Abstract
Resveratrol isa plant-derived polyphenol that promotes health and disease resistance inrodent models, and extends lifespan in lower organisms. A major challengeis to understand the biological processes and molecular pathways by whichresveratrol induces these beneficial effects. Autophagy is a criticalprocess by which cells turn over damaged components and maintain bioenergeticrequirements. Disruption of the normal balance between pro- andanti-autophagic signals is linked to cancer, liver disease, andneurodegenerative disorders. Here we show that resveratrol attenuatesautophagy in response to nutrient limitation or rapamycin in multiple celllines through a pathway independent of a known target, SIRT1. In alarge-scalein vitro kinase screen we identified p70 S6 kinase(S6K1) as a target of resveratrol. Blocking S6K1 activity by expression ofa dominant-negative mutant or RNA interference is sufficient to disruptautophagy to a similar extent as resveratrol. Furthermore,co-administration of resveratrol with S6K1 knockdown does not produce anadditive effect. These data indicate that S6K1 is important for the fullinduction of autophagy in mammals and raise the possibility that some ofthe beneficial effects of resveratrol are due to modulation of S6K1activity.
Introduction
Autophagy is an essential process by
which eukaryotic cells turn over long-lived cytosolic components, clear damaged
proteins and organelles, and maintain bio-energetic requirements during conditions
of nutrient and growth factor withdrawal
[1]. Degradation and recycling of
cellular components can involve the uptake of small amounts of cytoplasm at the
vacuole or lysosome surface (microautophagy) or, in response to a strong
stimulus such as starvation, the formation of specialized double
membraned-organelles termed autophagosomes, which engulf larger portions of
cytoplasm or organelles before fusing with a vacuole or lysosome (macroautophagy),
hereafter referred to simply as autophagy [1-3]. While this process is an important
part of the normal balance between anabolic and catabolic processes and can
prolong survival during nutrient limitation, autophagy is also an alternate
death pathway that facilitates type II programmed cell death [4-6]. For this
reason, imbalances in this pathway can contribute to
seemingly diverse pathologies.
Resveratrol is a small polyphenol that extends the
lifespan of simple model organisms, ostensibly by mimicking caloric restriction
[7,8]. In rodents, resveratrol protects from a variety of age-related diseases
including cancer, cardiovascular disease, neurodegeneration, obesity and diabetes
[9-14]. Although there is evidence that some of resveratrol's actions are
mediated by activation of the SIRT1 deacetylase,
the mechanisms underlying
the numerous beneficial effects of resveratrol remain to be elucidated.
It has previously been reported
that 24-48 hours of resveratrol treatment induces autophagy in cancer cells
grown in rich media, suggesting a mechanism by which resveratrol might enhance
cell death and suppress tumor growth [15-18]. Here we report the effect of
resveratrol treatment on the normal induction of autophagy over 4-6 hours
following nutrient withdrawal in tumor and non-tumor cell lines. In contrast to
the activation of the autophagic pathway observed in tumor cells in complete
media, we find that resveratrol markedly inhibits the starvation-induced
autophagic response. We show that this effect does not require SIRT1, and
identify p70 S6 kinase (S6K1) as a target of
resveratrol that is responsible for the inhibition of starvation-induced
autophagy.
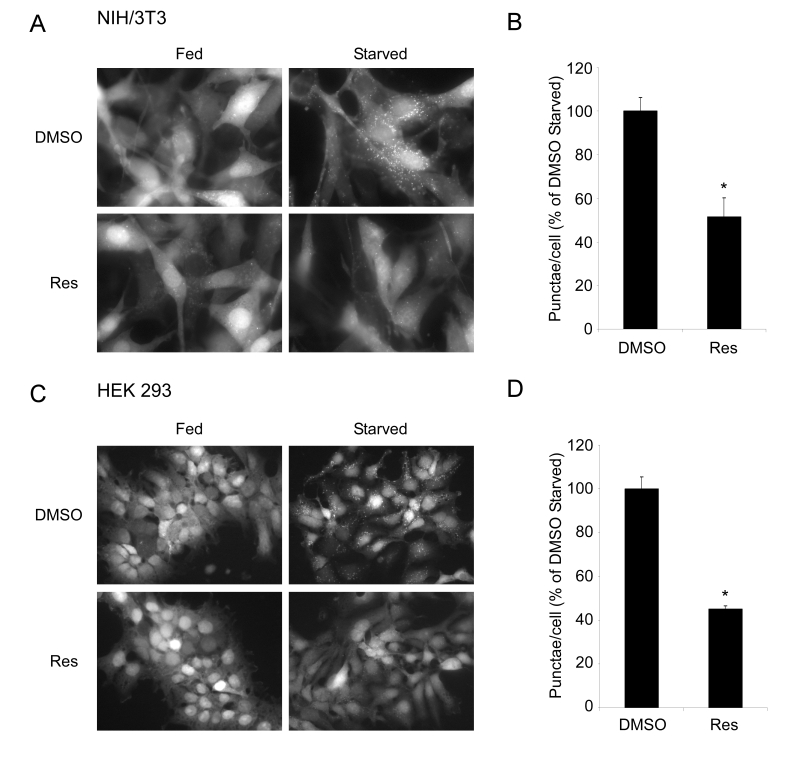
Figure 1. Resveratrol inhibits autophagy in mammalian cells. (A) NIH/3T3 cells stably expressing the GFP-LC3
fusion protein were subjected to nutrient withdrawal by
replacing growth media (Fed) with Earle's buffered saline
solution (Starved) and treated with either DMSO or 50 μM
resveratrol (Res) for 2 hours. Representative fields at
63X (oil immersion) magnification are shown. (B) Quantification
of punctae/cell in (A) of at least 4 fields per treatment
are represented as a percentage of the starved DMSO treated
cells. (C) HEK293 cells stably expressing the GFP-LC3 fusion
protein were subjected to starvation and either DMSO or 50 μM
Res for 6 hours. Representative fields at 40X magnification
are shown. (D) Quantification was performed on HEK293 cells
as in (B). Error bars represent s.e.m. * (p < 0.0022).
Results
Suppression of nutrient starvation- and
rapamycin-induced autophagy by resveratrol
Autophagy is an important
component of the cellular response to nutrient stress and growth factor
withdrawal. We therefore tested whether resveratrol treatment would influence
regulation of autophagy under these conditions. Autophagy was assessed by
monitoring the relocalization of a component of the autophagy machinery, LC3,
from the cytoplasm to the forming autophagosome [19]. NIH/3T3 cells or HEK293
cells stably expressing a GFP-LC3 fusion protein were generated and were
induced to undergo autophagy in the presence or absence of resveratrol.
Treatment with resveratrol resulted in a dramatic reduction in the number of
starvation-induced GFP-LC3 punctae (Figure 1). Similar inhibitory effects on
autophagy were observed in other cell lines, including human tumor cell lines (HeLa and U2OS), as well as mouse
embryonic fibroblasts (MEFs) using monodansylcadaverine (MDC), a fluorescent compound
that stains late autophagosomes (Supplementary Figures 1 and 3) [20]. Since previous
studies have described an increase in autophagy following 24 hours of
resveratrol treatment in nutrient rich
media, we also tested the effects of resveratrol under these conditions. Consistent with
these results, we observed an induction of autophagosome formation in cells treated
with resveratrol for 24 hours in complete media containing serum
(Supplementary Figure 2). Thus, the influence of resveratrol on autophagy is context
dependent, and in the case of autophagy induced by nutrient limitation,
resveratrol is inhibitory.
Rapamycin is an inhibitor of the nutrient sensing
mTOR-Raptor complex and has been shown to induce autophagy [21,22]. In yeast,
it had been shown that resveratrol can reverse some markers of autophagy
induced by rapamycin [23]. Consistent with these results and similar to the results
seen under nutrient limitation, rapamycin-induced autophagy is almost
completely abrogated by resveratrol treatment (Figure 2A and B). Decreased
GFP-LC3 punctae could be due either to increased flux or a block in autophagy. To
distinguish between these two possibilities, we examined LC3-II accumulation
with and without Bafilomycin A1, an inhibitor of lysosome degradation. Under
both conditions, resveratrol was able to block the accumulation of LC3-II, indicating
a suppression of autophagy rather than an enhancement of lysosomal clearance (Figure 2C).
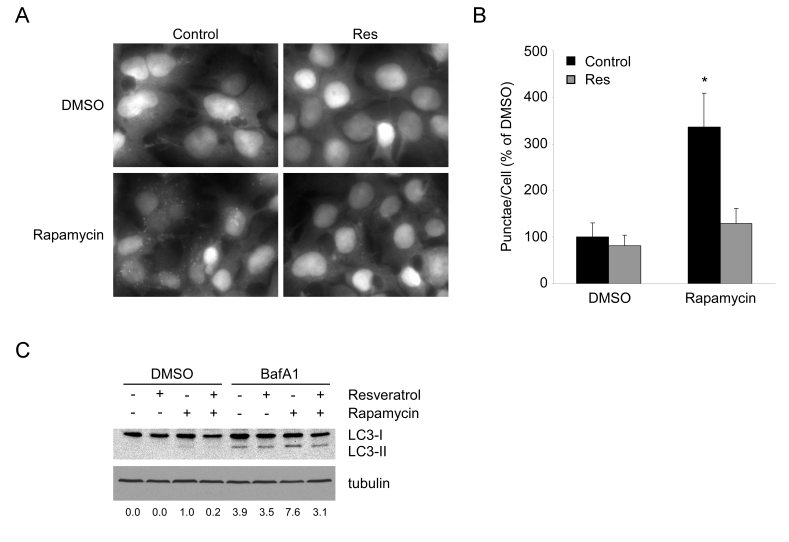
Figure 2. Resveratrol suppresses autophagy under TOR inhibition. (A) HEK293 cells stably
expressing GFP-LC3 growing in complete media were
pretreated with DMSO or 50 μM resveratrol (Res) for 1
hour, prior to addition of DMSO or 200 nM rapamycin
for 4 hours. 40X magnification fields have been
cropped and zoomed for ease of punctae visualization.
(B) Quantification of punctae/cell from (A) of 10 fields
per treatment are represented as a percentage of DMSO
treated cells. Error bars represent s.d.m. * (p < 0.0001) (C) HEK293 GFP-LC3 cells were pretreated for 1 hour with DMSO or resveratrol and subsequently treated with DMSO or 1 mM rapamycin in the presence or absence of 100 nM Bafilomycin A1 for 4 hours. A representative western blot of endogenous LC3 and tubulin are shown. Numbers represent the ratio of LC3-II to tubulin for each condition normalized to Rapamycin in the absence of BafA1.
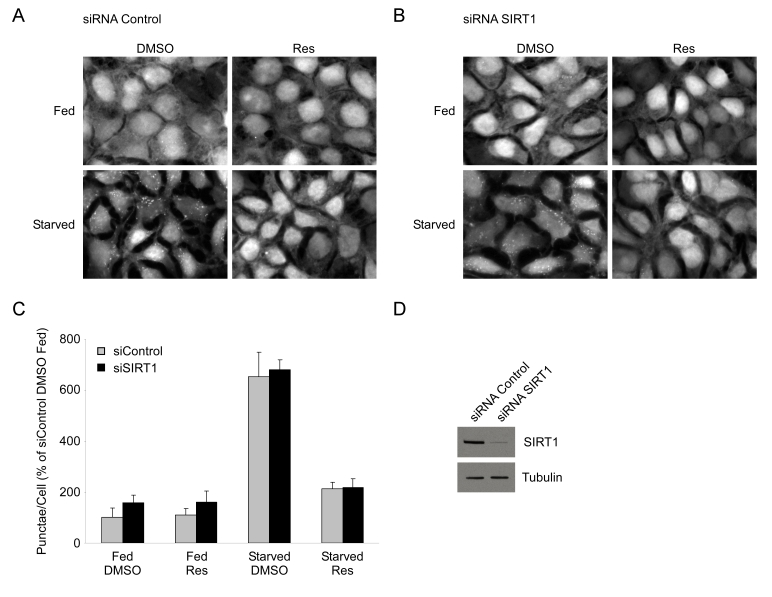
Figure 3. Resveratrol suppresses autophagy independently of SIRT1. HEK293 cells
stably expressing GFP-LC3 were transfected with either a control siRNA (A) or an siRNA
directed against SIRT1 (B) for 72 hours. Subsequently, cells were
subjected to nutrient starvation with or without 50 μM resveratrol
(Res) treatment for 4 hours. 40X magnification fields have been cropped and
zoomed for ease of punctae visualization. (C) Quantification
of punctae/cell from (A) and (B) of 4 fields are represented as a
percentage of fed DMSO treated control siRNA cells. Error bars represent
s.d.m. (D) Representative
western blot showing typical knockdown of SIRT1 by siRNA transfection in
HEK293 GFP-LC3 cells.
We next tested whether SIRT1, an NAD+-dependent
deacetylase that is activated by resveratrol [8], was required for this effect.
To test this hypothesis, HEK 293 GFP-LC3 cells were transfected with either
control siRNA or an siRNA directed against the SIRT1 deacetylase and
subsequently nutrient starved in the presence or absence of resveratrol. We
found that both control and SIRT1 knockdown cells
displayed a similar level of induction of GFP-LC3 punctae and that resveratrol
still produced an equivalent suppression of autophagy (Figure 3). Consistent with
these results, SIRT1+/+ and SIRT1-/- MEFs induced autophagy in response to nutrient withdrawal,
and in both cell lines the inhibitory effect of resveratrol on autophagy was comparable (Supplementary Figure 3). These data
indicate that inhibition of starvation-induced autophagy
by resveratrol is molecularly distinct from the induction seen in previous
studies and is not mediated by the SIRT1-dependent pathway that has previously
been described [24]. It will be interesting to explore the differences in these
systems that engage or disengage SIRT1 during autophagic induction.
Kinase profiling of resveratrol in vitro
Resveratrol has previously been shown to inhibit
several kinases including PKC and Src [25] and is structurally similar to the
flavanoid quercetin (Figure 4A), which is an inhibitor of PI 3-kinase [26].
Therefore, we hypothesized that the effect of resveratrol on autophagy could be
related to inhibition of one or more upstream kinases. To test this, we
performed an in vitro kinase screen and determined an inhibition profile
for resveratrol. Out of 100 kinases tested, Jak2, NLK, p70 S6 kinase (S6K1),
Pim-1, and Pim-2 emerged as potential targets of resveratrol
(Figure 4B, Supplementary Table 1).
With the exception of S6K1, these kinases play primary roles in the hematopoietic
system, and were thus viewed as unlikely to have been responsible for the
effect we were studying. Further-more, although JNK had previously been shown
to play a positive role in autophagy, its activity was not significantly
inhibited by resveratrol at this dose [27]. On the other hand, S6K1 is known
to play a requisite role in the regulation of autophagy in Drosophila [28],
making it a promising candidate for the in vivo target of resveratrol
that is responsible for inhibition of autophagy.
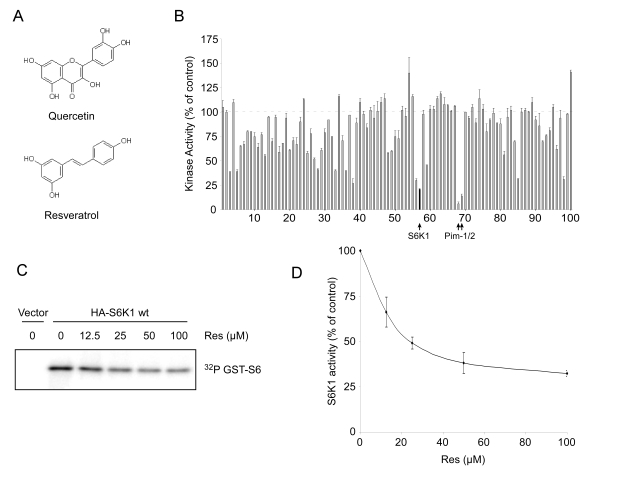
Figure 4. Resveratrol inhibits S6K1 in vitro. (A) Structural
similarity between resveratrol and quercetin, a known kinase inhibitor. (B) Kinase
inhibition profile for resveratrol at 20 μM obtained using
KinaseProfiler™ (Upstate). Dashed line represents 100% activity as compared
to control. Black filled-in bar on the graph indicates S6K1. Complete data
set is provided in Supplementary Table 1. Error bars represent s.d.m. (C) Phosphorylation
of recombinant GST-tagged S6 by immunoprecipitated HA-S6K1 under increasing
concentrations of resveratrol (Res). Autoradiograph depicts S6K1
phosphorylation of GST-S6. (D) Average of three separate kinase assay
experiments as performed in (C). Densitometry was performed using
NIH ImageJ. Error bars represent s.e.m.
To confirm the inhibition of S6 kinase by resveratrol in
vitro, we determined the effect of the compound on immunoprecipitated
HA-tagged S6K1 from HEK293 cells using purified GST-tagged full-length
recombinant S6 ribosomal protein (S6) as a substrate. In agreement with the primary
screen, we found that resveratrol inhibited the activity of S6 kinase in a
dose-dependent manner, exhibiting an IC50 of ~25 μM (Figure 4C and
D, and Supplementary Figure 4).
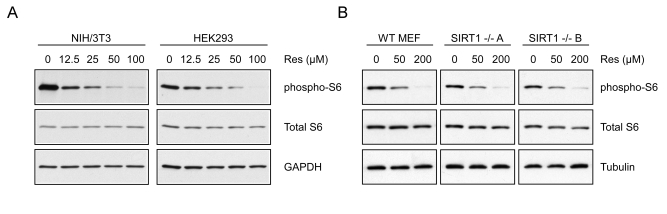
Figure 5. Resveratrol inhibits S6K1 in intact cells. (A) NIH/3T3 or
HEK293 cells were treated for 30 minutes with increasing doses of
resveratrol and whole cell extracts were western blotted for the indicated
proteins. (B)
WT
or two separate lines of SIRT1-/- MEFs (A and B) were treated
with increasing doses of resveratrol for 30 minutes and analyzed by western
blot.
Resveratrol-mediated inhibition of S6 kinase activity in
vivo
To test whether resveratrol alters S6 kinase activity
in cells, we treated NIH/3T3 and HEK293 cells with resveratrol and analyzed the
phosphorylation status of S6, a well-characterized downstream target of S6
kinase. After 30 minutes, phosphorylation of S6 was dramatically decreased in a
dose-dependent manner in both cell lines at concentrations consistent with the IC50 determined in
vitro (Figure 5A). These results were observed in a variety of different mammalian
cell lines and were independent of SIRT1, since treatment of either SIRT1-/- or wildtype MEFs
with resveratrol resulted in a similar decrease in S6 phosphorylation (Figure 5B). It is interesting to note that the doses required to inhibit
phosphorylation of S6 were significantly higher in MEFs than those in other
lines. Consistent with this observation, inhibition of autophagy in MEFs also
required a higher concentration than in other cell lines (Supplementary Figure 3).
Regulation of starvation-induced autophagy by S6
kinase 1
To test whether inhibition of S6K1 might
account for suppression of autophagy by resveratrol, GFP-LC3 expressing HEK293
cells were infected with retrovirus encoding a dominant-negative (K100R) mutant
of S6 kinase 1 or lentivirus encoding an shRNA against human S6K1. Cells expressing
K100R S6K1 or knocked down for S6K1 expression showed a significant reduction in
the number of GFP-LC3 punctae compared to control cells following nutrient withdrawal (Figure 6). These results demonstrate
that S6 kinase inhibition is sufficient
to suppress the induction of autophagy under nutrient-starved
conditions. Similar observations have been made previously in Drosophila [28],
but had not been extended to mammalian systems. Mieulet et al. have observed
that a normal basal level of autophagy still proceeds in S6K1;S6K2-/- muscle cells [29]; however, it is
not clear whether nutrient deprivation or rapamycin can induce an increase in autophagy
in this system. Moreover, studies in these mice suggest that mitogen signaling
through p90rsk might compensate for the loss of S6 kinase signaling [30]. Our
results support the view that under normal circumstances S6K1 plays a role in
the induction of autophagy in response to nutrient deprivation.
S6 kinase dependence of resveratrol-mediated
suppression of autophagy
To provide additional evidence that the effects of
resveratrol are mediated via S6K1, HEK293 cells infected with control virus or
a virus encoding shRNA against S6K1 were treated with resveratrol and subjected
to nutrient withdrawal. Individually, S6K1 shRNA and resveratrol both dramatically
reduced the number of GFP-LC3 punctae, and resveratrol treatment in the absence
of S6K1 produced no further decrease (Figure 7A). Quantification of punctae
revealed no statistically significant difference between any of the
experimental treatments, all of which were significantly different when
compared to control cells (Figure 7B). These data indicate that S6 kinase is
required for the full induction of autophagy in response to nutrient withdrawal
in mammals, and lend further support to the view that the reduced level of
autophagy in resveratrol-treated cells is due to inhibition of S6K1.
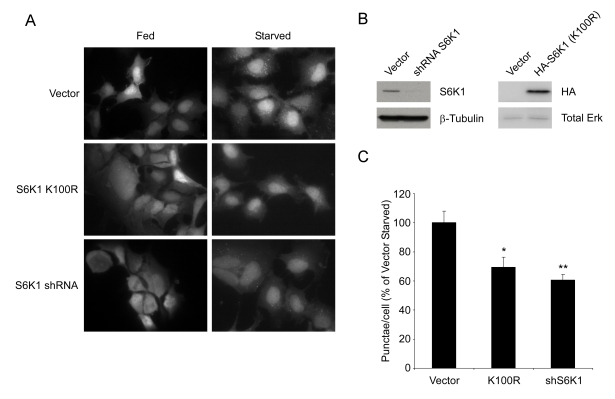
Figure 6. S6K1 is required for autophagy in mammalian cells. (A) HEK293 cells stably expressing GFP-LC3 were
infected with retrovirus encoding a dominant negative
S6K1 (K100R) or with lentivirus encoding a specific shRNA
directed against human S6K1 and subjected to nutrient
withdrawal by replacing supplemented media (Fed) with EBSS
for 4 hours (Starved). Representative fields at 63X (oil
immersion) magnification are shown. (B) Efficiency of S6K1
knockdown and expression of HA-tagged S6K1 (K100R) in HEK293
GFP-LC3 cells. (C) Quantification of punctae/cell from (A)
of at least 9 fields per treatment are represented as a percentage
of the starved vector control cells. Error bars represent
s.e.m. * (p < 0.0015) ** (p < 0.0002)
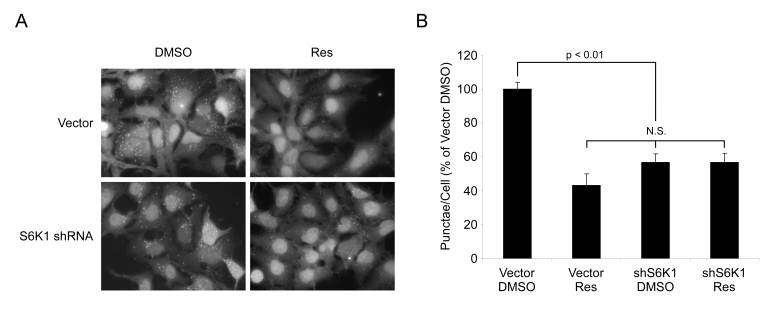
Figure 7. Resveratrol does not affect autophagy in the absence of S6K1. (A) HEK293 GFPLC3 cells were
infected with shRNA S6K1 lentivirus or control virus
and treated with EBSS (Starved) for 4 hrs ± 50 μM
resveratrol (Res). Representative fields at 63X
(oil immersion) magnification are shown. (B) Quantification
of punctae/cell from (A) of at least 4 fields per treatment
are represented as a percentage of DMSO treated starved vector
control cells. Error bars represent s.e.m. N.S. = not
significant.
Discussion
Resveratrol has been
characterized as an activator of SIRT1/Sir2, which are members of a family of
enzymes that promote longevity in lower organisms [7,8,31-33]. In rodents,
resveratrol has many beneficial effects including cancer prevention, cardio-
and neuroprotective effects, and improvements in insulin sensitivity, although
the extent to which these effects are mediated by SIRT1 is not yet clear
[9-14]. Here we identify a novel target of resveratrol activity, S6K1, that may
have important implications for understanding the mechanisms by which
resveratrol increases health in both lower organisms and mammals. We
suggest that inhibition of S6K1 may act in parallel to or in concert with
activation of SIRT1 to modulate lifespan and health.
A previous study described an important positive role
for SIRT1 in autophagy; however, we did not observe a noticeable change in the
rate of autophagy when altering SIRT1 activity in various assays. It was
recently shown that FK866, a NAMPT inhibitor, leads to reduced NAD+and is sufficient to induce autophagy. This suggests
that SIRT1 was also not required to induce autophagy, since NAD+is required for SIRT1 activity [34]. It will be
important to understand the differences in these studies in order to clarify
under what conditions SIRT1 can regulate autophagy.
The role of S6K in autophagy has
generated considerable confusion due to seemingly conflicting data. Inhibition
of the upstream kinase, mTOR, induces autophagy [3] and accordingly, ribosomal
protein S6 phosphorylation has been inversely correlated with autophagy [35]. In
addition, ATG1 has been shown to inhibit S6 kinase activity by blocking its
ability to be phosphorylated on Thr 389 [36]. However, in Drosophila, S6
kinase is required for induction of autophagy in response to starvation or
genetic manipulation of the insulin-signaling pathway [28]. These data suggest
that under some conditions, mTOR and S6K can oppose each other or that S6K may
be activated in an mTOR independent manner. Consistent with the work on Drosophila S6K, our data support a positive role for S6K1 in autophagic induction in
mammals, and raise important questions about how this could occur. It is
reasonable to envisage the existence of a novel substrate of mammalian S6
kinase that is required for initiation or maturation of autophagic vesicles,
which is targeted only in the absence of mTOR activity, and that this target's
phosphorylation may be required for autophagy to proceed. This mechanism would
provide a dual switch for the initiation of autophagy, facilitating tighter
control of a process that has both positive and negative implications for the
cell.
It is interesting to consider why resveratrol
treatment might have an inhibitory effect on starvation-induced autophagy, yet
stimulate the inappropriate induction of autophagy in nutrient rich media. One
possibility is that insulin signaling is the key difference. Under nutrient withdrawal,
where insulin signaling is minimal, inhibition of S6K1, leading to a reduction
in autophagy, might be the dominant effect of resveratrol. On the other hand,
when autophagy is held in check by robust signaling through
insulin-PI3K-Akt-mTOR (fed conditions), disrupting this pathway might lead to
the induction of autophagy over time. It will be interesting to test the effects
of resveratrol on autophagy in animals, especially under starvation or tumor
models, where we might observe a similar duality of function.
Negative regulation of homologs of S6 kinase in lower
organisms promotes beneficial effects on health and lifespan. In yeast,
deletion of Sch9, the homolog of mammalian S6K/Akt, protects against
age-dependent defects in a yeast model of aging and cancer and extends
chronological lifespan [37-39]. In Drosophila it has previously been
shown that expression of a dominant negative S6K can extend lifespan [40] and
results in increased resistance to oxidative stress [41]. Resveratrol's effects
on lifespan and resistance to oxidative stress are well established; therefore,
it would be exciting if some of these effects are due to suppression of S6K
activity.
Of considerable interest is the fact that
resveratrol-treated mice exhibit most of the phenotypes of S6K1-/- mice when
fed a high fat diet [12,13,42]. In comparison to control animals, both
resveratrol treated and S6K1-/- mice have significantly less body fat and their peripheral
tissues remain highly sensitive to insulin. Moreover, mitochondrial number and activity
are increased in both S6K1-/- and resveratrol-treated mice. It is therefore
interesting to speculate that resveratrol's ability to modulate S6K1 activity
might be responsible for at least some of the therapeutic effects observed in
recent studies.
A major unanswered question is how resveratrol can
mediate protection from a diverse range of disease processes such as cancer,
neurodegenerative disease, and liver disease. One possible explanation proposed
by Howitz and Sinclair to explain these effects is the Xenohormesis Hypothesis
[8,43]. The theory proposes that organisms have adapted to sense
stress-induced molecules produced by other species in their environment, and
use these cues to induce a protective response in preparation for adversity.
This may explain why many stress-induced phytochemicals, such as resveratrol,
quercetin, and pterostilbene are beneficial for health and seem to act through
multiple pathways [9]. In the case of resveratrol, this includes SIRT1 activation,
inhibition of cyclooxygenase and NFκB, induction of antioxidant enzymes,
and activation of AMPK in vivo [13,44], in addition to the inhibition
of the leukemia-related kinases and S6K1 reported here (Supplementary Table 1).
Taken together, these results suggest that the
requirement for S6 kinase in autophagy is evolutionarily conserved from flies
to mammals. Our observation that S6K1 is necessary to achieve full induction of
autophagy in response to nutrient withdrawal opens the door to future studies
to discover downstream targets of S6K1 that may be important regulators of
autophagy. Furthermore, we establish S6 kinase as a novel target for
resveratrol action which may play an important role in mediating its beneficial
effects on disease processes and aging in diverse organisms.
Methods
Cell lines, lysates, and antibodies.
HEK293, HEK293T, U2OS, HeLa, NIH/3T3, and mouse embryonic
fibroblast cells were maintained in DMEM (GIBCO, Carlsbad, CA) +10% FBS (Gemini
Bio-Products, West Sacramento, CA) + 100 units/ml penicillin/100 μg/ml
streptomycin (GIBCO, Carlsbad, CA) +2mM glutamine (GIBCO, Carlsbad, CA) at 37
ºC +5% CO2. SIRT1+/+ and SIRT1-/- MEFs were a gift from K. Chua, R. Mostoslavsky, and F.
Alt (Harvard Medical School, Boston, MA). HEK293 cells stably expressing
GFP-LC3 were maintained in media supplemented with 2 mg/ml puromycin
(InvivoGen, San Diego, CA). For S6 phosphorylation experiments, cells were
grown overnight and were subjected to various doses of resveratrol (LALILAB
Inc, Durham, NC) or DMSO control for the indicated times. Cells were
subsequently washed once in PBS and lysed in ice cold PBS +0.5% NP-40 (Sigma
Aldrich, St. Louis, MO) + Complete Mini EDTA-free protease inhibitor tablet
(Roche, Basel, Switzerland) + 1:100 phosphatase inhibitor cocktail 1 (Sigma
Aldrich, St. Louis, MO). Following normalization of protein by Bradford assay
(Bio-Rad, Hercules, CA), samples were resolved by SDS-PAGE and western blotted
with the indicated antibodies. Antibodies for p70 S6 kinase, Phospho-S6 ribosomal
protein (Ser240/244), S6 ribosomal protein, and total Erk antibodies were
obtained from Cell Signaling Technology (Beverley, MA). Polyclonal HA antibody
was obtained from Sigma Aldrich (St. Louis, MO). Monoclonal β-tubulin
antibody was obtained from Upstate (Lake Placid, NY). Monoclonal antibody for
GAPDH was obtained from Abcam (Cambridge, MA). Anti-LC3 peptide-based
polyclonal antibody was a gift from J. Brugge (Harvard Medical School, Boston,
MA).
Plasmids, RNAi, and virus infection.
The plasmids pBABE HA-S6K1 K100R, pRK7 HA-S6K1 wt,
pRK7 HA-S6K1 K100R, and pGEX-GST-S6 were gifts from J. Blenis (Harvard Medical School,
Boston, MA). Lentiviral based shRNAs directed against S6K1 were obtained from
B. Hahn and The RNAi Consortium (Harvard Medical School/Broad Institute).
pBABE-GFP-LC3 was a gift from J. Debnath (Harvard Medical School, Boston, MA).
RISC-free control siRNA and SIRT1 specific siGENOME siRNA (D-003540-05) (Dharmacon,
Chicago, IL) were transfected with Lipofectamine RNAiMAX (Invitrogen, Carlsbad,
CA) according to manufacturer's specifications. The shRNA sequence used for
human S6K1 was (AGCACAGCAA ATCCTCAGACA). Retrovirus and lentivirus were generated
by transient transfection of HEK293T cells with packaging plasmids and the
target plasmid using polyethyleneimine (PEI). Virus was harvested 48 and 72
hours post-transfection. For disruption of S6K1 function in GFP-LC3 expressing
HEK293 cells, cells were infected with retrovirus encoding S6K1 K100R or
lentivirus encoding an shRNA directed against S6K1. Cells were assayed for
autophagy and protein expression three days after infection.
Autophagy assays.
For autophagy studies, GFP-LC3 expressing cells were plated overnight
on coverslips. In the case of HEK293 cells glass coverslips were precoated
with a solution of 0.1 mg/ml poly-ornithine (Sigma Aldrich, St. Louis, MO) to
aid in attachment. Cells were washed twice with PBS and placed in either growth
medium or starvation medium, Earle's Balanced Salt Solution (Sigma Aldrich, St.
Louis, MO) for the indicated times. In resveratrol experiments, cells were
pretreated for 1 hour with either DMSO or resveratrol and then placed in
starvation media or treated with 200 nM rapamycin (Calbiochem, San Diego, CA) ±
resveratrol (where indicated). The dose of resveratrol used to assay autophagy
was 50 μM unless otherwise indicated. Following treatment cells were fixed
in 3.7% paraformaldehyde (Sigma Aldrich, St. Louis, MO) and mounted with
GEL/MOUNT (Biomedia corp., Foster City, CA). Cells were then visualized on a
Zeiss Axiovert with a 63X oil immersion lens and digital photomicrographs were
captured with a CCD camera. To quantify GFP-LC3 punctae, at least 4 random
fields were imaged and the average number of punctae/cell was calculated. Data
sets were compared using Student's t- Test (two-tailed assuming equal
variance). For autophagosome staining of SIRT1-/- or SIRT1+/+ MEFs, U2OS,
or HeLa, the cells were treated as described for GFP-LC3 samples and then subjected
to 30 μM monodansylcadaverine (Sigma Aldrich, St. Louis, MO) in the media
for 10 minutes at 37ºC +5% CO2 and then fixed with 3.7% paraformaldehyde and
visualized as described. For the LC3 flux assay, HEK293 GFP-LC3 cells
pretreated with 50 μM resveratrol for 1 hour followed by treatment with
either DMSO or 1 mM rapamycin (Calbiochem, San Diego, CA) in the presence or
absence of 100 nM bafilomycin A1 (Sigma Aldrich, St. Louis, MO). Cells were
washed once in 4ºC PBS and immediately lysed in 2X Lammeli sample dye and then
boiled for 10 minutes. Samples we subsequently run on a 15% polyacrylamide gel
and western blotted for LC3 and tubulin. LC3-II/tubulin ratios we quantified
with ImageJ (NIH).
Kinase profile and kinase assay.
A Kinase profile against 100 kinases listed for
resveratrol at 20 μM was generated utilizing Upstate's KinaseProfiler™
service. Assay protocols for each kinase are available from Upstate (Lake
Placid, NY). In vitro kinase assay was performed as described previously
[45]. Briefly, HEK293 cells were transfected with HA-S6K1 wt, HA-S6K1 K100R, or
vector as indicated. Cells were treated with 1 μg/ml insulin for 10
minutes prior to harvesting in PBS + 0.5% NP-40 + protease and phosphatase
inhibitors. 125 μg of lysate per sample was immunoprecipitated using 0.5
μg/sample of anti-HA High-Affinity (Roche, Basel Switzerland). Beads from
immunoprecipitations were washed three times in lysis buffer and once in kinase
buffer, and kinase assays were performed with recombinant GST-S6 as substrate
(1 μg per assay). In the indicated lanes varying doses of resveratrol or
DMSO control were added to the reaction prior to addition of the GST-S6
substrate. All samples were subjected to SDS-PAGE, and 32P incorporation was
quantified by using a Bio-Rad (Hercules, CA) Phosphor-Imager and subsequent
analysis with ImageJ (NIH).
Supplementary Materials
Resveratrol inhibits nutrient-starvation induced autophagy in multiple cell lines. (A) MDC staining of U2OS cells subjected to
nutrient limitation (Starved) ± 50 μM resveratrol (Res)
for 4 hours. (B) MDC staining of HeLa cells subjected to
nutrient limitation (Starved) ± 50 μM resveratrol (Res)
for 4 hours. An expansion of the area in the white box in
the far right panels is displayed for clarity.
Long-term resveratrol treatment induces autophagy in rich media. (A) HEK293
GFP-LC3 expressing cells incubated in complete media plus
serum were subjected to 50 or 100 μM resveratrol for 24
hours. (B) HEK293 GFP-LC3 cells were treated with EBSS
(Starved) ±50 μM resveratrol (Res) for punctae comparison.
Resveratrol mediated inhibition of autophagy is independent of SIRT1. (A) MDC staining of
wild-type (SIRT1+/+) MEFs subjected to nutrient limitation
(Starved) ± 200 μM resveratrol (Res) for 4 hours. (B) MDC
staining of SIRT1-/- MEFs subjected to EBSS (Starved) ± 200
μM Res for 4 hours.
Immunoprecipitated S6K1 phosphory-lates GST-S6 in vitro. HEK293 cells transfected
with vector control (vec), kinase dead (K100R), or wild-type
(WT) S6K1, were immunoprecipitated for HA-tagged S6K1, which
was subsequently used to phosphorylate full-length GST-S6
ribosomal protein. Top panel is an HA western blot (WB).
Middle panel is an autoradiogram indicating phosphorylated
GST-S6. The bottom panel is a coomassie stained gel indicating
the total GST-S6 in each lane. The black line indicates where
the gel was cropped to include only the positive and negative
controls for simplicity.
Acknowledgments
We would like to thank Dr. Bill Hahn and
the RNAi Consortium (Broad Institute) for providing lentiviral shRNA
constructs. We are grateful to K. Chua, R. Mostoslavsky, and F. Alt for providing
SIRT1-/- MEFs. DAS, JAB, and SMA were supported by grants from the NIH/NIA, P01
AG027916, R01 AG028730, R01 GM068072, The Ellison Medical Foundation, and the
Paul F. Glenn Foundation for Medical Research. JAB was supported by a grant
from the American Heart Association. SMT, SNH, and ABL are supported by a grant
from the American Cancer Society RSG CSM-107290. Thanks to members of Junying
Yuan's lab for advice and reagents.
Conflicts of Interest
David Sinclair is a consultant for Sirtris Pharmaceuticals,
a GSK company.
References
-
1.
Levine
B
and Klionsky
DJ.
Development by self-digestion: molecular mechanisms and biological functions of autophagy.
Dev Cell.
2004;
6:
463
-477.
[PubMed]
.
-
2.
Reggiori
F
and Klionsky
DJ.
Autophagy in the eukaryotic cell.
Eukaryot Cell.
2002;
1:
11
-21.
[PubMed]
.
-
3.
Lum
JJ
, DeBerardinis
RJ
and Thompson
CB.
Autophagy in metazoans: cell survival in the land of plenty.
Nat Rev Mol Cell Biol.
2005;
6:
439
-448.
[PubMed]
.
-
4.
Tsujimoto
Y
and Shimizu
S.
Another way to die: autophagic programmed cell death.
Cell Death Differ.
2005;
12 Suppl 2:
1528
-1534.
[PubMed]
.
-
5.
Shintani
T
and Klionsky
DJ.
Autophagy in health and disease: a double-edged sword.
Science.
2004;
306:
990
-995.
[PubMed]
.
-
6.
Rubinsztein
DC
, Gestwicki
JE
, Murphy
LO
and Klionsky
DJ.
Potential therapeutic applications of autophagy.
Nat Rev Drug Discov.
2007;
6:
304
-312.
[PubMed]
.
-
7.
Wood
JG
, Rogina
B
, Lavu
S
, Howitz
K
, Helfand
SL
, Tatar
M
and Sinclair
D.
Sirtuin activators mimic caloric restriction and delay ageing in metazoans.
Nature.
2004;
430:
686
-689.
[PubMed]
.
-
8.
Howitz
KT
, Bitterman
KJ
, Cohen
HY
, Lamming
DW
, Lavu
S
, Wood
JG
, Zipkin
RE
, Chung
P
, Kisielewski
A
, Zhang
LL
, Scherer
B
and Sinclair
DA.
Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan.
Nature.
2003;
425:
191
-196.
[PubMed]
.
-
9.
Baur
JA
and Sinclair
DA.
Therapeutic potential of resveratrol: the in vivo evidence.
Nat Rev Drug Discov.
2006;
5:
493
-506.
[PubMed]
.
-
10.
Anekonda
TS
Resveratrol--a boon for treating Alzheimer's disease.
Brain Res Rev.
2006;
52:
316
-326.
[PubMed]
.
-
11.
Jang
M
, Cai
L
, Udeani
GO
, Slowing
KV
, Thomas
CF
, Beecher
CW
, Fong
HH
, Farnsworth
NR
, Kinghorn
AD
, Mehta
RG
, Moon
RC
and Pezzuto
JM.
Cancer chemopreventive activity of resveratrol, a natural product derived from grapes.
Science.
1997;
275:
218
-220.
[PubMed]
.
-
12.
Lagouge
M
Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha.
Cell.
2006;
127:
1109
-1122.
[PubMed]
.
-
13.
Baur
JA
Resveratrol improves health and survival of mice on a high-calorie diet.
Nature.
2006;
444:
337
-442.
[PubMed]
.
-
14.
Parker
JA
, Arango
M
, Abderrahmane
S
, Lambert
E
, Tourette
C
, Catoire
H
and Neri
C.
Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons.
Nat Genet.
2005;
37:
349
-350.
[PubMed]
.
-
15.
Kueck
A
, Opipari
AW Jr
, Griffith
KA
, Tan
L
, Choi
M
, Huang
J
, Wahl
H
and Liu
JR.
Resveratrol inhibits glucose metabolism in human ovarian cancer cells.
Gynecol Oncol.
2007;
107:
450
-457.
[PubMed]
.
-
16.
Ohshiro
K
, Rayala
SK
, Kondo
S
, Gaur
A
, Vadlamudi
RK
, El-Naggar
AK
and Kumar
R.
Identifying the estrogen receptor coactivator PELP1 in autophagosomes.
Cancer Res.
2007;
67:
8164
-8171.
[PubMed]
.
-
17.
Ohshiro
K
, Rayala
SK
, El-Naggar
AK
and Kumar
R.
Delivery of cytoplasmic proteins to autophagosomes.
Autophagy.
2008;
4:
104
-106.
[PubMed]
.
-
18.
Opipari
AW Jr
, Tan
L
, Boitano
AE
, Sorenson
DR
, Aurora
A
and Liu
JR.
Resveratrol induced autophagocytosis in ovarian cancer cells.
Cancer Res.
2004;
64:
696
-703.
[PubMed]
.
-
19.
Kabeya
Y
, Mizushima
N
, Ueno
T
, Yamamoto
A
, Kirisako
T
, Noda
T
, Kominami
E
, Ohsumi
Y
and Yoshimori
T.
LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing.
Embo J.
2000;
19:
5720
-5728.
[PubMed]
.
-
20.
Biederbick
A
, Kern
HF
and Elsasser
HP.
Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles.
Eur J Cell Biol.
1995;
66:
3
-14.
[PubMed]
.
-
21.
Noda
T
and Ohsumi
Y.
Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast.
J Biol Chem.
1998;
273:
3963
-3966.
[PubMed]
.
-
22.
Meijer
AJ
and Codogno
P.
Regulation and role of autophagy in mammalian cells.
Int J Biochem Cell Biol.
2004;
36:
2445
-2462.
[PubMed]
.
-
23.
Kissova
I
, Deffieu
M
, Samokhvalov
V
, Velours
G
, Bessoule
JJ
, Manon
S
and Camougrand
N.
Lipid oxidation and autophagy in yeast.
Free Radic Biol Med.
2006;
41:
1655
-1661.
[PubMed]
.
-
24.
Lee
IH
, Cao
L
, Mostoslavsky
R
, Lombard
DB
, Liu
J
, Bruns
NE
, Tsokos
M
, Alt
FW
and Finkel
T.
A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy.
Proc Natl Acad Sci U S A.
2008;
105:
3374
-3379.
[PubMed]
.
-
25.
Yu
R
, Hebbar
V
, Kim
DW
, Mandlekar
S
, Pezzuto
JM
and Kong
AN.
Resveratrol inhibits phorbol ester and UV-induced activator protein 1 activation by interfering with mitogenactivated protein kinase pathways.
Mol Pharmacol.
2001;
60:
217
-224.
[PubMed]
.
-
26.
Matter
WF
, Brown
RF
and Vlahos
CJ.
The inhibition of phosphatidylinositol 3-kinase by quercetin and analogs.
Biochem Biophys Res Commun.
1992;
186:
624
-631.
[PubMed]
.
-
27.
Yu
L
, Alva
A
, Su
H
, Dutt
P
, Freundt
E
, Welsh
S
, Baehrecke
EH
and Lenardo
MJ.
Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8.
Science.
2004;
304:
1500
-1502.
[PubMed]
.
-
28.
Scott
RC
, Schuldiner
O
and Neufeld
TP.
Role and regulation of starvation-induced autophagy in the Drosophila fat body.
Dev Cell.
2004;
7:
167
-178.
[PubMed]
.
-
29.
Mieulet
V
, Roceri
M
, Espeillac
C
, Sotiropoulos
A
, Ohanna
M
, Oorschot
V
, Klumperman
J
, Sandri
M
and Pende
M.
S6 kinase inactivation impairs growth and translational target phosphorylation in muscle cells maintaining proper regulation of protein turnover.
Am J Physiol Cell Physiol.
2007;
293:
C712
-722.
[PubMed]
.
-
30.
Pende
M
, Um
SH
, Mieulet
V
, Sticker
M
, Goss
VL
, Mestan
J
, Mueller
M
, Fumagalli
S
, Kozma
SC
and Thomas
G.
S6K1(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycinsensitive 5'-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway.
Mol Cell Biol.
2004;
24:
3112
-3124.
[PubMed]
.
-
31.
Kaeberlein
M
, McVey
M
and Guarente
L.
The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms.
Genes Dev.
1999;
13:
2570
-2580.
[PubMed]
.
-
32.
Rogina
B
and Helfand
SL.
Sir2 mediates longevity in the fly through a pathway related to calorie restriction.
Proc Natl Acad Sci U S A.
2004;
101:
15998
-16003.
[PubMed]
.
-
33.
Tissenbaum
HA
and Guarente
L.
Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans.
Nature.
2001;
410:
227
-230.
[PubMed]
.
-
34.
Billington
RA
, Genazzani
AA
, Travelli
C
and Condorelli
F.
NAD depletion by FK866 induces autophagy.
Autophagy.
2008;
4:
385
-387.
[PubMed]
.
-
35.
Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes.
J Biol Chem.
1995;
270:
2320
-2326.
[PubMed]
.
-
36.
Lee
SB
, Kim
S
, Lee
J
, Park
J
, Lee
G
, Kim
Y
, Kim
JM
and Chung
J.
ATG1, an autophagy regulator, inhibits cell growth by negatively regulating S6 kinase.
EMBO Rep.
2007;
8:
360
-365.
[PubMed]
.
-
37.
Wei
M
, Fabrizio
P
, Hu
J
, Ge
H
, Cheng
C
, Li
L
and Longo
VD.
Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9.
PLoS Genet.
2008;
4:
e13
[PubMed]
.
-
38.
Madia
F
, Gattazzo
C
, Wei
M
, Fabrizio
P
, Burhans
WC
, Weinberger
M
, Galbani
A
, Smith
JR
, Nguyen
C
, Huey
S
, Comai
L
and Longo
VD.
Longevity mutation in SCH9 prevents recombination errors and premature genomic instability in a Werner/Bloom model system.
J Cell Biol.
2008;
180:
67
-81.
[PubMed]
.
-
39.
Longo
VD
The Ras and Sch9 pathways regulate stress resistance and longevity.
Exp Gerontol.
2003;
38:
807
-811.
[PubMed]
.
-
40.
Kapahi
P
, Zid
BM
, Harper
T
, Koslover
D
, Sapin
V
and Benzer
S.
Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway.
Curr Biol.
2004;
14:
885
-890.
[PubMed]
.
-
41.
Patel
PH
and Tamanoi
F.
Increased Rheb-TOR signaling enhances sensitivity of the whole organism to oxidative stress.
J Cell Sci.
2006;
119:
4285
-4292.
[PubMed]
.
-
42.
Um
SH
, Frigerio
F
, Watanabe
M
, Picard
F
, Joaquin
M
, Sticker
M
, Fumagalli
S
, Allegrini
PR
, Kozma
SC
, Auwerx
J
and Thomas
G.
Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity.
Nature.
2004;
431:
200
-5.
[PubMed]
.
-
43.
Lamming
DW
, Wood
JG
and Sinclair
DA.
Small molecules that regulate lifespan: evidence for xenohormesis.
Mol Microbiol.
2004;
53:
1003
-1009.
[PubMed]
.
-
44.
Dasgupta
B
and Milbrandt
J.
Resveratrol stimulates AMP kinase activity in neurons.
Proc Natl Acad Sci U S A.
2007;
104:
7217
-7222.
[PubMed]
.
-
45.
Roux
PP
, Ballif
BA
, Anjum
R
, Gygi
SP
and Blenis
J.
Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase.
Proc Natl Acad Sci U S A.
2004;
101:
13489
-13494.
[PubMed]
.