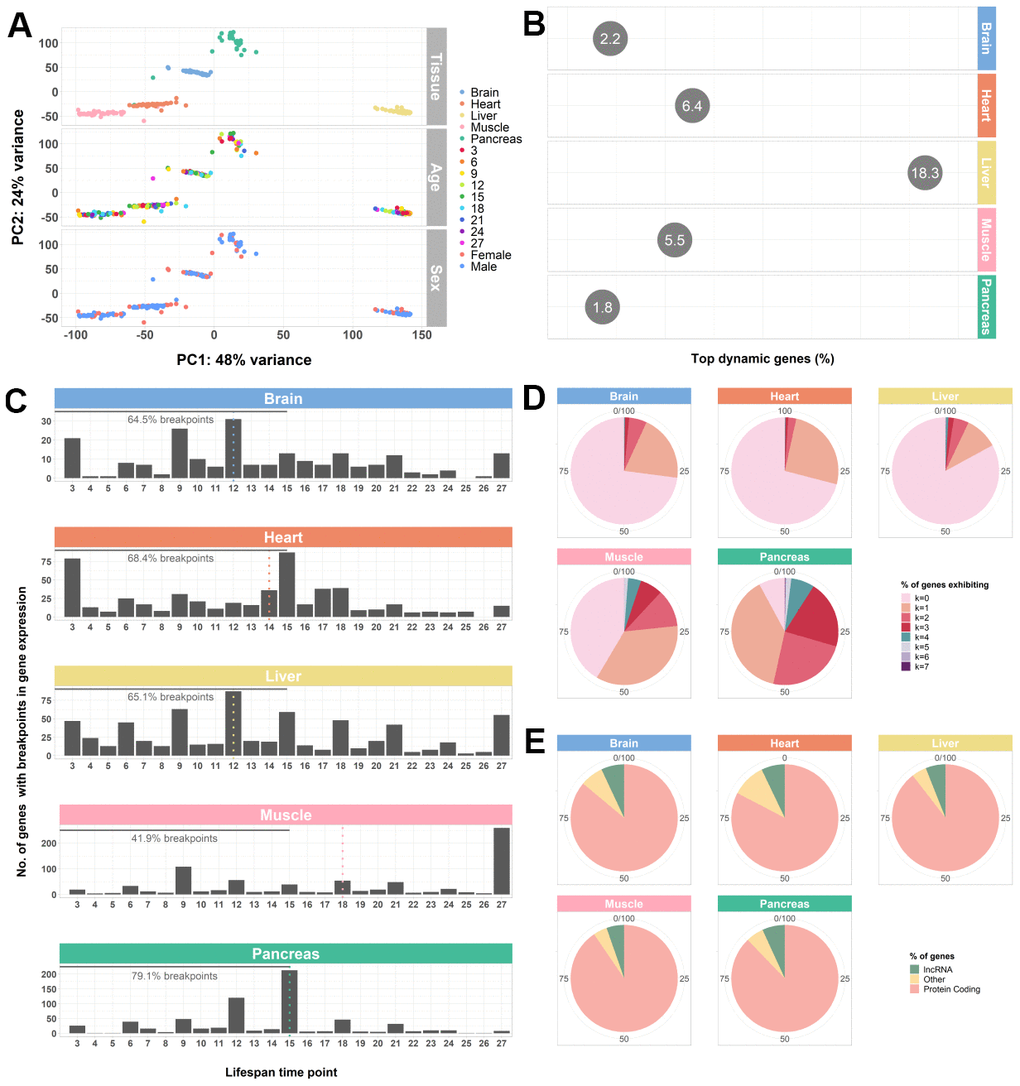
Figure 1.Whole-transcriptome characterization of different mouse tissues throughout the lifespan by segmented regression analysis. (A) PCA of all tissues performed on VST-normalized read counts of the 500 most variable genes and colored by all known effects highlights type of tissue as the main contributor to sample segregation. (B) Percentage of top dynamic (Trendy) genes across the lifespan for the mouse brain, heart, liver, muscle, and pancreas, highlighting the liver and the pancreas the ones with higher and lower dysregulation, respectively. The displayed values correspond to the percentages of the total number of Trendy genes found in each tissue relative to the total number of expressed genes per tissue (brain: 34164, heart: 28073, liver: 20157, muscle: 18978, pancreas: 18414). Top dynamic genes were selected based on tissue-specific adjusted R2 thresholds (brain: > 0.2; heart: > 0.2; liver: > 0.1; muscle: > 0.3; pancreas: > 0.3) and p-values < 0.1 in at least one segment; Supplemental File SF1). (C) Histograms of the distribution of breakpoints in gene expression of the top dynamic genes per tissue. Each bar depicts the sum of all breakpoints of all Trendy genes at that given time point. Monotonic behaviors (i.e., no breakpoints) are not included in the histograms. Dotted, vertical lines indicate the median breakpoint distribution for each tissue. Median breakpoint distribution in the muscle was significantly different from all the other tissues (Kruskal-Wallis Test followed by Dunn’s Test; Supplementary Table 1). (D) Percentage of top dynamic genes exhibiting 0 to 7 breakpoints (maximum number of breakpoints allowed in the Trendy regression model). In all tissues, except from the Pancreas, most Trendy genes exhibit monotonic expression patterns (continuously up or down). In the Pancreas, the majority of genes display one breakpoint. (E) Biotype distribution of the top dynamic genes per tissue. Biotype nomenclature based on Ensembl annotation. In all tissues, protein coding genes were significantly over-enriched relative to the reference genome’s annotation, whereas lncRNAs were significantly under-enriched (Fisher’s Exact Test; Supplementary Table 2).
Figure 1 — Integration of segmented regression analysis with weighted gene correlation network analysis identifies genes whose expression is remodeled throughout physiological aging in mouse tissues | Aging