiovascular prevention trials.
Limitations
Our study has limitations. First, analyses were restricted to individuals of European ancestry, which may limit generalizability to other populations with different genetic backgrounds. Second, while we assessed major cardiovascular outcomes, we did not examine other potential mediating pathways such as metabolic or inflammatory diseases that could contribute to IL6R’s mortality effects.
Conclusions
This Mendelian randomization study demonstrates that the IL6/IL6R axis has a causal impact on human survival through cardiovascular mechanisms: IL6R exerts a protective effect, whereas IL6 is detrimental. The protective influence of IL6R on atrial fibrillation, coronary artery disease, stroke and lung cancer provides a mechanistic basis for its mortality benefits. Concordant genetic and pharmacological evidence positions IL6R as a promising therapeutic target for cardiovascular disease prevention, while the neutral findings for CRP and GDF15 support their role as biomarkers rather than causal drivers. Collectively, these results identify the IL6 pathway as a key translational target for reducing cardiovascular disease and mortality.
Abbreviations
AF: Atrial Fibrillation; CAD: Coronary Artery Disease; CI: Confidence Interval; CRP: C-Reactive Protein; GDF-15: Growth Differentiation Factor-15; GWAS: Genome-Wide Association Study; IL6: Interleukin-6; IL6R: Interleukin-6 Receptor; MR: Mendelian Randomization; OR: Odds Ratio; SD: Standard Deviation; SNP: Single nucleotide polymorphism.
Author Contributions
Conception and design: EP Navarese, J Kubica. Analysis and interpretation of the data: EP Navarese, DJK Kereiakes, T Henry, M Farkouh. Drafting of the article: EP Navarese. Critical revision of the article for important intellectual content: EP Navarese, J Kubica, DJ Kereiakes, T Henry, ME Farkouh, G. Talanas, M Isgender, M Brouwer.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Funding
No funding was used for this paper.
References
- 1. Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000; 908:244–54. https://doi.org/10.1111/j.1749-6632.2000.tb06651.x [PubMed]
- 2. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013; 153:1194–217. https://doi.org/10.1016/j.cell.2013.05.039 [PubMed]
- 3. Swiatkiewicz I, Kozinski M, Magielski P, Fabiszak T, Sukiennik A, Navarese EP, Odrowaz-Sypniewska G, Kubica J. Value of C-reactive protein in predicting left ventricular remodelling in patients with a first ST-segment elevation myocardial infarction. Mediators Inflamm. 2012; 2012:250867. https://doi.org/10.1155/2012/250867 [PubMed]
- 4. Navarese EP, Podhajski P, Gurbel PA, Grzelakowska K, Ruscio E, Tantry U, Magielski P, Kubica A, Niezgoda P, Adamski P, Junik R, Przybylski G, Pilaczyńska-Cemel M, et al. PCSK9 Inhibition During the Inflammatory Stage of SARS-CoV-2 Infection. J Am Coll Cardiol. 2023; 81:224–34. https://doi.org/10.1016/j.jacc.2022.10.030 [PubMed]
- 5. Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, Collins R, Danesh J, and Emerging Risk Factors Collaboration. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet. 2010; 375:132–40. https://doi.org/10.1016/S0140-6736(09)61717-7 [PubMed]
- 6. Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014; 6:a016295. https://doi.org/10.1101/cshperspect.a016295 [PubMed]
- 7. Swerdlow DI, Holmes MV, Kuchenbaecker KB, Engmann JE, Shah T, Sofat R, Guo Y, Chung C, Peasey A, Pfister R, Mooijaart SP, Ireland HA, Leusink M, et al, and Interleukin-6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet. 2012; 379:1214–24. https://doi.org/10.1016/S0140-6736(12)60110-X [PubMed]
- 8. Levin MG, Burgess S. Mendelian Randomization as a Tool for Cardiovascular Research: A Review. JAMA Cardiol. 2024; 9:79–89. https://doi.org/10.1001/jamacardio.2023.4115 [PubMed]
- 9. Skrivankova VW, Richmond RC, Woolf BAR, Yarmolinsky J, Davies NM, Swanson SA, VanderWeele TJ, Higgins JPT, Timpson NJ, Dimou N, Langenberg C, Golub RM, Loder EW, et al. Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization: The STROBE-MR Statement. JAMA. 2021; 326:1614–21. https://doi.org/10.1001/jama.2021.18236 [PubMed]
- 10. Folkersen L, Gustafsson S, Wang Q, Hansen DH, Hedman ÅK, Schork A, Page K, Zhernakova DV, Wu Y, Peters J, Eriksson N, Bergen SE, Boutin TS, et al. Genomic and drug target evaluation of 90 cardiovascular proteins in 30,931 individuals. Nat Metab. 2020; 2:1135–48. https://doi.org/10.1038/s42255-020-00287-2 [PubMed]
- 11. Said S, Pazoki R, Karhunen V, Võsa U, Ligthart S, Bodinier B, Koskeridis F, Welsh P, Alizadeh BZ, Chasman DI, Sattar N, Chadeau-Hyam M, Evangelou E, et al. Genetic analysis of over half a million people characterises C-reactive protein loci. Nat Commun. 2022; 13:2198. https://doi.org/10.1038/s41467-022-29650-5 [PubMed]
- 12. Kurki MI, Karjalainen J, Palta P, Sipilä TP, Kristiansson K, Donner KM, Reeve MP, Laivuori H, Aavikko M, Kaunisto MA, Loukola A, Lahtela E, Mattsson H, et al, and FinnGen. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature. 2023; 613:508–18. https://doi.org/10.1038/s41586-022-05473-8 [PubMed]
- 13. Nielsen JB, Thorolfsdottir RB, Fritsche LG, Zhou W, Skov MW, Graham SE, Herron TJ, McCarthy S, Schmidt EM, Sveinbjornsson G, Surakka I, Mathis MR, Yamazaki M, et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat Genet. 2018; 50:1234–9. https://doi.org/10.1038/s41588-018-0171-3 [PubMed]
- 14. Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC, Webb TR, Zeng L, Dehghan A, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015; 47:1121–30. https://doi.org/10.1038/ng.3396 [PubMed]
- 15. Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A, Rutten-Jacobs L, Giese AK, van der Laan SW, Gretarsdottir S, Anderson CD, Chong M, Adams HHH, et al, and AFGen Consortium, and Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium, and International Genomics of Blood Pressure (iGEN-BP) Consortium, and INVENT Consortium, and STARNET, and BioBank Japan Cooperative Hospital Group, and COMPASS Consortium, and EPIC-CVD Consortium, and EPIC-InterAct Consortium, and International Stroke Genetics Consortium (ISGC), and METASTROKE Consortium, and Neurology Working Group of the CHARGE Consortium, and NINDS Stroke Genetics Network (SiGN), and UK Young Lacunar DNA Study, and MEGASTROKE Consortium. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018; 50:524–37. https://doi.org/10.1038/s41588-018-0058-3 [PubMed]
- 16. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013; 37:658–65. https://doi.org/10.1002/gepi.21758 [PubMed]
- 17. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018; 50:693–8. https://doi.org/10.1038/s41588-018-0099-7 [PubMed]
- 18. GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013; 45:580–5. https://doi.org/10.1038/ng.2653 [PubMed]
- 19. Rose-John S. IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int J Biol Sci. 2012; 8:1237–47. https://doi.org/10.7150/ijbs.4989 [PubMed]
- 20. Wensley F, Gao P, Burgess S, Kaptoge S, Di Angelantonio E, Shah T, Engert JC, Clarke R, Davey-Smith G, Nordestgaard BG, Saleheen D, Samani NJ, Sandhu M, et al, and C Reactive Protein Coronary Heart Disease Genetics Collaboration (CCGC). Association between C reactive protein and coronary heart disease: mendelian randomisation analysis based on individual participant data. BMJ. 2011; 342:d548. https://doi.org/10.1136/bmj.d548 [PubMed]
- 21. Kang S, Kishimoto T. Interplay between interleukin-6 signaling and the vascular endothelium in cytokine storms. Exp Mol Med. 2021; 53:1116–23. https://doi.org/10.1038/s12276-021-00649-0 [PubMed]
- 22. Ferreira RC, Freitag DF, Cutler AJ, Howson JM, Rainbow DB, Smyth DJ, Kaptoge S, Clarke P, Boreham C, Coulson RM, Pekalski ML, Chen WM, Onengut-Gumuscu S, et al. Functional IL6R 358Ala allele impairs classical IL-6 receptor signaling and influences risk of diverse inflammatory diseases. PLoS Genet. 2013; 9:e1003444. https://doi.org/10.1371/journal.pgen.1003444 [PubMed]
- 23. Navarese EP, Lansky AJ, Kereiakes DJ, Kubica J, Gurbel PA, Gorog DA, Valgimigli M, Curzen N, Kandzari DE, Bonaca MP, Brouwer M, Umińska J, Jaguszewski MJ, et al. Cardiac mortality in patients randomised to elective coronary revascularisation plus medical therapy or medical therapy alone: a systematic review and meta-analysis. Eur Heart J. 2021; 42:4638–51. https://doi.org/10.1093/eurheartj/ehab246 [PubMed]
- 24. Navarese EP, Andreotti F. Cardiac mortality, adequate power, and objective inclusion of the entire evidence are key to accurately define the long-term effect of revascularisation vs. medical therapy alone in stable coronary syndromes. Eur Heart J. 2021; 42:4699–700. https://doi.org/10.1093/eurheartj/ehab677 [PubMed]
- 25. Yudkin JS, Kumari M, Humphries SE, Mohamed-Ali V. Inflammation, obesity, stress and coronary heart disease: is interleukin-6 the link? Atherosclerosis. 2000; 148:209–14. https://doi.org/10.1016/s0021-9150(99)00463-3 [PubMed]
- 26. Mehta NN, deGoma E, Shapiro MD. IL-6 and Cardiovascular Risk: A Narrative Review. Curr Atheroscler Rep. 2024; 27:12. https://doi.org/10.1007/s11883-024-01259-7 [PubMed]
- 27. Rosas IO, Bräu N, Waters M, Go RC, Hunter BD, Bhagani S, Skiest D, Aziz MS, Cooper N, Douglas IS, Savic S, Youngstein T, Del Sorbo L, et al. Tocilizumab in Hospitalized Patients with Severe Covid-19 Pneumonia. N Engl J Med. 2021; 384:1503–16. https://doi.org/10.1056/NEJMoa2028700 [PubMed]
- 28. Stone JH, Tuckwell K, Dimonaco S, Klearman M, Aringer M, Blockmans D, Brouwer E, Cid MC, Dasgupta B, Rech J, Salvarani C, Schett G, Schulze-Koops H, et al. Trial of Tocilizumab in Giant-Cell Arteritis. N Engl J Med. 2017; 377:317–28. https://doi.org/10.1056/NEJMoa1613849 [PubMed]