



Introduction
Since Hayflick's discovery of the phenomenon of cellular (replicative) senescence [1], the contribution or even relevance of this phenomenon to organismal aging has been a subject for continuous debates [2-5]. Although the question still remains open, an increasing amount of evidence, especially from recent years, indicates that cellular senescence (CS) could have a role in aging and age-related diseases (ARDs), rather than being just a laboratory phenomenon [3, 6-11]. In fact, the current situation in the field could be defined as an attempt to understand to what extent and how is CS involved in aging and ARDs.
Apart from an irreversible growth arrest (“Hayflick's limit” — a finite number of cell divisions), the CS phenotype is characterized by cell hypertrophy, an increased metabolic activity including synthesis of macromolecules (RNA, protein, lipid) and organelles [12, 13], increased secretion of pro-inflammatory substances and resistance to apoptosis [7, 8, 11]. After being initially discovered in primary cultures of human fibroblasts, CS has also been found in other cell types such as keratinocytes, endothelial cells, lymphocytes, adrenocortical cells, vascular smooth muscle cells, chondrocytes, etc., both for in vitro and in vivo conditions [3, 11, 14, 15], and in cell cultures derived from many other organisms examined thus far (e.g., mice, monkeys, chickens, Galapagos tortoise, etc.) [16-18]. Moreover, it appears that CS is not restricted only to dividing cells. At least some features of CS were also found in classical post-mitotic cells such as neurons, myocardiocytes and adipocytes (reviewed by Tchkonia et al. [19]).
The complex nature of aging and aging-associated phenomena including CS requires a holistic view with a focus on the interplay between their components [11, 12, 20, 21]. Here we consider the potential molecular links between CS, longevity, ARDs, oxidative stress, and chronic inflammation from a systems biology perspective. Highlighting the common genes, interactions, regulatory molecules (miRNAs) and common pathways may help in understanding how CS interplays with and contributes to other aging-associated conditions.
Results and Discussion
1. CS genes share common features with LAGs and ARD genes
A comprehensive data mining of scientific literature brought about a list of 262 human genes identified as being associated with CS (see Suppl. Table 1). These genes possess diverse functions, with the majority falling into three categories: regulation of cell cycle and proliferation, biosynthesis and programmed cell death (for GO functional analysis, see Suppl. Table 2). We have previously shown that longevity-associated (LAGs) and ARD genes also show functional diversity. Besides that, they display a number of distinct features including higher connectivity and interconnectivity, evolutionary conservation, and essentiality to growth and development [22-24]. This combination makes many of them putative candidates as antagonistic pleiotropy genes, i.e., genes which may have undesirable effects later in life, potentially linking aging, longevity and ARDs [25, 26]. Therefore, one of the first questions that arise in this context is whether CS genes share any common features with LAGs and ARD genes.
1.1. Connectivity and interconnectivity: miRNA-regulated PPI networks
To what extent are the CS genes/proteins working in a cooperative manner? In most cases, proteins do not act on their own but rather together with their partners through protein-protein interactions (PPIs). Currently, the human interactome includes approximately 10,000 genes with more than 35,000 physical PPIs ([27], http://thebiogrid.org). Most of CS genes (231 of the 262) as well as LAGs and ARD genes can be found in the human interactome [24]. As shown in Fig. 1 (insert), they have a much higher average connectivity (number of first-order protein partners) compared to all interactome proteins. This is in accordance with observations demonstrating that disease proteins have higher average connectivity than other proteins, and that highly connected proteins are more likely to be disease-associated [28]. It was particularly evident for cancer genes [22] and for genes common to major human ARDs [23, 25].
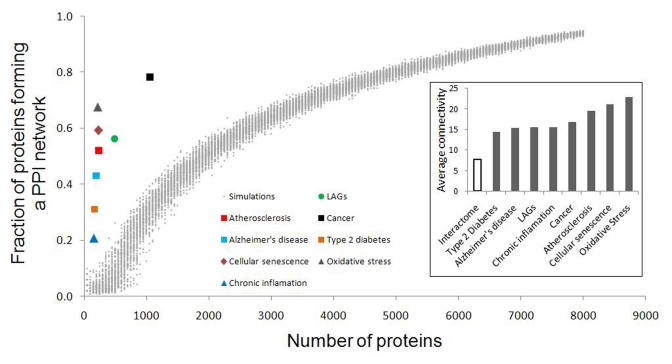
Figure 1. Fraction of CS, longevity and ARD proteins forming a continuous PPI network. Values obtained from simulations with sets of randomly selected proteins are presented as dots. For all the sets of interest, the fraction of interconnected proteins was significantly higher than expected by chance (p < E-25). Insert: average connectivity (number of first-order protein partners) of the sets analyzed in this study. For more details, see Materials and Methods.
Not only are CS genes, LAGs, and ARD genes more connected, but they are also highly interconnected. Indeed, when compared with randomly generated sets, the above genes display a significantly higher interconnectivity (the fraction of genes that form a continuous network) (Fig. 1).
For example, 59% of the CS genes are connected between themselves and eventually form a continuous network (Fig. 2), whereas only 4 ± 2% (mean ± SD) genes form a network by chance (p < E−25). The percent of interconnected CS genes would be even higher if other (regulatory) interactions are considered. In particular, if we also take into account post-transcriptional regulation of gene expression by microRNAs (miRNAs), almost two thirds (64%) of the CS genes become connected, either through PPIs or common miRNAs (Fig. 2). Thus, CS genes together with their regulatory miRNAs might work in a cooperative manner by forming a miRNA-regulated PPI network. Such networks are also formed by LAGs and ARD genes (currently available in the NetAge database: [24], http://www.netage-project.org).
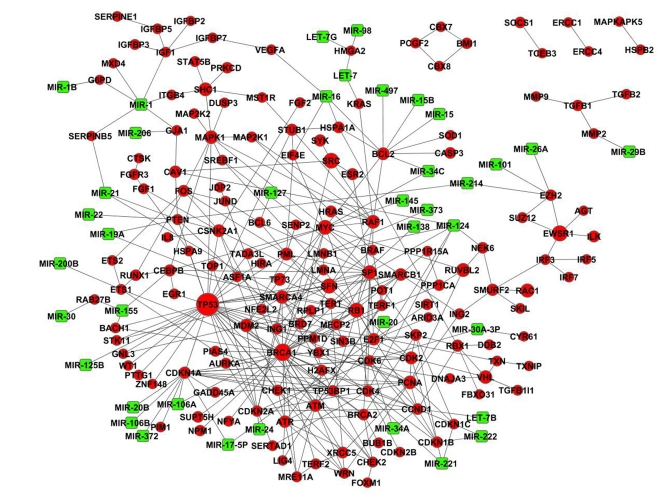
Figure 2. MicroRNA-regulated cellular senescence PPI network. Genes are depicted as red circles and miRNAs as green squares.
1.2. Essentiality
Genes with multiple PPIs have a higher probability to be essential, just because the deletions of these genes may result in the disruption of function of a larger number of proteins [28-30]. In line with this assumption, the portion of essential genes among the CS genes, LAGs and ARD genes is much higher than that in the whole genome or interactome (Fig. 3). Moreover, there is a significant correlation between connectivity and essentiality of different sets examined in this study (Fig. 3, insert). It is important to stress that many genes essential for development and growth tend to have detrimental effects at the later stages of life as suggested by the theory of antagonistic pleiotropy [31].
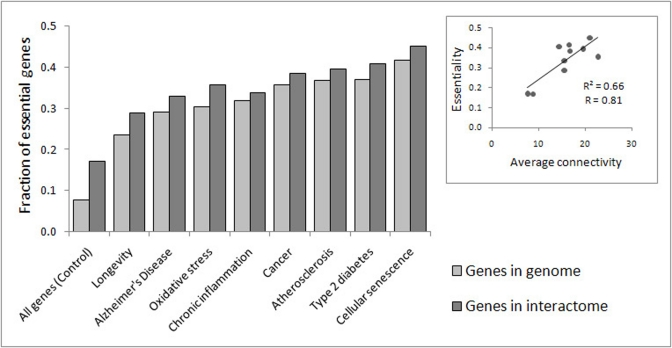
Figure 3. Fraction of genes which are essential to growth and development in each of the gene sets under analysis. The difference between each set and all genes (control) was highly significant (p < E-25). Insert: The correlation between essentiality and average connectivity (R – Pearson's coefficient of correlation; p = 0.004).
Remarkably, the percent of essential CS genes (42%) is considerably higher than that for all genes (p < E-25) and even higher than the percent of essential LAGs (p = 2.5E-10 and p = 6.2E-06 for entire genome and interactome, respectively), genes involved in Alzheimer's disease (p = 1.5E-04 and p = 0.001) and aging-associated processes (p < 0.002) (Fig. 3). This could at least in part be explained by the fact that the CS genes are highly enriched with genes involved in the regulation of basic, housekeeping processes such as cell cycle, cell growth, programmed cell death, DNA repair, and cellular response to stress (Suppl. Table 2).
1.3. Evolutionary conservation
Essential genes are generally more evolutionary conserved than non-essential ones [32]. In support of this notion are recent findings of Waterhouse et al. [33] who demonstrated that the essential genes from model organisms are significantly enriched in orthologs across the vertebrate, arthropod and fungal lineages. The high percentage of essential CS genes led us to explore the possibility that CS genes are highly evolutionary conserved as a whole. With this in mind, we have examined the frequency of orthologs for the human CS genes in over 100 species found currently in the InParanoid database ([34], http://inparanoid.sbc.su.se). We found that CS genes are significantly more conserved than are the genes in the whole human genome. This is clearly noted across the vertebrate species but the difference is insignificant in lower organisms. In Fig. 4, this is shown for a selected set of well-studied model organisms, from yeast to mouse. The same observation was true for the whole InParanoid set (data not shown). Notably, the pattern of evolutionary conservation of CS genes is almost identical to that of cancer-associated genes (see Fig. 1B in [22]). It would be then tempting to speculate that this similarity is a result of the co-evolution between these two phenomena. Indirectly supporting this assumption are observations on the naked mole rat, a species with an extraordinary resistance to cancer [35], whose fibroblasts do not undergo CS. Instead, they display early contact inhibition, an anti-cancer mechanism based on cell division arrest before reaching a high cell density [36]. Another example is chinchilla, a rodent with a low cancer incidence [37] that also does not develop CS, but evolved other anti-cancer adaptations such as continuous slow cell proliferation [18].
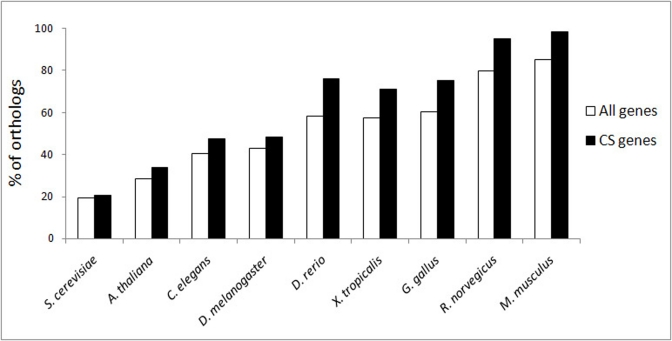
Figure 4. Evolutionary conservation of human CS genes. The difference between CS genes and all genes in InParanoid was significant for D. rerio (p = 0.0001), X. tropicalis (p = 0.003), G. gallus (p = 0.002), R. norvegicus (p = 0.006) and M. musculus (p = 0.02).
2. Molecular links between CS genes, LAGs and ARD genes
For a more specific analysis, we further addressed the following questions: (i) Are there genes common for CS and other aging/longevity-related categories? (ii) Could genes involved in CS, ARDs and in the control of life span interact via direct PPIs or their common partners? (iii) Is the set of CS genes enriched in genes associated with ARDs and age-related conditions? (iv) Could the expression of these genes be under the control of common regulatory molecules, more specifically, miRNAs? (v) Are there common pathways for CS, ARDs and aging-associated processes?
2.1. Common genes
The analysis revealed that 19% of the CS genes are also orthologous to LAGs from model organisms, and 53% of the CS genes are involved in at least one ARD (Table 1). The highest overlap was observed for cancer (53%); lesser values were observed for atherosclerosis (20%), Alzheimer's disease (9%) and type 2 diabetes (9%). The overlap of CS genes with oxidative stress and chronic inflammation associated genes reached 21% and 8%, respectively. In all the above cases, the overlap was significantly greater than expected by chance (p < E-25). Notably, among overlapping genes there are many which are essential for growth and development (from 38% to 55%, depending on the gene set). This high percentage of essential genes is comparable to that found in the Common Gene Signature of longevity and major ARD networks [25].
Table 1. Molecular links between CS, longevity, ARDs and aging-associated processes
| AS – Antherosclerosis, AD – Alzheimer's disease | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
2.2. Protein-protein interactions and common protein partners
Apart from common genes, a great number of CS proteins directly interact with LAGs and ARD proteins through PPIs (Table 1). As such, the majority of CS genes fall either in the category of common genes or in that of genes directly connected to LAGs or ARD genes. In total, the genes in these two categories exceed 80% of the entire CS set. In addition, there are many common external protein partners, the number of which is more than one order of magnitude higher than that of common genes. As a result, almost all CS genes are linked to longevity and/or ARDs in one of the following ways - as common molecules, by forming protein complexes via PPIs, or through common partners.
2.3. Common miRNA regulators
Another important possibility by which CS genes, LAGs and ARD genes could be linked is the post-transcriptional co-regulation of their expression through common miRNAs. Among the CS genes, 40 have thus far been experimentally validated as being the targets of 39 miRNAs. Of these miRNAs, almost all have targets reported to be involved in cancer and atherosclerosis and many have targets associated with other age-related conditions and longevity (Table 1). Notably, a large number of these miRNAs were indeed found to be directly involved in ARDs (Table 2). For example, out of the 38 miRNAs regulating the expression of both CS and cancer genes, miRNAs belonging to the miR-17, miR-19, miR-21, miR-24, miR-155, miR-214, miR-221, miR-372, and miR-373 families are oncogenic (oncomirs), while miRNAs of the let-7, miR-1, miR-8, miR-15, miR-29, miR-34, miR-101, miR-124, miR-125, miR-127, miR-145 families have a tumor suppressor activity [38, 39, 40, 41]. In fact, as seen in Table 2, all the miRNAs common to CS and atherosclerosis, AD or type 2 diabetes are also cancer-associated.
Table 2. Families of common miRNAs reported as being involved in CS and ARDs
2.4. Cancer genes and miRNAs bridge CS with other ARDs
There are many common genes for the major human ARDs [22, 23, 25]. What are these genes and how do they contribute to the overlaps between CS and ARD genes? As already mentioned (see section 2.1), the CS genes are highly over-represented among LAGs and in all major ARDs and aging-associated conditions (Table 1). This also follows from the enrichment analysis as shown in Fig. 5.
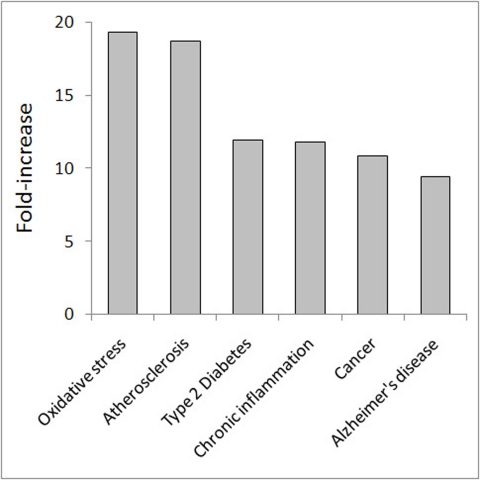
Figure 5. Enrichment of genes involved in ARDs and aging-associated conditions among CS genes. The fold-increase was computed as the ratio between the number of observed genes vs. the expected value. In all cases, the fold-increase was highly significant (p < E-25).
The highest fold-increase (19.3-fold vs. the expected value; p < E-25) was found for oxidative stress. This was quite expected as oxidative stress is one of the major CS inducers [42, 43]. The unexpected observation, however, was that the second highest fold-increase value was found for atherosclerosis (18.7-fold; p < E-25), which is almost twice as high as that for cancer (10.9-fold; p < E-25) and other ARDs (10-12-fold; p < E-25). However, further analysis revealed that cancer genes are the primary determinants of the links between CS and ARDs. Indeed, when the cancer genes were removed from the other ARD sets, no significant enrichment was found for any of these sets in CS. In contrast, after the removal of atherosclerosis, diabetes or Alzheimer's disease genes from the cancer set, the enrichment value for cancer genes in CS remained almost unchanged. Thus, the cancer genes are central in linking CS with other ARDs. Of note, the enrichment for CS genes with cancer genes increases when they are also represented in another or several other ARDs (Table 3). In particular, this can explain the high fold-increase in the case of atherosclerosis, since almost all atherosclerosis genes in CS are also cancer-associated (51 of 52). Though the impact of CS on the development of other ARDs is only beginning to be unveiled, accumulating evidence suggests a role of vascular cell senescence in atherosclerosis [44] and a clear cellular senescence component in type 2 diabetes [45] and Alzheimer's disease [46]. Altogether, our findings indicate that (i) cancer genes (together with miRNAs) determine the links between CS and other major human ARDs, and (ii) CS is particularly enriched in genes which are common to both cancer and other ARD(s).
Table 3. Enrichment of different sets of ARD genes in CS genes
3. Common signaling pathways
While analyzing the PPI networks of longevity and major human ARDs, we found that about half of the common proteins are related to signal transduction [23]. Moreover, we showed that the vast majority of these proteins are hubs, thus playing a central role in linking different ARDs. Therefore, our next question was whether there are common signaling pathways to CS and other sets examined in this study. Enrichment analysis could serve as a tool for answering this question. We found that several pathways are particularly enriched across CS, ARDs and aging-associated processes (Suppl. Table 3). Surprisingly, among them are many growth-promoting pathways such as the MAPK signaling, insulin signaling, mTOR signaling, ErbB signaling, neurotrophin signalling, etc (Fig. 6). This might seem paradoxical, since an irreversible growth arrest is the major (though not the only) feature of CS. However, recent findings shed light on this apparent discrepancy, clearly demonstrating that stimulation of cells, which have ceased proliferation, with growth-promoting mediators induces CS. In other words, growth-promoting pathways convert reversible quiescence into senescence [12, 47, 48]. Such an activation eventually leads to an enhanced secretion of cytokines, chemokines, proteases and ROS by senescent cells [49], collectively termed as the senescence-associated secretory phenotype (SASP) [9, 50]. This could be especially relevant to aging organisms since they display a persistent activation of growth-promoting pathways [13, 51]. As such, senescent cells are likely to create a pro-inflammatory/tumorigenic microenvironment which promotes the development of aging-associated conditions (chronic inflammation and oxidative stress) and ARDs [6, 51, 53]. In addition, the formation of the “senescent” microenvironment could be greatly attributed to the pathways ensuring cell-cell and cell-ECM (extracellular matrix) interactions, such as “Focal adhesion” and “Regulation of actin cytoskeleton”, which we also found to be significantly enriched across CS and ARDs (Fig. 6, Suppl. Table 3). In particular, the importance of these pathways in CS is highlighted by the fact that the downregulation of caveolin-1, a central regulator of focal adhesion kinase activity and actin stress fiber formation [54] resulted in the re-entry of senescent human fibroblasts into cell cycle and restoration of their clonogenic potential [55]. Remarkably, focal adhesion is among the most enriched pathways in the common signaling network for the major human ARDs and longevity [23].
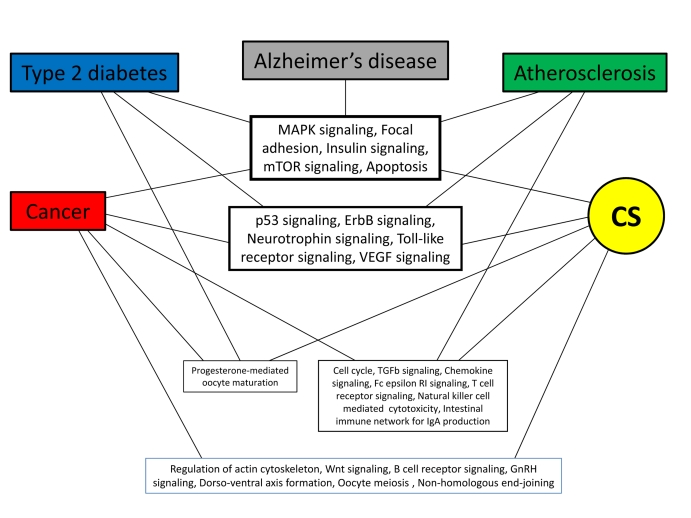
Figure 6. Common pathways enriched across CS and ARDs. Pathways directly involved in specific pathologies were excluded in order to remove bias. See also Suppl. Table 3.
Given the over-representation of growth-promoting and cell-cell/ECM contact pathways in CS, it was expected to see cancer-associated pathways in the enrichment list, since cell proliferation and growth promotion are intrinsic properties of the cancer cells. Indeed, the most over-represented category in CS is “Pathways in cancer” followed by the pathways for specific forms of cancer, including “Bladder cancer”, “Prostate cancer”, “Colorectal cancer”, “Chronic myeloid leukemia”, “Glioma”, “Melanoma”, and others and tumor suppressor “p53 signaling” (Suppl. Table 3). It should however be kept in mind that the final outcome (CS or cancer) of activation or inhibition of these pathways depends on many additional factors, discussed in details elsewhere [21].
All the above mentioned signaling pathways are also involved in cellular response to stress, further linking CS with aging and ARDs. For example, the CS-associated Gadd45 proteins, prominent stress sensors, are involved in the determination of the stressed cell fate via interactions with p53, MAPK, mTOR and other growth-promoting signaling pathways (reviewed by Moskalev et al. [56]). An age-related decrease in Gadd45 inducibility could promote tumorigenesis, immune disorders and insulin resistance [56]. Depending on the severity of stress and activated gene modules, the CS-associated proteins could either mediate DNA repair with subsequent cell survival (quiescence or re-entry to cell cycle), or induce CS or apoptosis (for details see: [57]). A common event in the stressed cells is a down-regulation of insulin/IGF1-Akt-mTOR axis by up-regulated p53 [58-64]. The resultant temporary quiescence period allows the repair of the DNA damage and the return to the routine cell cycle. However, if the level of stress is too high and/or its duration is too long, the cell cycle arrest turns into CS or apoptosis [64-67]. The induction of CS is mediated by the reactivation of the initially repressed PI3K/Akt-mTOR pathway [68, 69]. Of note, its activation is also considered a hallmark of organismal aging [8, 70, 71]. Accordingly, the inhibition of mTOR signaling with rapamycin decreased the hypertrophic phenotype of senescent cells in vitro [47, 72], extended the lifespan and delayed cancer in mice, even when the treatment was initiated later in life [73]. Thus, the common signaling pathways and the very mechanisms of CS induction link it to aging and ARDs.
Concluding remarks
Our study shows that CS is tightly interconnected to aging, longevity and ARDs, either by sharing common genes and regulators or by PPIs and eventually by common pathways. The identification of a common molecular basis is an important step towards understanding the relationships between all these conditions. The next natural step would be the integration of these data with gene/miRNA expression profiles. Such integration could further highlight the key players in linking CS and ARDs. However, this is not a trivial task as the vast majority of data concerning CS derives from in vitro studies on fibroblasts while ARDs are well studied in a variety of cells and in vivo systems. Broadening the CS investigation by including more cell types, 3D in vitro models and in vivo studies will help in developing a more holistic view on the CS phenomenon.
Our analysis also provides the basis for new predictions since the genes common to both cancer and other ARD(s) are highly likely candidates to be involved in CS and vice versa. In addition, a higher connectivity, evolutionary conservation among vertebrates and essentiality may increase the probability for CS genes to be found as being involved in ARD(s).
Another interesting finding is the similarity between the patterns of evolutionary conservation of CS and cancer genes, which suggests the co-evolution of these two phenomena and calls for a wide comparative study. Of special interest would be the investigation of CS in species with exceptional longevity and resistance to tumors, such as bowhead whales. The comparative studies could shed more light on the links between cancer and CS, and may help in understanding why cancer genes stand at the crossroad of CS and ARDs.
An important point for future investigations is examining the role of CS in the formation of an aging microenvironment and its impact on the pathogenesis of ARDs. In turn, the CS phenotype could be modulated by microenvironment. As demonstrated by Choi et al. [74], the interaction of senescent cells with ECM from young cells is sufficient to restore their replicative potential and youthful morphotype. Our present and previous [23] in silico analyses together with experimental data [55] indicate that a special emphasis should be put on focal adhesion and its interactions with growth-promoting pathways.
The principal questions remain as to when it is worthwhile to induce and when to inhibit CS. What strategy is preferable, anti- or pro-CS? These questions stem, in particular, from the suggested antagonistic nature of CS in cancer [6, 75]. The same dual role of CS could also be true for other ARDs but yet has to be established. Whatever the case, it seems quite plausible that the links between CS and ARDs may vary at different stages of disease and life. Induction of CS was proposed as a potential anti-cancer therapy [76, 77]. Alternatively, the recent study of Baker et al. [10] showed that the drug-induced clearance of senescent cells from an early age delayed the onset of several age-related conditions such as sarcopenia, cataracts and loss of adipose tissue in progeroid mice, and these beneficial effects were also pronounced when the elimination of the senescent cells was initiated in the adults. As a feasible tool for enhancing the clearance of senescent cells, Krizhanovsky et al. [78] suggested immunostimulatory therapy. To some extent, in favor of the anti-CS strategy are the results of our preliminary analysis hinting that the pro-longevity genes are rather anti-CS while the pro-CS genes dominate among the anti-longevity genes (unpublished data). Given the potential benefits of the anti-CS approach, an intriguing possibility could be based on iPS technology. Using this technology, Lapasset et al. [79] demonstrated that senescent cells and the cells derived from centenarians could be reprogrammed and eventually rejuvenated. As a perspective for an in vivo application of this method, “a possible scenario may be that after several rounds of iP, the microenvironment itself would also assume a younger phenotype” [80]. Future studies of the different aspects of the links between CS and ARDs will help in selecting the most adequate therapeutic strategy.
Materials and Methods
A list of genes that have been established as being involved in CS was compiled from scientific literature and manually curated. The selection of genes was based on two lines of evidence: 1) genetic or RNA interference (RNAi) interventions (gene knockout, partial or full loss-of-function mutations, RNAi-induced gene silencing, overexpression) which reportedly cause cells to either induce, inhibit or reverse CS, and 2) genes shown to be markers of CS. The lists of LAGs and the genes involved in ARDs and aging-associated processes (oxidative stress and chronic inflammation) were obtained from databases and scientific literature as described in detail elsewhere [23, 24, 81]. The list of differentially expressed miRNAs in CS and of those which have been shown to affect CS was gathered and manually curated from the scientific literature [82-86]. Annotations regarding the involvement of miRNAs in different ARDs were taken from the Human MicroRNA Disease Database ([87], http://cmbi.bjmu.cn/hmdd). Oncogene/Tumor Suppressor classification of cancer-associated miRNAs was done according to Wang et al. [38]. Experimentally validated targets of miRNAs have been obtained from the TarBase database ([88], http://diana.cslab.ece.ntua.gr/tarbase/) and updated by data mining from the scientific literature.
Evolutionary conservation of CS genes was analyzed using the InParanoid7 – Eukaryotic Ortholog Groups database ([34], http://inparanoid.sbc.su.se). To exclude inparalogs, we have used the default threshold score of 0.05. Essentiality of human genes was evaluated based on the data on mouse lethal phenotypes, which were retrieved from the Mouse Genome Informatics database ([89], www.informatics.jax.org). Functional and pathway analyses were performed with the tools provided by DAVID Bioinformatics Resources 6.7 ([90, 91], http://david.abcc.ncifcrf.gov) using data from the KEGG database ([92], http://www.genome.jp/kegg/pathway.html) and Gene Ontology ([93], http://www.geneontology.org). Protein-protein interacttion (PPI) data from the BioGRID database ([27], http://thebiogrid.org), human interactome release 3.1.71, was used for the analysis of connectivity and interconnectivity. The largest fraction of proteins which forms a continuous network was used as a measure of interconnectivity. To generate control data, simulations with sets of randomly selected proteins from the interactome were performed. The size of protein sets ranged from 50 to 8000 genes, with a step of 50. In each case, one hundred simulations were run and the relation between the size of the set and the fraction of genes interconnected by chance was quantified. The simulations and the creation of the microRNA-regulated PPI CS network were done using YABNA (Yet Another Biological Networks Analyzer). The YABNA software program and the algorithm for the construction of miRNA-regulated PPI networks have been previously described in detail ([24, 25], http://www.netage-project.org). The graphical output of the CS miRNA-regulated PPI network was generated using Cytoscape 2.8.0 ([94], http://www.cytoscape.org/).
The statistical package for the social sciences (SPSS, Inc., Chicago, IL) software was used for the statistical evaluation of the results. Significance of the difference between mean values was calculated using the Student's t-test. The difference between observed and expected values was evaluated using the chi-square test. Pearson's coefficient of correlation was used for pair-wise correlative analysis. Differences were considered significant at p-value less than 0.05. Statistical analysis of the enrichment of GO categories and KEGG pathways was carried out using DAVID Bioinformatics Resources 6.7 ([90, 91], http://david.abcc.ncifcrf.gov), with Bonferroni correction.
Acknowledgments
This work was funded by the European Union FP7 Health Research Grant number HEALTH-F4-2008-202047 (to V.E.F.) and the Israel Ministry of Science and Technology (to A.B.).
Conflicts of Interest
The authors of this manuscript have no conflict of interest to declare.
References
- 1. Hayflick L and Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961; 25: 585 -621. .
- 2. Gershon H and Gershon D. Paradigms in aging research: a critical review and assessment. Mech Ageing Dev. 2000; 117: 21 -28. [PubMed] .
- 3. Jeyapalan JC and Sedivy JM. Cellular senescence and organismal aging. Mech Ageing Dev. 2008; 129: 467 -474. [PubMed] .
- 4. Rubin H. Cell aging in vivo and in vitro. Mech. Ageing Dev.. 1997; 98: 1 -35. [PubMed] .
- 5. Phipps SM, Berletch JB, Andrews LG, Tollefsbol TO. Aging cell culture: methods and observations. Methods Mol Biol. 2007; 371: 9 -19. [PubMed] .
- 6. Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005; 120: 513 -522. [PubMed] .
- 7. Campisi J. Cellular senescence: putting the paradoxes in perspective. Curr Opin Genet Dev. 2011; 21: 107 -112. [PubMed] .
- 8. Blagosklonny MV. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006; 5: 2087 -2102. [PubMed] .
- 9. Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009; 11: 973 -979. [PubMed] .
- 10. Baker DJ, Wijshake T, Tchkonia T, Lebrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16(Ink4a)-positive senescent cells delays ageing-associated disorders. Nature. 2011; doi: 10.1038/nature10600 .
- 11. Sikora E, Arendt T, Bennett M, Narita M. Impact of cellular senescence signature on ageing research. Ageing Res Rev. 2011; 10: 146 -52. [PubMed] .
- 12. Blagosklonny MV. Cell cycle arrest is not senescence. Aging (Albany NY). 2011; 3: 94 -101. [PubMed] .
- 13. Blagosklonny MV and Hall MN. Growth and aging: a common molecular mechanism. Aging (Albany NY). 2009; 1: 357 -362. [PubMed] .
- 14. Muller M. Cellular senescence: molecular mechanisms, in vivo redox considerations. Antioxid Redox Signal. 2009; 11: 59 -98. [PubMed] .
- 15. Jun JI and Lau LF. Cellular senescence controls fibrosis in wound healing. Aging (Albany NY). 2010; 2: 627 -631. [PubMed] .
- 16. Hayflick L. 1994; How and Why We Age. Ballantine Books New York .
- 17. Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006; 311: 1257 [PubMed] .
- 18. Seluanov A, Hine C, Bozzella M, Hall A, Sasahara TH, Ribeiro AA, Catania KC, Presgraves DC, Gorbunova V. Distinct tumor suppressor mechanisms evolve in rodent species that differ in size and lifespan. Aging Cell. 2008; 7: 813 -823. [PubMed] .
- 19. Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, Khosla S, Jensen MD, Kirkland JL. Fat tissue, aging, and cellular senescence. Aging Cell. 2010; 9: 667 -684. [PubMed] .
- 20. Rattan SI and Ali RE. Hormetic prevention of molecular damage during cellular aging of human skin fibroblasts and keratinocytes. Ann N Y Acad Sci. 2007; 1100: 424 -430. [PubMed] .
- 21. Rodier F and Campisi J. Four faces of cellular senescence. J Cell Biol. 2011; 192: 547 -556. [PubMed] .
- 22. Budovsky A, Tacutu R, Yanai H, Abramovich A, Wolfson M, Fraifeld V. Common gene signature of cancer and longevity. Mech Ageing Dev. 2009; 130: 33 -39. [PubMed] .
- 23. Wolfson M, Budovsky A, Tacutu R, Fraifeld V. The signaling hubs at the crossroad of longevity and age-related disease networks. Int J Biochem Cell Biol. 2009; 41: 516 -520. [PubMed] .
- 24. Tacutu R, Budovsky A, Fraifeld VE. The NetAge database: a compendium of networks for longevity, age-related diseases and associated processes. Biogerontology. 2010; 11: 513 -522. [PubMed] .
- 25. Tacutu R, Budovsky A, Wolfson M, Fraifeld VE. MicroRNA-regulated protein-protein interaction networks: how could they help in searching for pro-longevity targets? Rejuvenation Res. 2010; 13: 373 -377. [PubMed] .
- 26. Curran SP and Ruvkun G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007; 3: e56 [PubMed] .
- 27. Stark C, Breitkreutz BJ, Chatr-Aryamontri A, Boucher L, Oughtred R, Livstone MS, Nixon J, Van Auken K, Wang X, Shi X, Reguly T, Rust JM, Winter A, Dolinski K, Tyers M. The BioGRID Interaction Database: 2011 update. Nucleic Acids Res. 2011; 39: Database issue D698 -704. [PubMed] .
- 28. Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabási AL. The human disease network. Proc Natl Acad Sci U S A. 2007; 104: 8685 -8690. [PubMed] .
- 29. Jeong H, Mason SP, Barabási AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001; 411: 41 -42. [PubMed] .
- 30. Zotenko E, Mestre J, O'Leary DP, Przytycka TM. Why do hubs in the yeast protein interaction network tend to be essential: reexamining the connection between the network topology and essentiality. PLoS Comput Biol. 2008; 4: e1000140 [PubMed] .
- 31. Williams GC. Pleiotropy, natural selection, and the evolution of senescence. Evolution. 1957; 11: 398 -411. .
- 32. Jordan IK, Rogozin IB, Wolf YI, Koonin EV. Essential genes are more evolutionarily conserved than are nonessential genes in bacteria. Genome Res. 2002; 12: 962 -968. [PubMed] .
- 33. Waterhouse RM, Zdobnov EM, Kriventseva EV. Correlating traits of gene retention, sequence divergence, duplicability and essentiality in vertebrates, arthropods, and fungi. Genome Biol Evol. 2011; 3: 75 -86. [PubMed] .
- 34. Ostlund G, Schmitt T, Forslund K, Köstler T, Messina DN, Roopra S, Frings O, Sonnhammer EL. InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res. 2010; 38: (Database issue) D196 -203. [PubMed] .
- 35. Buffenstein R. The naked mole-rat: a new long-living model for human aging research. J Gerontol A Biol Sci Med Sci. 2005; 60: 1369 -1377. [PubMed] .
- 36. Seluanov A, Hine C, Azpurua J, Feigenson M, Bozzella M, Mao Z, Catania KC, Gorbunova V. Hypersensitivity to contact inhibition provides a clue to cancer resistance of naked mole-rat. Proc Natl Acad Sci U S A. 2009; 106: 19352 -19357. [PubMed] .
- 37. Greenacre CB. Spontaneous tumors of small mammals. Vet Clin North Am Exot Anim Pract. 2004; 7: 627 -651. [PubMed] .
- 38. Wang D, Qiu C, Zhang H, Wang J, Cui Q, Yin Y. Human microRNA oncogenes and tumor suppressors show significantly different biological patterns: from functions to targets. PLoS One. 2010; 5: pii -e13067. .
- 39. Vo DT, Qiao M, Smith AD, Burns SC, Brenner AJ, Penalva LO. The oncogenic RNA-binding protein Musashi1 is regulated by tumor suppressor miRNAs. RNA Biol. 2011; 8: 817 -828. .
- 40. Fabbri M, Ivan M, Cimmino A, Negrini M, Calin GA. Regulatory mechanisms of microRNAs involvement in cancer. Expert Opin Biol Ther. 2007; 7: 1009 -1019. [PubMed] .
- 41. Boominathan L. The guardians of the genome (p53, TA-p73, and TA-p63) are regulators of tumor suppressor miRNAs network. Cancer Metastasis Rev. 2010; 29: 613 -639. [PubMed] .
- 42. Toussaint O, Royer V, Salmon M, Remacle J. Stress-induced premature senescence and tissue ageing. Biochem Pharmacol. 2002; 64: 1007 -1009. [PubMed] .
- 43. Ben-Porath I and Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005; 37: 961 -976. [PubMed] .
- 44. Minamino T and Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ Res. 2007; 100: 15 -26. [PubMed] .
- 45. Erusalimsky JD. Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol. 2009; 106: 326 -332. [PubMed] .
- 46. Flanary BE, Sammons NW, Nguyen C, Walker D, Streit WJ. Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res. 2007; 10: 61 -74. [PubMed] .
- 47. Demidenko ZN and Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008; 7: 3355 -3361. [PubMed] .
- 48. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011; 12: 21 -35. [PubMed] .
- 49. Krtolica A, Parrinello S, Lockett S, Desprez P, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc Natl Acad Sci U S A. 2001; 98: 12072 -12077. [PubMed] .
- 50. Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008; 6: 2853 -2868. [PubMed] .
- 51. Piper MD, Selman C, McElwee JJ, Partridge L. Separating cause from effect: how does insulin/IGF signalling control lifespan in worms, flies and mice? J Intern Med. 2008; 263: 179 -191. [PubMed] .
- 52. Vasile E, Tomita Y, Brown LF, Kocher O, Dvorak HF. Differential expression of thymosin beta-10 by early passage and senescent vascular endothelium is modulated by VPF/VEGF: evidence for senescent endothelial cells in vivo at sites of atherosclerosis. FASEB J. 2001; 15: 458 -466. [PubMed] .
- 53. Price JS, Waters JG, Darrah C, Pennington C, Edwards DR, Donell ST, Clark IM. The role of chondrocyte senescence in osteoarthritis. Aging Cell. 2002; 1: 57 -65. [PubMed] .
- 54. Cho KA, Ryu SJ, Oh YS, Park JH, Lee JW, Kim HP, Kim KT, Jang IS, Park SC. Morphological adjustment of senescent cells by modulating caveolin-1 status. J Biol Chem. 2004; 279: 42270 -42278. [PubMed] .
- 55. Cho KA, Ryu SJ, Park JS, Jang IS, Ahn JS, Kim KT, Park SC. Senescent phenotype can be reversed by reduction of caveolin status. J Biol Chem. 2003; 278: 27789 -27795. [PubMed] .
- 56. Moskalev AA, Smit-McBride Z, Shaposhnikov MV, Plyusnina EN, Zhavoronkov A, Budovsky A, Tacutu R, Fraifeld VE. Gadd45 proteins: Relevance to aging, longevity and age-related pathologies. Ageing Res Rev. 2011; doi:10.1016/j.arr.2011.09.003 .
- 57. Erol A. Deciphering the intricate regulatory mechanisms for the cellular choice between cell repair, apoptosis or senescence in response to damaging signals. Cell Signal. 2011; 23: 1076 -1081. [PubMed] .
- 58. Levine AJ, Feng Z, Mak TW, You H, Jin S. Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev. 2006; 20: 267 -275. [PubMed] .
- 59. Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005; 102: 8204 -8209. [PubMed] .
- 60. Budanov AV and Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008; 134: 451 -460. [PubMed] .
- 61. Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010; 107: 9660 -9664. [PubMed] .
- 62. Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY). 2010; 2: 344 -352. [PubMed] .
- 63. Maki CG. Decision-making by p53 and mTOR. Aging (Albany NY). 2010; 2: 324 -326. [PubMed] .
- 64. Serrano M. Shifting senescence into quiescence by turning up p53. Cell Cycle. 2010; 9: 4256 -4257. [PubMed] .
- 65. Leontieva OV, Gudkov AV, Blagosklonny MV. Weak p53 permits senescence during cell cycle arrest. Cell Cycle. 2010; 9: 4323 -4327. [PubMed] .
- 66. Long JS and Ryan KM. p53 and senescence: a little goes a long way. Cell Cycle. 2010; 9: 4050 -4051. .
- 67. Galluzzi L, Kepp O, Kroemer G. TP53 and MTOR crosstalk to regulate cellular senescence. Aging (Albany NY). 2010; 2: 535 -537. [PubMed] .
- 68. Leontieva OV and Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY). 2010; 2: 924 -935. [PubMed] .
- 69. Wesierska-Gadek J. mTOR and its link to the picture of Dorian Gray - re-activation of mTOR promotes aging. Aging (Albany NY). 2010; 2: 892 -893. [PubMed] .
- 70. Blagosklonny MV. Aging: ROS or TOR. Cell Cycle. 2008; 7: 3344 -3354. [PubMed] .
- 71. McCormick MA, Tsai SY, Kennedy BK. TOR and ageing: a complex pathway for a complex process. Philos Trans R Soc Lond B Biol Sci. 2011; 366: 17 -27. [PubMed] .
- 72. Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009; 8: 1888 -1895. [PubMed] .
- 73. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009; 460: 392 -395. [PubMed] .
- 74. Choi HR, Cho KA, Kang HT, Lee JB, Kaeberlein M, Suh Y, Chung IK, Park SC. Restoration of senescent human diploid fibroblasts by modulation of the extracellular matrix. Aging Cell. 2011; 10: 148 -157. [PubMed] .
- 75. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010; 5: 99 -118. [PubMed] .
- 76. Xue J, Zender L, Miething C, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumor clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007; 445: 656 -660. [PubMed] .
- 77. Nardella C, Clohessy JG, Alimonti A, Pandolfi PP. Pro-senescence therapy for cancer treatment. Nat Rev Cancer. 2011; 11: 503 -511. [PubMed] .
- 78. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008; 134: 657 -667. [PubMed] .
- 79. Lapasset L, Milhavet O, Prieur A, Besnard E, Babled A, Aït-Hamou N, Leschik J, Pellestor F, Ramirez JM, De Vos J, Lehmann S, Lemaitre JM. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011; 25: 2248 -2253. [PubMed] .
- 80. Abramovich A, Muradian KK, Fraifeld VE. Have we reached the point for in vivo rejuvenation? Rejuvenation Res. 2008; 11: 489 -492. [PubMed] .
- 81. de Magalhaes JP, Budovsky A, Lehmann G, Costa J, Li Y, Fraifeld V, Church GM. The Human Ageing Genomic Resources: online databases and tools for biogerontologists. Aging Cell. 2009; 8: 65 -72. [PubMed] .
- 82. Maes OC, Sarojini H, Wang E. Stepwise up-regulation of microRNA expression levels from replicating to reversible and irreversible growth arrest states in WI-38 human fibroblasts. J Cell Physiol. 2009; 221: 109 -119. [PubMed] .
- 83. Li G, Luna C, Qiu J, Epstein DL, Gonzalez P. Alterations in microRNA expression in stress-induced cellular senescence. Mech Ageing Dev. 2009; 130: 731 -741. [PubMed] .
- 84. Bonifacio LN and Jarstfer MB. MiRNA profile associated with replicative senescence, extended cell culture, and ectopic telomerase expression in human foreskin fibroblasts. PLoS One. 2010; 5: pii -e12519. .
- 85. Borgdorff V, Lleonart ME, Bishop CL, Fessart D, Bergin AH, Overhoff MG, Beach DH. Multiple microRNAs rescue from Ras-induced senescence by inhibiting p21(Waf1/Cip1). Oncogene. 2010; 29: 2262 -2271. [PubMed] .
- 86. Marasa BS, Srikantan S, Martindale JL, Kim MM, Lee EK, Gorospe M, Abdelmohsen K. MicroRNA profiling in human diploid fibroblasts uncovers miR-519 role in replicative senescence. Aging (Albany NY). 2010; 2: 333 -343. [PubMed] .
- 87. Lu M, Zhang Q, Deng M, Miao J, Guo Y, Gao W, Cui Q. An analysis of human microRNA and disease associations. PLoS One. 2008; 3: e3420 [PubMed] .
- 88. Papadopoulos GL, Reczko M, Simossis VA, Sethupathy P, Hatzigeorgiou AG. The database of experimentally supported targets: a functional update of TarBase. Nucleic Acids Res. 2009; 37: D155 -158. [PubMed] .
- 89. Ringwald M and Eppig JT. Mouse mutants and phenotypes: Accessing information for the study of mammalian gene function. Methods. 2011; 53: 405 -410. [PubMed] .
- 90. Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003; 4: P3 [PubMed] .
- 91. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4: 44 -57. [PubMed] .
- 92. Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010; 38: Database issue D355 -360. [PubMed] .
- 93. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000; 25: 25 -29. [PubMed] .
- 94. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13: 2498 -2504. [PubMed] .