



Introduction
Aging of multicellular and unicellular eukaryotic organisms is a multifactorial biological phenomenon that has various causes and affects a plethora of cellular activities [1]. These numerous activities are modulated by only a few nutrient- and energy-sensing signaling pathways that are conserved across phyla and include the insulin/insulin-like growth factor 1 (IGF-1), AMP-activated protein kinase/target of rapamycin (AMPK/ TOR) and cAMP/protein kinase A (cAMP/PKA) pathways [2-5]. By sharing a compendium of protein kinases and adaptor proteins, the insulin/IGF-1, AMPK/TOR and cAMP/PKA pathways in yeast, worms, fruit flies and mammals converge into a network regulating longevity [2-4],6,7]. This network may also include several proteins that currently are not viewed as being in any of these three pathways [2,3,8,9]. Moreover, this network responds to the age-related partial mitochondrial dysfunction and is modulated by mitochondrially produced reactive oxygen species (ROS) [3,8,10,11]. By sensing the nutritional status of the whole organism as well as the intracellular nutrient and energy status, functional state of mitochondria, and concentration of ROS produced in mitochondria, the longevity network regulates life span across species by coordinating information flow along its convergent, divergent and multiply branched signaling pathways.
By defining the organismal and intracellular nutrient and energy status, nutrient intake plays an important role in modulating life span and influences age-related pathologies [12,13]. Two dietary regimens are known to have the most profound life-extending effects across species and to improve overall health by delaying the onset of age-related diseases. They include: 1) caloric restriction (CR), a diet in which only calorie intake is reduced but the supply of amino acids, vitamins and other nutrients is not compromised [13-15]; and 2) dietary restriction (DR), in which the intake of nutrients (but not necessarily of calories) is reduced by limiting food supply without causing malnutrition [16-18]. In a "TOR-centric" view of longevity regulation, TOR alone governs the life-extending and health-improving effects of CR/DR by: 1) integrating the flow of information on the organismal and intracellular nutrient and energy status from the protein kinases AMPK, PKA, PKB/AKT (the insulin/IGF-1 pathway) and ERK1/2 (the PKA-inhibited Raf/MEK/ERK protein kinase cascade) as well as from the mitochondrial redox protein P66Shc; 2) sensing the intracellular levels of amino acids in an AMPK-independent manner; and 3) operating as a control center which, based on the information it has gathered and processed, modulates many longevity-related processes in a sirtuin-independent fashion [19-21]. The inability of CR to increase the replicative life span (RLS) of yeast mutants lacking components of the TOR pathway [22] and the lack of the beneficial effect of DR on life span in worms with reduced TOR signaling [23,24] support the proposed central role for TOR in orchestrating the life-extending effect of CR/DR in these two longevity paradigms. Moreover, while the postulated by the TOR-centric model dispensability of sirtuins for the longevity benefit associated with DR has been confirmed in worms [24], the importance of the sirtuin Sir2p in mediating the life-extending effect of CR in replicatively aging yeast is debated [22,25-[27]. Noteworthy, while TOR is a central regulator of the life-extending effect of CR in replicatively aging yeast, the longevity benefit associated with CR in chronologically aging yeast is mediated by a signaling network that includes: 1) the TOR and cAMP/PKA pathways converged on Rim15p, which therefore acts as a nutritional integrator; and 2) some other, currently unknown pathways that are not centered on Rim15p [6]. Considering the likely convergence of the insulin/IGF-1, AMPK/TOR and cAMP/PKA signaling pathways into a network regulating longevity in worms, fruit flies and mammals (see above), it is conceivable that - akin to TOR - the insulin/IGF-1 and cAMP/PKA pathways may contribute to the beneficial effect of CR/DR on their longevity. Although some findings in worms, fruit flies and mammals support the involvement of the insulin/IGF-1 pathway in the longevity benefit associated with CR/DR, other data imply that such benefit is independent of insulin/IGF-1 (reviewed by Narasimhan et al. [3]). The role of cAMP/PKA signaling in the life-extending effect of CR/DR in these multicellular eukaryotes remains to be tested. Importantly, the recently reported in worms involvement of both independent and overlapping pathways in life span extension by different DR regimens [28] supports the notion that the longevity benefit associated with nutrient limitation is mediated by a signaling network that integrates several pathways.
Akin to CR and DR regimens, certain pharmacological interventions can extend longevity across phyla and improve health by beneficially influencing age-related pathologies. Noteworthy, all of the currently known anti-aging compounds increase life span under non-CR or non-DR conditions (Supplementary Table 1). Under such conditions, these compounds have been shown to: 1) provide the longevity and health benefits associated with CR and DR, but without restricting caloric and nutrient intake; and 2) mimic numerous life-extending effects of CR and DR on gene expression pattern, metabolic and physiological processes, and stress response pathways. Therefore, the names "CR mimetics" and "DR mimetics" have been coined for them [29,30]. Importantly, most CR mimetics and DR mimetics target signaling pathways that modulate longevity in response to the organismal and intracellular nutrient and energy status, including the insulin/IGF-1 and AMPK/TOR pathways as well as the sirtuin-governed protein deacetylation module of the longevity signaling network integrating these pathways (Supplementary Table 1). Furthermore, such compounds as resveratrol, metformin and mianserin increase life span only under non-CR or non-DR conditions, but are unable to do so if the supply of calories or nutrients is limited [31-35]. Hence, one could envision that most, if not all, longevity pathways are "adaptable" by nature, i.e., that they modulate longevity only in response to certain changes in the extracellular and intracellular nutrient and energy status of an organism. However, Li+ in worms and rapamycin in fruits flies extend life span even under DR conditions [36,37]. It is likely therefore that some longevity pathways could be "constitutive" or "housekeeping" by nature, i.e., that they: 1) modulate longevity irrespective of the organismal and intra-cellular nutrient and energy status; and 2) do not overlap (or only partially overlap) with the adaptable longevity pathways that are under the stringent control of calorie and/or nutrient availability. The challenge is to identify such housekeeping longevity pathways, perhaps by using a chemical screen for compounds that can extend longevity even under CR/DR conditions. Because under such conditions the adaptable pro-aging pathways are fully suppressed and the adaptable anti-aging pathways are fully activated, a compound that can increase life span is expected to target the housekeeping longevity pathways.
Noteworthy, two anti-aging compounds alter lipid levels in mammals and fruit flies under non-DR conditions. In fact, resveratrol treatment reduces the levels of the neutral lipids triacylglycerols (TAG) and increases free fatty acid (FFA) levels in mouse adipocytes [38]. Furthermore, feeding rapamycin to fruit flies results in elevated TAG levels [37]. Although it remains to be seen if such effects of resveratrol and rapamycin on lipid levels play a casual role in their anti-aging action under non-DR conditions, it should be stressed that lipid metabolism has been shown to be involved in longevity regulation in yeast [39,40], worms [9,41-43], fruit flies [41,44] and mice [38,41,45-48]. We recently proposed a mechanism linking yeast longevity and lipid dynamics in the endoplasmic reticulum (ER), lipid bodies and peroxisomes. In this mechanism, a CR diet extends yeast chronological life span (CLS) by activating FFA oxidation in peroxisomes [39-40]. It is conceivable that the identification of small molecules targeting this mechanism could yield novel anti-aging compounds. Such compounds can be used as research tools for defining the roles for different longevity pathways in modulating lipid metabolism and in integrating lipid dynamics with other longevity-related processes. Furthermore, the availability of such compounds would enable a quest for housekeeping longevity assurance pathways that do not overlap (or only partially overlap) with the adaptable TOR and cAMP/PKA pathways. Moreover, such compounds would have a potential to be used as pharmaceutical agents for increasing life span and promoting healthy aging by delaying the onset of age-related diseases, regardless of an organism's dietary regimen.
We sought to identify small molecules that increase the CLS of yeast under CR conditions by targeting lipid metabolism and modulating housekeeping longevity assurance pathways. Our chemical genetic screen identified lithocholic acid (LCA) as one of such small molecules. We provide evidence that LCA extends longevity of chronologically aging yeast through two different mechanisms. In one mechanism, this bile acid targets - regardless of the number of available calories - housekeeping longevity assurance pathways that do not overlap with the adaptable TOR and cAMP/PKA pathways and modulate a compendium of pro- and anti-aging processes. In the other mechanism, LCA targets the adaptable cAMP/PKA pathway under non-CR conditions by unmasking the previously unknown anti-aging potential of PKA.
Results
Our rationale for choosing a mutant strain and growth conditions to screen compound libraries for anti-aging small molecules
To perform a chemical genetic screen for small molecules that increase the CLS of yeast by targeting lipid metabolism, we chose the single-gene-deletion mutant strain pex5Δ. Because pex5Δ lacks a cytosolic shuttling receptor for peroxisomal import of Fox1p and Fox2p, these two enzymes of the β-oxidation of FFA reside in the cytosol of pex5Δ cells [49] (Figure 1A). In contrast, the Pex5p-independent peroxisomal import of Fox3p, the third enzyme of the FFA β-oxidation pathway, sorts it to the peroxisome in pex5Δ cells [49]. By spatially separating Fox1p and Fox2p from Fox3p within a cell, the pex5Δ mutation impairs FFA oxidation (Figure 1A). In chronologically aging yeast grown under CR conditions on 0.2% or 0.5% glucose, peroxisomal FFA oxidation regulates longevity by 1) efficiently generating acetyl-CoA to synthesize the bulk of ATP in mitochondria; and 2) acting as a rheostat that modulates the age-related dynamics of FFA and diacylglycerol (DAG), two regulatory lipids known to promote longevity-defining cell death [39,40,50]. Unlike CR yeast, chronologically aging non-CR yeast grown on 1% or 2% glucose are unable to generate significant quantities of ATP by oxidizing peroxisome-derived acetyl-CoA in mitochondria and, instead, produce the bulk of ATP via glycolytic oxidation of glycogen- and trehalose-derived glucose [39,40]. Consistent with the essential role of peroxisomal FFA oxidation as a longevity assurance process only under CR, the pex5Δ mutation substantially shortened the CLS of CR yeast but caused a significantly lower reduction of longevity in non-CR yeast, especially in yeast grown on 2% glucose (Figures 1B to F).
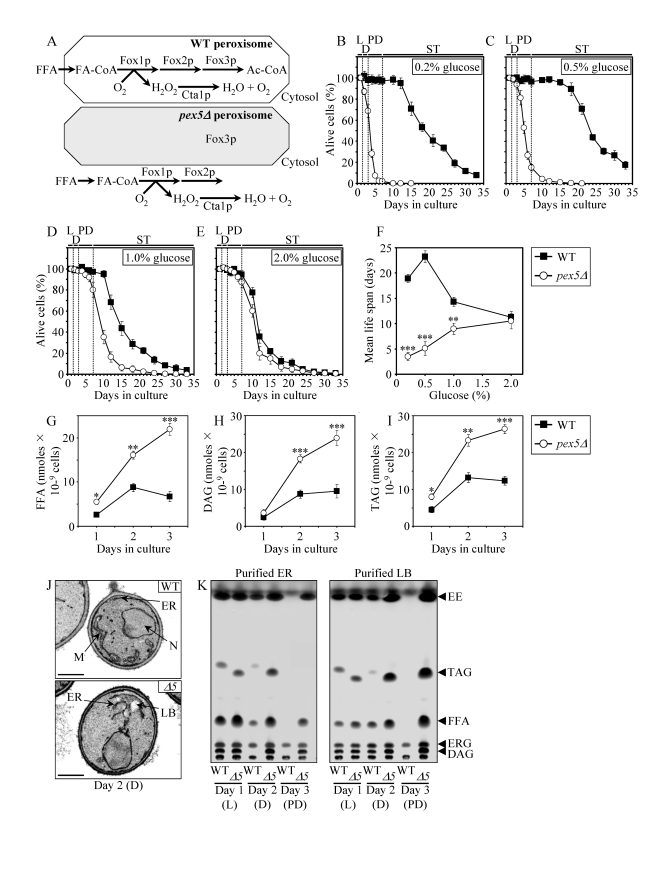
Figure 1. The
pex5Δ mutation shortens chronological life span (CLS), alters cell morphology and remodels lipid metabolism
in CR yeast. (A) Outline of subcellular
localization of the Fox1p, Fox2p and Fox3p enzymes of fatty acid
ß-oxidation in WT and pex5Δ cells. (B - F)
Survival and the mean life spans of chronologically aging WT and pex5Δ
yeast cultured in medium initially containing 0.2%, 0.5%, 1% or 2%
glucose. Data are presented as means ± SEM (n = 16-38; ***p < 0.001; **p
< 0.01). (G - I) Levels of free fatty acids
(FFA), diacylglycerols (DAG) and triacylglycerols (TAG) in WT and pex5Δ
cells grown on 0.2% glucose and taken for analyses at the indicated
time-points. FFA and TAG were measured by quantitative mass spectrometry.
The levels of DAG were quantitated by densitometric analysis of TLC plates.
Data are presented as means ± SEM (n = 3-8; ***p < 0.001; **p < 0.01;
*p < 0.05). (J and K)
Transmission electron micrographs (J) and spectra of lipids
extracted from purified endoplasmic reticulum (ER) and lipid bodies (LB)
and analyzed by TLC (K) for WT and pex5Δ (Δ5)
yeast grown on 0.2% glucose and taken for analyses at the
indicated time-points. Abbreviations: Cta1p, peroxisomal catalase; D,
diauxic growth phase; EE, ethyl esters; ERG, ergosterol; FA-CoA, CoA esters
of fatty acids; L, logarithmic growth phase; M, mitochondrion; N, nucleus;
PD, post-diauxic growth phase; ST, stationary growth phase.
In chronologically aging CR yeast, peroxisomal FFA oxidation modulates, perhaps via several negative feedback loops, the following three processes: 1) the ER-confined biosynthesis of TAG from FFA and DAG; 2) the subsequent deposition of TAG, the major neutral lipid reserves, in lipid bodies; and 3) the consequent lipolysis of deposited TAG and the resulting formation of FFA and DAG [39,40]. By impairing the ability of peroxisomal FFA oxidation to act as a rheostat that regulates cellular aging by modulating the age-related dynamics of FFA, DAG and TAG in the ER and lipid bodies, the pex5Δ mutation caused the accumulation of the closely apposed ER membranes and ER-originated lipid bodies in CR yeast (Figure 1J). Of note, these morphological features of pex5Δ yeast under CR were similar to those observed in a mouse model for the peroxisome biogenesis disorder Zellweger syndrome with hepatocyte-specific elimination of the PEX5 gene [51]. Furthermore, the pex5Δ mutation increased the concentrations of FFA, DAG and TAG in CR yeast (Figures 1G to I), promoting their buildup in the ER and lipid bodies (Figure 1K). CR yeast carrying the pex5Δ mutation also accumulated the ER-derived and lipid bodies-deposited ergosteryl esters (EE) neutral lipid species (Figure 1K).
Following a short-term exposure to exogenous FFA (palmitoleic acid or oleic acid) or DAG, wild-type (WT) cells grown under CR conditions died (Supplementary Figure 1A). The vast majority of these WT cells displayed propidium iodide (PI) positive staining characteristic of the loss of plasma membrane integrity, a hallmark event of necrotic cell death (Supplementary Figure 1B and 1C). In contrast, only a minor portion of these WT cells displayed Annexin V positive staining used to visualize the externalization of phosphatidylserine, a hallmark event of apoptotic cell death (Supplementary Figure 1B and 1C). Thus, a brief exposure of WT cells grown under CR conditions to exogenous FFA or DAG caused their necrotic, not apoptotic, death. Importantly, we found that the pex5Δ mutation enhances the susceptibility of CR yeast to necrotic death caused by a short-term exposure to exogenous FFA or DAG (Supplementary Figure 1A and 1C), perhaps due to the increased concentrations of endogenous FFA and DAG seen in pex5Δ cells under CR (Figures 1G and H).
In addition to its effect on lipid metabolism and lipid-induced necrotic cell death, the pex5Δ mutation also altered mitochondrial morphology and oxidation-reduction processes in mitochondria of CR yeast. In fact, this mutation caused the fragmentation of a tubular mitochondrial network into individual mitochondria under CR conditions (Figures S2A and S2B). Furthermore, in CR yeast the pex5Δ mutation 1) greatly reduced the rate of oxygen consumption by mitochondria (Supplementary Figure 2C); 2) substantially decreased the mito-chondrial membrane potential (Supplementary Figure 2D); and 3) diminished the level of intracellular ROS (Supplementary Figure 2E), known to be generated mostly as by-products of mitochondrial respiration [10,52]. Interestingly, all these mitochondrial abnormalities in pex5Δ yeast under CR were reminiscent of changes in mitochondrial morpholo-gy and functions seen in mice with hepatocyte-specific elimination of the PEX5 gene, a model for the peroxi-some biogenesis disorder Zellweger syndrome [51].
Besides its profound effect on lipid metabolism, lipid-induced necrosis, mitochondrial morphology and functions, the pex5Δ mutation also 1) reduced the resistance of chronologically aging CR yeast to chronic oxidative, thermal and osmotic stresses (Supplementary Figure 3A); 2) sensitized CR yeast to death that was initiated in response to a short-term exposure to exogenous hydrogen peroxide or acetic acid (Supplementary Figure 3B) and that is known to be caused by mitochondria-controlled apoptosis [53,54]; and 3) elevated the frequencies of deletion and point mutations in mitochondrial and nuclear DNA of CR yeast (Figures S3C to S3E).
The profound changes in cell morphology and physiology, stress resistance, susceptibility to lipid-induced necrosis and mitochondria-controlled apoptosis, and stability of nuclear and mitochondrial DNA seen in pex5Δ yeast under CR conditions coincided with considerable changes in their proteome. Indeed, our mass spectrometry-based quantitative proteomic analysis of proteins recovered in total cell lysates as well as in purified ER and mitochondria revealed that the pex5Δ mutation altered the abundance of many proteins (Figure 2A). Protein species that were depleted or enriched in the total cell lysate, ER and mitochondria of pex5Δ yeast grown under CR conditions included proteins involved in a number of cellular processes (Figure 2B). Importantly, lack of 91 of these proteins increased the CLS of yeast under CR (Figure 2B), suggesting their essential pro-aging role in longevity regulation when calorie supply is limited. Noteworthy, 58 of the genes encoding these proteins and termed gerontogenes (i.e., the genes whose mutant alleles extend life span; [55]) have not been previously known as being critical for defining the CLS of yeast. The identities of protein species that were depleted or enriched in pex5Δ yeast grown under CR conditions, the extent to which their levels were altered and the names of gerontogenes identified in our functional analysis will be reported elsewhere (Goldberg et al., manuscript in preparation). Importantly, for most of these proteins (with some exceptions, see Figures S4C and S4D) the fold increase or decrease in the level of a protein enriched or depleted in pex5Δ was found to be in good correlation with the fold increase or decrease (respectively) in the mean CLS of a mutant strain lacking it (Figures S4A and S4B).
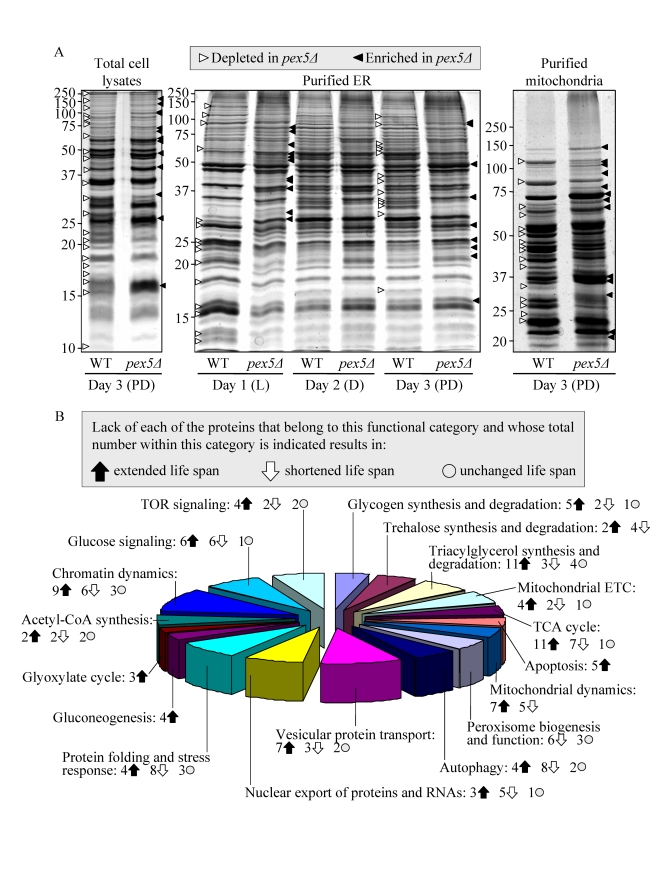
Figure 2. The pex5Δ mutation alters the abundance of many proteins recovered in total cell lysates, purified ER and mitochondria of CR yeast. (A) The spectra of
proteins recovered in total cell lysates, purified ER and mitochondria of
WT and pex5Δ cells that were grown under CR on 0.2% glucose and
taken for analyses at the indicated time-points. (B) Functional
categories of proteins that were enriched or depleted in the total cell
lysate, ER and mitochondria of pex5Δ cells (as compared to WT
cells) under CR conditions. Lack of 91 of these proteins increased the CLS
of yeast under CR, suggesting their essential pro-aging role in longevity
regulation when calorie supply is limited.
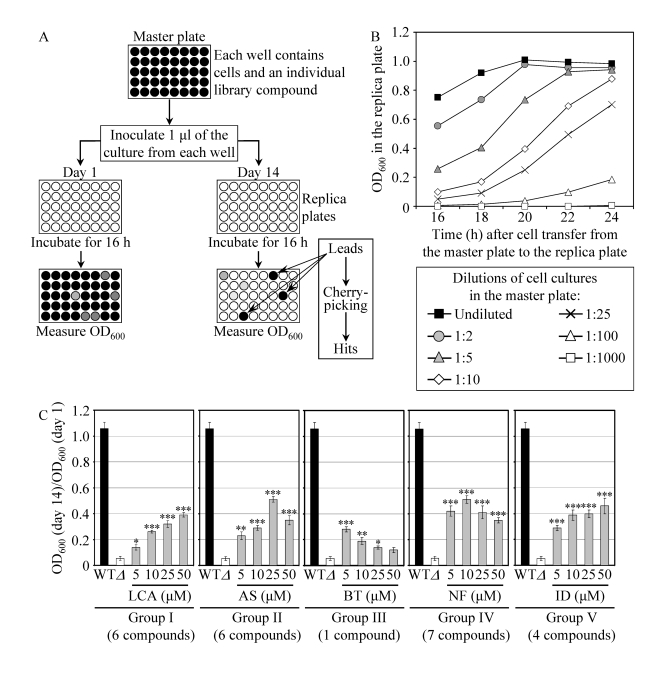
Figure 3. A high-throughput screen of compound libraries for small molecules that extend the CLS of yeast under CR conditions. (A) A microplate assay for
measuring yeast CLS by monitoring optical density at 600 nm (OD600)
was used for screening representative compounds from several commercial
libraries for small molecules that extend the CLS of pex5Δ cells grown
under CR on 0.5% glucose. (B) The OD600 of a cell culture
in the replica microplate following incubation for 16 to 24 hours
correlates with the number of viable cells present in this culture before
it was taken from the master microplate for replica plating. (C) The
effect of various concentrations of the identified anti-aging small
molecules on the CLS of the pex5Δ (Δ) strain under CR
conditions. The "OD600 at day 14/OD600 at day 1"
ratio was used as a measure of CLS. Data are
presented as means ± SEM (n = 3-5; ***p < 0.001; **p < 0.01; *p <
0.05). The anti-aging small molecules LCA, AS, BT, NF and ID belong
to five chemical groups.
Altogether, these findings imply that, by impairing peroxisomal FFA oxidation and affecting lipid metabolism in the ER and lipid bodies, the pex5Δ mutation alters the levels of numerous pro- and anti-aging proteins and impacts many longevity-related processes, thereby shortening the CLS of yeast when calorie supply is limited. We therefore chose the short-lived pex5Δ strain to carry out a chemical genetic screen for anti-aging compounds that target lipid metabolism to extend CLS in yeast placed on a CR diet.
A chemical genetic screen for small molecules that extend the CLS of yeast under CR conditions
To facilitate a high-throughput screen of compound libraries for anti-aging small molecules, we adopted a previously described microplate assay [56] for measuring CLS by monitoring optical density at 600 nm (OD600) (Figure 3A). In our assay, a small aliquot of the pex5Δ culture grown in a nutrient-rich medium containing 0.5% glucose and recovered from mid-logarithmic phase was transferred into each well of a 96-well master microplate containing the same growth medium and a compound from a commercially available library. At days 1, 7, 10 and 14 of the incubation of master microplates, a small aliquot of each culture was transferred into individual wells of a new (replica) microplate containing growth medium only. Following incubation of replica microplates for 16 hours, the OD600 of the culture in each well of the replica microplate was measured. Importantly, we found that under such conditions the OD600 of a cell culture in a well of the replica microplate correlates with the number of viable cells in the corresponding well of the master microplate (Figure 3B). To calculate survival at each time point, the OD600 at a particular time point was divided by the OD600 at day 1. By translating our microplate assay into high-throughput format and screening representative compounds from the NIH Clinical Collection, Prestwick Chemical Inc. and Sigma-LOPAC commercial libraries, we identified "lead" compounds. The subsequent "cherry-picking" analysis of these small molecules revealed "hit" compounds that in our microplate assay reproducibly extended the CLS of pex5Δ. Using the web-based eMolecules searching engine, we identified commercially available structural analogs of the hit compounds and then tested their life-extending efficacy in our microplate assay for measuring the CLS of pex5Δ. By screening the total of approximately 19,000 representative compounds from seven commercial libraries, we identified 24 small molecules that greatly extend the CLS of pex5Δ under CR and belong to 5 chemical groups (Figure 3C). Group I consisted of 6 bile acids, including lithocholic acid (LCA), deoxycholic acid (DCA), chenodeoxycholic acid (CDCA), cholic acid (CA), dehydrocholic acid (DHCA) and hyodeoxycholic acid (HDCA) (Figures 3C and S5). Noteworthy, the anti-aging efficacy of these bile acids correlated with their hydrophobicity. In fact, LCA - the most hydrophobic bile acid species [57] - displayed the highest ability to delay chronological aging of pex5Δ under CR conditions in the microplate assay (Supplementary Figure 5). The identities of small molecules that belong to groups II to V (Figure 3C) of the anti-aging compounds identified in our screen and the structure-activity analysis of their life-extending potential will be reported elsewhere (Goldberg et al., manuscript in preparation).
Noteworthy, none of the small molecules that has been shown to extend CLS (i.e., caffeine, methionine sulfoximine, rapamycin and spermidine; Supplementary Table 1; [56,58,59]) and/or RLS (i.e., rapamycin and resveratrol; Supplementary Table 1; [27,31]) in yeast has been identified in our screen for compounds capable of increasing the CLS of pex5Δ under CR. Furthermore, none of these currently known life-extending molecules is structurally related to the anti-aging compounds that we revealed. Thus, it is likely that LCA and all other novel anti-aging compounds identified in our screen target longevity-related cellular processes that are not modulated by the presently known anti-aging small molecules. Because our screen was aimed at identifying compounds that extend yeast longevity by targeting lipid metabolism, it is conceivable that the age-related dynamics of TAG, FFA and DAG is one of such cellular processes.
Pharmacophore modeling of the anti-aging potential of bile acids
Similar to their effect on pex5Δ, some of the group I anti-aging compounds extended the CLS of WT strain under CR conditions. Specifically, LCA and two other bile acids - DCA and CDCA - increased both the mean and maximum CLS of WT yeast grown under CR on 0.2% glucose (Figures 4A to 4D). Moreover, DHCA increased only the mean CLS of WT yeast under CR at 0.2% glucose, whereas HDCA increased only their maximum CLS (Figures 4A to 4D). Akin to its highest life-extending efficacy in pex5Δ under CR, the most hydrophobic bile acid - LCA [57] - provided WT cells with the greatest longevity benefit when calorie supply was limited. In fact, LCA increased the mean CLS of WT strain under CR at 0.2% glucose by almost 250% and its maximum CLS by more than 200% (Figures 4A to D). Our comparative analysis of the structural differences between various bile acids and their relative life-extending efficacies revealed that the positions 6, 7 and 12 in the six-member rings B and C of the steroid nucleus are important for the anti-aging potential of a bile acid. Indeed, the ability of LCA to extend both the mean and maximum CLS of WT yeast under CR can be: 1) eliminated (with respect to the mean CLS) or greatly reduced (with respect to the maximum CLS) by attaching an α-oriented hydroxyl group at the position 6 (as in HDCA); 2) greatly reduced (with respect to both the mean and maximum CLS) by attaching an α-oriented hydroxyl group at the position 7 (as in CDCA); and 3) greatly reduced (with respect to both the mean and maximum CLS) by attaching an α-oriented hydroxyl group at the position 12 (as in DCA) (Figures 4B to E). All these modifications to the structure of LCA increase polarity of the hydrophilic (concave) side [α-face] of the steroid nucleus by positioning a hydroxyl group below the nucleus and axially to its plane (Figure 4E). Furthermore, the anti-aging potential of LCA can be abolished by attaching a β-oriented hydroxyl group at the position 7 (as in UDCA), thereby conferring polarity to the hydrophobic (convex) side [β-face] of the steroid nucleus by positioning a hydroxyl group above the nucleus and equatorially to its plane (Figures 4B to E).
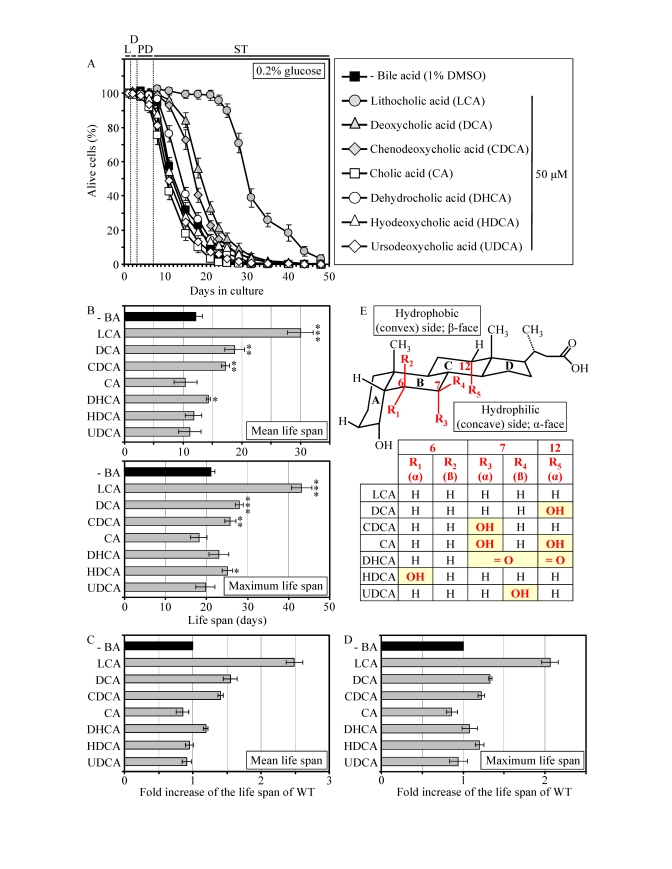
Figure 4. LCA and some other bile acids extend the CLS of WT strain under CR conditions. (A
- D) Effect of various bile acids on survival (A) and on the
mean and maximum life spans (B - D)
of chronologically aging WT strain grown under CR conditions on 0.2%
glucose. Data are presented as means ± SEM (n =
3-28; ***p < 0.001; **p < 0.01; *p < 0.05). (E)
Structure and hydrophilic/hydrophobic properties of bile acids. The R1
(α), R3 (α) and R5 (α) hydroxyl groups at the positions 6, 7
and 12 in the six-member rings B and C of the steroid nucleus
increase polarity of the hydrophilic (concave) side [α-face] of the
nucleus by being located below the nucleus and axially to its plane. The R4
(β) hydroxyl group at the position 7 in the six-member ring B of the
steroid nucleus confers polarity of the hydrophobic (convex) side
[β-face] of the nucleus by being located above the nucleus and
equatorially to its plane.
Moreover, the simultaneous attachments of two α-oriented hydroxyl groups (as in CA) or two keto groups (as in DHCA) at the positions 7 and 12 eliminated the ability of LCA to extend both the mean and maximum CLS of WT yeast under CR (Figures 4B to E). Altogether, the results of our pharmacophore modeling of the anti-aging potential of bile acids imply that the maintenance of the minimal polarity of both the hydrophilic (concave) and hydrophobic (convex) sides of the steroid nucleus - by avoiding the presence of polar substituents at the positions 6, 7 and 12 - is mandatory for the extreme life-extending efficacy of LCA under CR conditions. Such stringent structural requirements are consistent with a target specificity of LCA action as an anti-aging small molecule.
LCA extends the CLS of WT yeast under both CR and non-CR conditions, although to a different extent
If added to growth medium at the time of cell inoculation, LCA increased both the mean and maximum CLS of WT strain not only under CR at 0.2% or 0.5% glucose (Figures 5A, 5B and 5G - 5I) but also under non-CR conditions administered by culturing yeast in medium initially containing 1% or 2% glucose (Figures 5C, 5D and 5G - 5I). At any tested concentration of glucose in growth medium, LCA displayed the greatest beneficial effect on both the mean and maximum CLS of WT strain if used at a final concentration of 50 μM (Figures 5E and 5F). It should be stressed that the life-extending efficacy of 50 μM LCA under CR exceeded that under non-CR conditions, being inversely proportional to the concentration of glucose in growth medium and thus in correlation with the extent of calorie supply limitation (Figures 5G to 5I). Importantly, although 50 μM LCA displayed a profound effect on CLS, it did not cause significant changes in growth of WT strain at any tested concentration of glucose in medium. In fact, both growth rate in logarithmic phase and time prior to entry into stationary (ST) phase were similar for WT cells cultured in medium with or without LCA (Supplementary Figure 6).
LCA extends yeast CLS independent of TOR, by modulating housekeeping longevity assurance pathways
Our chemical genetic screen was aimed at identifying small molecules that can increase the CLS of yeast under CR by modulating housekeeping longevity pathways. Such pathways may regulate yeast longevity irrespective of the number of available calories and may not necessarily overlap (or may only partially overlap) with the adaptable longevity pathways that are under the stringent control of calorie availability. In chronologically aging yeast, the TOR and cAMP/PKA signaling pathways are the two adaptable longevity pathways that govern the life-extending effect of CR (Figure 10A) [5,6,60-62].
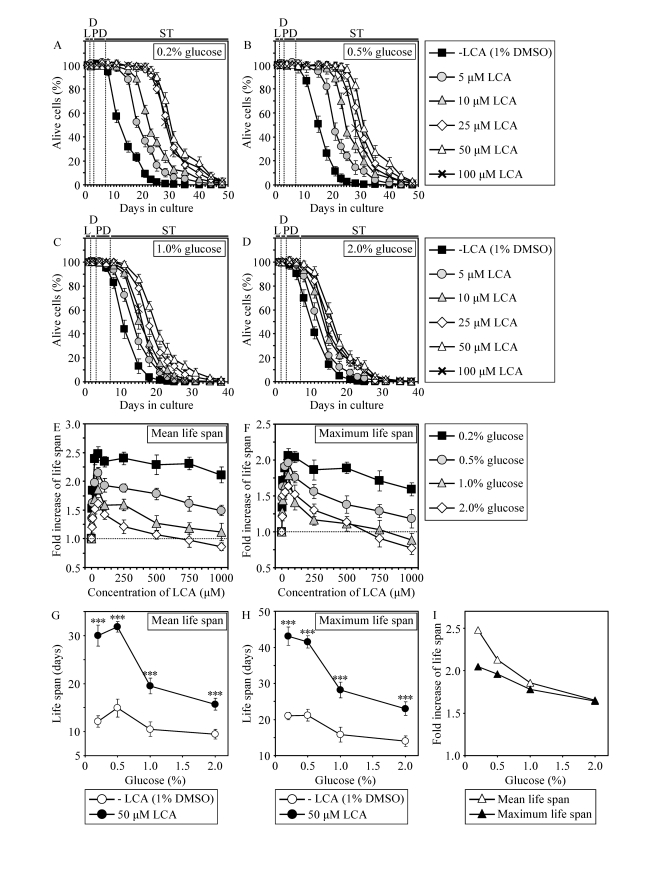
Figure 5. In chronologically aging WT yeast, the life-extending efficacy of LCA under CR exceeds that under non-CR conditions. (A - F)
Effect of various concentrations of LCA on survival (A - D)
and on the fold increase in the mean (E) or
maximum (F) life span of chronologically aging WT strain
cultured in medium initially containing 0.2%, 0.5%, 1% or 2% glucose. Data are presented as means ± SEM (n = 3-28). (G
- I) Effect of 50 μM LCA on the mean or
maximum CLS of WT yeast cultured in
medium initially containing 0.2%, 0.5%, 1% or 2% glucose. Data are presented as means ± SEM (n = 12-28; ***p
< 0.001).
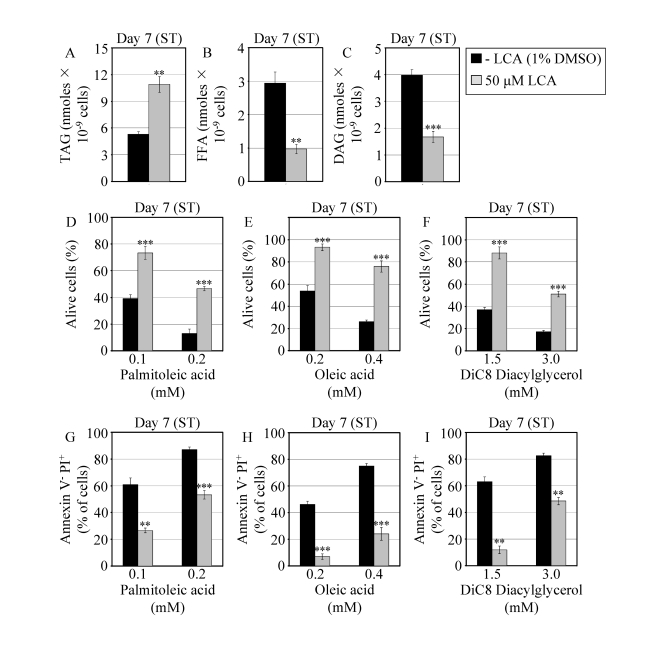
Figure 6. In chronologically aging WT yeast that entered the non-proliferative stationary (ST) phase under CR, LCA alters the levels of lipids and protects cells from lipid-induced necrotic death. (A - C)
Levels of triacylglycerols (TAG) and free fatty acids (FFA) measured by
quantitative mass spectrometry (A and B, respectively) and of
diacylglycerols (DAG) monitored by TLC (C) in WT cells grown in
medium with or without LCA. (D - F) Viability of WT cells
pre-grown in medium with or without LCA and then treated for 2 h with
palmitoleic acid (D), oleic acid (E) or DiC8 diacylglycerol (F).
(G - I) Percent of WT cells (pre-grown in medium with or
without LCA) that following their treatment with palmitoleic acid (G),
oleic acid (H) or DiC8 diacylglycerol (I) displayed Annexin V
negative and PI positive (Annexin V- and PI+)
staining characteristic of necrotic cell death. Data are presented as means
± SEM (n = 3-9; ***p < 0.001; **p < 0.01).
WT cells grown on 0.2% glucose in the presence or absence of LCA were taken
for analyses at day 7, when they reached reproductive maturation by
entering into ST phase.
Reduction of the Tor1p protein kinase activity in yeast placed on a CR diet or exposed to rapamycin prevents inhibitory phosphorylation of Atg13p, a key positive regulator of autophagy, thereby activating this essential anti-aging process (Figure 10A) [63,64]. Under CR conditions or in response to rapamycin, Tor1p is also unable to phosphorylate and activate the nutrient-sensory protein kinase Sch9p [60,65]. The resulting inhibition of the Sch9p kinase activity suppresses its ability to attenuate protein synthesis in mitochondria, thus turning on this essential anti-aging process [61].
Furthermore, by inhibiting the Sch9p kinase activity, CR restrains Sch9p from activating protein synthesis in the cytosol, thereby slowing down this essential pro-aging process [60,62,65]. Moreover, the attenuation of the Sch9p kinase activity in CR yeast prevents the retention of Rim15p in the cytosol, hence allowing this nutrient-sensory protein kinase to enter the nucleus where it orchestrates an anti-aging transcriptional program by activating the stress response transcriptional activators Msn2p, Msn4p and Gis1p [58,62]. The longevity benefit associated with CR in chronologically aging yeast is also due to the attenuation of signaling through the cAMP/PKA pathway, which is driven by glucose deprivation [5,6,62]. By preventing inhibitory phosphorylation of Atg13p, the reduction of the PKA kinase activity in CR yeast results in activation of autophagy (Figure 10A) [63,66]. In addition, by inhibiting the PKA kinase activity, CR suppresses the ability of PKA to activate protein synthesis in the cytosol [62]. Moreover, reduced PKA kinase activity in CR yeast enables nuclear import of Msn2p and Msn4p, thus turning on an anti-aging transcriptional program driven - in a Rim15p-dependent fashion - by these two transcriptional activators [27,62,67]. Noteworthy, the kinase activity of the cytosolic pool of Rim15p is inactivated through PKA-dependent phosphorylation (Figure 10A) [62]. Although some of the Rim15p phosphorylation targets are involved in longevity regulation and reside outside the nucleus [68], a role of such phosphorylation in the life-extending effect of CR in yeast remains to be established.
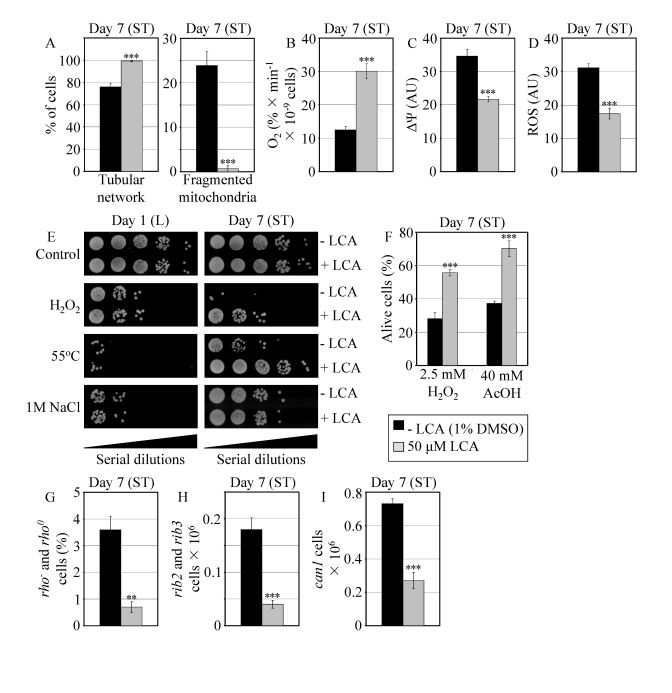
Figure 7. In reproductively mature WT yeast that entered the non-proliferative stationary (ST) phase under CR, LCA modulates mitochondrial morphology and functions, enhances stress resistance, attenuates mitochondria-controlled apoptosis, and increases stability of nuclear and mitochondrial DNA. (A)
Percent of WT cells grown in medium with or without LCA and exhibiting a
tubular mitochondrial network or fragmented mitochondria. Mitochondria were
visualized by indirect immunofluorescence microscopy using monoclonal
anti-porin primary antibodies and Alexa Fluor 568-conjugated goat
anti-mouse IgG secondary antibodies. (B - D) Oxygen
consumption by WT cells grown in medium with or without LCA (B),
their mitochondrial membrane potential ΔΨ (C) and their
ROS levels (D). ΔΨ and ROS were visualized in living cells by
fluorescence microscopy using fluorescent dyes Rhodamine 123 or
Dihydrorhodamine 123, respectively. (E) The resistance of WT cells
pre-grown in medium with or without LCA to chronic oxidative, thermal and
osmotic stresses. (F) Viability of WT cells pre-grown in medium with
or without LCA and then treated for 1 h with hydrogen peroxide or acetic
acid (AcOH) to induce mitochondria-controlled apoptosis. (G- I)
The frequencies of rho- and rho0mutations
in mitochondrial DNA (G), rib2 and rib3 mutations in
mitochondrial DNA (H), and of can1 (Canr)
mutations in nuclear DNA (I) of WT cells grown in medium with or without
LCA. Data in A - D and F - I are presented as
means ± SEM (n = 4-17; ***p < 0.001; **p <
0.01). WT cells grown on 0.2% glucose in the presence or absence of
LCA were taken for analyses at day 7, when they reached reproductive
maturation by entering into ST phase.
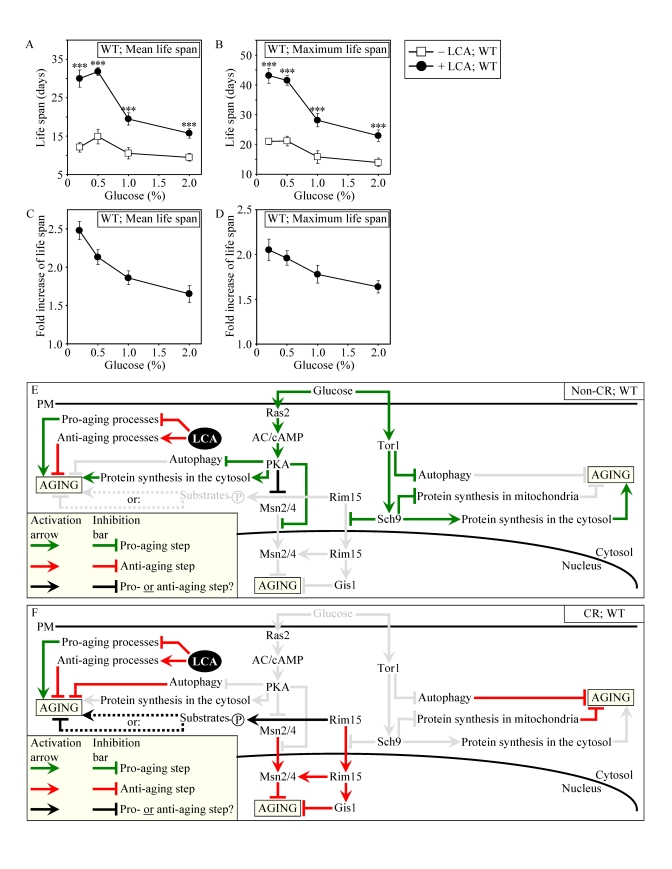
Figure 8. LCA increases the CLS of WT strain to the highest extent under CR conditions. (A and B)
Effect of LCA on the mean (A) and maximum (B) life spans of
chronologically aging WT strain. Data are
presented as means ± SEM (n = 12-28; ***p < 0.001). (C - E) Effect of LCA on the
fold increase in the mean (C) or maximum (D)
life span of chronologically aging WT strain. Data are presented as means ± SEM (n = 12-28). Cells
in A to D were cultured in medium initially containing 0.2%,
0.5%, 1% or 2% glucose in the presence of LCA (50 μM) or in its
absence. Survival data are provided in Supplementary Figure 9. (E
and F) Outline of pro- and anti-aging processes that are controlled
by the TOR and/or cAMP/PKA signaling pathways and are modulated by LCA in
WT cells grown under non-CR (E) or CR (F) conditions.
Activation arrows and inhibition bars denote pro-aging processes (displayed
in green color), anti-aging processes (displayed in red color) or processes
whose role in longevity regulation is presently unknown (displayed in black
color). Doted lines denote hypothetical processes. Abbreviations:
PM, plasma membrane.
Our evaluation of the life-extending efficacy of LCA in WT strain on a high- or low-calorie diet revealed that this compound increased CLS irrespective of the number of available calories (Figures 8A and 8B). Intriguingly, the extent to which LCA extended longevity was highest under CR conditions (Figures 8C and 8D), when the pro-aging processes modulated by the adaptable TOR and cAMP/PKA pathways are suppressed and the anti-aging processes are activated (Figure 8F). The life-extending efficacy of LCA in CR yeast significantly exceeded that in yeast on a high-calorie diet (Figures 8C and 8D), in which the adaptable TOR and cAMP/PKA pathways greatly activate the pro-aging processes and suppress the anti-aging processes (Figure 8E). Altogether, these findings suggest that, consistent with its sought-after effect on a longevity signaling network, LCA mostly targets certain housekeeping longevity assurance pathways that do not overlap (or only partially overlap) with the adaptable TOR and cAMP/PKA pathways modulated by calorie availability (Figures 8E and 8F).
Consistent with our assumption that LCA extends longevity not by modulating the adaptable TOR pathway (Figures 9E and 9F), lack of Tor1p did not impair the life-extending efficacy of LCA under CR (Figures 9A to 9D). Importantly, by eliminating a master regulator of this key adaptable pathway that shortens the CLS of yeast on a high-calorie diet, the tor1Δ mutation abolished the dependence of the anti-aging efficacy of LCA on the number of available calories. In fact, LCA extended longevity of the tor1Δ mutant strain to a very similar degree under CR and non-CR conditions (Figures 9C and 9D).
We next assessed how the adaptable cAMP/PKA pathway influences the life-extending efficacy of LCA in yeast on a high- or low-calorie diet. Although the ras2Δ mutation greatly decreases the PKA protein kinase activity by eliminating a GTP-binding protein that activates adenylate cyclase responsible for the synthesis of the PKA activator cAMP (Figures S7E and S7F) [62], it did not abolish the ability of LCA to extend CLS under CR and non-CR conditions (Figures S7A and S7B). However, the life-extending efficacy of LCA was decreased by the ras2Δ mutation, as compared to that seen in WT cells exposed to this compound (Figures S7C and S7D). In spite of such partial reduction of the anti-aging potential of LCA in ras2Δ, LCA still significantly increased its CLS under CR and non-CR conditions (Figures S7C and S7D).
Thus, it seems that LCA extends longevity of chronologically aging yeast through two different mechanisms. Firstly, irrespective of the number of available calories, this bile acid targets certain house-keeping longevity assurance pathways that 1) inhibit some pro-aging processes and/or activate some anti-aging processes; and 2) do not overlap with the adaptable cAMP/PKA pathway modulated by calorie availability (Figure 10B). Secondly, we propose that LCA unmasks the anti-aging potential of PKA by activating PKA-dependent phosphorylation of the cytosolic pool of Rim15p (Figure 10B). Because such phosphorylation of Rim15p is known to inactivate its protein kinase activity (Figure 10A) [62], we hypothesize that, while the nuclear pool of Rim15p has a well established anti-aging function [5,6,27,62], the cytosolic pool of this nutrient-sensory protein kinase plays an essential pro-aging role by phosphorylating a compendium of proteins that promote aging only if phosphorylated (Figure 10B). Noteworthy, some of the Rim15p phosphorylation targets are involved in longevity regulation and reside outside the nucleus [68]. In our hypothesis, LCA can unmask the anti-aging potential of PKA only when PKA is activated by cAMP, i.e., under non-CR conditions (Figures 8E and F). Consistent with our hypothesis on the two mechanisms underlying the anti-aging effect of LCA, lack of Ras2p only partially and to the same extent reduced the life-extending potential of LCA under both CR and non-CR conditions (Figures S7C and S7D), likely by impairing the mechanism in which LCA unmasks the anti-aging potential of PKA. The resulting inability of PKA to inhibit the proposed pro-aging role of the cytosolic pool of Rim15p in ras2Δ cells would make the Rim15p-dependent pro-aging mechanism constitutively active in these cells, regardless of the number of available calories or presence of LCA (Figures S7E and S7F).
The TOR and cAMP/PKA pathways converge on Rim15p whose nuclear pool plays a pivotal role in governing the life-extending effect of CR by enabling the establishment of an anti-aging transcriptional program driven by Msn2p, Msn4p and Gis1p (Figure 10A) [5,6,27,62]. Our evaluation of the life-extending efficacy of LCA in yeast lacking Rim15p further supported the notion that one of the two mechanisms underlying the anti-aging effect of this bile acid involves its ability to modulate certain housekeeping longevity assurance pathways that are not centered on Rim15p and do not overlap with the adaptable TOR and cAMP/PKA pathways. In fact, although the life-extending potential of LCA in rim15Δ was partially reduced (Figures S8C and S8D) due to the impairment of the Rim15p-centered mechanism of its anti-aging action (Figures S8E and S8F), LCA still significantly increased the CLS of rim15Δ under CR and non-CR conditions (Figures S8A to S8D). Importantly, by eliminating a key nutrient-sensory protein kinase on which the adaptable TOR and cAMP/PKA pathways converge to regulate longevity in a calorie availability-dependent fashion, the rim15Δ mutation abolishedthe dependence of the anti-aging efficacy of LCA on the number of available calories (Figures S8C and S8D).
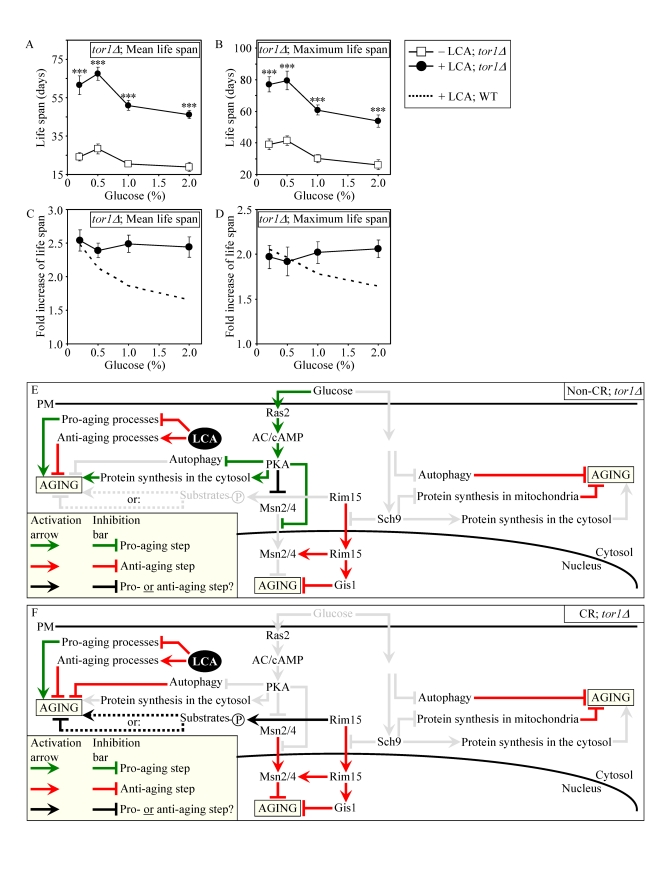
Figure 9. Lack of Tor1p does not impair the life-extending effect of LCA and abolishes the dependence of the anti-aging efficacy of LCA on the number of available calories. (A
and B) Effect of LCA on the mean (A) and maximum (B)
life spans of chronologically aging tor1Δ strain. Data are presented as means ± SEM (n = 4-7; ***p
< 0.001). (C and D) Effect
of LCA on the fold increase in the mean (C) or
maximum (D) life spans of chronologically aging tor1Δ and WT strains.
Data are presented as means ± SEM (n = 4-7). Cells
in A to D were cultured in medium initially containing 0.2%,
0.5%, 1% or 2% glucose in the presence of LCA (50 μM) or in its
absence. Survival data are provided in Supplementary Figure 10. (E
and F) Outline of pro- and anti-aging processes that are controlled
by the TOR and/or cAMP/PKA signaling pathways and are modulated by LCA in tor1Δcells grown under non-CR (E) or CR (F)
conditions.
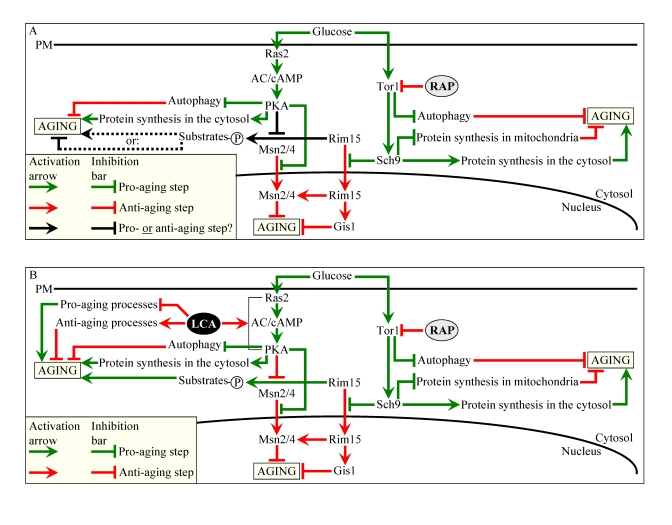
Figure 10. Outline of pro- and anti-aging processes that are controlled by the TOR and/or cAMP/PKA signaling pathways and are modulated by LCA or rapamycin (RAP) in chronologically aging yeast. The
currently accepted (A) and updated, based on this study (B),
outlines of pro- and anti-aging processes are shown. Activation
arrows and inhibition bars denote pro-aging processes (displayed in green
color), anti-aging processes (displayed in red color) or processes whose
role in longevity regulation was unknown (displayed in black color). Doted
lines denote hypothetical, until this study, processes. See text for
details.
Discussion
In this study, we designed a chemical genetic screen for small molecules that increase the CLS of yeast under CR conditions by targeting lipid metabolism and modulating housekeeping longevity pathways that regulate longevity irrespective of the number of available calories. Our screen identifies LCA as one of such molecules. Our analysis of how LCA influences various longevity-related processes and how it affects the CLS of yeast mutants impaired in the adaptable TOR and cAMP/PKA longevity pathways provided important new insights into mechanisms of longevity regulation, as outlined below.
LCA extends yeast CLS by modulating housekeeping longevity assurance processes that are not regulated by the adaptable TOR and cAMP/PKA signaling pathways
Our findings imply that LCA extends longevity of chronologically aging yeast by targeting two different mechanisms. One mechanism extends longevity regardless of the number of available calories. This mechanism involves the LCA-governed modulation of certain housekeeping longevity assurance pathways that do not overlap with the adaptable TOR and cAMP/PKA pathways (Figure 10B). We identify a compendium of processes that compose LCA-targeted housekeeping longevity assurance pathways. Our data provide evidence that LCA modulates these pathways by 1) suppressing the pro-aging process [39,40,50] of lipid-induced necrotic cell death, perhaps due to its observed ability to reduce the intracellular levels of FFA and DAG that trigger such death; 2) attenuating the pro-aging process [69,70] of mitochondrial fragmentation, a hallmark event of age-related cell death; 3) altering oxidation-reduction processes in mitochondria - such as oxygen consumption, the maintenance of membrane potential and ROS production - known to be essential for longevity regulation [8,10,11,71]; 4) enhancing cell resistance to oxidative and thermal stresses, thereby activating the anti-aging process [11,39,40,72,73] of stress response; 5) suppressing the pro-aging process [69,70] of mitochondria-controlled apoptosis; and 6) enhancing stability of nuclear and mitochondrial DNA, thus activating the anti-aging process [74,75] of genome maintenance. The observed pleiotropic effect of LCA on a compendium of housekeeping longevity assurance processes implies that this bile acid is a multi-target life-extending compound that increases CLS in yeast by modulating a network of the highly integrated processes that are not controlled by the adaptable TOR and cAMP/PKA pathways. The major challenge now is to define the molecular mechanisms by which LCA modulates each of these pro- and anti-aging housekeeping processes and integrates them in chronologically aging yeast.
The other mechanism underlying the life-extending effect of LCA in chronologically aging yeast increases life span only under non-CR conditions. This mechanism consists in LCA-driven unmasking of the previously unknown anti-aging potential of PKA, a key player in the adaptable cAMP/PKA pathway. We propose that LCA unveils the anti-aging potential of PKA by activating PKA-dependent phosphorylation of the cytosolic pool of Rim15p, a key nutrient-sensory protein kinase on which the adaptable TOR and cAMP/PKA pathways converge to regulate longevity in a calorie availability-dependent fashion (Figure 10B). Of note, the nuclear pool of Rim15p is well known for its anti-aging role in governing the life-extending effect of CR by enabling a pro-longevity transcriptional program driven by Msn2p, Msn4p and Gis1p (Figure 10B) [6,62]. In our hypothesis 1) unlike its nuclear pool, the cytosolic pool of Rim15p has an essential pro-aging function in phosphorylating a compendium of its cytosolic target proteins [68] some of which promote aging only if phosphorylated (Figure 10B); 2) under non-CR conditions LCA activates the PKA-dependent phosphorylation of Rim15p (Figure 10B); and 3) because the phosphorylation of Rim15p inactivates its protein kinase activity [62], the dephosphorylation of pro-aging target proteins of Rim15p in the cytosol by phosphatases inhibits the ability of these target proteins to promote aging (Figure 10B). To test the validity of our hypothesis, we are currently evaluating how genetic manipulations that alter the abundance of various extra-nuclear target proteins of Rim15p or affect their phosphorylation status influence the life-extending efficacy of LCA.
Bile acids are beneficial to health and longevity across phyla
It should be stressed that, although we found that LCA greatly extends yeast longevity, yeast do not synthesize this or any other bile acid found in mammals [57,76]; our mass spectrometry-based analysis of the total yeast lipidome has confirmed lack of endogenous bile acids. One could envision that during evolution yeast have lost the ability to synthesize bile acids but have maintained the life-extending response to these biologically active molecules by retaining certain longevity-related processes that are sensitive to regulation by bile acids. Alternatively, one could think that during evolution yeast have developed the ability to sense bile acids produced by mammals (and/or bile acid-like lipids synthesized by worms), recognize these mildly toxic molecules as environmental stressors providing hormetic benefits and/or as indicators of the state of the environment or food supply, and then to respond by undergoing certain life-extending changes to their physiology that ultimately increase their chances of survival. It is conceivable therefore that the life-extending potential of LCA and other bile acids as well as, probably, the mechanisms underlying their anti-aging action are evolutionarily conserved.
In fact, following their synthesis from cholesterol in the intestine, hypodermis, spermatheca and sensory neurons of worms, bile acid-like dafachronic acids (including 3-keto-LCA) are delivered to other tissues where they activate the DAF-12/DAF-16 signaling cascade that in turn orchestrates an anti-aging transcriptional program, thereby increasing the life span of the entire organism [41]. Bile acids also provide health benefits to mammals. Synthesized from cholesterol in hepatocytes of the liver, these amphipathic molecules have been for a long time considered to function only as trophic factors for the enteric epithelium and as detergents for the emulsification and absorption of dietary lipids and fat-soluble vitamins [57,76,77]. Recent years have been marked by a significant progress in our understanding of the essential role that bile acids play as signaling molecules regulating lipid, glucose and energy homeostasis and activating detoxification of xenobiotics [57,77,78]. By stimulating the G-protein-coupled receptor TGR5, bile acids activate the cAMP/PKA signaling pathway that 1) enhances energy expenditure in brown adipose tissue and muscle by stimulating mitochondrial oxidative phosphorylation and un-coupling; 2) improves liver and pancreatic function by activating the endothelial nitric oxide synthase; and 3) enhances glucose tolerance in obese mice by inducing intestinal glucagon-like peptide-1 release [57,76,78]. Furthermore, by activating the farnesoid X receptor (FXR) and several other nuclear hormone receptors inside mammalian cells, bile acids 1) modulate the intracellular homeostasis of cholesterol, neutral lipids and fatty acids; 2) regulate glucose metabolism by enhancing glycogenesis and attenuating gluconeo-genesis; and 3) stimulate clearance of xenobiotic and endobiotic toxins by activating transcription of numerous xenobiotic detoxification genes [57,76-78]. All these health-improving, beneficial metabolic effects of bile acids prevent the development of obesity following administration of high-fat diet [57,76,77]. Thus, bile acids have a great potential as pharmaceutical agents for the treatment of diabetes, obesity and various associated metabolic disorders, all of which are age-related [57,76]. Moreover, bile acids have been shown to inhibit neuronal apoptosis in experimental rodent models of neurodegenerative disorders by promoting mitochondrial membrane stability, preventing the release of cytochrome c from mitochondria, reducing activities of various caspases, and activating the NF-κB, PI3K and MAPK survival pathways [79,80].
It should be stressed that many of the metabolic, stress response and apoptotic processes modulated by bile acids in mammals are essential for healthy aging and longevity regulation. Importantly, we found that, by modulating several of these health- and longevity-related processes in chronologically aging yeast, LCA increases their life span. Moreover, the long-lived Ghrhrlit/lit mice displayed elevated levels of several bile acids and exhibited increased FXR-dependent transcription of numerous xenobiotic detoxification genes; if administered to food consumed by wild-type mice, cholic acid - one of these bile acids - mimicked the FXR-governed gene expression pattern observed in Ghrhrlit/lit mice [81,82]. It has been therefore proposed that, by promoting chemical hormesis in mammals, these mildly toxic molecules with detergent-like properties may extend their longevity by acting as endobiotic regulators of aging [73,82,83].
Altogether, these findings support the notion that bile acids act as endobiotic and xenobiotic regulators of aging that are beneficial to health and longevity across phyla. A comparative analysis of the mechanisms underlying such health-improving and life-extending action of bile acids implies that these mechanisms are likely to be evolutionarily conserved.
Methods
Yeast strains and growth conditions. The WT strain BY4742 (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) and single-gene-deletion mutant strains in the BY4742 genetic background (all from Open Biosystems) were grown in YP medium (1% yeast extract, 2% peptone) containing 0.2% 0.5%, 1% or 2% glucose as carbon source. Cells were cultured at 30oC with rotational shaking at 200 rpm in Erlenmeyer flasks at a "flask volume/medium volume" ratio of 5:1.
Chemical genetic screen for compounds that increase chronological life span (CLS). The screen was conducted at the High Throughput/Content Screening Facility at McGill University. The single-gene-deletion mutant strain pex5Δ (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 pex5Δ::kanMX4) from Open Biosystems was grown in YPA0.5D medium (1% yeast extract, 2% peptone, 50 μg/ml ampicillin, 0.5% glucose). 3-μl aliquots of the pex5Δculture recovered from mid-logarithmic phase at a cell titre of 2 Í 107 cells/ml were aliquoted into 96-well master microplates using a Beckman Coulter high density Biomek FXII replica pinning robot. Each well of a master microplate contained 96 μl of YPA0.5D medium. 1 μl of a compound stock solution from a commercially available library (each compound at 5 mM in dimethylsulfoxide (DMSO)) was added to each well using a Beckman Coulter high density Biomek FXII replica pinning robot. Wells of a master microplate supplemented with 1% DMSO (1 μl of DMSO per a well containing 3 μl of the pex5Δculture and 96 μl of YPA0.5D medium) were used as negative controls. Each master plate was created in duplicate. The master microplates were sealed and incubated without shaking at 30oC in a moist chamber. At days 1, 7, 10 and 14 of the incubation of master microplates, a 3-μl aliquot of each culture was transferred into individual wells of a new (replica) microplate containing 97 μl of YPA0.5D medium. Following incubation of sealed replica microplates in a moist chamber for 16 hours at 30oC (to allow for growth of cells that were still viable), the optical density at 600 nm (OD600) of the culture in each well of the replica microplate was measured using a Molecular Devices Analyst HT plate reader. To calculate survival at each time point, the OD600 at a particular time point was divided by the OD600 at day 1. "Cherry-picking" of the identified "lead" com-pounds for possible "hits" was carried out as described above, with each lead compound being used at a final concentration of 5, 10, 25 or 50 μM and assessed in triplicate for validation. Commercially available structural analogs of hit compounds were identified using the web-based eMolecules searching engine. In total, approximately 19,000 representative compounds from the BIOMOL, Chembridge, Maybridge, MicroSource Discovery, NIH Clinical Collection, Prestwick Chemical Inc. and Sigma-LOPAC commercial libraries were tested using the screen for chemical modulators of longevity.
Pharmacological manipulation of CLS. CLS analysis was performed as previously described [39]. The chenodeoxycholic (C9377), cholic (C1129), dehydrocholic (D3750), deoxycholic (D2510), hyodeoxycholic (H3878), lithocholic (L6250) and ursodeoxycholic (U5127) bile acids were from Sigma. Their stock solutions in DMSO were made on the day of adding each of these compounds to cell cultures. Compounds were added to growth medium at the indicated concentration immediately following cell inoculation. The final concentration of DMSO in yeast cultures supplemented with a bile acid (and in the corresponding control cultures supplemented with drug vehicle) was 1% (v/v).
Miscellaneous procedures. Fluorescence [39], immuno-fluorescence [39] and electron [84] microscopies followed by morphometric analyses of the resulting images have been described elsewhere. Extraction of lipids and their separation, identification and quantitation with the help of TLC were performed according to established procedures [84]. Mass spectrometric identification and quantitation of various lipid species were carried as previously described [85]. Subcellular fractionation and organelle purification,cell viability and stress resistance assays, oxygen consumption assay, the measurement of the frequencies of spontaneous point and deletion mutations in mitochondrial and nuclear DNA, total cell lysates preparation, and mass spectrometric identification and quantitation of proteins were performed according to established procedures [39].
Supplementary Materials
Acknowledgments
This work was supported by grants from the CIHR, NSERC of Canada, Canada Foundation for Innovation, and Concordia University Chair Fund. AME is a Concordia University Research Chair in Bioinorganic Chemistry. DYT is a Canada Research Chair in Molecular Genetics. VIT is a Concordia University Research Chair in Genomics, Cell Biology and Aging.
Conflicts of Interest
The authors of this manuscript have no conflict of interests to declare.
References
- 1. Guarente LP , Partridge L and Wallace DC. Cold Spring Harbor Cold Spring Harbor Laboratory Press Molecular Biology of Aging. 2008; .
- 2. Greer EL and Brunet A. Signaling networks in aging. J Cell Sci. 2008; 121: 407 -412. [PubMed] .
- 3. Narasimhan SD , Yen K and Tissenbaum HA. Converging pathways in lifespan regulation. Curr Biol. 2009; 19: R657 -R666. [PubMed] .
- 4. Shaw RJ LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol. 2009; 196: 65 -80. .
- 5. Fontana L , Partridge L and Longo VD. Extending healthy life span - from yeast to humans. Science. 2010; 328: 321 -326. [PubMed] .
- 6. Wei M , Fabrizio P , Hu J , Ge H , Cheng C , Li L and Longo VD. Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9. PLoS Genet. 2008; 4: e13 [PubMed] .
- 7. Laplante M and Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009; 122: 3589 -3594. [PubMed] .
- 8. Finley LW and Haigis MC. The coordination of nuclear and mitochondrial communication during aging and calorie restriction. Ageing Res Rev. 2009; 8: 173 -188. [PubMed] .
- 9. Soukas AA , Kane EA , Carr CE , Melo JA and Ruvkun G. Rictor/TORC2 regulates fat metabolism, feeding, growth, and life span in Caenorhabditis elegans. Genes Dev. 2009; 23: 496 -511. [PubMed] .
- 10. Giorgio M , Trinei M , Migliaccio E and Pelicci PG. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals. Nat Rev Mol Cell Biol. 2007; 8: 722 -728. [PubMed] .
- 11. Lapointe J and Hekimi S. When a theory of aging ages badly. Cell Mol Life Sci. 2010; 67: 1 -8. [PubMed] .
- 12. Mair W and Dillin A. Aging and survival: the genetics of life span extension by dietary restriction. Annu Rev Biochem. 2008; 77: 727 -754. [PubMed] .
- 13. Colman RJ , Anderson RM , Johnson SC , Kastman EK , Kosmatka KJ , Beasley TM , Allison DB , Cruzen C , Simmons HA , Kemnitz JW and Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009; 325: 201 -204. [PubMed] .
- 14. Masoro EJ Amsterdam Elsevier Caloric Restriction: A Key to Understanding and Modulating Aging. 2002; .
- 15. >Min KJ , Flatt T , Kulaots I and Tatar M. Counting calories in Drosophila diet restriction. Exp Gerontol. 2007; 42: 247 -251. [PubMed] .
- 16. Weindruch R and Walford RL. Springfield Thomas The Retardation of Aging and Disease by Dietary Restriction. 1998; .
- 17. Zimmerman JA , Malloy V , Krajcik R and Orentreich N. Nutritional control of aging. Exp Gerontol. 2003; 38: 47 -52. [PubMed] .
- 18. Piper MD , Mair W and Partridge L. Counting the calories: the role of specific nutrients in extension of life span by food restriction. J Gerontol A Biol Sci Med Sci. 2005; 60: 549 -555. [PubMed] .
- 19. Blagosklonny MV Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006; 5: 2087 -2102. [PubMed] .
- 20. Blagosklonny MV Aging: ROS or TOR. Cell Cycle. 2008; 7: 3344 -3354. [PubMed] .
- 21. Blagosklonny MV TOR-driven aging: speeding car without brakes. Cell Cycle. 2009; 8: 4055 -4059. [PubMed] .
- 22. Kaeberlein M , Powers RW 3rd , Steffen KK , Westman EA , Hu D , Dang N , Kerr EO , Kirkland KT , Fields S and Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005; 310: 1193 -1196. [PubMed] .
- 23. Meissner B , Boll M , Daniel H and Baumeister R. Deletion of the intestinal peptide transporter affects insulin and TOR signaling in Caenorhabditis elegans. J Biol Chem. 2004; 279: 36739 -36745. [PubMed] .
- 24. Hansen M , Taubert S , Crawford D , Libina N , Lee SJ and Kenyon C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell. 2007; 6: 95 -110. [PubMed] .
- 25. Chen D and Guarente L. SIR2: a potential target for calorie restriction mimetics. Trends Mol Med. 2007; 13: 64 -71. [PubMed] .
- 26. Kaeberlein M and Powers RW 3rd. Sir2 and calorie restriction in yeast: a skeptical perspective. Ageing Res Rev. 2007; 6: 128 -140. [PubMed] .
- 27. Medvedik O , Lamming DW , Kim KD and Sinclair DA. MSN2 and MSN4 link calorie restriction and TOR to sirtuin-mediated lifespan extension in Saccharomyces cerevisiae. PLoS Biol. 2007; 5: e261 [PubMed] .
- 28. Greer EL and Brunet A. Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell. 2009; 8: 113 -127. [PubMed] .
- 29. Ingram DK , Zhu M , Mamczarz J , Zou S , Lane MA , Roth GS and deCabo R. Calorie restriction mimetics: an emerging research field. Aging Cell. 2006; 5: 97 -108. [PubMed] .
- 30. Lane MA , Roth GS and Ingram DK. Caloric restriction mimetics: a novel approach for biogerontology. Methods Mol Biol. 2007; 371: 143 -149. [PubMed] .
- 31. Howitz KT , Bitterman KJ , Cohen HY , Lamming DW , Lavu S , Wood JG , Zipkin RE , Chung P , Kisielewski A , Zhang LL , Scherer B and Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003; 425: 191 -196. [PubMed] .
- 32. Wood JG , Rogina B , Lavu S , Howitz K , Helfand SL , Tatar M and Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004; 430: 686 -689. [PubMed] .
- 33. Baur JA , Pearson KJ , Price NL , Jamieson HA , Lerin C , Kalra A , Prabhu VV , Allard JS , Lopez-Lluch G , Lewis K , Pistell PJ , Poosala S and Becker KG. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006; 444: 337 -342. [PubMed] .
- 34. Petrascheck M , Ye X and Buck LB. An antidepressant that extends lifespan in adult Caenorhabditis elegans. Nature. 2007; 450: 553 -556. [PubMed] .
- 35. Onken B and Driscoll M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans healthspan via AMPK, LKB1, and SKN-1. PLoS ONE. 2010; 5: e8758 [PubMed] .
- 36. McColl G , Killilea DW , Hubbard AE , Vantipalli MC , Melov S and Lithgow GJ. Pharmacogenetic analysis of lithium-induced delayed aging in Caenorhabditis elegans. J Biol Chem. 2008; 283: 350 -357. [PubMed] .
- 37. Bjedov I , Toivonen JM , Kerr F , Slack C , Jacobson J , Foley A and Partridge L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010; 11: 35 -46. [PubMed] .
- 38. Picard F , Kurtev M , Chung N , Topark-Ngarm A , Senawong T , Machado De Oliveira R , Leid M , McBurney MW and Guarente L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature. 2004; 429: 771 -776. [PubMed] .
- 39. Goldberg AA , Bourque SD , Kyryakov P , Gregg C , Boukh-Viner T , Beach A , Burstein MT , Machkalyan G , Richard V , Rampersad S , Cyr D , Milijevic S and Titorenko VI. Effect of calorie restriction on the metabolic history of chronologically aging yeast. Exp Gerontol. 2009; 44: 555 -571. [PubMed] .
- 40. Goldberg AA , Bourque SD , Kyryakov P , Boukh-Viner T , Gregg C , Beach A , Burstein MT , Machkalyan G , Richard V , Rampersad S and Titorenko VI. A novel function of lipid droplets in regulating longevity. Biochem Soc Trans. 2009; 37: 1050 -1055. [PubMed] .
- 41. Russell SJ and Kahn CR. Endocrine regulation of ageing. Nat Rev Mol Cell Biol. 2007; 8: 681 -691. [PubMed] .
- 42. Wang MC , O'Rourke EJ and Ruvkun G. Fat metabolism links germline stem cells and longevity in C. elegans. Science. 2008; 322: 957 -960. [PubMed] .
- 43. Narbonne P and Roy R. Caenorhabditis elegans dauers need LKB1/AMPK to ration lipid reserves and ensure long-term survival. Nature. 2009; 457: 210 -214. [PubMed] .
- 44. Grönke S , Mildner A , Fellert S , Tennagels N , Petry S , Müller G , Jäckle H and Kühnlein RP. Brummer lipase is an evolutionary conserved fat storage regulator in Drosophila. Cell Metab. 2005; 1: 323 -330. [PubMed] .
- 45. Blüher M , Kahn BB and Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003; 299: 572 -574. [PubMed] .
- 46. Chiu CH , Lin WD , Huang SY and Lee YH. Effect of a C/EBP gene replacement on mitochondrial biogenesis in fat cells. Genes Dev. 2004; 18: 1970 -1975. [PubMed] .
- 47. Haemmerle G , Lass A , Zimmermann R , Gorkiewicz G , Meyer C , Rozman J , Heldmaier G , Maier R , Theussl C , Eder S , Kratky D , Wagner EF and Klingenspor M. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006; 312: 734 -737. [PubMed] .
- 48. Gerhart-Hines Z , Rodgers JT , Bare O , Lerin C , Kim SH , Mostoslavsky R , Alt FW , Wu Z and Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 2007; 26: 1913 -1923. [PubMed] .
- 49. Léon S , Goodman JM and Subramani S. Uniqueness of the mechanism of protein import into the peroxisome matrix: transport of folded, co-factor-bound and oligomeric proteins by shuttling receptors. Biochim Biophys Acta. 2006; 1763: 1552 -1264. [PubMed] .
- 50. Low CP , Liew LP , Pervaiz S and Yang H. Apoptosis and lipoapoptosis in the fission yeast Schizosaccharomyces pombe. FEMS Yeast Res. 2005; 5: 1199 -1206. [PubMed] .
- 51. Dirkx R , Vanhorebeek I , Martens K , Schad A , Grabenbauer M , Fahimi D , Declercq P , Van Veldhoven PP and Baes M. Absence of peroxisomes in mouse hepatocytes causes mitochondrial and ER abnormalities. Hepatology. 2005; 41: 868 -878. [PubMed] .
- 52. D'Autréaux B and Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007; 8: 813 -824. [PubMed] .
- 53. Eisenberg T , Büttner S , Kroemer G and Madeo F. The mitochondrial pathway in yeast apoptosis. Apoptosis. 2007; 12: 1011 -1023. [PubMed] .
- 54. Pereira C , Silva RD , Saraiva L , Johansson B , Sousa MJ and Côrte-Real M. Mitochondria-dependent apoptosis in yeast. Biochim Biophys Acta. 2008; 1783: 1286 -1302. [PubMed] .
- 55. Sinclair DA Toward a unified theory of caloric restriction and longevity regulation. Mech Ageing Dev. 2005; 126: 987 -1002. [PubMed] .
- 56. Powers RW 3rd , Kaeberlein M , Caldwell SD , Kennedy BK and Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006; 20: 174 -184. [PubMed] .
- 57. Thomas C , Pellicciari R , Pruzanski M , Auwerx J and Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008; 7: 678 -693. [PubMed] .
- 58. Wanke V , Cameroni E , Uotila A , Piccolis M , Urban J , Loewith R and De Virgilio C. Caffeine extends yeast lifespan by targeting TORC1. Mol Microbiol. 2008; 69: 277 -285. [PubMed] .
- 59. Eisenberg T , Knauer H , Schauer A , Büttner S , Ruckenstuhl C , Carmona-Gutierrez D , Ring J , Schroeder S , Magnes C , Antonacci L , Fussi H , Deszcz L and Hartl R. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009; 11: 1305 -1314. [PubMed] .
- 60. Huber A , Bodenmiller B , Uotila A , Stahl M , Wanka S , Gerrits B , Aebersold R and Loewith R. Characterization of the rapamycin-sensitive phosphoproteome reveals that Sch9 is a central coordinator of protein synthesis. Genes Dev. 2009; 23: 1929 -1943. [PubMed] .
- 61. Pan Y and Shadel GS. Extension of chronological life span by reduced TOR signaling requires down-regulation of Sch9p and involves increased mitochondrial OXPHOS complex density. Aging. 2009; 1: 131 -145. [PubMed] .
- 62. Smets B , Ghillebert R , De Snijder P , Binda M , Swinnen E , De Virgilio C and Winderickx J. Life in the midst of scarcity: adaptations to nutrient availability in Saccharomyces cerevisiae. Curr Genet. 2010; 56: 1 -32. [PubMed] .
- 63. Stephan JS , Yeh YY , Ramachandran V , Deminoff SJ and Herman PK. The Tor and PKA signaling pathways independently target the Atg1/Atg13 protein kinase complex to control autophagy. Proc Natl Acad Sci USA. 2009; 106: 17049 -17054. [PubMed] .
- 64. Kamada Y , Yoshino K , Kondo C , Kawamata T , Oshiro N , Yonezawa K and Ohsumi Y. Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol Cell Biol. 2010; 30: 1049 -1058. [PubMed] .
- 65. Urban J , Soulard A , Huber A , Lippman S , Mukhopadhyay D , Deloche O , Wanke V , Anrather D , Ammerer G , Riezman H , Broach JR , De Virgilio C and Hall MN. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol Cell. 2007; 26: 663 -674. [PubMed] .
- 66. Yorimitsu T , Zaman S , Broach JR and Klionsky DJ. Protein kinase A and Sch9 cooperatively regulate induction of autophagy in Saccharomyces cerevisiae. Mol Biol Cell. 2007; 18: 4180 -4189. [PubMed] .
- 67. Lee P , Cho BR , Joo HS and Hahn JS. Yeast Yak1 kinase, a bridge between PKA and stress-responsive transcription factors, Hsf1 and Msn2/Msn4. Mol Microbiol. 2008; 70: 882 -895. [PubMed] .
- 68. Ptacek J , Devgan G , Michaud G , Zhu H , Zhu X , Fasolo J , Guo H , Jona G , Breitkreutz A , Sopko R , McCartney RR , Schmidt MC and Rachidi N. Global analysis of protein phosphorylation in yeast. Nature. 2005; 438: 679 -684. [PubMed] .
- 69. Fabrizio P and Longo VD. Chronological aging-induced apoptosis in yeast. Biochim Biophys Acta. 2008; 1783: 1280 -1285. [PubMed] .
- 70. Hamann A , Brust D and Osiewacz HD. Apoptosis pathways in fungal growth, development and ageing. Trends Microbiol. 2008; 16: 276 -283. [PubMed] .
- 71. Skulachev VP , Anisimov VN , Antonenko YN , Bakeeva LE , Chernyak BV , Erichev VP , Filenko OF , Kalinina NI , Kapelko VI , Kolosova NG , Kopnin BP , Korshunova GA and Lichinitser MR. An attempt to prevent senescence: a mitochondrial approach. Biochim Biophys Acta. 2009; 1787: 437 -461. [PubMed] .
- 72. Schulz TJ , Zarse K , Voigt A , Urban N , Birringer M and Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007; 6: 280 -293. [PubMed] .
- 73. Gems D and Partridge L. Stress-response hormesis and aging: "that which does not kill us makes us stronger". Cell Metab. 2008; 7: 200 -203. [PubMed] .
- 74. Pang CY , Ma YS and Wei YU. MtDNA mutations, functional decline and turnover of mitochondria in aging. Front Biosci. 2008; 13: 3661 -3675. [PubMed] .
- 75. Sinclair DA and Oberdoerffer P. The ageing epigenome: damaged beyond repair. Ageing Res Rev. 2009; 8: 189 -198. [PubMed] .
- 76. Lefebvre P , Cariou B , Lien F , Kuipers F and Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009; 89: 147 -191. [PubMed] .
- 77. Hylemon PB , Zhou H , Pandak WM , Ren S , Gil G and Dent P. Bile acids as regulatory molecules. J Lipid Res. 2009; 50: 1509 -1520. [PubMed] .
- 78. Vallim TQ and Edwards PA. Bile acids have the gall to function as hormones. Cell Metab. 2009; 10: 162 -164. [PubMed] .
- 79. Ramalho RM , Viana RJ , Low WC , Steer CJ and Rodrigues CM. Bile acids and apoptosis modulation: an emerging role in experimental Alzheimer's disease. Trends Mol. Med. 2008; 14: 54 -62. .
- 80. Amaral JD , Viana RJ , Ramalho RM , Steer CJ and Rodrigues CM. Bile acids: regulation of apoptosis by ursodeoxycholic acid. J Lipid Res. 2009; 50: 1721 -1734. [PubMed] .
- 81. Amador-Noguez D , Yagi K , Venable S and Darlington G. Gene expression profile of long-lived Ames dwarf mice and Little mice. Aging Cell. 2004; 3: 423 -441. [PubMed] .
- 82. Amador-Noguez D , Dean A , Huang W , Setchell K , Moore D and Darlington G. Alterations in xenobiotic metabolism in the long-lived Little mice. Aging Cell. 2007; 6: 453 -470. [PubMed] .
- 83. Gems D Long-lived dwarf mice: are bile acids a longevity signal. Aging Cell. 2007; 6: 421 -423. [PubMed] .
- 84. Guo T , Gregg C , Boukh-Viner T , Kyryakov P , Goldberg A , Bourque S , Banu F , Haile S , Milijevic S , San KH , Solomon J , Wong V and Titorenko VI. A signal from inside the peroxisome initiates its division by promoting the remodeling of the peroxisomal membrane. J Cell Biol. 2007; 177: 289 -303. [PubMed] .
- 85. Bourque SD and Titorenko VI. A quantitative assessment of the yeast lipidome using electrospray ionization mass spectrometry. J Vis Exp. 2009; 30: doi: 10 .