



Introduction
Apoptosis, the programmed cell death, is finely regulated at gene level resulting in the orderly and efficient removal of damaged cells such as those occurring following DNA damage or during development [1]. The machinery of apoptosis is complex and involves many signaling pathways. Apoptosis can be triggered in a cell through either the caspase-mediated extrinsic or intrinsic pathways. Both pathways converge to activate the effector apoptotic caspases resulting ultimately in morphological and biochemical cellular alterations, characteristics of apoptosis [2]. Usually, the balance between the pro-apoptotic and anti-apoptotic protein regulators is a critical key point to determine if a cell undergoes apoptosis. The induction of apoptosis as result of DNA damage in precancerous lesions can remove potentially harmful cells, thereby blocking tumor growth. Deregulation of this death process is associated with unchecked cell proliferation, development and progression of cancer and cancer resistance to drug therapies [3,4]. For that reason, deregulation of apoptosis is considered one of the hallmarks of cancer [5]. Therapeutic strategies targeting molecules involved in apoptotic resistance therefore represent a valid approach to be pursued in order to restore cancer cells sensitivity to apoptosis and overcome the ineffectiveness of the treatments [6,7]. This article focuses on the mechanisms of apoptosis, how defects along the apoptotic pathway contribute to cancer development and drug resistance and, briefly, how apoptosis can be used as a vehicle of targeted treatment in cancer.
Morphological and biochemical changes in apoptosis
From the morphological point of view apoptotic cells show a characteristic cytoplasmic cell shrinkage, budding of plasma membrane, membrane exposure of phosphatidylserine (PS) on extracellular side, chromatin condensation and DNA fragmentation [8,9]. The plasma membrane is intact throughout the total process. The expression of PS in the outer layers of the cell membrane allows early recognition of dead cells by macrophages, resulting in phagocytosis without the release of proinflammatory cellular components [10]. At the later stage of apoptosis some of the morphological features include membrane blebbing, ultrastructural modification of cytoplasmic organelles and loss of membrane integrity [11]. Usually phagocytic cells engulf apoptotic cells before apoptotic bodies occur [12]. Apoptosis is primarily executed by a family of proteases known as the caspases (cysteinyl, aspartate-specific proteases) [13]. Caspases are central to the mechanism of apoptosis as they are both the initiators (caspase-2, -8, -9 and -10, primarily responsible for the beginning of the apoptotic pathway) and the executors (caspase-3, -6 and -7, responsible for the definite cleavage of cellular components) of cell death [14]. After being produced as inactive proteins (zymogens or pro-caspases), the initiator caspases auto-activate through auto-proteolysis, a process that is facilitated by their interaction with specific adapter molecules [15]. Once activated, the initiator caspases cleave off the executors caspases that perform critical cleavage of specific cellular substrates resulting in the final apoptotic cell death [16]. This caspases activity is responsible of the apoptotic hallmarks, such as chromatin condensation, plasma membrane asymmetry and cellular blebbing. The extensive and irreversible proteolytic activity mediated by executor caspases represents the ultimate outcome of both the extrinsic and the intrinsic apoptotic pathways (see below). Thus, both pathways converge on caspases-3, 6, or -7 that allow disruption of DNA and cellular components inducing the typical morphological changes in apoptosis [17]. Of note, caspases activity has been also extended to non-apoptotic functions such as cell differentiation/maturation suggesting that the caspase cascade may become activated independently of– or without inducing- an apoptotic cascade [18–20].
Extrinsic apoptotic pathway
The extrinsic apoptotic pathway (death receptor-dependent) is initiated by the interaction of cell surface exposed death receptors, belonging to the superfamily of tumor necrosis factor receptor (TNFR), with their respective protein TNF family ligands [21]. Death receptors are structurally defined by an intracellular protein-protein interaction domain, called the death domain (DD), which is critically involved in apoptosis-inducing signalling [22]. The more broadly characterized signaling systems of death receptor-ligands include TNFR1-TNFα, FAS (CD95, APO-1)-FasL, TRAILR1 (DR4)-TRAIL, TRAILR2 (DR5)-TRAIL. Upon death receptor stimulation by its corresponding ligand, the same receptor undergoes oligomerization and a conformational change to reveal its cytoplasmic DD to support homotypic interactions with other DD-containing proteins [21]. The role of adapter proteins (FADD/TRADD) is to sequester, at level of this protein complex, the initiator pro-caspase-8 and/or -10 resulting in the formation of the so-called death-inducing signaling complex (DISC), increasing the local concentration of pro-caspase and promoting the mutual auto-activation [23]. The activation of initiator caspases results in the processing of the downstream effector caspases-3, -6 and -7 whose activation leads to the cleavage of essential substrates for cell viability, inducing cell death (Figure 1) [17]. Some cells do not die in response to the extrinsic pathway alone and require an amplification step that is induced by caspase-8. In this situation, capase-8 targets the BH3-only protein Bid (BH3-interacting-domain death agonist) for cleavage and generate the activated fragment t-Bid; t-Bid then directly activates pro-apoptotic multi-domain proteins to induce mitochondrial outer membrane permeability (MOMP), so this co-engages the intrinsic pathway [3] (Figure 1) (see below).
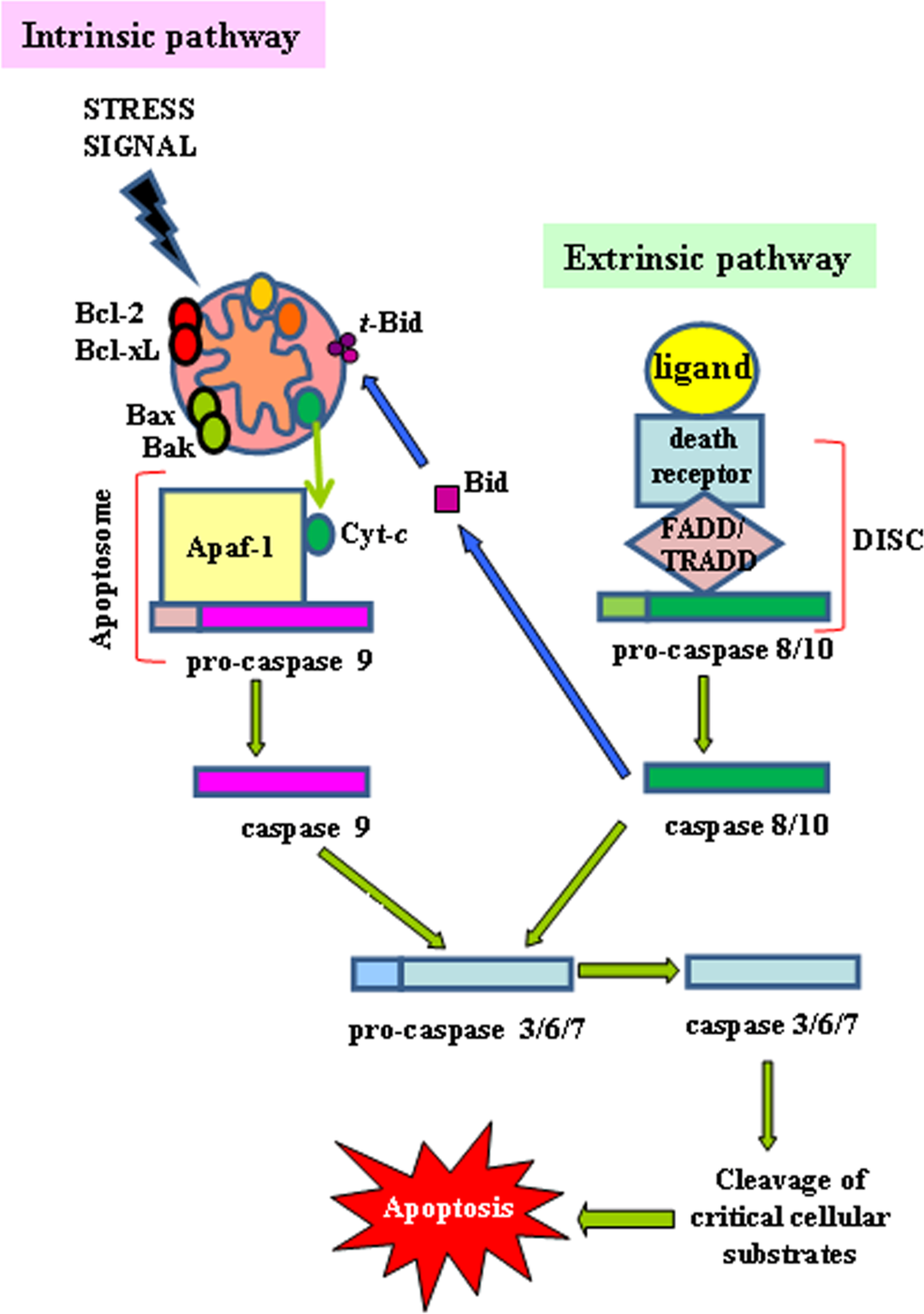
Figure 1. Intrinsic and extrinsic apoptotic pathways The intrinsic (mitochondrial) and the extrinsic (ligands/death receptors) cell death pathways and their convergence through t-Bid are depicted (see text for details)
Intrinsic apoptotic pathway
The intrinsic apoptotic pathway (mitochondria-dependent) is mediated by intracellular signals that converge at the mitochondrial level in response to different stress conditions (i..e, irradiation, treatment with chemotherapeutic agents, etc.) [24]. Internal stimuli such as irreparable genetic damage, hypoxia, extremely high concentrations of cytosolic Ca+ and severe oxidative stress are some triggers of the initiation of the intrinsic mitochondrial pathway [25]. Subsequent activation of pro-apoptotic BH3-only members of the Bcl-2 family (Bax, Bak) neutralizes the antiapoptotic proteins Bcl-2, Bcl-xL, and Mcl-1, leading to disruption of mitochondrial membrane outer membrane permeability (MOMP) so that proteins normally confined in the intermembrane space spread into the cytosol. These proteins include the so-called apopto-genic factors, such as cytochrome-c, which plays a crucial role in activating the mitochondrial-dependent death in the cytosol [26]. Cytochrome-c binds to the cytosolic Apaf-1 (apoptosis protease activating factor-1) and triggers the formation of a complex named apoptosome, which recruits initiator pro-caspase-9 to its caspase recruitment domain (CARD), allowing auto-activation and then proteolysis. The process in turn activates downstream executor caspases-3, -6 and -7 for cleavage of cellular substrates leading to apoptotic cell death (Figure 1) [27,28].
The B-cell lymphoma 2 (Bcl-2) family proteins
The intrinsic pathway is closely regulated by the B-cell lymphoma 2 (Bcl-2) family of intracellular proteins. This proteins family regulates both pro-apoptotic and anti-apoptotic intrinsic pathways controlling the alteration of MOMP [29]. Therefore, by mediating per-meabilization of the mitochondrial membrane, the Bcl-2 proteins serve as an “apoptotic switch” [30]. The Bcl-2 proteins are classified into three subgroups, one group with anti-apoptotic and two with pro-apoptotic function, depending on the composition of typical BH (Bcl-2 Homology) domains, listed from BH1 to BH4 [31,32] (Figure 2). Whereas the BH1 and BH2 domains of bcl-2 are required for dimerization with pro-apoptotic proteins, BH3 domain is crucially important to the interaction between pro-apoptotic and anti-apoptotic proteins and is contained by all family members. The amino-terminal BH4 domain is mainly found in the bcl-2 family members with death-repressing activity, but is also present in some pro-apoptotic molecules. The anti-apoptotic multi-domain group includes Bcl-2, Bcl-xL, Bcl-W, Mcl-1, A1, and Bcl-B, containing from three to four BH domains; the pro-apoptotic multi-domain group includes Bax, Bak and Bok proteins, containing three BH-domains (BH1, BH2 and BH3); and the pro-apoptotic BH3-only proteins group includes Bid (BH3 interacting-domain death agonist), Bim (Bcl-2-like protein 11), Bad (Bcl-2-associated death promoter), Puma (p53 upregulated modulator of apoptosis), Noxa, BMF, HRK and BIK (Figure 3) [33]. While the anti-apoptotic proteins regulate apoptosis by blocking the mitochondrial release of cytochrome-c, the pro-apoptotic proteins act by promoting such release.
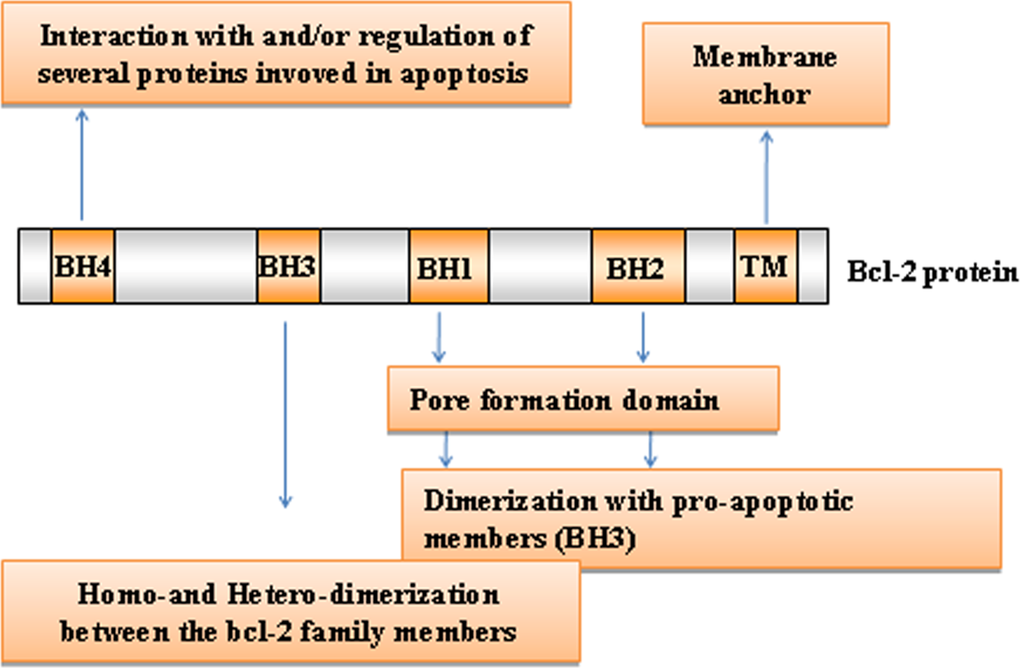
Figure 2. Bcl-2 family members domain composition and function Typical BH (Bcl-2 Homology) domains, listed from BH1 to BH4, are shown. TM: transmembrane domain.
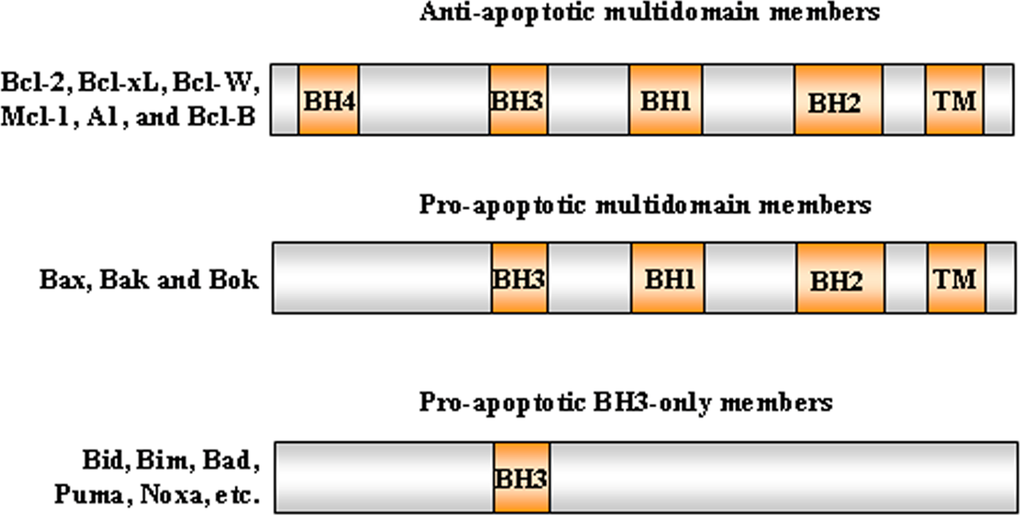
Figure 3. Bcl-2 protein subgroups The Bcl-2 proteins are classified into three subgroups, one group with anti-apoptotic and two with pro-apoptotic function, depending on the composition of the typical BH domains, listed from BH1 to BH4. Representative members of each subfamily are shown.
The balance and protein-protein interactions between Bcl-2 family members is required to determine whether a cell undergoes cell survival or apoptosis. The activation of Bax (cytosolic protein that translocates into mitochondria during induction of apoptosis), and Bak (integral membrane protein located in the mitochondria and endoplasmic reticulum) involves conformational changes that trigger the formation of homo-oligomeric protein complexes that end up altering the mitochondrial membrane permeability [34,35]. The pro-apoptotic BH3-only proteins act upstream of this event binding with high affinity to anti-apoptotic Bcl-2 family members thereby allowing Bax/Bak to elicit MOMP and activation of the caspase cascade [36,37]. Anti-apoptotic multidomain members of the Bcl-2 protein family not only counteract the pore-forming activity of Bax and Bak by engaging in direct inhibitory interactions, but also prevent the generation of pro-apoptotic cytosolic Ca2+ waves either by reducing capacity of endoplasmic reticulum (ER) Ca2+ storage, an effect that is antagonized by Bax and Bak or by interacting with inositol 1,4,5-trisphosphate (IP3) receptor [38,39]. Other apoptotic factors that are released from the mitochondrial intermembrane space into the cytoplasm include apoptosis inducing factor (AIF), second mitochondria-derived activator of caspase (Smac), direct IAP Binding protein with Low pI (DIABLO) and Omi/high temperature requirement protein A (HtrA2) [40].
The inhibitors of apoptosis proteins (IAPs)
Considering that proteolysis is an irreversible process, strict control of caspases-mediated proteolytic cleavage is imperative to prevent inappropriate cell destruction [41]. Negative regulation of caspases function is achieved by IAP proteins family whose principal members in humans are NAIP (BIRC1), cIAP1 (BIRC2), cIAP2 (BIRC3), X-linked IAP (XIAP, BIRC4), Survivin (BIRC5), Apollon (BRUCE, BIRC6), Livin/ML-IAP (BIRC7), and IAP-like protein 2 (ILP2 – BIRC8) [42]. Their characteristic BIR (baculovirus IAP repeat) domain mediates the interaction with various proteins and gives them the ability to bind and inactivate caspases [43]. The activities of IAPs, however, may be suppressed by mitochondrial proteins, such as Omi/HtrA2 and Smac/DIABLO, released into the cytosol during apoptosis (Figure 4). These endogenous IAPs antagonists are able to bind to the BIR domain of IAPs reducing their ability to interact with caspase-3 or -9 thereby restoring their activity [44]. XIAP is the best characterized IAP so far and is generally recognized as the most potent endogenous caspase inhibitor. XIAP anti-apoptotic activity involves inhibition of active executor capsases as well as prevention of initiator caspase-9 activation [45].
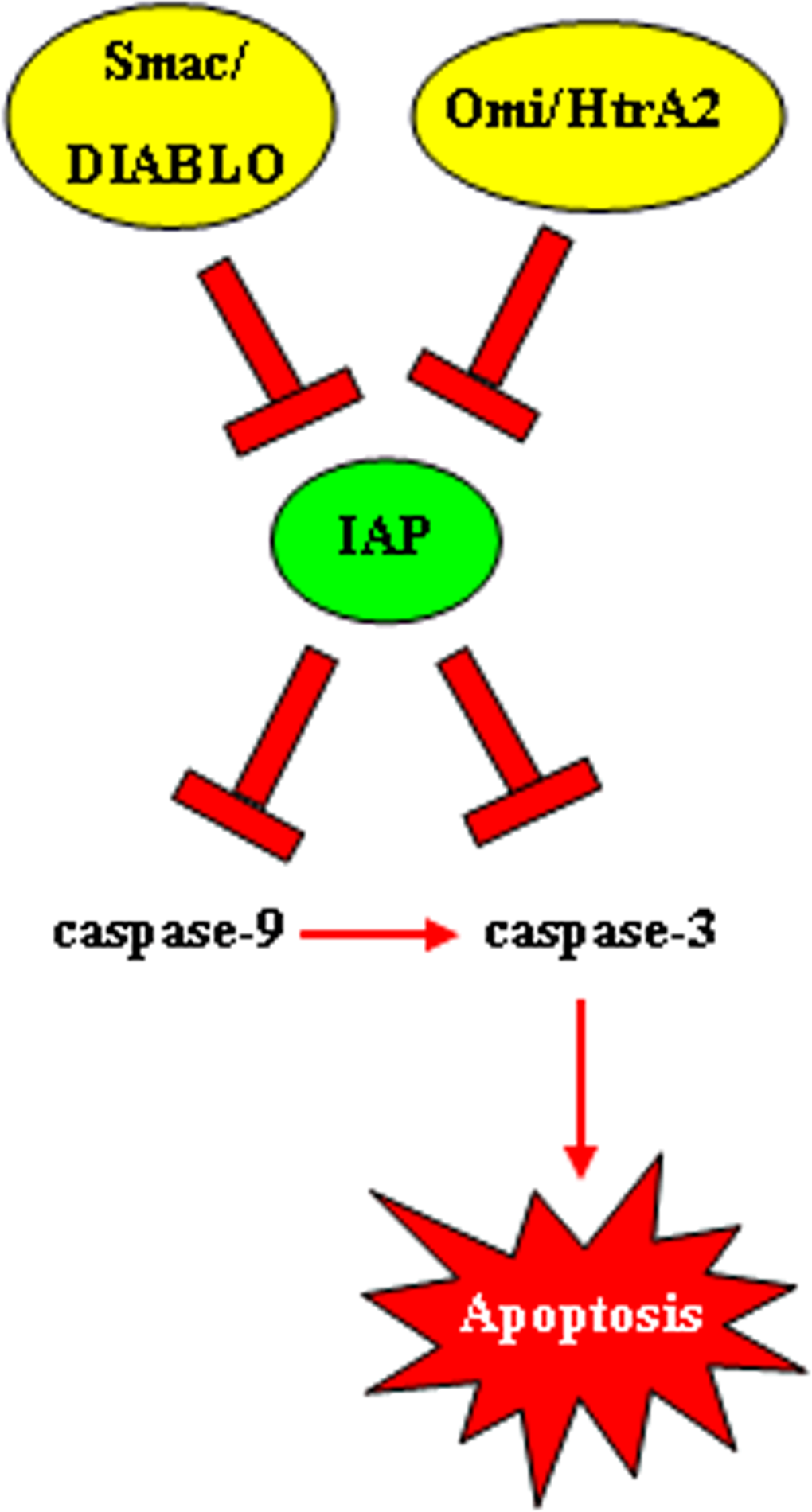
Figure 4. Function of inhibitors of apoptosis proteins (IAPs) IAPs are often overexpressed in cancer and they have the ability to bind and inactivate caspases 9 and 3. The activities of IAPs, on the other hand, may be suppressed by mitochondrial proteins, such as Omi/HtrA2 and Smac/DIABLO, released into the cytosol during apoptosis.
Alterations of the apoptotic pathways
There are many ways through which both the extrinsic and the intrinsic apoptotic pathways may be altered, resulting in reduction of apoptosis or acquisition of apoptosis resistance. They include impaired death receptor signaling, disrupted balance between pro-apoptotic and anti-apoptotic proteins, reduced caspase function and impaired p53 function (Figure 5). Alteration of extrinsic apoptotic signaling has been associated with different types of human tumors, underscoring how the loss of activity of Fas-FasL system [46] or the aberrant expression of cytosolic components of this death receptor apoptotic pathway (i.e., FADD) [47] can contribute to the tumor transformation. Several genetic defects have been proven to contribute to the resistance of tumor cells to Fas-mediated apoptosis. Fas transcriptional silencing is a common oncogenic event in the epithelial transformation, while its mutation has been often associated with B-cell germinal center-derived lymphomas [48]. In acute myelogenous leukemia (AML) reduced or absent expression of FADD has been frequently observed, resulting in resistance to chemotherapy and poor patient prognosis [47,49]. Moreover, in several cancers including neuroblastoma, medulloblastoma, and small cell lung cancer (SCLC), absent or reduced expression of caspase-8 was reported [50–52]. Another resistance mechanism reported in a variety of human tumors is the overexpression of anti-apoptotic protein c-Flip, recruited at the DISC level, that prevents the pro-caspase-8 auto-activation thereby rendering cell resistant to death receptor-mediated apoptosis [53–55].
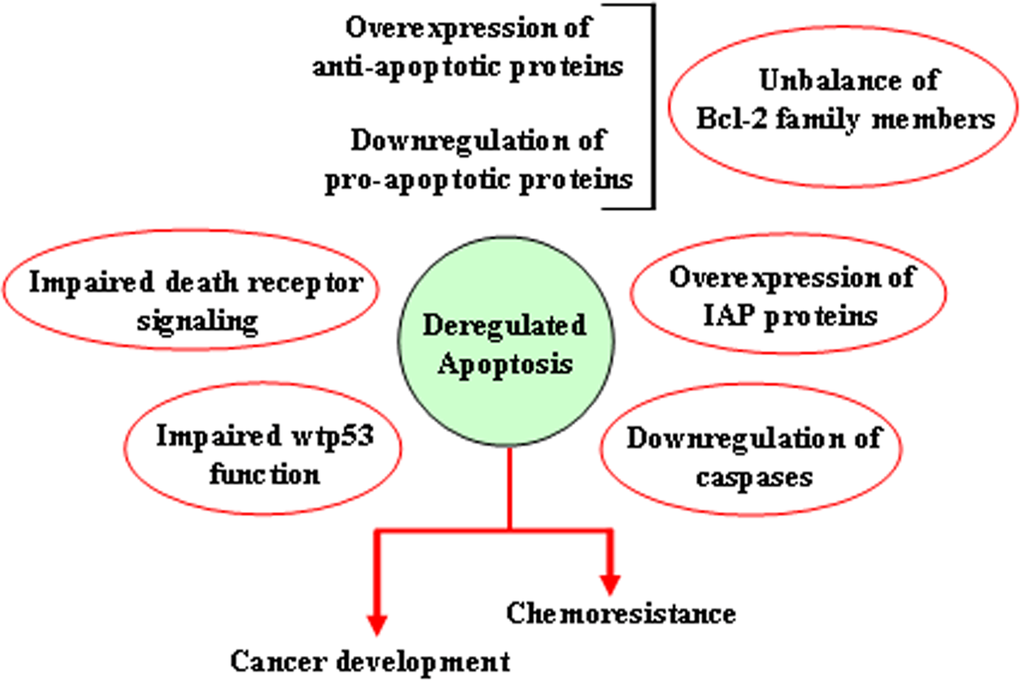
Figure 5. Mechanisms leading to deregulation of apoptosis Schematic representation of the different ways through which both the extrinsic and the intrinsic apoptotic pathways may be altered, resulting in reduction of apoptosis or acquisition of apoptosis resistance.
As for the extrinsic pathway, alteration of some components of the intrinsic apoptotic pathway can play a fundamental role in the development of resistance to chemotherapy in different types of tumors. Disruption in the balance of anti-apoptotic and pro-apoptotic members of the Bcl-2 family results in deregulated apoptosis in the affected cells. This can be due to overexpression of one or more anti-apoptotic proteins or downregulation of one or more pro-apoptotic proteins or a combination of both. Anti-apoptotic Bcl-2 over-expression has been reported in several human cancers, including prostate cancer, diffuse large B-cell lymphoma (DLBCL), melanoma, etc. [56–58], resulting in protection of cancer cells from apoptosis or inhibition of TRAIL-induced apoptosis [59,60]. Overexpression of Bcl-xL has also been reported in colorectal cancer and Kaposi's sarcoma [61,62]. Such overexpression confers a multi-drug resistance phenotype in tumor cells and prevents them from undergoing apoptosis [63]. Thus, high expression levels of anti-apoptotic proteins Bcl-2 and Bcl-xL have been reported to correlate with cisplatin resistance and tumor recurrence in different cancers including non-small cell lung cancer (NSCLC), head and neck, ovarian, and breast [64–68]. On the other hand, mutations in the pro-apoptotic Bax gene have been reported in colorectal cancers and contribute to resistance to anticancer treatments [69]. Increased Bcl-2/Bax ratio has been reported in chronic lymphocytic leukaemia (CLL) patients. [70]. Other examples of alteration of the intrinsic pathway include reduced expression of the basic component of the apoptosome, Apaf-1, in melanomas [71,72], as result of promoter aberrant methylation. In addition, tumor cells resistance to apoptosis also occurs as a result of alteration of mediators that control the intrinsic apoptotic pathway downstream from the apoptosome formation, i.e. acting on caspase activity. In this regard, high level of IAPs expression has been found in different types of cancers, and this evidence is considered a marker of poor prognosis for patients [73,74].
Pharmacological targeting of the apoptotic pathways
Based on this evidence, restoration of apoptotic pathway by drugs targeting both apoptotic pathways constitutes a promising anticancer therapeutic approach. Regarding the extrinsic pathway, the down-regulation of c-Flip by metabolic inhibitors and the promotion of caspase-8 activation by interferon, are some examples of strategies aimed at making tumors responsive to death receptor-induced apoptosis, and more generally, to chemotherapy-induced apoptosis [55,75,76]. The therapeutic importance of inducing apoptosis through the extrinsic pathway also extends to cancer cells that do not show defects in components of that pathway. Indeed, inducing the apoptosis by stimulating the extrinsic pathway can overcome the resistance to therapeutic agents that act by causing DNA damage, as death receptor-dependent apoptosis may occur regardless of the stress response. An example of such therapeutic strategy is represented by the ligand TRAIL known to induce apoptosis in different tumor cell lines [77]. The preferential destructive effect against tumor cells and the apparent absence of systemic toxicity through TRAIL-induced apoptosis, led to the development of antibodies with agonistic activity against the TRAIL death receptors (DR4 and DR5) or soluble recombinant derivatives of TRAIL (sTRAIL) as promising chemotherapeutic agents [78]. An attractive strategy to sensitize resistant malignancies to TRAIL-induced cell death is the design of small molecules that target and promote caspase-8 activation. Through an in silico screening some authors successfully found a small molecule activator of caspase-8 [79]. Experimental validation performed in multiple cell lines, such as leukemic and prostate cells, revealed that CaspPro small molecule promotes caspase-8 activation, caspase-3 activation and PARP cleavage, in the presence of TRAIL, leading to cell death [79]. Owing to its different toxicity for transformed as opposed to normal cells, Apo2/TRAIL shows promise as potential cancer therapy agent [80,81].
As in the extrinsic pathway, mediators of the intrinsic pathway involved both in tumorigenesis and chemo-resistance, are targeted for therapeutic approaches. These anticancer strategies attempt to develop drug-designed inhibitors of anti-apoptotic proteins typically overexpressed in cancer cells, such as Bcl-2, Bcl-xL and IAPs [82]. Efforts to target Bcl-2 proteins involve the development of agents that disrupt Bcl-2 complexes. BH3 mimetics bind to the hydrophobic groove of antiapoptotic proteins mimicking the action of BH3-only proteins in binding to pro-survival proteins, leading to the release of BH3-only proteins from complexes and activation of BAX and BAK. So far, nearly a dozen BH3 mimetics are under investigation as Bcl-2 inhibitors in different phases of human clinical trials such as AT-101 (R-(−)-gossypol) [83,84], ABT-199 (venetoclax) [85], ABT-737 [86], ABT-263 (navitoclax, orally available derivative of ABT-737) [87,88], GX15-070 (obatoclax) [89,90] and TW37 [91]. The field of inhibitors of Bcl-2 family members is in continuous development [92,93], underscoring the importance of these molecules as potent anticancer agents. Moreover, targeting the specific BH4 domain of Bcl-2 is also emerging as a novel strategy for anticancer therapy [94]. Thus, Bcl-2, via its BH4 domain, cooperates with numerous proteins regulating different cellular pathways involved in tumor progression and chemoresistance such as hypoxia and angiogenesis [95–97]. Recently, a small molecule namely BDA-366 was discovered as a potent and effective BH4 domain antagonist, displaying remarkable anticancer activity in vitro and in vivo, thus providing the proof-of-concept of this approach [98]. Another innovative approach to inhibit Bcl-2 comes from the evidence that human bcl-2 gene contains a GC-rich sequence located in its promoter with the potential to form G-quadruplex structures [99] and functions as a transcriptional repressor element. Therefore, G-quadruplex-specific ligands can regulate the transcription of bcl-2 through stabilization of quadruplex structure [100,101].
Interestingly, it has been shown that the tumor suppressor p53, at least in part by transcription independent mechanisms, contributes to cell death induction by BH3 mimetic inhibitors of BCL-xL. In addition to mildly facilitating the ability of compounds to derepress BAX from BCL-xL, p53 also provides a death signal downstream of anti-apoptotic proteins inhibition that is independent from PUMA, as enhanced p53 can substitute for PUMA to promote BAX activation in response to BH3 mimetics [102]. It is thus of particular relevance that p53, even when expressed constitutively under conditions where it does not influence the expression of its pro-apoptotic transcription targets, enhances cell death induced by BCL-xL inhibition [102]. Such results suggest on one hand that BH3 mimetics may not totally substitute for the lack of an active p53 tumor suppressor in cancer cells; on the other hand, they imply that healthy tissues may be more harmed than anticipated when BCL-xL inhibitors are combined with chemotherapeutic agents that even mildly affect p53.
Among the therapeutic strategies targeting IAPs two approaches have being developed, that is the use of antisense oligonucleotides and of small-molecule inhibitors. The XIAP down-regulation through administration of antisense agents carried by an adenoviral vector has been proven effective in inducing apoptosis in chemoresistant ovarian cancer cells [103] and sensitizing lung cancer cells to the radiation treatment [104]. Similarly, the inhibition of XIAP expression with specific oligomers has been shown to induce caspase-3 activity, boosting the apoptotic effect of cisplatin and TRAIL in human prostate androgen-insensitive cancer cells [105]. Moreover, preclinical studies have shown that Smac mimetics can directly trigger cancer cell death or sensitize tumor cells to various cytotoxic therapies, including conventional chemotherapy, radiotherapy, or novel agents. They promote activation of caspases by neutralizing XIAP-mediated caspase inhibition [106]. Therefore, the success of each therapeutic strategy depends mainly on the ability of the therapeutic tool to induce apoptosis either by targeting the overexpressed anti-apoptotic proteins or by stimulating the expression of the pro-apoptotic molecules.
However, it is worth to mention that the cancer genetic background may induce failure of apoptosis by drugs. In this regard, KRAS and the PI3K/AKT/mTOR pathway are frequently dysregulated in cancer and, for such reason, are the most critical targets in clinical oncology. Thus, direct targeting of KRAS has not been successful so far and, similarly, inhibition of the PI3K/AKT/mTOR pathway often results in apoptosis resistance. Using a panel of 20 human KRAS-mutant NSCLC (non-small cell lung cancer) cell lines Hata and collaborators show that most human KRAS-mutant cell lines fail to undergo marked apoptosis in response to AZD6244 (Selumetinib, a potent, selective, and ATP-uncompetitive inhibitor of MEK1/2 kinases) [107] in combination with GDC-0941 (an orally bioavailable inhibitor of class I PI3K) [108], thus suggesting that failure to induce apoptosis may limit the efficacy of combined MEK and PI3K inhibition for KRAS-mutant NSCLCs. This differential apoptotic response induced by MEKi/PI3Ki is not simply explained by variable inhibition of RAS effector pathways but results from differential ability of the MEK and PI3K pathways to modulate the BCL-2 family of apoptotic regulatory proteins [109]. Another recent study reveals that Bcl-xL upregulation is an important mechanism of apoptosis resistance in mutant KRAS cells. Concurrent induction of pro-apoptotic Noxa/Bik and antagonism of Bcl-xL have been shown to synergistically interact to overcome KRAS-mediated apoptosis resistance [110]. These findings highlight a promising therapeutic strategy to overcome apoptosis resistance in KRAS-mutant colorectal cancer cells. Moreover, Corcoran and collaborators identified, by a pooled shRNA-drug screen, a synthetic, lethal interaction of combined Bcl-xL and MEK inhibition to promote tumor regressions in KRAS mutant cancer models [111]. Therefore, a dual-targeted or multitargeted strategy may be more efficient to overcome the resistance due to cancer genetic background.
Oncosuppressor p53 and apoptosis
The tumor suppressor p53 is a transcription factor that, upon DNA damage, is activated to induce sequence-specific target genes involved in either cancer cell growth arrest or apoptosis [112]. Activation of wild-type (wt) p53 occurs in response to genotoxic stress essentially through posttranslational modifications, such as acetylation and phosphorylation, resulting in protein stabilization (by escape from proteasome-mediated degradation) and nuclear localization leading to binding to sequence-specific promoters of target genes as final outcome of its function as transcription factor [113]. The induction of apoptosis by p53 in response to cellular stress is its most conserved function and is crucial for p53 tumor suppression [114]. The apoptotic activation of p53 is central not only for preventing tumor transformation but also for efficient response to therapies aiming at tumor eradication. In response to cellular stress p53 regulates molecules involved in both the death receptor (extrinsic) and mitochondria-dependent (intrinsic) apoptotic pathways [115]. In response to multiple chemotherapeutic drugs two pro-apoptotic members of the TNFR superfamily, Fas/Apo1 and Killer/DR5, are regulated in a p53-dependent manner [116,117]. In addition to stimulating Fas transcription, activated p53 may enhance levels of Fas at the cell surface promoting trafficking of the Fas receptor from the Golgi [118]. Another membrane-bound protein that was identified as a p53 target gene is p53 apoptosis effector related PMP-22 (PERP), although the precise mechanism by which its induction occurs has not being fully elucidated [119] (Figure 6). Regarding the apoptotic function of the intrinsic pathway, p53 seems to play a pivotal role because it modulates both pro-survival and pro-apoptotic Bcl-2 family members. Indeed, a key subset of the Bcl-2 family genes are p53 targets, including Bax, Noxa, PUMA and Bid [120–122] (Figure 6). PUMA gene is extremely effective in inducing apoptotic cell death within few hours and, more importantly, knockout experiments in human colorectal cancer cells showed that PUMA is required for p53-induced apoptosis [123]. Moreover, p53 appears to promote the convergence of the intrinsic and extrinsic pathways through Bid regulation. Indeed, Bid gene has been found to be transcriptionally induced by p53 in response to γ-irradiation [124]. Interestingly, cellular chemo-sensitivity to the DNA-damaging agents doxorubicin and 5-fluorouracil appears to be critically dependent on the presence of wtp53 and Bid. Therefore, the induction of Bid by p53 helps to sensitize the cells to the toxic effects of chemotherapeutic drugs [124]. While the induction of some p53 target genes appears to be sufficient to initiate apoptosis, another class of p53 target genes (i.e., Apaf-1, caspase-6, and Bid) does not efficiently induce apoptosis per se but rather sensitizes cancer cells to the effects of chemotherapeutic agents, improving the apoptotic outcome [124–127]. Moreover, p53 also participates in apoptosis induction in a transcription-independent way by acting directly at mitochondria [128]; mechanistically, p53 interacts with anti-apoptotic Bcl-xL as well as pro-apoptotic Bcl-2 family proteins resulting in releasing of the pro-apoptotic effectors Bax/Bak that elicit cytochrome-c release and procaspase-3 activation [129].
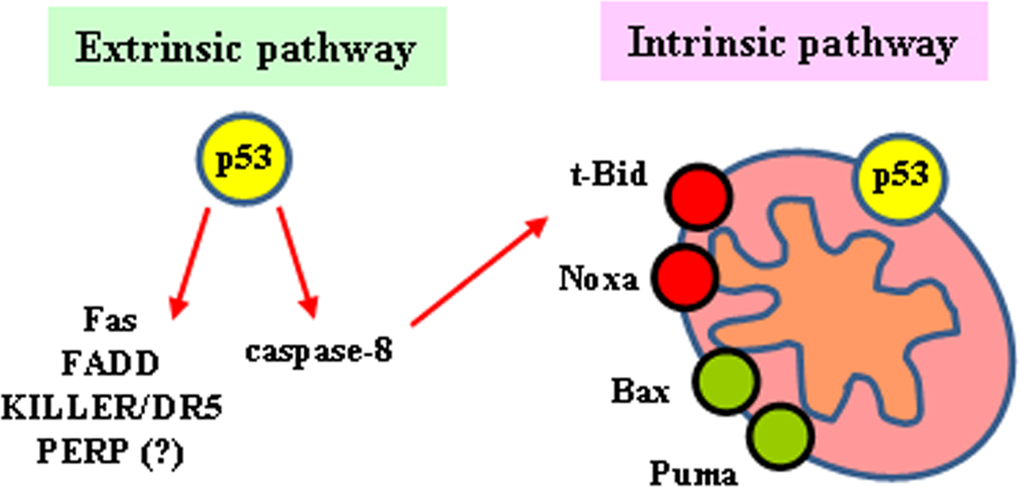
Figure 6. p53-mediated apoptosis Role of p53 in both the extrinsic and the intrinsic pathway and their convergence through t-Bid.
Waking up the guardian: p53 as a druggable target
Because of its critical antitumor function, p53 is frequently targeted for inactivation and suffers disabling mutations or deletions in about 50% of all malignant tumors. The other half of human cancers express wild-type p53 protein that, however, can be inactivated by deregulation of regulatory proteins [130]. Stimulation of disabled p53 pathways has been suggested as a valuable anticancer strategy and, interestingly, activated wtp53 may target cancer cells though sparing the normal ones [131] which is an important concern in clinical studies. The p53 oncosuppressor protein is normally kept at low level because subject to negative regulation by MDM2-dependent proteasome degradation [132]; in response to genotoxic stress, however, p53 undergoes post-translational modifications that allow the protein to escape MDM2 control, accumulate, and become active [133]. The mdm2 gene is amplified in a significant proportion of human tumor types, thereby contributing to tumor development by efficiently reducing the availability of a functional p53 protein [134]. The MDM2-negative regulation of p53 protein can be neutralized by specific protein modifications such as serine 46 (Ser46) phosphorylation [135], a key determinant in shifting the p53 pro-apoptotic transcription in response, for instance, to UV irradiation and chemotherapy [136,137]. In particular, p53-Ser46 phosphorylation by kinase HIPK2 is able to neutralize MDM2-mediated p53 inhibition rescuing p53 transcriptional activity of pro-apoptotic factors such as p53AIP1, PIG3, Bax, Noxa, Puma and Killer/DR5 [138–142]. The interaction between p53 and MDM2 is a promising target in anticancer therapy [143]. To this aim, various peptidomimetic small molecules have been developed as protein-protein interaction blockers [144]. Among these is Nutlin-3, an imidazoline-based MDM2 antagonist that potentially inhibits the p53/MDM2 interaction though maintaining MDM2 E3 ligase activity [145]. The pharmacological action of Nutlin-3 is through both the transcription-dependent and -independent p53 apoptotic pathways [128,146,147]. MDM2 can also trigger, in response to low genotoxic damage, the downregulation of p53 apoptotic activator HIPK2 [148]. In agreement, the use of Nutlin-3 has been shown to mainly induce mitotic arrest rather than apoptosis [149]. Interestingly, co-treatment of cancer cells with zinc ion in the presence of Nutlin-3 can interfere with the interplay between HIPK2, p53 and MDM2 favoring HIPK2 stabilization and induction of p53 apoptotic activity through inhibition of MDM2 ligase activity [150]. In addition, p53 apoptotic activation can be achieved by zinc combination with low-dose doxorubicin (ADR) that used alone does not achieve such effect; mechanistically, zinc supplementation reduces the p53 binding to MDM2, improving the low-dose drug-induced cytotoxic effect and cancer cell apoptosis [151]. Additionally, zinc ion restores the HIPK2/p53 apoptotic pathway that is inhibited by hypoxia [152]. Co-treatment with Nutlin-3 and Bcl-2 inhibitor ABT-737 has been shown to greatly enhance the sensitivity to apoptosis of cancer cells with high MDM2 levels [153], suggesting that the combined inhibition of MDM2 and Bcl-2 could be a multi-target-based anticancer strategy to trigger tumor death [154]. Some p53 activators as small-molecules MDM2 antagonist are in clinical trials [155] ( https://clinicaltrials.gov). In contrast with the majority of the approaches that target the interaction between p53 and MDM2, a new method has been developed aimed at inhibiting the activity of the MDM2/MDM4 complexes by interfering with their heterodimerization [156]. The binding of the peptide mimicking the MDM4 C-terminus tail to MDM2 impairs MDM2-mediated p53 ubiquitination and activates p53-dependent transcription and oncosuppressive activities [156]. MDM4 (also known as HDM4, MDMX or HDMX) is a cytoplasmic protein with p53-activating function under DNA damage conditions. Particularly, MDM4 promotes mitochondrial localization of p53 phosphorylated at Ser46 through MDM4/HIPK2/p53 cytoplasmic assembly, uncovering coordinated repression of molecules with anti-apoptotic activity such as Bcl-2, release of cytochrome-c and apoptosis [157,158]. The existence of nuclear and cytoplasmic complexes able to stimulate the same p53 modification, that is Ser46P, may indicate the presence of overlapping pathways to ensure the proper realization of a crucial process as the apoptosis. These findings highlight the potential therapeutic value of targeting the MDM2/MDM4 heterodimers for p53 apoptotic function.
Pharmacological reactivation of mutant (mut) p53 is an interesting field of research under continuous development aimed at designing new drugs. Some of them exploit the intrinsically unstable nature of mutp53 and therefore the possibility to stabilize the wild-type conformation thus restoring wild-type function and tumor response to therapies. Numerous findings about this subject have been shown and summarized in different reviews [159–161].
MicroRNA and apoptosis
MicroRNAs (miRNAs) are highly conserved, small noncoding RNA molecules, which post-transcriptionally regulate gene expression via inhibition of mRNA translation or inducing degradation of target mRNAs [162]. They are key regulators of many cell processes often deregulated in cancer, including apoptosis. Indeed, it is becoming clear that miRNAs might act as both anti-apoptotic and pro-apoptotic by directly targeting, respectively, pro- or anti-apoptotic mRNAs or their positive regulators [163]. The currently known apoptosis-regulating miRNAs list is expected to expand quickly and hopefully also their therapeutic use; therefore, we just highlight here some miRNAs involved in apoptosis regulation. Among the microRNAs involved in regulating the extrinsic apoptotic pathway, miR-20a, miR-21, miR-196b and miR-590 were reported as potential modulator of Fas/FasL system in different cancer types [164–167], while MiR-34a, miR-181c and miR-187 were shown to directly target TNF-α mRNA [168–170]. Among the microRNAs involved in regulating the intrinsic pathway there are the let-7 family, miR-15a, miR-16-1, miR-204, and miR-608, just to mention a few. The Let-7 family is highly conserved in sequence across animal species and is one of the first identified miRNA families. Let-7 miRNAs have been shown to negatively regulate Bcl-xL expression in human hepatocellular carcinomas and induce apoptosis in cooperation with anti-cancer drug targeting Mcl-1 [171]. Bcl-2 mRNA may be targeted by miR-204 with consequent increase in cells responsiveness to both 5-fluorouracil and oxaliplatin treatments and therefore to apoptotic cell death [172]. MiR-608 has been reported to target Bcl-xL in chordoma malignancy and lung cancer [173]. Notably, numerous miRNAs are also transcriptionally modulated by wtp53 [174] and among them is miR-34a [175,176], a member of the MiR-34 family implicated in cell death/survival signaling. MiR-34a is associated with G1 cell cycle arrest, senescence and apoptosis, thereby possessing a tumor suppressor activity. Deregulation of MiR-34a has been reported in several types of cancers [175,176]. Mutant (mut) p53 was also found to play a role in the regulation of miRNA processing. Garibaldi and collaborators demonstrate that mutp53 proteins modulate the biogenesis of several miRNAs in cancer cells directly interfering with Drosha-p72 association and promoting cell survival and cell migration [177]. They demonstrate a global impact of mutp53 on miRNA biogenesis and suggest that miRNAs are downregulated by mutp53 in order to inactivate tumor suppressive pathways. Of note they found that the endogenous wtp53 has an opposite effect on the expression of mutp53 repressed miRNAs on colon cancer cell lines confirming the contribution of mutp53 gain of oncogenic function (GOF) on miRNA repression [177]. Additional studies on a large scale would help in identifying the entire repertoire of miRNAs negatively downregulated by different mutp53 in different tumor models. According to the authors, the characterization of the entire gene-regulatory networks governed by mutp53-miRNA cross-talks will offer a molecular basis for diagnostic and therapeutic strategies based on miRNA biology. In the meanwhile, developing strategies to block mutp53 GOF may have clinical impact in cancer treatment.
Delivery of miRNAs as synthetic miRNA mimics or antagomirs has emerged as a promising approach to treat cancer. Although different miRNAs are currently in the preclinical stage and ready to enter Phase 1 clinical trials, to date, only two miRNA therapeutics are registered for the treatment of cancers [https://clinicaltrials.gov]. The first therapeutic trial began in 2013 and is a Phase I, open-label, multicenter, dose-escalation study to investigate the safety, pharmacokinetics and pharmacodynamics of MRX34 in patients with unresectable primary liver cancer or advanced/metastatic cancer with or without liver involvement or in patients with hematologic malignancies (Mirna Therapeutics). MRX34 is based on the formulation of miR-34 mimic and the liposomal delivery technology SMARTICLES (Marina Biotech). The second one, began in early 2015, and is an early stage clinical trial of a new therapeutical approach for selected patients with malignant pleural mesothelioma or non-small cell lung cancer. The trial aims to test optimal dose of TargomiRs, an experimental medication consisting of three components, that is, miR-16-based microRNA mimic, a nanoparticle drug delivery system using nonliving bacterial minicells, and an anti-epidermal growth factor receptor antibody as a targeting moiety. The trial is being carried out in three different hospitals in Australia and the study is expected to be completed in mid 2016.
Concluding remarks
Intensive investigation in the last decades on the molecular mechanisms of apoptosis in cancer cells has led to the identification of the several molecules involved in both the intrinsic and the extrinsic apoptotic pathways. This is underscored by the extensive literature that those studies have produced in the last years. Those findings also reported how the many different alterations of key players of the apoptotic mechanisms are responsible of evasion from apoptosis and therefore of tumor development and resistance to therapies. For that reason, evasion from apoptosis is an hallmark of cancer and apoptotic proteins are interesting therapeutic targets. Therefore, this insight into the deregulation of apoptosis has focused the research attention towards the development of apoptosis-reactivating strategies, to be used in the treatment of various types of cancer, that hold great promise for the benefit of patients, although the mechanisms defining their mode of action still need to be unveiled, as recently highlighted [178]. Some molecules or therapeutic strategies are in preclinical trial, others are already in clinical trials, though underscoring the usefulness of such discoveries.
However, the study of apoptosis still presents challenges that should be addressed in future studies. They include, for instance, the study of 3-D cellular models, since most of the findings have been so far produced in 2-D cell culture systems. Knowing that the tumor is a three-dimensional entity and that the environment plays a big role in the cross-talk with cancer cells, it is likely that more physiological studying approach for the manipulation of the apoptotic machinery might give us novel insight into the mechanisms of tumor development and response to therapies. Moreover, additional studies on the development of drugs aiming at targeting, for instance, IAP proteins or mutp53 should take in consideration also the in vivo toxicity and the fact that they should selectively induce apoptosis in malignant and not in normal cells. In conclusion, there is little doubt that drugs that target the deregulated apoptotic pathways could have wide application in the treatment of cancer. The intense effort devoted lately to target the apoptotic pathway is encouraging and supportive for the development of new approaches to drug discovery and therapy.
Funding
This work was supported by AIRC Grant (IG 2015 Id.16742) to GDO.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1. Fuchs Y and Steller H. Programmed cell death in animal development and disease.Cell.2011;147:742-758..
- 2. Wong RSY. Apoptosis in cancer: from pathogenesis to treatment.JECCR.2011;30:87.
- 3. Plati J, Bucur O, Khosravi-Far R. Dysregulation of apoptotic signaling in cancer. Molecular mechanisms and therapeutic opportunities.J Cell Biochem.2008;104:1124-1149..
- 4. Fulda S. Tumor resistance to apoptosis.Int J Cancer.2009;124:515-515..
- 5. Hanahan D and Weinberg RA. Hallmarks of cancer: The next generation.Cell.2011;144:646-674..
- 6. Gimenez-Bonafe P, Tortosa A, Perez-Tomas R. Overcoming drug resistance by enhancing apoptosis of tumor cells.Curr Cancer Drug Targ.2009;9:320-340..
- 7. Fulda S. Targeting apoptosis for anticancer therapy.Sem Cancer Biol.2015;31:84-88..
- 8. Hacker G. The morphology of apoptosis.Cell Tissue Res.2000;301:5-17..
- 9. Saraste A and Pulkki K. Morphologic and biochemical hallmarks of apoptosis.Cardiovasc Res.2000;45:528-537..
- 10. Hengartner MO. Apoptosis: corralling the corpses.Cell.2000;104:325-328..
- 11. Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P, Zhivotovsky B, Blagosklonny MV, Malorni W, Knight RA, Piacentini M, Nagata S, Melino G. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death.Cell Death Diff.2005;12:1463-1467..
- 12. Bucur O, Nat R, Cretoiu D, Popescu LM. Phagocytosis of apoptotic cells by microglia in vitro.J Cell Mol Med.2001;5:438-441..
- 13. Li J and Yuan J. Caspases in apoptosis and beyond.Oncogene.2008;27:6194-6206..
- 14. Thomberry NA and Laxebnik Y. Caspases: enemies within.Science.1998;281:1312-1316..
- 15. Nicholson DW. Caspase structure, proteolytic substrates, and function during apoptotic cell death.Cell Death Diff.1999;6:1028-1042..
- 16. Stennicke HR and Salvesen GS. Caspases - controlling intracellular signals by protease zymogen activation.Biochim Biophys Acta-Port Struct Mol Enzimol.2000;1477:299-306..
- 17. Degterev A, Boyce M, Yuan JY. A decade of caspases.Oncogene.2003;22:8543-8567..
- 18. Pistritto G, Jost M, Srinivasula SM, Baffa R, Poyet JL, Kari C, Lazebnik Y, Rodeck U, Alnemri ES. Expression and transcriptional regulation of caspase-14 in simple and complex epithelia.Cell Death Diff.2002;9:995-1006..
- 19. Pistritto G, Papaleo V, Sanchez P, Ceci C, Barbaccia ML. Divergent modulation of neuronal differentiation by caspase-2 and-9.PLoS ONE.2012;7:e36002.
- 20. Shalini S, Dorstyn L, Dawar S, Kumar S. Old, new and emerging functions of caspases.Cell Death Diff.2015;22:526-39..
- 21. Guicciardi ME, Gores, Gregory J. Life and death by death receptors.FASEB J.2009;23:1625-1637..
- 22. Fulda S and Debatin KM. Death receptor signaling in cancer therapy.Curr Med Chem Anticancer Agents.2003;3:253-262..
- 23. Boatright KM and Salvesen GS. Mechanisms of caspase activation.Curr Opin Cell Biol.2003;6:725-731..
- 24. Green DR and Kroemer G. The pathophysiology of mitochondrial cell death.Science.2004;305:626-629..
- 25. Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilisation in cell death.Physiol Rev.2007;87:99-163..
- 26. Danial NN and Korsmeyer SJ. Cell death: critical control points.Cell.2004;116:205-219..
- 27. Slee EA, Adrain C, Martin SJ. Serial killers: ordering caspase activation events in apoptosis.Cell Death Diff.1999;6:1067-1074..
- 28. Kuribayashi K, Mayes PA, El-Dery WS. What are caspases 3 and 7 doing upstream of the mitochondria?Cancer Biol Ther.2006;5:763-765..
- 29. Giam M, Huang DC, Bouillet P. BH3-only proteins and their roles in programmed cell death.Oncogene.2008;27:S128-36..
- 30. Adams JM and Cory S. The Bcl-2 apoptotic switch in cancer development and therapy.Oncogene.2007;26:1324-1337..
- 31. Danial NN. BCL-2 family proteins: Critical checkpoints of apoptotic cell death.Clin Cancer Res.2007;13:7254-7263..
- 32. Lomonosova E and Chinnadurai G. BH3-only proteins in apoptosis and beyond: an overview.Oncogene.2009;27:S2-S19..
- 33. Youle RJ and Strasser A. The BCL-2 protein family: opposing activities that mediate cell death.Nat Rev Mol Cell Biol.2008;9:47-59..
- 34. Dewson G, Kratina T, Sim HW, Puthalakath H, Adams JM, Colman PM, Kluck RM. To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3 : Groove interactions.Mol Cell.2008;30:369-380..
- 35. Bleicken St, Classen M, Padmavathi PVL, Ishikawa T, Zeth K, Steinhoff HJ, Bordignon E. Molecular details of Bax activation, oligomerization, and membrane insertion.J Biol Chem.2010;285:6636-6647..
- 36. Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu HC, Kim H, Cheng EH, Tjandra N, Walensky LD. BAX activation is initiated at a novel interaction site.Nature.2008;455:1076-U6..
- 37. Brunelle JK and Letai A. Control of mitochondrial apoptosis by the Bcl-2 family.J Cell Sci.2009;122:437-441..
- 38. Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, Korsmeyer SJ. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum.Proc Natl Acad Sci USA.2005;102:105-110..
- 39. Distelhorst CW and Bootman MD. Bcl-2 interaction with the inositol 1,4,5-trisphosphate receptor: Role in Ca2+ signaling and disease.Cell Calcium.2011;50:234-241..
- 40. Hegde R, Srinivasula SM, Zhang Z, Wassell R, Mukattash R, Cilenti L, DuBois G, Lazebnik Y, Zervos AS, Fernandes-Alnemri T, Alnemri ES. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction.J Biol Chem.2002;277:432-438..
- 41. Pop C and Salvesen GS. Human caspases: activation, specificity, and regulation.J Biol Chem.2009;284:21777-21781..
- 42. Salvesen GS and Duckett CS. IAP proteins: Blocking the road to death's door.Nat Rev Mol Cell Biol..2002;3:401-410..
- 43. Berthelet J and Dubrez L. Regulation of apoptosis by inhibitors of apoptosis (IAPs).Cells.2013;2:163-87..
- 44. LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG. IAP-targeted therapies for cancer.Oncogene.2008;27:6252-6275..
- 45. Mace PD, Shirley S, Day CL. Assembling the building blocks: structure and function of inhibitor of apoptosis proteins.Cell Death Differ.2010;17:46-53..
- 46. Müschen M and Beckmann MW. CD95 ligand expression as a criterion of malignant transformation in breast cancer.J Pathol.2000;191:468-470..
- 47. Tourneur L, Buzyn A, Chiocchia G. FADD adaptor in cancer.Med Immunol.2005;4:1.
- 48. Müschen M, Rajewsky K, Krönke M, Küppers R. The origin of CD95-gene mutations in B-cell lymphoma.Trends Immunol.2002;23:75-80..
- 49. Tourneur L, Delluc S, Levy V, Valesi F, Radford-Weiss I, Legrand O, Vargftig J, Boix C, Macintyre EA, Varet B, Chiocchia G, Buzyn A. Absence or low expression of fas-associated protein with death domain in acute myeloid leukemia cells predicts resistance to chemotherapy and poor outcome.Cancer Res.2004;64:8101-8108..
- 50. Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA, Behm FG, Look AT, Lahti JM, Kidd VJ. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN.Nat Med.2000;6:529-535..
- 51. Shivapurkar N, Toyooka S, Eby MT, Huang CX, Sathyanarayana UG, Cunningham HT, Reddy JL, Brambilla E, Takahashi T, Minna JD, Chaudhary PM, Gazdar AF. Differential inactivation of caspase-8 in lung cancers.Cancer Biol Ther.2002;1:65-69..
- 52. Zuzak TJ, Steinhoff DF, Sutton LN, Phillips PC, Eggert A, Grotzer MA. Loss of caspase-8 mRNA expression is common in childhood primitive neuroectodermal brain tumor/medulloblastoma.Eur J Cancer.2002;38:83-91..
- 53. Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schröter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular FLIP.Nature.1997;388:190-195..
- 54. Bagnoli M, Canevari S, Mezzanzanica D. Cellular FLICE-inhibitory protein (c-FLIP) signalling: A key regulator of receptor-mediated apoptosis in physiologic context and in cancer.Int J Biochem Cell Biol.2010;42:210-213..
- 55. Shirley S and Micheau O. Targeting c-FLIP in cancer.Cancer Lett.2013;332:141-150..
- 56. Gandour-Edwards R, Mack PC, Devere-White RW, Gumerlock PH. Abnormalities of apoptotic and cell cycle regulatory proteins in distinct histopathologic components of benign prostatic hyperplasia.Prost Cancer Prost Dis.2004;7:321-326..
- 57. Abramson JS and Shipp MA. Advances in the biology and therapy of diffuse large B-cell lymphoma: moving toward a molecularly targeted approach.Blood.2005;106:1164-1174..
- 58. Watanabe A, Yasuhira S, Inoue T, Kasai S, Shibazaki M, Takahashi K, Akasaka T, Masuda T, Maesawa C. BCL2 and BCLxL are key determinants of resistance to antitubulin chemotherapeutics in melanoma cells.Exp Dermatol.2013;22:518-523..
- 59. Raffo AJ, Perlman H, Chen MW, Day ML, Streitman JS, Buttyan R. Overexpression of bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo.Cancer Res.1995;55:4438.
- 60. Fulda S, Meyer E, Debatin KM. Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression.Oncogene.2000;21:2283-2294..
- 61. Foreman KE, Wrone-Smith T, Boise LH, Thompson CB, Polverini PJ, Simonian PL, Nunez G, Nickoloff BJ. Kaposi's sarcoma tumor cells preferentially express Bcl-xL.Am J Pathol.1996;149:795-803..
- 62. Krajewska M, Moss SF, Krajewski S, Song K, Holt PR, Reed JC. Elevated expression of Bcl-X and reduced Bak in primary colorectal adenocarcinomas.Cancer Res.1996;56:2422-2427..
- 63. Minn AJ, Rudin CM, Boise LH, Thompson CB. Expression of Bcl-xL can confer a multidrug resistance phenotype.Blood.1995;86:1903-1910..
- 64. Han JY, Hong EK, Choi BG, Park JN, Kim KW, Kang JH, Jin JY, Park SY, Hong YS, Lee KS. Death receptor 5 and Bcl-2 protein expression as predictors of tumor response to gemcitabine and cisplatin in patients with advanced non-small-cell lung cancers.Med Oncol.2003;20:355-362..
- 65. Erovic BM, Pelzmann M, Grasl MCh, Pammer J, Kornek G, Brannath W, Selzer E, Thurnher D. Mcl-1, vascular endothelial growth factor-R2, and 14-3-3sigma expression might predict primary response against radiotherapy and chemotherapy in patients with locally advanced squamous cell carcinomas of the head and neck.Clin Cancer Res.2005;11:8632-8636..
- 66. Williams J, Lucas PC, Griffith KA, Choi M, Fogoros S, Hu YY, Liu JR. Expression of Bcl-xL in ovarian carcinoma is associated with chemoresistance and recurrent disease.Gynecol Oncol.2005;96:287-295..
- 67. Michaud WA, Nichols AC, Mroz EA, Faquin WC, Clark JR, Begum S, Westra WH, Wada H, Busse PM, Ellisen LW, Rocco JW. Bcl-2 blocks cisplatin-induced apoptosis and predicts poor outcome following chemoradiation treatment in advanced oropharyngeal squamous cell carcinoma.Clin Cancer Res.2009;15:1645-1654..
- 68. Yang D, Chen MB, Wang LQ, Yang L, Liu CY, Lu PH. Bcl-2 expression predicts sensitivity to chemotherapy in breast cancer: a systematic review and meta-analysis.JECCR.2013;32:105.
- 69. Miquel C, Borrini F, Grandjouan S, Aupérin A, Viguier J, Velasco V, Duvillard P, Praz F, Sabourin JC. Role of bax mutations in apoptosis in colorectal cancers with microsatellite instability.Am J Clin Pathol.2005;23:562-570..
- 70. Pepper C, Hoy T, Bentley DP. Bcl-2/Bax ratios in chronic lymphocytic leukaemia and their correlation with in vitro apoptosis and clinical resistance.Br J Cancer.1997;76:935-938..
- 71. Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M, Opitz-Araya X, McCombie R, Herman JG, Gerald WL, Lazebnik YA, Cordón-Cardó C, Lowe SW. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma.Nature.2001;409:207-211..
- 72. Baldi A, Santini D, Russo P, Catricala' C, Amantea A, Picardo M, Tatangelo F, Botti G, Dragonetti E, Murace R, Tonini G, Natali PG, Baldi F, Paggi MG. Analysis of APAF-1 expression in human cutaneous melanoma progression.Exp Dermatol.2004;13:93-97..
- 73. Schimmer AD. Inhibitor of apoptosis proteins: translating basic knowledge into clinical practice.Cancer Res.2004;64:7183-7190..
- 74. Fulda S. Inhibitor of apoptosis proteins in hematological malignancies.Leukemia.2009;23:467-476..
- 75. Fulda S and Debatin KM. IFNgamma sensitizes for apoptosis by upregulating caspase-8 expression through the Stat1 pathway.Oncogene.2002;21:2295-2308..
- 76. Subramanian K, Hirpara JL, Tucker-Kellogg L, Tucker-Kellogg G, Pervaiz S. FLIP: A flop for execution signals.Cancer Lett.2013;332:151-155..
- 77. Almasan A and Ashkenazi A. Apo2L/TRAIL: apoptosis signaling, biology, and potential for cancer therapy.Cytok Growth Fact Rev.2003;14:337-348..
- 78. Amarante-Mendes GP and Griffith TS. Therapeutic applications of TRAIL receptor agonists in cancer and beyond.Pharmacol Ther.2015;155:117-131..
- 79. Bucur O, Gaidos G, Yatawara A, Pennarum B, Rupasinghe C, Roux J, Andrei S, Guo B, Panaititu A, Pellegrini M, Mierke DF, Khosravi-Far R. A novel caspase 8 selective small molecule potentiates TRAIL-induced cell death.Sci Rep.2015;5:9893.
- 80. Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, Blackie C, Chang L, McMurtrey AE, Hebert A, DeForge L, Koumenis IL, Lewis D, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand.J Clin Invest.1999;104:155-162..
- 81. LeBlanc H and Ashkenazi A. Apo2/TRAIL and its death and decoy receptors.Cell Death Diff.2003;10:66-75..
- 82. Kang MH and Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy.Clin Cancer Res.2009;15:1126-1132..
- 83. Sonpavde G, Matveev V, Burke JM, Caton JR, Fleming MT, Hutson TE, Galsky MD, Berry WR, Karlov P, Holmlund JT, Wood BA, Brookes M, Leopold L. Randomized phase II trial of docetaxel plus prednisone in combination with placebo or AT-101, an oral small molecule Bcl-2 family antagonist, as first-line therapy for metastatic castration-resistant prostate cancer.Annal Oncol.2012;23:1803-1808..
- 84. Stein MN, Hussain M, Stadler WM, Liu G, Tereshchenko IV, Goodin S, Jeyamohan C, Kaufman HL, Mehnert J, Di Paola RS. A Phase II study of AT-101 to overcome Bcl-2-mediated resistance to androgen deprivation therapy in patients with newly diagnosed castration-sensitive metastatic prostate cancer.Clin Genitour Cancer.2016;14:22-27..
- 85. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DCS, Hymowitz SG, Jin S, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets.Nat Med.2013;19:202-208..
- 86. Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumors.Nature.2005;435:677-681..
- 87. Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, Roberts L, Tahir SK, Mao Y, Yang X, Zhang HC, Fesik S, Rosenberg SH, Elmore SW. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor.Cancer Res.2008;68:3421-3428..
- 88. Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, Carney DA, He SZ, Huang DCS, Xiong H, Cui Y, Busman TA, McKeegan EM, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease.J Clin Oncol.2012;30:488-496..
- 89. Chiappori A, Williams C, Northfelt DW, Adams JW, Malik S, Edelman MJ, Rosen P, Van Echo DA, Berger MS, Haura EB. Obatoclax mesylate, a pan-bcl-2 inhibitor, in combination with docetaxel in a phase 1/2 trial in relapsed non-small-cell lung cancer.J Thorac Oncol.2014;9:121-125..
- 90. Theile D, Allendorf D, Koehler BC, Jassowicz A, Weiss J. Obatoclax as a perpetrator in drug-drug interactions and its efficacy in multidrug resistance cell lines.J Pharm Pharmacol.2015;67:1575-1584..
- 91. Zeitlin BD, Joo E, Dong Z, Warner K, Wang G, Nikolovska-Coleska Z, Wang S, Nor JE. Antiangiogenic effect of TW37, a small-molecule inhibitor of Bcl-2.Cancer Res.2006;66:8698-8706..
- 92. Tao ZF, Hasvold L, Wang L, Wang X, Petros AM, Park CH, et al. Discovery of a potent and selective Bcl-xL inhibitor with in vivo activity.ACS Med Chem Lett.2014;5:1088-1093..
- 93. Bruncko M, Wang L, Sheppard GS, Phillips DC, Tahir SK, Xue J, et al. Structure-guided design of a series of MCL-1 inhibitors with high affinity and selectivity.J Med Chem.2015;58:2180-2194..
- 94. Vervloessem T, La Rovere R, Bultynck G. Antagonizing Bcl-2's BH4 domain in cancer.Aging (Albany NY).2015;7:748-749. https://doi.org/10.18632/aging.100828
- 95. Trisciuoglio D, Gabellini C, Desideri M, Ragazzoni Y, De Luca T, Ziparo E, Del Bufalo D. Involvement of BH4 domain of bcl-2 in the regulation of HIF-1-mediated VEGF expression in hypoxic tumor cells.Cell Death Diff.2011;18:1024-1035..
- 96. Trisciuoglio D, De Luca T, Desideri M, Passeri D, Gabellini C, Scarpino S, Liang C, Orlandi A, Del Bufalo D. Removal of the BH4 domain from Bcl-2 protein triggers an autophagic process that impairs tumor growth.Neoplasia.2013;15:315-327..
- 97. Gabellini C, De Luca T, Trisciuoglio D, Desideri M, Di Martile M, Passeri D, Candiloro A, Biffoni M, Rizzo MG, Orlandi A, Del Bufalo D. BH4 domain of bcl-2 protein is required for its proangiogenic function under hypoxic condition.Carcinogenesis.2013;34:2558-2567..
- 98. Han B, Park D, Li R, Xie M, Owonikoko T, Zhang G, Sica GL, Ding C, Zhou J, Magis AT, Chen ZG, Shin DM, Ramalingam S, Khuri FR, Curran WJ, Deng X. Small-molecule Bcl-2 BH4 antagonist for lung cancer therapy.Cancer Cell.2015;27:852-863..
- 99. Dexheimer TS, Sun D, Hurley LH. Deconvoluting the structural and drug-recognition complexity of the G-quadruplex-forming region upstream of the bcl-2 P1 promoter.J Am Chem Soc.2006;128:5404-5415..
- 100. Lin J, Hou JQ, Xiang HD, Yan YY, Gu YC, Tan JH, Li D, Gu LQ, Ou TM, Huang ZS. Stabilization of G-quadruplex DNA by C-5-methyl-cytosine in bcl-2 promoter:implications for epigenetic regulation.Biochem Biophys Res Commun.2013;433:368-373..
- 101. Sun H, Xiang J, Shi Y, Yang Q, Guan A, Li Q, Yu L, Shang Q, Zhang H, Tang Y, Xu G. A newly identified G-quadruplex as a potential target regulating Bcl-2 expression.Biochim Biophys Acta.2014;1840:3052-3057..
- 102. Le Pen J, Laurent M, Sarosiek K, Vuillier C, Gautier F, Montessuit S, Martinou JC, Letaï A, Braun F, Juin PP. Constitutive p53 heightens mitochondrial apoptotic priming and favors cell death induction by BH3 mimetic inhibitors of BCL-xL.Cell Death Dis.2016;7:e2083.
- 103. Sasaki H, Sheng Y, Kotsuji F, Tsang BK. Down-regulation of X-linked inhibitor of apoptosis protein induces apoptosis in chemoresistant human ovarian cancer cells.Cancer Res.2000;60:5659-5666..
- 104. Holcik M, Yeh C, Korneluk RG, Chow T. Translational upregulation of X-linked inhibitor of apoptosis (XIAP) increases resistance to radiation induced cell death.Oncogene.2000;19:4174-4177..
- 105. Amantana A, London CA, Iversen PL, Devi GR. X-linked inhibitor of apoptosis protein inhibition induces apoptosis and enhances chemotherapy sensitivity in human prostate cancer cells.Mol Cancer Ther.2004;3:699-707..
- 106. Fulda S. Promises and challenges of Smac mimetics as cancer therapeutics.Clin Cancer Res.2015;21:5030-5036..
- 107. Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, Cockerill M, Cartlidge S, Smith PD. AZD6244 (ARRY-142886), a potent inhibitor of mitogen activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models.Mol Cancer Ther.2007;6:2209-2219..
- 108. Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, Friedman LS, Hayes A, Hancox TC, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d] pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer.J Med Chem.2008;51:5522-5532..
- 109. Hata AN, Yeo A, Faber AC, Lifshits E, Chen Z, Cheng KA, Walton Z, Sarosiek KA, Letai A, Heist RS, Mino-Kenudson M, Wong KK, Engelman JA. Failure to induce apoptosis via BCL-2 family proteins underlies lack of efficacy of combined MEK and PI3K inhibitors for KRAS-mutant lung cancers.Cancer Res.2014;74:3146-3156..
- 110. Okamoto K, Zaanan A, Kawakami H, Huang S, Sinicrope FA. Reversal of mutant KRAS-mediated apoptosis resistance by concurrent Noxa/Bik induction and Bcl-2/Bcl-xL antagonism in colon cancer cells.Mol Cancer Res.2015;13:659-669..
- 111. Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, Greninger P, Brown RD, Godfrey JT, Cohoon TJ, Song Y, Lifshits E, Hung KE, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models.Cancer Cell.2013;23:121-128..
- 112. Vousden KH and Lane DP. P53 in health and disease.Nat Rev Mol Cell Biol.2007;8:275-283..
- 113. Brooks CL and Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation.Curr Opin Cell Biol.2003;15:164-171..
- 114. Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis - the p53 network.J Cell Sci.2003;116:4077-4085..
- 115. Vousden KH. P53: Death Star.Cell.2000;103:691-694..
- 116. Wu GS, Burns TF, McDonald III ER, Jiang W, Meng R, Krantz ID, Kao G, Gan DD, Zhou JY, Muschel R, Hamilton SR, Spinner NB, Matkowitz S, et al. KILLER/DR5 is a DNA damage-induced p53-regulated death receptor gene.Nat Genet.1997;17:141-143..
- 117. Muller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M, Krammer PH. P53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs.J Exp Med.1998;188:2033-2045..
- 118. Bennet M, Macdonald K, Chan SW, Luzio JP, Simari R, Weissberg. Cell surface trafficking of Fas: a rapid mechanisms of p53-mediated apoptosis.Science.1998;282:290-293..
- 119. Attardi LD, Reczek EE, Cosmas C, Demicco EG, McCurrach ME, Lowe SW, Jacks T. PERP, an apoptosi-associated target of p53, is a novel member of the PMP-22/gas3 family.Genes Dev.2000;14:704-718..
- 120. Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis.Science.2000;288:1053-1058..
- 121. Nakano K and Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53.Mol Cell.2001;7:683-694..
- 122. Thornborrow EC, Patel S, Mastropietro AE, Schwartzfarb EM, Manfredi JJ. A conserved intronic response element mediates direct p53-dependent transcriptional activation of both the human and murine bax gene.Oncogene.2002;21:990-999..
- 123. Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L. PUMA mediates the apoptotic response to p53 in colorectal cancer cells.Proc Natl Acad Sci U S A.2003;100:1931-1936..
- 124. Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ, El-Deiry WS. BID regulation by p53 contributes to chemosensitivity.Nat Cell Biol.2002;4:842-849..
- 125. Moroni MC, Hickman ES, Denchi EL, Caprara G, Colli E, Cecconi F, Muller H, Helin K. Apaf-1 is a transcriptional target for E2F and p53.Nat Cell Biol.2001;3:553-558..
- 126. MacLachlan TK and El-Deiry WS. Apoptotic threshold is lowered by p53 transactivation of caspase-6.Proc Natl Acad Sci.2002;99:9492-9497..
- 127. Sax JK and El-Deiry WS. P53 downstream targets and chemosensitivity.Cell Death Diff.2003;10:413-417..
- 128. Chi SW. Structural insights into the transcription-independent apoptotic pathway of p53.BMB Rep.2014;47:167-172..
- 129. Tomita Y, Marchenko N, Erster S, Nemajerova A, Dehner A, Klein C, Pan H, Kessler H, Pancoska P, Moll UM. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization.JBC.2006;281:8600-8606..
- 130. Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway.Nat Rev Cancer.2009;9:862-873..
- 131. D'Orazi G, Marchetti A, Crescenzi M, Coen S, Sacchi A, Soddu S. Exogenous wt-p53 protein is active in transformed cells but not in their nontransformed counterparts: implications for cancer gene therapy without tumor targeting.J Gene Med.2000;2:11-21..
- 132. Momand J, Wu HH, Dasgupta G. MDM2 - master regulator of the p53 tumor suppressor protein.Gene.2000;242:15-29..
- 133. Harris SL and Levine AJ. The p53 pathway: positive and negative feedback loops.Oncogene.2005;24:2899-2908..
- 134. Momand J, Jung D, Wilczynski S, Niland J. The MDM2 gene amplification database.Nucleic Acids Res.1998;26:3453-3459..
- 135. Di Stefano V, Blandino G, Sacchi A, Soddu S, D'Orazi G. HIPK2 neutralizes MDM2 inhibition rescuing p53 transcriptional activity and apoptotic function.Oncogene.2004;23:5185-5192..
- 136. Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y, Taya Y. p53AlP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53.Cell.2000;102:849-862..
- 137. Mayo LD, Seo YR, Jackson MW, Smith ML, Guzman JR, Korgaonkar CK, Donner DB. Phosphorylation of human p53 at serine 46 determines promoter selection and whether apoptosis is attenuated or amplified.J Biol Chem.2005;280:25953-25959..
- 138. D'Orazi G, Cecchinelli B, Bruno T, Manni I, Higashimoto Y, Saito S, Gostissa M, Coen S, Marchetti A, Del Sal G, Piaggio G, Fanciulli M, Appella E, et al. Homeodomain-interacting protein kinase 2 phosphorylates p53 at Ser46 and mediates apoptosis.Nat Cell Biol.2002;4:11-19..
- 139. Di Stefano V, Rinaldo C, Sacchi A, Soddu S, D'Orazi G. Homeodomain-interacting protein kinase-2 activity and p53 phosphorylation are critical events for cisplatin-mediated apoptosis.Exp Cell Res.2004;293:311-320..
- 140. Pistritto G, Puca R, Nardinocchi L, Sacchi A, D'Orazi G. HIPK2-induced p53Ser46 phosphorylation activates the KILLER/DR5-mediated caspase-8 extrinsic apoptotic pathway.Cell Death Diff.2007;14:1837-1839..
- 141. Puca R, Nardinocchi L, Sacchi A, Rechavi G, Givol D, D'Orazi G. HIPK2 modulates p53 activity towards pro-apoptotic transcription.Mol Cancer.2009;8:85.
- 142. Garufi A and D'Orazi G. High glucose dephosphorylates serine 46 and inhibits p53 apoptotic activity.JECCR.2014;33:79.
- 143. Jin ZG, Shen JF, He JY, Hu CQ. Combination therapy with p53-MDM2 binding inhibitors for malignancies.Med Chem Res.2015;24:1369-1379..
- 144. Vassilev LT. MDM2 inhibitors for cancer therapy.Trends Mol Med.2007;13:23-31..
- 145. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlot U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2.Science.2004;303:844-848..
- 146. Kojima K, Konopleva M, McQueen T, O'Brien S, Plunkett W, Andreeff M. Mdm2 inhibitor Nutlin-3a induces p53-mediated apoptosis by transcription-dependent and transcription-independent mechanisms that may overcome Atm-mediated resistance to fluodarabine in chronic lymphocytic leukemia.Blood.2006;108:993-1000..
- 147. Vaseva AV and Moll UM. The mitochondrial p53 pathway.Biochim Biophys Acta.2009;1787:414-420..
- 148. Rinaldo C, Prodosmo A, Mancini F, Iacovelli S, Sacchi A, Moretti F, Soddu Silvia. MDM2-regulated degradation of HIPK2 prevents p53Ser46 phosphorylation and DNA damage-induced apoptosis.Mol Cell.2007;25:739-750..
- 149. Rinaldo C, Prodosmo A, Siepi F, Moncada A, Sacchi A, Selivanova G, Soddu S. HIPK2 regulation by MDM2 determines tumor cell response to the p53-reactivating drugs Nutlin-3 and RITA.Cancer Res.2009;69:6241-6248..
- 150. Nardinocchi L, Puca R, Givol D, D'Orazi G. Counteracting MDM2-induced HIPK2 downregulation restores the HIPK2/p53 apoptotic signaling in cancer cells.FEBS Lett.2010;584:4253-4258..
- 151. Garufi A, Ubertini V, Mancini F, D'Orazi V, Baldari S, Moretti F, Bossi G, D'Orazi G. The beneficial effect of Zinc(II) on low-dose chemotherapeutic sensitivity involves p53 activation in wild-type p53-carrying colorectal cancer cells.JECCR.2015;34:87.
- 152. Nardinocchi L, Puca R, D'Orazi G. HIF-1( antagonizes p53-mediated apoptosis by triggering HIPK2 degradation.Aging (Albany NY).2011;3:33-43. https://doi.org/10.18632/aging.100254
- 153. Kojima K, Konopleva M, Samudio IJ, Schober WD, Bornmann WG, Andreeff M. Concomitant inhibition of MDM2 and Bcl-2 protein function synergistically induces mitovhondrial apoptosis in AML.Cell Cycle.2006;5:2778-2786..
- 154. Wade M, Rodewald LW, Espinosa JM, Wahl G. BH3 activation blocks Hdmx suppression of apoptosis and cooperates with Nutlin to induce cell death.Cell Cycle.2008;7:1973-1982..
- 155. Zhao YJ, Aguilar A, Bernard D, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment.J Med Chem.2015;58:1038-1052..
- 156. Pellegrino M, Mancini F, Lucà R, Coletti A, Giacchè N, Manni I, Arisi I, Florenzano F, Teveroni E, Buttarelli M, Fici L, Brandi R, Bruno T, et al. Targeting the MDM2/MDM4 interaction interface as a promising approach for p53 reactivation.Cancer Res.2015;75:4560-4572..
- 157. Mancini F, Di Conza G, Pellegrino M, Rinaldo C, Prodosmo A, Giglio S, D'Agnano I, Florenzano F, Felicioni L, Buttitta F, Marchetti A, Sacchi A, Pontecorvi A, et al. MDM4 (MDMX) localizes at the mitochondria and facilitates the p53-mediated intrinsic-apoptotic pathway.EMBO J.2009;28:1926-1939..
- 158. Mancini F, Pieroni L, Monteleone V, Luca R, Fici L, Luca E, Urbani A, Xiong S, Soddu S, Masetti R, Lozano G, Pontecorvi A, Moretti F. MDM4/HIPK2/p53 cytoplasmic assembly uncovers coordinated repression of molecules with anti-apoptotic activity during early DNA damage response.Oncogene.2016;35:228-240..
- 159. Hoe KK, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy.Nat Rev Drug Discov.2014;13:217-236..
- 160. Yu X, Narayanan S, Vazquez A, Carpizo DR. Small molecule compounds targeting the p53 pathway: are we finally making progress?Apoptosis.2014;19:1055-1068..
- 161. Zawancka-Pankau and Selivanova G. Pharmacological reactivation of p53 as a strategy to treat cancer.J Intern Med.2015;277:248-259..
- 162. Lujambio A and Lowe SW. The microcosmos of cancer.Nature.2012;482:347-355..
- 163. Lima RT, Busacca S, Almeida GM, Gaudino G, Fennell DA, Vasconcelos M Helena. MicroRNA regulation of core apoptosis pathways in cancer.Eur J Cancer.2011;47:163-174..
- 164. Huang G, Nishimoto K, Zhou Z, Hughes D, Kleinerman ES. miR-20a encoded by the miR-17-92 cluster increases the metastatic potential of osteosarcoma cells by regulating Fas expression.Cancer Res.2012;72:908-916..
- 165. Wang P, Zhuang L, Zhang J, Fan J, Luo J, Chen H, Wang K, Liu L, Chen Z, Meng Z. The serum miR-21 level serves as a predictor for the chemosensitivity of advanced pancreatic cancer, and miR-21 expression confers chemoresistance by targeting FasL.Mol Oncol.2013;7:334-345..
- 166. Li Z, Huang H, Chen P, He M, Li Y, Arnovitz S, Jiang X, He C, Hyjek E, Zhang J. miR-196b directly targets both HOXA9/MEIS1 oncogenes and FAS tumour suppressor in MLL-rearranged leukaemia.Nat Commun.2012;2:688.
- 167. Shaffiey F, Cross E, Sathyanarayana P. Mir-590 is a novel STAT5 regulated oncogenic miRNA and targets FasL in acute myeloid leukemia.Blood.2013;122:3811-3811..
- 168. Guennewig B, Roos M, Dogar AM, Gebert LF, Zagalak JA, Vongrad V, Metzner KJ, Hall J. Synthetic pre-microRNAs reveal dual-strand activity of miR-34a on TNF-α.RNA.2014;20:61-75..
- 169. Zhang L, Dong L, Li Y, Hong Z, Wei W. The microRNA miR-181c controls microglia-mediated neuronal apoptosis by suppressing tumor necrosis factor.J Neuroinflamm.2012;9:211.
- 170. Rossato M, Curtale G, Tamassia N, Castellucci M, Mori L, Gasperini S, Mariotti B, De Luca M, Mirolo M, Cassatella MA. IL-10-induced microRNA-187 negatively regulates TNF-α, IL-6, and IL-12p40 production in TLR4-stimulated monocytes.Proc Natl Acad Sci USA.2012;109:E3101-E3110..
- 171. Shimizu S, Takehara T, Hikita H, Kodama T, Miyagi T, Hosui A, Tatsumi T, Ishida H, Noda T, Nagano H, Doki Y, Mori M, Hayashi N. The let-7 family of microRNAs inhibits Bcl-xL expression and potentiates sorafenib-induced apoptosis in human hepatocellular carcinoma.J Hepatol.2010;52:698-704..
- 172. Sacconi A, Biagioni F, Canu V, Mori F, Di Benedetto A, Lorenzon L, Ercolani C, Di Agostino S, Cambria AM, Germoni S, Grasso G, Blandino R, Panebianco V, et al. miR-204 targets Bcl-2 expression and enhances responsiveness of gastric cancer.Cell Death Dis.2012;3:e423.
- 173. Zhang Y, Schiff D, Park D, Abounader R. MicroRNA-608 and MicroRNA-34a regulate chordoma malignancy by targeting EGFR, Bcl-xL and MET.PloS ONE.2014;9:e91546.
- 174. Kurataka O and Takahiro O. Genetic networks lead and follow tumor development: MicroRNA regulation of cell cycle and apoptosis in the p53 pathways.Bio Med Res Intern..2014;2014ID749724.
- 175. Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis.Mol Cell.2007;26:731-743..
- 176. Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis.Mol Cell.2007;26:745-752..
- 177. Garibaldi F, Falcone E, Trisciuoglio D, Colombo T, Lisek K, Walerych D, Del Sal G, Paci P, Bossi G, Piaggio G, Gurtner A. Mutant p53 inhibits miRNA biogenesis by interfering with the microprocessor complex.Oncogene.2016; https://doi.org/10.1038/onc.2016.51[Epub ahead of print].
- 178. Baig S, Seevasant I, Mohamad J, Mukheem A, Huri HZ, Kamarul T. Potential of apoptotic pathway-targeted cancer thereapeutic research: Where do we stand?Cell Death Dis.2016;7:e2058.