



Introduction
Ni is considered to be an essential element in microorganisms, plants, and animals, and has been a constituent of enzymes and proteins [1]. Due to its chemical properties, gloss, and low price, Ni has a wide variety of industrial applications [1]. Exposure to Ni has largely increased in industrial societies due to the environmental pollution by heavy metals at all stages of production, use, and disposal [2]. Human and animals contact Ni via several different pathways: ingestion, dermal contact, and inhalation [3]. After entering the body, Ni penetrates all organs and accumulates in various tissues and induces the tissue damage [4]. Ni can cause allergy, contact dermatitis, and toxicity of organ systems [4]. Ni may cause neurotoxicity, hepatotoxicity, nephrotoxicity, gene toxicity, reproductive toxicity, and increased risk of cancer [1,4].
It has been reported that NiSO4 induces DNA damage, apoptosis and oxidative damage in the liver and testes of mouse and the liver of Carassius auratus [5-7]. Our studies have also shown that dietary NiCl2 in excess of 300 mg/kg can cause histopathological lesions, immunotoxicity, oxidative damage, apoptosis and cell cycle arrest in the kidney, thymus, spleen, small intestine, cecal tonsil and bursa of Fabricius of broiler chickens [8-22]. Also, previous research have demonstrated that Ni can induce apoptosis in B cells [23], human T hybridoma cells [24], HEp-2 and MCF-7 cells [25], CHO cells [26], and normal rat kidney cells [27]. So far, the extrinsic and intrinsic apoptotic pathways are widely explored in the apoptosis [28]. The intrinsic signaling pathways are known as mitochondria- and ER-initiated apoptosis [29,30]. The extrinsic apoptotic pathway is activated by the binding of extracellular death ligands to cell-surface death receptors, and Fas-mediated apoptosis is the main pathway [31]. These references have demonstrated that Ni and Ni compounds induce apoptosis through mitochondria-mediated apoptosis, which are companied with the disruption of MMP, increase in cyt c and AIF translocation from mitochondria to cytoplasm and activation of caspase-9 and caspase-3 [25,32,33]. The results of Zhao et al. [33] suggest that metallic nickel particles can induce Fas-mediated apoptosis in JB6 cells. Recent studies have shown that the ER also plays an important role in the intrinsic pathway [34,35]. Up to now, there is only a report that nickel acetate can induce ER stress and increase CHOP protein expression in NRK52E cells and Hepa-1c1c7 Cells [36]. It has also been reported that nickel acetate can induce G2/M phase arrest in Chinese hamster ovary cells [26], but the mechanisms is still unclear. Inflammatory response also induced by Ni and Ni compounds [37,38]. Li et al. [39] have reported that NiCl2 increases the secretion of a pro-inflammatory cytokine (IL-1β) in bone marrow-derived macrophages and bone marrow dendritic cells. NiCl2 (20uM, 72h) increases pro-inflammatory cytokines (TNF-α, IL-β, and ICAM-1) mRNA expression levels in A549 cells [40].
Previous studies have suggested that Ni can induce apoptosis, cell cycle arrest and inflammatory response, however, the precise mechanism is still indistinct. There are no investigations on the Ni- or Ni compounds-induced Fas-mediated apoptotic pathway in vitro. Limited studies show the mechanism of NiCl2-induce ER stress, cell cycle arrest and inflammatory response at present. Therefore, the objective of this study was to determine potential mechanisms of NiCl2-induced apoptosis, cell cycle arrest and inflammatory response. We measured the gene expression which involved in the apoptosis pathway, cell cycle G2/M phase and inflammatory response.
Results
Clinical observation
The clinical observation was shown in the reference [22]. From 14 to 42 days of age during the experiment, broiler feed intake in the three NiCl2-treated groups began to decline when compared with those in the control group, except the 300 mg/kg group at 14 days of age. From 21 to 42 days of age during the experiment, broilers in the three NiCl2-treated groups showed inappetence, growth retardation and depression. A few broilers showed polypnea. No unexpected deaths occurred during the experiment.
Histopathological changes in the liver
In Figures 1, NiCl2 resulted in dose-dependent histopathological changes in the liver, including granular degeneration, vacuolar degeneration, and fatty degeneration.
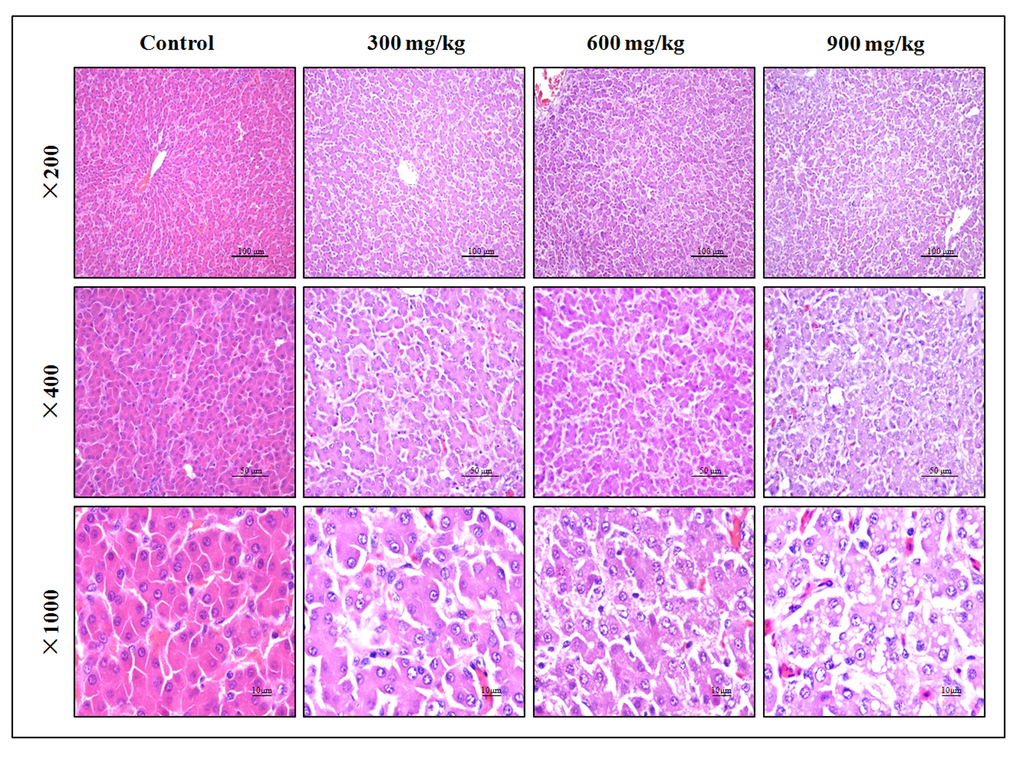
Figure 1. Histopathological changes in the liver at 42 days of age. Control group: No changes are observed; 300 mg/kg group: Hepatic cells show slight granular and vacuolar degeneration; 600 mg/kg group: Hepatic cells show granular, vacuolar and fatty degeneration. 900 mg/kg group: Hepatic cells show marked granular, vacuolar and fatty degeneration. (H·E).
Changes of hepatic function parameters
The activities of serum AST and ALT can directly reflect the damage of hepatic function. The results showed that AST and ALT activities were significantly higher (P < 0.05 or P < 0.01) in the 900 mg/kg group at 14 days of age and in the 600 and 900 mg/kg groups at 28 days of age and in the three NiCl2-treated groups at 42 days of age than those in the control group. ALP activity was higher (P < 0.05 or P < 0.01) in the 300, 600 and 900mg/kg groups than that in the control group from 14 to 42 days of age. GGT activity was also increased (P < 0.05 or P < 0.01) in the 600 and 900mg/kg groups at 28 and 42 days of age.
ALB, GLB and TP contents were lower (P < 0.05 or P < 0.01) in the 600 and 900mg/kg groups than those in the control group at 28 and 42 days of age. Also, TP content was decreased (P<0.05) in the 900mg/kg group at 14 days of age. TBIL content was increased (P < 0.05 or P < 0.01) in the 600 and 900mg/kg groups at 14 days of age and in the 300, 600 and 900mg/kg groups from 28 to 42 days of age when compared with that in the control group.
The results were shown in Figure 2.
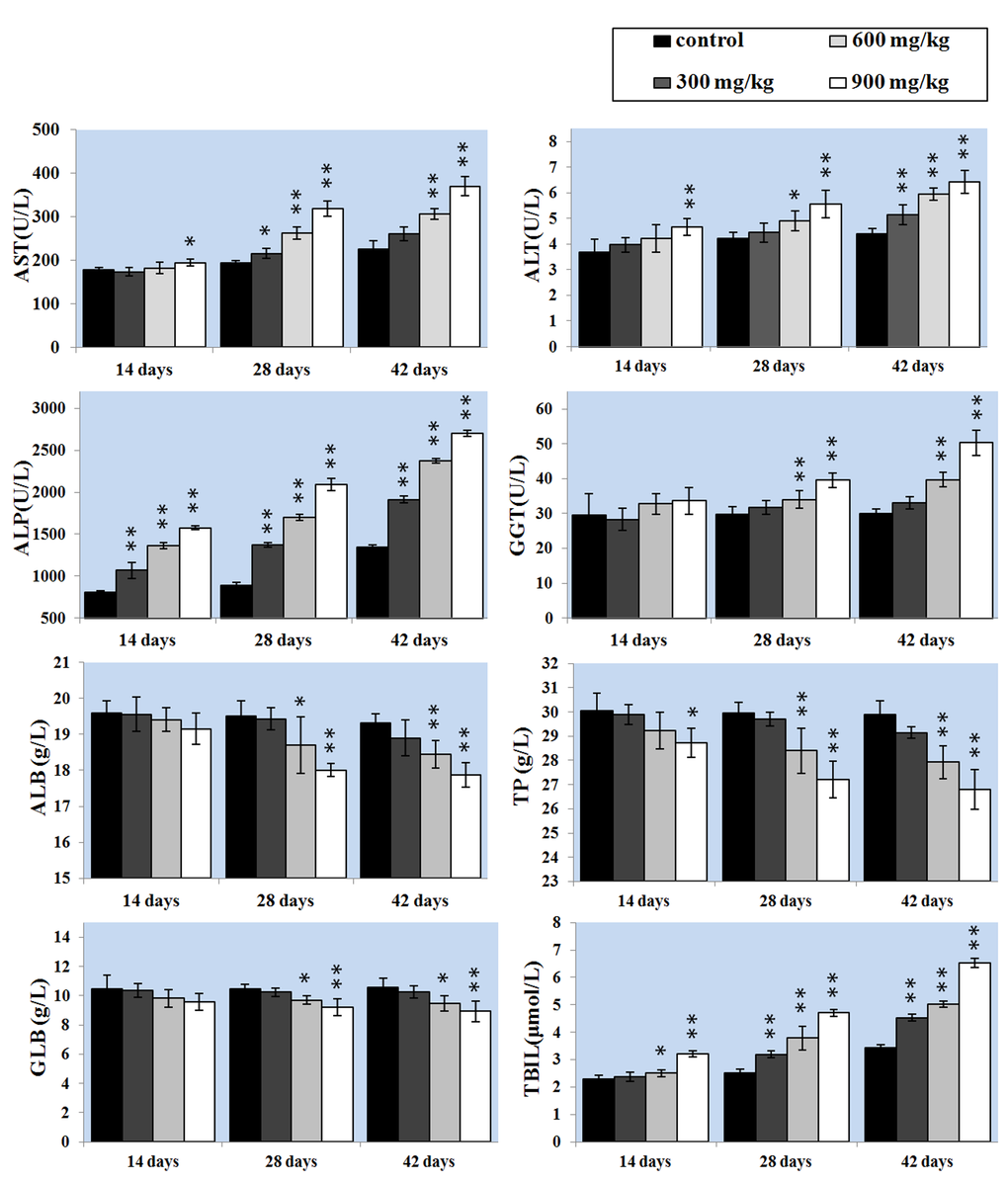
Figure 2. Changes of the hepatic function parameters. Data are presented with the mean ± standard deviation (n=5). *P<0.05, compared with the control group . **P<0.01, compared with the control group.
Effects of NiCl2 on apoptosis in the liver
The effects of dietary NiCl2 on the apoptosis in the liver were observed with methods of the TUNEL and flow cytometry assay. The results presented in Figure 3B showed that the number of apoptotic cells was significantly greater (P < 0.05 or P < 0.01) in the 600 and 900 mg/kg groups at 14 days of age than in the control group. Apoptotic cells were significantly increased (P < 0.05 or P < 0.01) also in the three NiCl2-treated groups from 28 to 42 days of age.
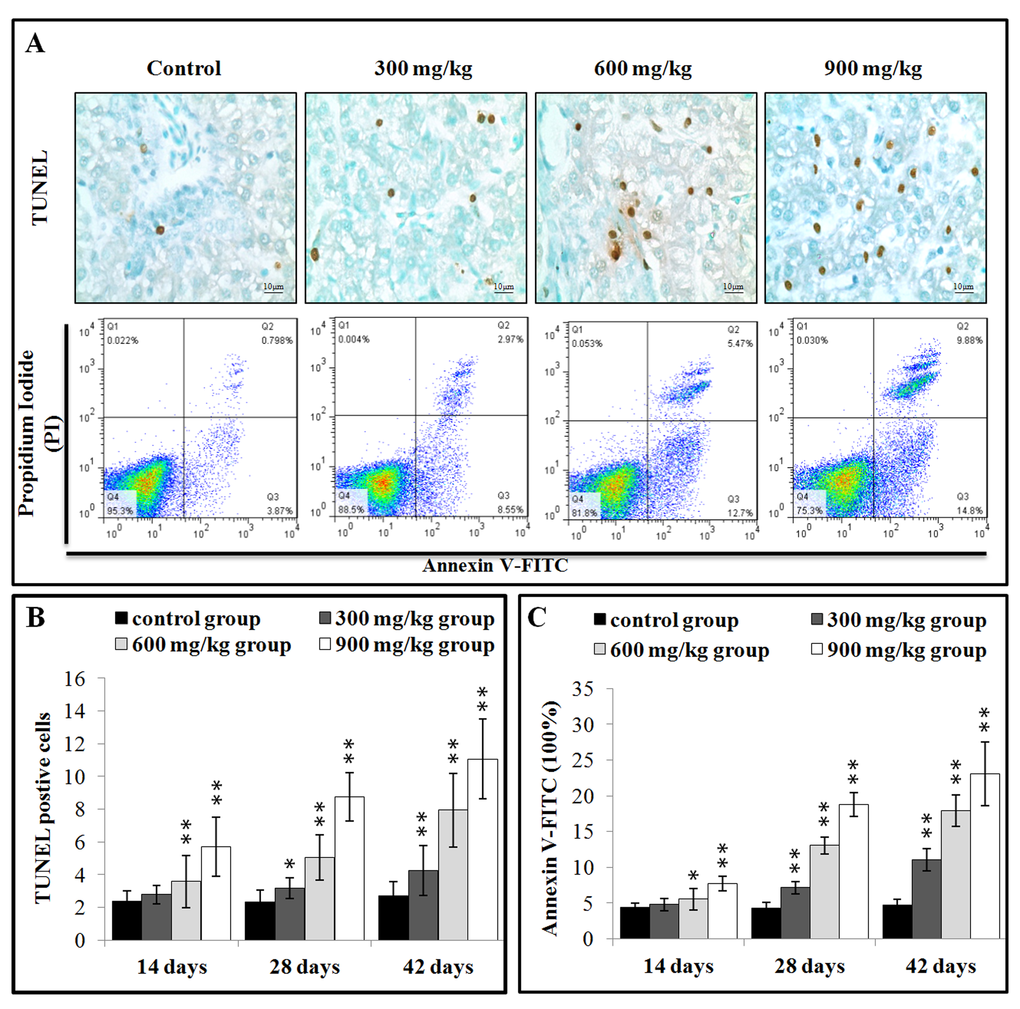
Figure 3. NiCl2 induces apoptosis in the liver. (A) Representative TUNEL and flow cytometric diagram of apoptosis analysis. (B) Changes of the TUNEL positive cells in the liver. (C) Changes of the percentages of annexin V positive cells in the liver. Data are presented with the mean ± standard deviation B:(n=5×5), C: (n=5). *P<0.05, compared with the control group. **P<0.01, compared with the control group.
After treated with NiCl2, hepatic cells were labeled using Annexin V-FITC and propidium iodide (PI) to discriminate live (Annexin V-FITC- and PI-), early apoptotic (Annexin V-FITC+ and PI-), late apoptotic (Annexin V-FITC+ and PI+) and primary/ secondary necrotic cells (Annexin V-FITC¬and PI+). The results presented in Figure 3C showed that the apoptotic cells (early apoptotic + late apoptotic cells) were significantly higher (P < 0.05 or P < 0.01) in the 600 mg/kg and 900 mg/kg groups at 14 days of age, and in the three NiCl2-treated groups from 28 to 42 days of age than those in the control group.
Effects of NiCl2 on apoptotic gene in the liver
The Bcl-2 family proteins have been shown to regulate the MMP. Therefore, the mRNA expressions of Bcl-2, Bax, Bcl-xL, Bak and Mcl-1 were detected and then the mRNA expression ratios of Bax/Bcl-2 were calculated. In Figure 4A, Bcl-2 mRNA expression was significantly decreased (P < 0.05 or P < 0.01) in the 900 mg/kg group at 14 days of age and in the 600 and 900 mg/kg groups at 28 days of age, and in the three NiCl2-treated groups at 42 days of age. Bax mRNA expression was significantly increased (P < 0.05 or P < 0.01) in the three NiCl2-treated groups from 28 to 42 days of age and in the 600 and 900 mg/kg groups at 14 days of age. Also, the Bax/Bcl-2 ratio was significantly higher (P < 0.05 or P < 0.01) in the three NiCl2-treated groups from 28 to 42 days of age and in the 900 mg/kg groups from 14 days of age than those in the control group. Bcl-xL and Mcl-1 mRNA expressions were significantly lower (P < 0.05 or P < 0.01) in the three NiCl2-treated groups from 28 to 42 days of age and in the 900 mg/kg group at 14 days of age than that in the control group. The mRNA expression of Bak was significantly increased (P < 0.05 or P < 0.01) in the three NiCl2-treated groups from 28 to 42 days of age and in the 600 and 900 mg/kg groups at 14 days of age.
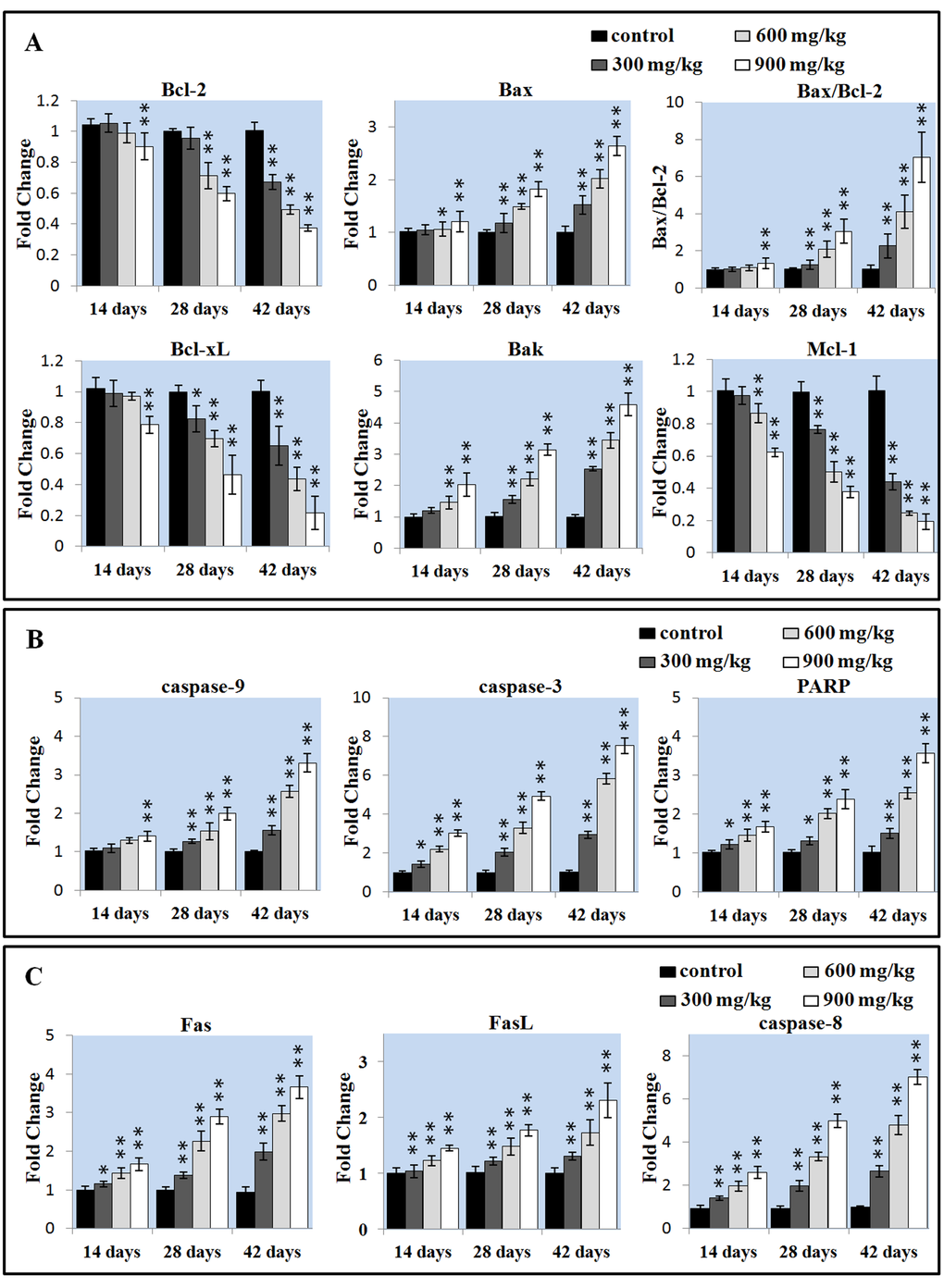
Figure 4. Changes of the apoptotic gene mRNA expression levels in the liver. (A) Changes of the Bcl-2 family proteins mRNA expression levels. (B) Changes of the caspase-9, caspase-3 and PARP mRNA expression levels. (C) Changes of the Fas, FasL, caspase-8 proteins mRNA expression levels. Data are presented with the mean ± standard deviation (n=5). *P<0.05, compared with the control group. **P<0.01, compared with the control group.
The disruption of MMP results in the release of apoptosis-inducing protein cytochrome c (cyt-c) that promotes caspase-9, caspase-3 and PARP activation and apoptosis [41]. We detected the mRNA expression of caspase-3, caspase-9 and PARP in the liver. The caspase-9 mRNA expression was significantly increased (P < 0.05 or P < 0.01) in the three NiCl2-treated groups from 28 to 42 days of age and in the 900 mg/kg groups at 14 days of age when compared with those in the control group. The mRNA expressions of caspase-3 and PARP was significantly higher (P < 0.05 or P < 0.01) in the three NiCl2-treated groups from 14 to 42 days of age than that in the control group, as shown in Figure 4B.
We investigated whether Fas-mediated apoptosis pathway was involved in NiCl2-induced mediated apoptosis. As shown in Figure 4C, the Fas, FasL and caspase-8 mRNA expressions were significantly increased (P < 0.05 or P < 0.01) in three NiCl2-treated groups from 14 to 42 days of age when compared with those in the control group.
We also detected whether ER stress-mediated apoptosis pathway was involved in NiCl2-induced mediated apoptosis. We first examined whether NiCl2 could induce ER stress in the kidney. The extended ER stress can induce apoptosis. The results showed that NiCl2 significantly increased the mRNA expression of GRP78 and GRP94, which were the markers of ER stress. The GRP78 and GRP94 mRNA expression was significantly higher (P < 0.05 or P < 0.01) in the 600 and 900 mg/kg groups at 14 days of age and in the three NiCl2-treated groups from 28 to 42 days of age than that in the control group.
To further confirm that UPR pathways were involved in NiCl2-induced ER stress, we examined all the three UPR pathways: PERK pathway, IRE1 pathway and ATF6 pathway. The results indicated that NiCl2 induced the ER stress through activating PERK, IRE1 and ATF6 UPR pathways. In the PERK pathway, the eIF2α and ATF4 mRNA expressions were significantly higher (P < 0.05 or P < 0.01) in the three NiCl2-treated groups from 28 to 42 days of age than those in the control group. In the IRE1 pathway, the IRE1 mRNA expression was significantly increased (P < 0.05 or P < 0.01) in the 900 groups at 14 days of age, and in the 600 and 900 groups from 28 to 42 days of age when compared with that in the control group, and the XBP1 mRNA expression was significantly increased (P < 0.05 or P < 0.01) in the 900 groups at 14 days of age, in the 600 and 900 groups at 28 days of age, and in the three NiCl2-treated groups at 42 days of age when compared with that in the control group. In the ATF6 pathway, the ATF6 mRNA expression was significantly higher (P < 0.05 or P < 0.01) in the 600 and 900 mg/kg groups at 42 days of age than that in the control group.
Effects of NiCl2 on cell cycle in the liver
Cell cycle includes S (DNA replication), M (nuclear division and cell division), G1 (the cell-cycle gap phase between M phase and S phase), G2 (the cell-cycle gap phase between S phase and M phase) phases, which is central to maintain homeostasis in the multicellular organisms [42]. We measured the percentage of G0/G1 phase (a prolonged nondividing state), S phase (DNA replication) and G2/M phase (the completed of DNA replication) cells in the liver.
As shown in Figure 5A and B, NiCl2 induced a dose-and time-dependent increase in G2/M phase cells and a corresponding decrease in cells at other stages of the cell cycle. The cell percentages in G0/G1 phase were significantly decreased (P < 0.05 or P < 0.01) in the 600 and 900 mg/kg groups at 28 days of age and in the three NiCl2-treated groups at 42 days of age when compared with those in the control group. The cell percentages in G2/M phase were significantly increased (P < 0.05 or P < 0.01) in the 900 mg/kg groups at 14 days of age and in the three NiCl2-treated groups from 28 to 42 days of age in comparison with those in the control group.
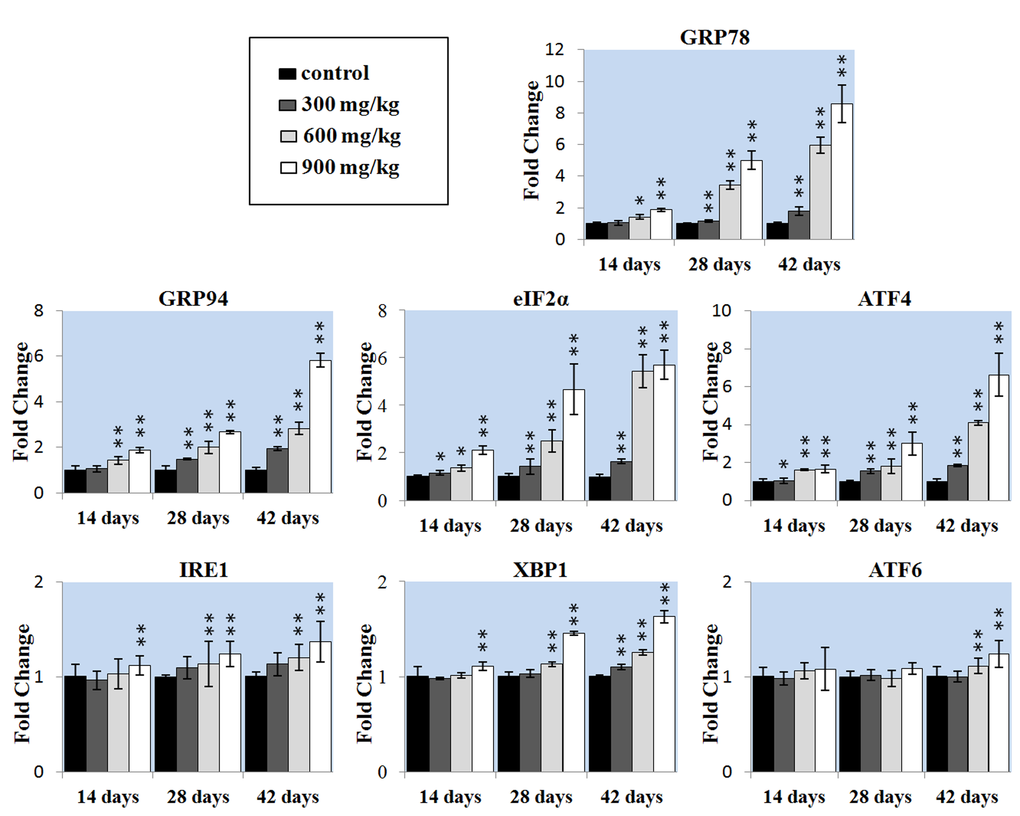
Figure 5. Changes of the ER stress gene mRNA expression levels in the liver. Data are presented with the mean ± standard deviation (n=5). *P<0.05, compared with the control group. **P<0.01, compared with the control group.
The mRNA expression changes of G2/M cell cycle regulatory molecule protein p53, p21, cdc2 and cyclin B were shown in Figures 5C. The p53 mRNA expression was significantly higher (P < 0.05 or P < 0.01) in the three NiCl2-treated groups from 14 to 42 days of age than that in the control group. The p21 mRNA expression significantly increased (P < 0.05 or P < 0.01) in the 900 mg/kg groups at 14 days of age and in the three NiCl2-treated groups from 28 to 42 days of age in comparison with those in the control group. The cdc2 mRNA expression was significantly lower (P < 0.05 or P < 0.01) in the 900 mg/kg group at 14 days of age, in the 600 and 900 mg/kg groups at 28 days of age and in the three NiCl2-treated groups at 42 days of age than that in the control group. The cyclin B mRNA expression was significantly decreased (P < 0.05 or P < 0.01) in the 600 and 900 mg/kg groups at 14 days of age and in the three NiCl2-treated groups from 28 to 42 days of age when compared with that in the control group.
Effects of NiCl2 on inflammatory response in the liver
As shown in Figure 6, the mRNA expressions of pro-inflammatory genes (NF-κB, TNF-α, IL-1β, IL-6 and IL-8) were significantly increased (P < 0.01) in the three NiCl2-treated groups from 28 to 42 days of age when compared with those in the control group. The TNF-α mRNA expression was significantly higher (P < 0.05 or P < 0.01) in the 600 and 900 mg/kg groups at 14 days of age than those in the control group. The IL-6 mRNA expression was significantly higher (P < 0.05 or P < 0.01) in the three NiCl2-treated groups at 14 days of age than those in the control group. The IL-8 mRNA expression was significantly increased (P < 0.01) in the 900 mg/kg groups at 14 days of age when compared with that in the control group (Figure 7).
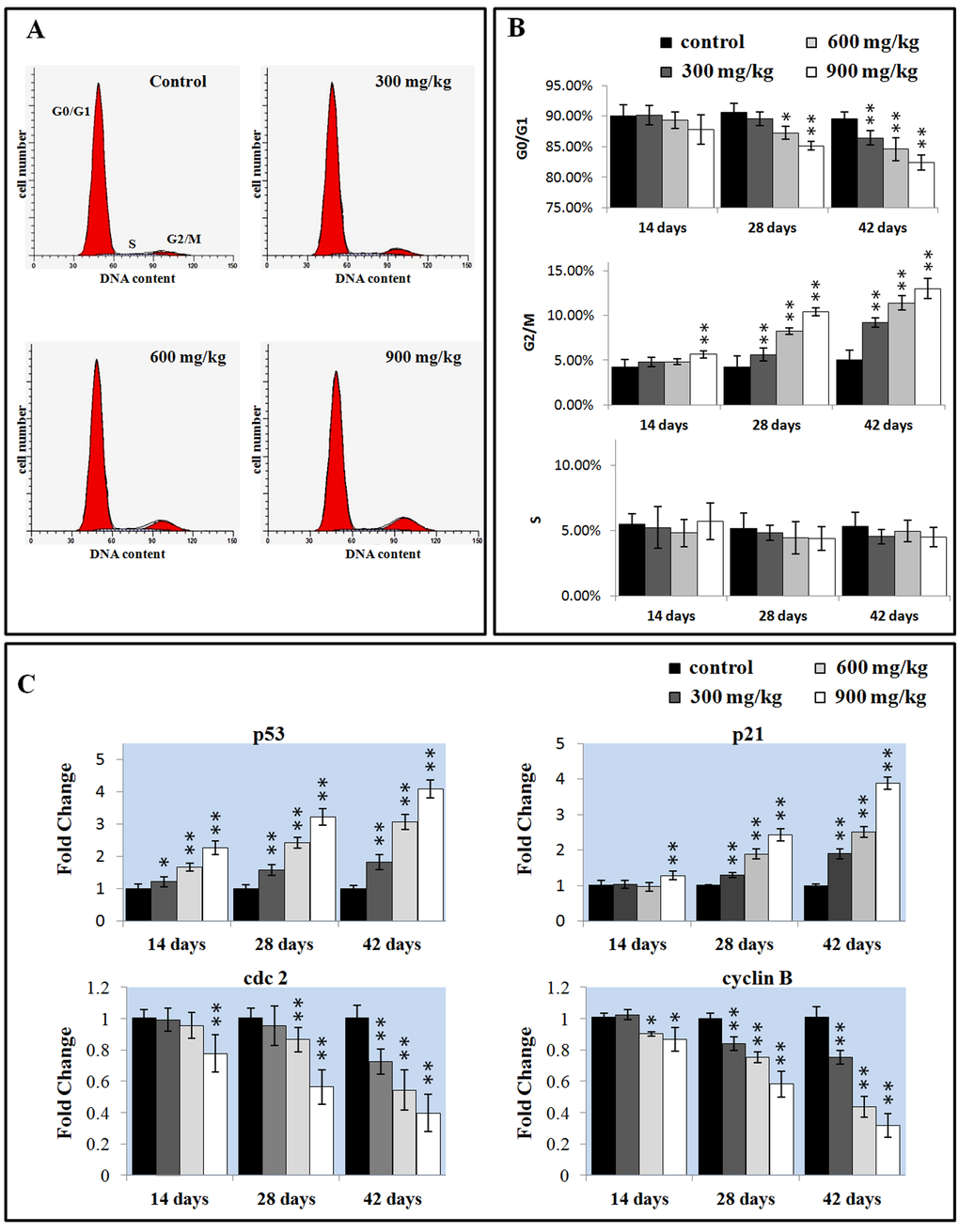
Figure 6. Changes of the cell cycle in the liver. (A) Representative flow cytometric diagram of cell cycle analysis. (B) Changes of the percentage of G0/G1, G2/M, and S phase in the liver. (C) Changes of the G2/M phase regulators mRNA expression levels. Data are presented with the mean ± standard deviation (n=5). *P<0.05, compared with the control group. **P<0.01, compared with the control group.
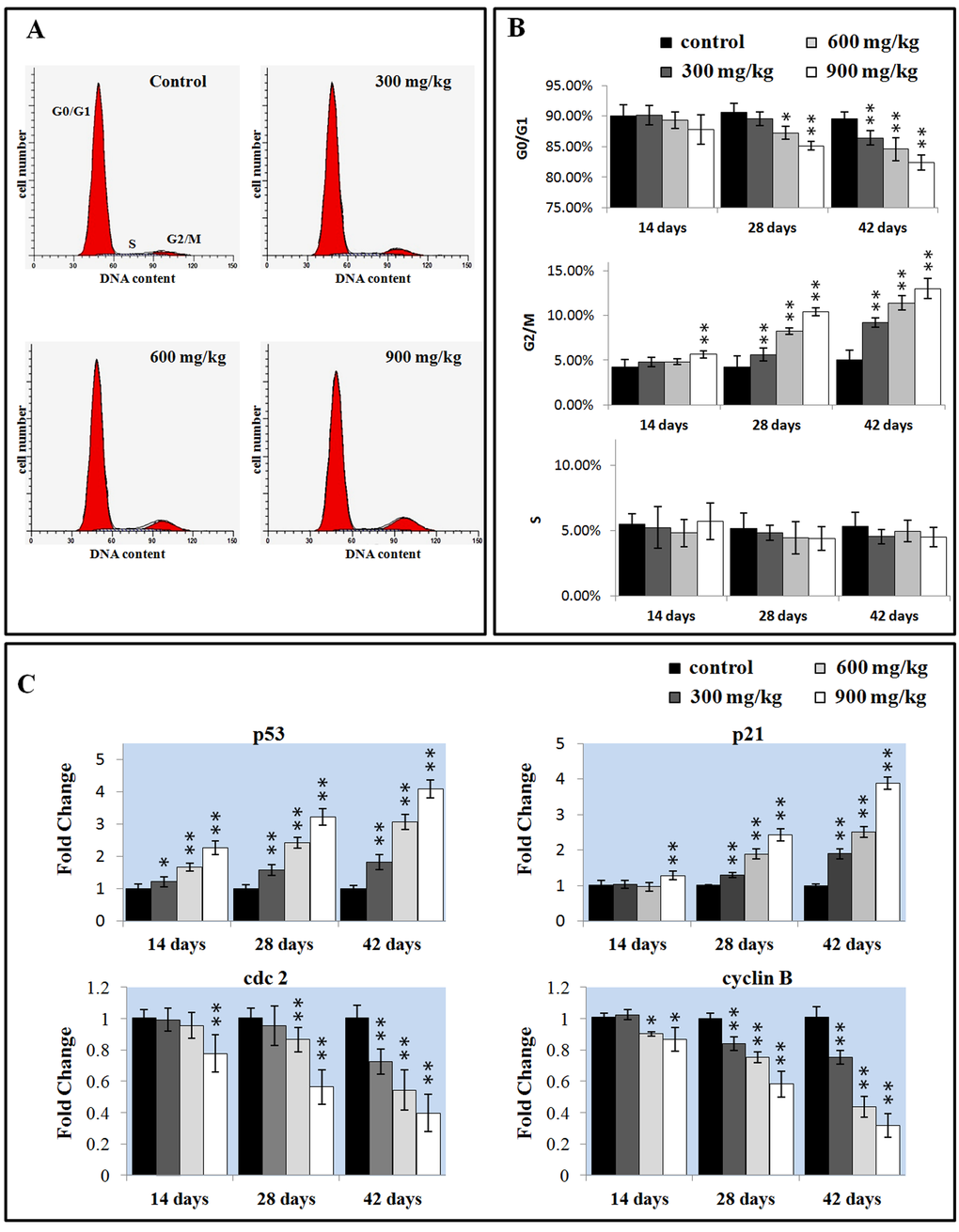
Figure 7. Changes of the inflammatory mediators mRNA expression levels in the liver. Data are presented with the mean ± standard deviation (n=5). *P<0.05, compared with the control group. **P<0.01, compared with the control group.
Changes of Ni contents in the liver
The Ni contents were increased in the three NiCl2-treated groups at 42 days of age in comparison with those in the control group (P < 0.05 or P < 0.01), as shown in Figure 8.
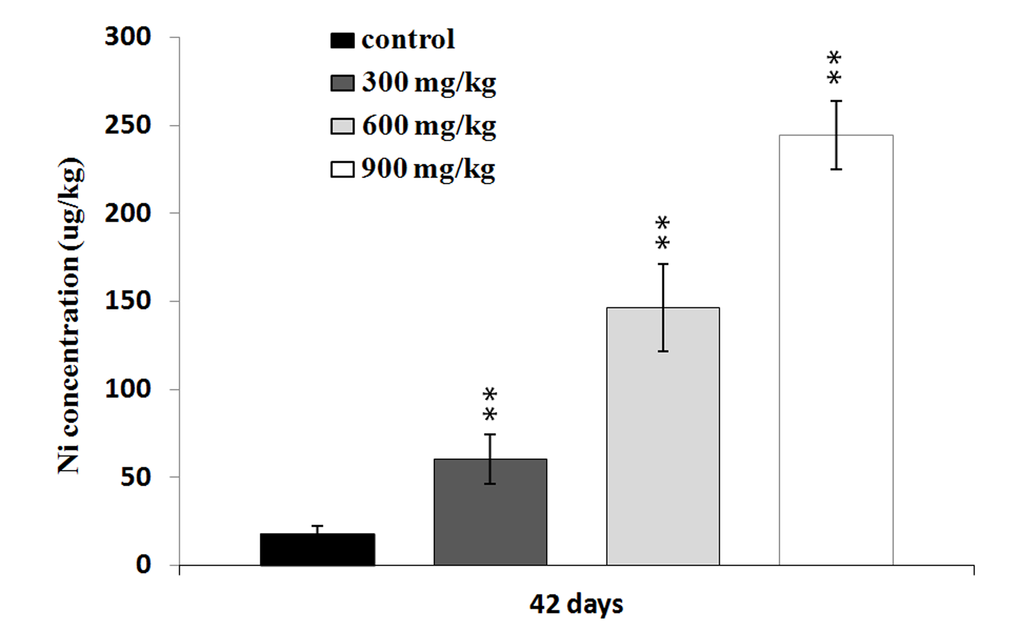
Figure 8. Changes of the Ni concentrations in the liver at 42 days. Data are presented with the mean ± standard deviation (n=5). *P<0.05, compared with the control group. **P<0.01, compared with the control group.
Discussion
The liver is a primary site for xenobiotic metabolism and is the most common target organ for chemicals-induced injuries. Previous studies have shown that Ni exposure caused liver injuries and dysfunction [7,43]. The present investigation, the decrease of serum ALB, GLB, TP contents and increase of serum AST, ALT, ALP, GGT activities and TBIL contents demonstrate that excessive NiCl2 induces hepatic damage. Moreover, histopathological changes of the liver also have been observed in NiCl2-treated broiler chickens. The histopathological injury and the alteration of hepatic function parameters are consistent with Ni accumulation in the liver, indicating that Ni accumulation is main or/and direct reason of the hepatic injury.
Many studies have suggested that one possible molecular mechanism involved in Ni toxicity is the excess apoptosis. Our previous studies have proved that NiCl2 induced apoptosis in the thymus, cecal tonsil, spleen, bursa of Fabricius, and kidney [8,10,17,19,44]. In this study, we found consistent evidence that dietary NiCl2 in excess of 300 mg/kg had adverse effects on the hepatic cells. The results of TUNEL and flow cytometry showed that NiCl2 significantly increased the percentage of apoptosis in the liver. In order to reveal the possible mechanism of NiCl2-induced apoptosis, we analyzed the mRNA expression of apoptotic genes which involved in mitochondria-, Fas- and ER stress-mediated apoptotic pathway. In the mitochondria-mediated apoptotic pathway, the balance between pro-apoptotic and anti-apoptotic Bcl-2 protein family members controls the mitochondrial apoptotic pathway [45]. The Bcl-2 family protein is consisting of apoptosis repressors (Bcl-2, Bcl-xL, and Mcl-1) and inducers (Bax and Bak) [45]. Loss of MMP is a prerequisite for mitochondrial-mediated apoptosis as it is associated with the reshuffling of Bcl-2 family proteins [46]. In this study, NiCl2 increased pro-apoptotic Bax and Bak mRNA expressions, and concomitantly decreased anti-apoptotic Bcl-2, Bcl-xL and Mcl-1 mRNA expressions. And, the Bax/Bcl-2 ratio was significantly increased. Our previous studies have also shown that NiCl2 increases Bax mRNA expression levels and decreases Bcl-2 mRNA expression levels in the thymus, cecal tonsil, kidney and spleen [8,10,17,44]. Consistent with our findings, reduction of Bcl-2 and Bcl-xL protein expressions and enhancement of Bad, Bcl-Xs, Bax protein expressions have been observed after human proximal tubule cells and human bronchial epithelial cells have been cultured with nickel acetate and NiONPs [47,48]. Ni3S2 down-regulates several anti-apoptotic proteins Bcl-2 and Bcl-xL in human BEAS-2B cells [49]. Changes of Bcl-2 family proteins disrupt the MMP, which translocates cyt-c from the mitochondrial intermembrane space to the cytosol [46]. Cyt-c can cleave and activate caspase-9, which then activate the downstream caspase-3. Caspase-3 is one of the key executioners of apoptosis, and is capable of cleaving or degrading many key proteins such as nuclear lamins, fodrin, and the nuclear enzyme PARP, which then induces apoptosis [50]. Our results showed that NiCl2 increased the mRNA expressions of caspase-3, caspase-9 and PARP, indicating that NiCl2 induced hepatic apoptosis through mitochondria-mediated apoptotic pathway. Our results are agreement with the results of Ahamed et al. [32] in which nickel ferrite nanoparticles increase activation and gene expression of caspase-3 and caspase-9 in HepG2 and MFC-7 cancer cells. An increase in the cleavage of procaspase-3 and PARP proteins has been also observed in nickel acetate-treated nasal epithelium [51].
In this study, the increased transcription levels of Fas, FasL, caspase-8 gene indicate the activation of Fas-mediated apoptotic pathway. In the Fas-mediated apoptotic pathway, FasL interacts with the Fas receptor, which then leads to caspase-8 activation [52]. Activated caspase-8 can directly cleave and activate caspase-3, which then causes apoptosis [52]. Zhao et al. [33] has also reported that nickel particles can increase Fas and caspase-8 expressions in JB6 cells.
Proper ER function is essential to cell survival, and perturbation of its function induces cellular damage and results in apoptosis [35]. Under prolonged and irreversible ER stress, cells that become irreversibly damaged are eliminated by apoptosis [35]. Our findings showed that NiCl2 induced ER stress, which was characterized by increasing ER stress markers GRP78 and GRP94 mRNA expressions. UPR is a defense mechanism against various cellular stress which causes accumulation of unfolded proteins in the ER [53]. In this study, NiCl2 increased transcription levels of eIF2a, ATF4, IREI, XBP1 and ATF6, implying that three UPR pathways: PERK pathway, IRE1 pathway and ATF6 pathway were involved in NiCl2-induced ER stress. Our findings are consistent with the results of our previous in the kidney [54]. PERK activates eIF2a, and then the phosphorylated eIF2a induces the translation of ATF4 mRNA [55]. IRE1 can activate XBP1 kinase and RNase activities to initiate XBP1 mRNA splice, which produces a potent transcriptional activator [56]. ATF6 is transported from ER to the golgi compartment, where it is cleaved to a cytosolic fragment that migrates to the nucleus to further activate the transcription of UPR genes [35]. Under prolonged ER stress, PERK pathway can trigger cell death. ATF4 can promote apoptosis through up-regulating the pro-apoptotic transcriptional factor CHOP [35]. CHOP induces the expression of several pro-apoptotic proteins such as GADD34, ERO1α and BH3-only proteins (BIM, PUMA and NOXA), thereby promoting apoptosis [57-59]. The IRE1 pathway has a pro-survival function, but it may cause apoptosis under prolonged ER stress [60]. IRE1 forms a complex with TRAF2 inducing the activation of ASK1/JNK, which triggers cell death [61,62]. Furthermore, IRE1 also can activate NF-κB signaling that might lead to apoptosis [30].
The cell cycle analysis can give another insight into the toxicology mechanisms of NiCl2 in the liver. In the present study, a significant increase in the G2/M phase was observed in the liver of NiCl2-treated broiler chickens. A similar activation of G2/M phase arrest has been also observed in the nickel acetate-treated Chinese hamster ovary cells [26]. Our previous studies have suggested that NiCl2 induces G2/M phase arrest in the kidney, but G0/G1 phase arrest in the thymus and bursa of Fabricius [8,18,19]. The p53 protein is considered to be the most widespread inhibitor of cell proliferation, and p53-dependent G2/M cell cycle arrest is an important component of the cellular response to stress [63]. The cell cycle transition from G2 to M phase is strictly regulated by cdc2-cyclin B complex [64]. p53 can induce p21 up-regulation, which inhibits cdc2 activation, then causes cells to arrest in G2/M phase [65]. In this study, the results showed that the G2/M phase arrest process was associated with the increased p53 and p21mRNA expressions, and decreased cdc2 and cyclin B mRNA expressions. All of the present evidence suggests that NiCl2 may induce G2/M phase accumulation by inhibiting the cdc2-cyclinB complexes through directly stimulating the p53 and p21 expressions. Lee et al. [51] has demonstrated that nickel acetate-induced G2/M arrest is associated with up-regulation of p53, p21 expressions, decrease in phosphorylation of cdc2, and down-regulation of cyclin B1 expression in nasal epithelium. Salnikow et al. [66,67] also has observed that NiCl2 and nickel sulfide (Ni3S2) up-regulate p53 protein levels in human lung cells, MCF-7 and A549 cells. The p53 protein not only leads to cell cycle arrest in the G2/M phase of the cell cycle by directly stimulating the p21expression, but also promotes apoptosis through up-regulation of Bax and down-regulation of Bcl-2 protein expression [63].
In this study, we also found that NiCl2 induced hepatic inflammatory responses, which is company with the up-regulation of pro-inflammatory genes (TNF-a, IL-1β, IL-6 and IL-8) mRNA expression levels and the increased NF-κB transcription level. The excessive mRNA expressions of proinflammatory cytokines further caused cell functional disorder and apoptosis [68]. NF-κB is highly activated at sites of inflammation in diverse diseases and can induce transcription of proinflammatory cytokines, chemokines, adhesion molecules [68]. Our results indicated that NiCl2 amplified proinflammatory mediators secretion by activating the NF-κB signaling pathway in this inflammatory response, which was in agreement with the results obtained by Freitas et al. [69]. Capasso et al. [38] also suggest that nickel oxide nanoparticles induce inflammation through activation of NF-κB pathway in lung epithelial cells.
In conclusion, dietary NiCl2 induces apoptosis through mitochondria, Fas, ER stress-mediated apoptotic pathway and causes cell cycle G2/M phase arrest via p53-dependent pathway and generates inflammatory response by NF-κB pathway activation. The prolonged cell cycle G2/M phase arrest and inflammatory response also can induce apoptosis.
Materials and Methods
Experimental design
Two hundred and eighty one-day-old healthy broiler chickens (Chia Tai Group, Wenjiang, Sichuan, China) were divided into four groups (N = 70), and were housed in cages with electrical heaters and provided with feed and water as well as the under-mentioned experimental diets ad libitum for 42 days. The growth cycle of commercial broilers is about 42 days, after which they are used for consumption. In this rapid growth period food consumption is high, and broilers will easily be affected by diet containing metal pollutants (such as Ni). The aim of our study is to evaluate the effect of dietary NiCl2 on the broilers in this period of rapid growth.
To observe the time-dependent dynamic change, we chose three time points (14, 28, and 42 days of age) for examining hepatic function, the alterations of apoptosis, cell cycle and inflammatory response.
A corn-soybean basal diet formulated by the National Research Council [70] was the control diet, and NiCl2 (NiCl2·6H2O, Cheng Du Kelong Chemical Co., Ltd., Chengdu, China) was mixed into this basal diet to produce experimental diets containing 300, 600 and 900 mg/kg NiCl2, respectively.
The basis of doses (300, 600 and 900 mg/kg NiCl2) selection: Ling and Leach reported that dietary NiCl2 concentrations of 300 mg/kg and over resulted in significant reduction in growth rate. Mortality and anemia were observed in chicks receiving 1100 mg/kg nickel [71]. Weber and Reid found a significant growth reduction at 700 mg/kg NiSO4 and nickel acetate and over [72]. Chicks fed more than 250-300 mg/kg Ni in the diet exhibited depressed growth and reduced feed intake [73]. Bersenyi et al. [74] reported that supplementation of 500 mg/kg NiCl2 reduced weight gain (by 10%), feed intake (by 4%) and worse feed FCE (by 5%) in growing broiler cockerels. According to the above-mentioned research results and our preliminary experiment, we chose the doses of 300, 600 and 900 mg/kg NiCl2 in this study for observing the does-dependent changes.
The animal protocols and all procedures of the experiment were performed in compliance with the laws and guidelines of Animal Care and Use Committee, Sichuan Agricultural University (Approval No: 2012-024).
Histopathological examination of liver
Five broiler chickens in each group were humanely killed at 42 days of age. Livers were removed, fixed in 4% paraformaldehyde, dehydrated in ethanol and embedded in paraffin. Serial slices at 5 μm thickness were prepared and stained with haematoxylin and eosin (H·E), and examined by light microscopy.
Determination of the hepatic function parameters
At 14, 28, and 42 days during the experiment, five broiler chickens in each group were phlebotomized from jugular vein to collect serum. Non-anticoagulative blood samples were clotted for 15 min at room temperature and then centrifuged at 3000 rpm for 15 min. The serum ALT and AST, ALP, GGT and the contents of TP, ALB, GLB, TBIL were detected by biochemical methods following the instruction of the reagent kits (these kits purchased from Nanjing Jiancheng Bioengineering Institute of China, Nanjing, China).
Apoptosis analysis by flow cytometry
At 14, 28, and 42 days of age, five broiler chickens in each group were taken for determination of the changes of apoptosis in the liver by flow cytometry.
The broiler chickens in each subsample were humanely killed, and their livers were immediately taken and ground to form a cell suspension, which was filtered through a 300-mesh nylon screen. The cells were washed twice with ice-cold phosphate buffer saline (PBS, pH 7.2-7.4), and then suspended in PBS at a concentration of 1×106 cells/mL. A total of 100 µL of the cell suspension was transferred to a 5-mL culture tube. The cells were respectively stained with 5 μL Annexin V-FITC (Cat: 51-65874X, BD, USA) and 5 μL of PI (Cat: 51-66211E, BD, USA) at 25ºC in the dark. Finally, 400 μL of 1×binding buffer were added to each tube after 15 minutes, and cells were analyzed by flow cytometry (BD FACSCalibur) within 40 min of preparation. The results were analyzed using the Mod Fit LT for Mac V3.0 computer program.
Detection of hepatic apoptosis by TUNEL
Five broiler chickens in each group were humanely sacrificed at 14, 28, and 42 days of age. Livers were removed, fixed in 4% paraformaldehyde, dehydrated in ethanol and embedded in paraffin.
TUNEL analysis was carried out according to the manual of In Situ Cell Death Detection Kit (Cat: 11684817980, Roche, Mannheim, Germany). Briefly, tissue slices (5 μm thick) were rehydrated in a series of xylene and ethanol solutions and then rinsed in ddH2O, digested with 50 μL proteinase K (diluted in Tris•HCl pH 7.8) for 15 min, and incubated with 3% H2O2 in methanol for 15 min at room temperature to inactivate endogenous peroxidase. The slices were transferred to a reaction mixture containing biotin-dUTP terminal deoxynucleotidyl and incubated in a humidified chamber for 1 h at 37 °C, followed by washing in PBS (pH 7.2-7.4). Slices were incubated in Converter-POD (HRP) for 30 min at 37 °C. Reaction product was visualized with DAB kit (AR1022, Boster, Wuhan, China). After final washing in ddH2O, slices were lightly counterstained with methyl green, dehydrated in ethanol, cleared in xylene and mounted.
Cells were observed with light microscopy (Olympus, Shimadzu, Japan). The nuclei of apoptotic cells containing DNA strand breaks were stained brown. The TUNEL positive cells (apoptotic cells) were counted by use of a computer-supported imaging system connected to a light microscope with an objective magnification of ×1000. Apoptotic cells were quantified by use of Image-Pro Plus 5.1 (Madia Cybernetics, Bethesda, MD, USA) image analysis software. Five slices in each chicken and five fields in each slice were measured and averaged.
Cell cycle analysis by flow cytometry
At 14, 28, and 42 days of age, five broiler chickens in each group were taken for determination of the cell cycle stages in the liver by flow cytometry.
The broiler chickens in each subsample were humanely killed, and their livers were immediately taken and ground to form a cell suspension, which was filtered through a 300-mesh nylon screen. The cells were washed twice with ice-cold PBS (pH 7.2-7.4), and then suspended in PBS at a concentration of 1 × 106 cells/mL. A total of 500 µL of the cell suspension was transferred to a 5-mL culture tube. After centrifugation (600 rpm, 5 min), the supernatant was decanted, the cells were incubated for 30 min at room temperature in the dark with 5 µL 0.25% Triton X-100 and 5 μL PI (Cat. No.51-66211E). Finally, 500 μL of PBS were added to each tube, and cells were analyzed by flow cytometry (BD FACSCalibur) within 45 min of preparation. The results were analyzed using the Mod Fit LT for Mac V3.0 computer program.
Determination of the mRNA expression by quantitative real-time PCR
The livers from five broiler chickens in each group were taken at 14, 28, and 42 days of age and stored in liquid nitrogen. They were then homogenized in liquid nitrogen using a mortar and pestle. The total RNA was isolated using RNAiso Plus (9108/9109, Takara, Japan). Subsequently, RNA was transferred to cDNA using a Prim-Script™ RT reagent Kit (RR047A, Takara, Japan) according to the manufacture’s protocol. The cDNA product was used as a template for qRT-PCR analysis. Sequences for target genes were obtained from the NCBI database. Oligonucleotide primers were designed using Primer 5 software and synthesized at Takara (Dalian, China; see Table 1, 2 and 3).
Table 1. A list of primers of the apoptotic genes in qRT-PCR analysis
| Gene symbol | Accession number | Primer | Primer sequence(5’-3’) | Product size | Tm (°C) |
| Bcl-2 | NM205339 | Forward Reverse | GATGACCGAGTACCTGAACC CAGGAGAAATCGAACAAAGGC | 114bp | 61 |
| Bax | XM422067 | Forward Reverse | TCCTCATCGCCATGCTCAT CCTTGGTCTGGAAGCAGAAGA | 169bp | 62 |
| Bcl-xL | GU230783 | Forward Reverse | ATGAGTTTGAGCTGAGGTACCGG AGAAGAAAGCCACGATGCGC | 150bp | 59 |
| Bak | NM001030920 | Forward Reverse | TCTACCAGCAAGGCATCACGG ATCGAGTGCAGCCACCCATC | 122bp | 60 |
| Mcl-1 | XM001233734 | Forward Reverse | GGATCATCACGGACGCATTG TGATGCTGCTTTCCAGGTCC | 111bp | 60 |
| caspase-9 | AY057940 | Forward Reverse | CGAAGGAGCAAGCACGACAG CCGCAGCCCTCATCTAGCAT | 130bp | 61 |
| caspase-3 | NM204725 | Forward Reverse | TGGCCCTCTTGAACTGAAAG TCCACTGTCTGCTTCAATACC | 139bp | 62 |
| PARP | NM205263 | Forward Reverse | AAGCTCCGAACTGATATTAAGGTGG GCTTAAATGGCTTGTAACGCTGA | 172bp | 56 |
| Fas | NM001199487 | Forward Reverse | TGTTCGTCATCACCGTCTATCG TTCGTAGGCTCCTCCCATCC | 133bp | 60 |
| FasL | AJ890143 | Forward Reverse | AGATCGCATCCCTCCAGCTC GAGACAGGTTCCCACTCCAATG | 135bp | 59 |
| Caspase-8 | NM204592 | Forward Reverse | TGGGAAAGTGGACAAGAGCCT CCACAGATGATGCCAGCCAA | 146bp | 59 |
| GRP78 | NM205491 | Forward Reverse | GAATCGGCTAACACCAGAGGA CGCATAGCTCTCCAGCTCATT | 118bp | 59 |
| GRP94 | NM204289 | Forward Reverse | CTTCGCTTCCAGTCTTCCCATC AGAAGGCGTTCAACAAATGGTG | 149bp | 58 |
| Eif2a | NM001031323 | Forward Reverse | GCTGCGAGTCAGTAATGGGTATAA CTGCCAGGAAACTTGCCACA | 103bp | 59 |
| ATF4 | AB013138 | Forward Reverse | TTGATGCCCTGTTAGGTATGGAA GGTATGAGTGGAGGTTCTTTGTTGT | 139bp | 60 |
| IRE1 | NM001285499 | Forward Reverse | TGAGGGCAATGAGAAATAAGAAGC TGTAGGAGCAGGTGAGGGAAGC | 127bp | 61 |
| XBP1 | NM001006192 | Forward Reverse | GCGAGTCTACGGATGTGAAGGA TGTGGAGGTTGTCAGGAATGGT | 140bp | 61 |
| ATF6 | XM422208 | Forward Reverse | GATTGTGGGCGTCACTTCTCG TGGGATGCCAATGTTAGCCTG | 142bp | 57 |
| β-actin | L08165 | Forward Reverse | TGCTGTGTTCCCATCTATCG TTGGTGACAATACCGTGTTCA | 178bp | 62 |
Table 2. A list of primers of the cell cycle regulator genes in qRT-PCR analysis
| Gene symbol | Accession number | Primer | Primer sequence(5’-3’) | Product size | Tm (°C) |
| p53 | NM205264.1 | Forward Reverse | ACCTGCACTTACTCCCCGGT TCTTATAGACGGCCACGGCG | 127bp | 59 |
| p21 | AF513031.1 | Forward Reverse | TCCCTGCCCTGTACTGTCTAA GCGTGGGCTCTTCCTATACAT | 123bp | 60 |
| cdc2 | NM205314.1 | Forward Reverse | TCTGCTCTGTATTCCACTCCTG ATTGTTGGGTGTCCCTAAAGC | 144bp | 60 |
| cyclinB | NM205239.2 | Forward Reverse | ATCACCAACGCTCACAAGAAC AGGCTCCACAGGAACATCTG | 171bp | 59 |
| β-actin | L08165 | Forward Reverse | TGCTGTGTTCCCATCTATCG TTGGTGACAATACCGTGTTCA | 178bp | 62 |
Table 3. A list of primers of the inflammatory mediator genes in qRT-PCR analysis
| Gene symbol | Accession number | Primer | Primer sequence(5’-3’) | Product size | Tm (°C) |
| NF-κB | NM205134 | Forward Reverse | CTGAAACTACTGATTGCTGCTGGA GCTATGTGAAGAGGCGTTGTGC | 179bp | 62 |
| TNF-α | NM204267 | Forward Reverse | CCCCTACCCTGTCCCACAA TGAGTACTGCGGAGGGTTCAT | 100bp | 58 |
| IL-1β | Y15006 | Forward Reverse | CAGCCTCAGCGAAGAGACCTT CACTGTGGTGTGCTCAGAATCC | 106bp | 60 |
| IL-6 | AJ309540 | Forward Reverse | AATCCCTCCTCGCCAATCTG GCCCTCACGGTCTTCTCCATA | 106bp | 60 |
| IL-8 | HM179639 | Forward Reverse | GCCCTCACGGTCTTCTCCATA CTGGCCCTCCTCCTGGTT | 105bp | 60 |
| β-actin | L08165 | Forward Reverse | TGCTGTGTTCCCATCTATCG TTGGTGACAATACCGTGTTCA | 178bp | 62 |
For qRT-PCR reactions, 25μL mixtures were made containing 12.5μL SYBR® Premix Ex TaqTMII system (DRR820A, Takara, Japan), 1μL of forward and 1μL reverse primer, 8.5μL of RNAase-free water (RT12102, Tiangen, China) and 1μL of cDNA. Bio Rad C1000 Thermal Cycler (Bio Rad, USA) was used to perform qRT-PCR reactions. The PCR procedure consisted of 95 °C for 3 min followed by 44 cycles 95 °C for10 s, Tm of a specific primer pair for 30 s, and then 95 °C for 10 s, 72 °C for 10 s. The melting curve analysis showed only one peak for each PCR product.
Chicken β-actin expression was used as an internal reference housekeeping gene. Gene expression values from control group subsamples at 14, 28, and 42 days of age were used to calibrate gene expression in subsamples from corresponding experimental subsamples. All Ddata output from the qRT-PCR experiments were analyzed using the 2-ΔΔCT method [75].
Determination of the hepatic Ni contents by GFAAS
After five broilers in each group were humanely killed at 42 days of age, livers were immediately removed, weighed, dried, and collected for determination of Ni contents.
Ni concentrations in the liver were measured by GFAAS according to the reference [16].
Statistical analysis
The significance of difference among four groups was analyzed by variance analysis, and results presented as mean ± standard deviation (M±SD). The variation was measured by one-way analysis of variance (ANOVA) test of SPSS 16.0 for windows. Statistical significance was considered at P < 0.05.
Abbreviations
Ni: nickel; NiCl2: nickel chloride; NiSO4: nickel sulfate; HEp-2: human airway epithelial; MCF-7: human breast cancer; CHO: Chinese hamster ovary; ER: endoplasmic reticulum; MMP: mitochondrial membrane potential; cyt c: cytochrome c; AIF: apoptosis inducing factor; CHOP: C/EBP homologous protein; IL-1β: interleukin-1β; TNF-α: tumor necrosis factor α; ICAM-1: intercellular cell adhesion molecule-1; AST: aspartate aminotransferase; ALT: alanine aminotransferase; ALP: Alkaline phosphatase; GGT: Glutamyltransferase; ALB: albumin; GLB: globulin; TP: total protein; TBIL: total bilirubin; TUNEL: terminal deoxynucleotidyl transferase 2ʹ-deoxyuridine 5ʹ-triphosphate dUTP nick end-labeling; Bcl-xL: Bcl-extra long; Mcl-1: myeloid cell leukemia factor-1; PARP: poly ADP-ribose polymerase; FasL: Fas ligand; GRP78: glucose-regulated protein 78; UPR: unfolded protein response; PERK: protein kinase RNA (PKR)-like ER kinase; IRE1: inositol-requiring enzyme 1; ATF6: activated transcription factor 6; eIF2α: elongation initiation factor 2α; XBP1: X-boxbinding protein 1; NF-κB: nuclear factor-κB; NiONPs: NiO nanoparticles; Ni3S2: nickel sulfide; GADD34: growth arrest and DNA-damage-inducible 34; ERO1α: endoplasmic reticulum oxidoreductase 1α; TRAF2: TNF receptor-associated factor 2; ASK1/JNK: apoptosis signal-regulating kinase 1/c-Jun amino terminal kinase; FCE: feed conversion efficiency; PI: propidium iodide; GFAAS: graphite furnace atomic absorption spectrometry.
Funding
The study was supported by the program for Changjiang scholars and innovative research team in university (IRT 0848) and the Shuangzhi project of Sichuan Agricultural University (03570327; 03571189).
Conflicts of Interest
The authors declare no competing financial interests.
References
- 1. Poonkothai M, Vijayavathi BS. Nickel as an essential element and a toxicant. Inter J Environ Sci. 2012; 1:285–88.
- 2. Martinez-Zamudio R, Ha HC. Environmental epigenetics in metal exposure. Epigenetics. 2011; 6:820–27. https://doi.org/10.4161/epi.6.7.16250 [PubMed]
- 3. Cempel M, Nikel G. Nickel: A Review of Its Sources and Environmental Toxicology. Polish J Environ Stud. 2006; 15:375–82.
- 4. Das KK, Das SN, Dhundasi SA. Nickel, its adverse health effects & oxidative stress. Indian J Med Res. 2008; 128:412–25. [PubMed]
- 5. Zheng GH, Liu CM, Sun JM, Feng ZJ, Cheng C. Nickel-induced oxidative stress and apoptosis in Carassius auratus liver by JNK pathway. Aquat Toxicol. 2014; 147:105–11. https://doi.org/10.1016/j.aquatox.2013.12.015 [PubMed]
- 6. Su L, Deng Y, Zhang Y, Li C, Zhang R, Sun Y, Zhang K, Li J, Yao S. Protective effects of grape seed procyanidin extract against nickel sulfate-induced apoptosis and oxidative stress in rat testes. Toxicol Mech Methods. 2011; 21:487–94. https://doi.org/10.3109/15376516.2011.556156 [PubMed]
- 7. Liu CM, Zheng GH, Ming QL, Chao C, Sun JM. Sesamin protects mouse liver against nickel-induced oxidative DNA damage and apoptosis by the PI3K-Akt pathway. J Agric Food Chem. 2013; 61:1146–54. https://doi.org/10.1021/jf304562b [PubMed]
- 8. Tang K, Guo H, Deng J, Cui H, Peng X, Fang J, Zuo Z, Wang X, Wu B, Li J, Yin S. Inhibitive effects of nickel chloride (NiCl2) on thymocytes. Biol Trace Elem Res. 2015; 164:242–52. https://doi.org/10.1007/s12011-014-0219-x [PubMed]
- 9. Wu B, Cui H, Peng X, Fang J, Zuo Z, Deng J, Huang J. Toxicological effects of nickel chloride on IgA+ B Cells and sIgA, IgA, IgG, IgM in the intestinal mucosal immunity in broilers. Int J Environ Res Public Health. 2014; 11:8175–92. https://doi.org/10.3390/ijerph110808175 [PubMed]
- 10. Wu B, Cui H, Peng X, Fang J, Zuo Z, Deng J, Huang J. Dietary nickel chloride induces oxidative stress, apoptosis and alters Bax/Bcl-2 and caspase-3 mRNA expression in the cecal tonsil of broilers. Food Chem Toxicol. 2014; 63:18–29. https://doi.org/10.1016/j.fct.2013.10.033 [PubMed]
- 11. Tang K, Li J, Yin S, Guo H, Deng J, Cui H. Effects of Nickel Chloride on Histopathological Lesions and Oxidative Damage in the Thymus. Health. 2014; 6:2875–82. https://doi.org/10.4236/health.2014.621326
- 12. Huang J, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Wu B. Effect of dietary nickel chloride on splenic immune function in broilers. Biol Trace Elem Res. 2014; 159:183–91. https://doi.org/10.1007/s12011-014-0003-y [PubMed]
- 13. Guo H, Wu B, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Deng J, Yin S, Li J, Tang K. NiCl2-down-regulated antioxidant enzyme mRNA expression causes oxidative damage in the broiler(')s kidney. Biol Trace Elem Res. 2014; 162:288–95. https://doi.org/10.1007/s12011-014-0132-3 [PubMed]
- 14. Wu B, Cui H, Peng X, Fang J, Zuo Z, Huang J, Luo Q, Deng Y, Wang H, Liu J. Changes of the serum cytokine contents in broilers fed on diets supplemented with nickel chloride. Biol Trace Elem Res. 2013; 151:234–39. https://doi.org/10.1007/s12011-012-9554-y [PubMed]
- 15. Wu B, Cui H, Peng X, Fang J, Zuo Z, Deng J, Huang J. Dietary nickel chloride induces oxidative intestinal damage in broilers. Int J Environ Res Public Health. 2013; 10:2109–19. https://doi.org/10.3390/ijerph10062109 [PubMed]
- 16. Guo H, Deng H, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Wu B, Chen K. Nickel chloride (NiCl2)-caused inflammatory responses via activation of NF-κB pathway and reduction of anti-inflammatory mediator expression in the kidney. Oncotarget. 2015; 6:28607–20. [PubMed]
- 17. Guo H, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Wu B, Chen K, Deng J. Modulation of the PI3K/Akt Pathway and Bcl-2 Family Proteins Involved in Chicken’s Tubular Apoptosis Induced by Nickel Chloride (NiCl2). Int J Mol Sci. 2015; 16:22989–3011. https://doi.org/10.3390/ijms160922989 [PubMed]
- 18. Guo H, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Wu B, Chen K, Deng J. Dietary NiCl2 causes G2/M cell cycle arrest in the broiler’s kidney. Oncotarget. 2015; 6:35964–77. [PubMed]
- 19. Yin S, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Wu B, Guo H. Toxic effect of NiCl2 on development of the bursa of Fabricius in broiler chickens. Oncotarget. 2016; 7:125–39. [PubMed]
- 20. Wu B, Guo H, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Huang J. Pathway underlying small intestine apoptosis by dietary nickel chloride in broiler chickens. Chem Biol Interact. 2016; 243:91–106. https://doi.org/10.1016/j.cbi.2015.11.010 [PubMed]
- 21. Wu B, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Huang J. Toxicological effects of nickel chloride on the cytokine mRNA expression and protein levels in intestinal mucosal immunity of broilers. Environ Toxicol. 2015; 30:1309–21. https://doi.org/10.1002/tox.22001 [PubMed]
- 22. Yin S, Guo H, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Tang K, Li J. Nickel Chloride (NiCl2) Induces Histopathological Lesions via Oxidative Damage in the Broiler’s Bursa of Fabricius. Biol Trace Elem Res. 2016; 171:214–23. https://doi.org/10.1007/s12011-015-0528-8 [PubMed]
- 23. Nowak M, Kopp F, Roelofs-Haarhuis K, Wu X, Gleichmann E. Oral nickel tolerance: fas ligand-expressing invariant NK T cells promote tolerance induction by eliciting apoptotic death of antigen-carrying, effete B cells. J Immunol. 2006; 176:4581–89. https://doi.org/10.4049/jimmunol.176.8.4581 [PubMed]
- 24. Guan F, Zhang D, Wang X, Chen J. Nitric oxide and bcl-2 mediated the apoptosis induced by nickel(II) in human T hybridoma cells. Toxicol Appl Pharmacol. 2007; 221:86–94. https://doi.org/10.1016/j.taap.2007.01.029 [PubMed]
- 25. Siddiqui MA, Ahamed M, Ahmad J, Majeed Khan MA, Musarrat J, Al-Khedhairy AA, Alrokayan SA. Nickel oxide nanoparticles induce cytotoxicity, oxidative stress and apoptosis in cultured human cells that is abrogated by the dietary antioxidant curcumin. Food Chem Toxicol. 2012; 50:641–47. https://doi.org/10.1016/j.fct.2012.01.017 [PubMed]
- 26. Shiao YH, Lee SH, Kasprzak KS. Cell cycle arrest, apoptosis and p53 expression in nickel(II) acetate-treated Chinese hamster ovary cells. Carcinogenesis. 1998; 19:1203–07. https://doi.org/10.1093/carcin/19.7.1203 [PubMed]
- 27. Chen C-Y, Lin T-K, Chang Y-C, Wang Y-F, Shyu H-W, Lin K-H, Chou M-C. Nickel(II)-induced oxidative stress, apoptosis, G2/M arrest, and genotoxicity in normal rat kidney cells. J Toxicol Environ Health A. 2010; 73:529–39. https://doi.org/10.1080/15287390903421250 [PubMed]
- 28. Guo H, Chen L, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Wu B. Research Advances on Pathways of Nickel-Induced Apoptosis. Int J Mol Sci. 2015; 17:E10. https://doi.org/10.3390/ijms17010010 [PubMed]
- 29. Mishra NC, Kumar S. Apoptosis: a mitochondrial perspective on cell death. Indian J Exp Biol. 2005; 43:25–34. [PubMed]
- 30. Logue SE, Cleary P, Saveljeva S, Samali A. New directions in ER stress-induced cell death. Apoptosis. 2013; 18:537–46. https://doi.org/10.1007/s10495-013-0818-6 [PubMed]
- 31. Schmitz I, Kirchhoff S, Krammer PH. Regulation of death receptor-mediated apoptosis pathways. Int J Biochem Cell Biol. 2000; 32:1123–36. https://doi.org/10.1016/S1357-2725(00)00048-0 [PubMed]
- 32. Ahamed M, Akhtar MJ, Alhadlaq HA, Khan MA, Alrokayan SA. Comparative cytotoxic response of nickel ferrite nanoparticles in human liver HepG2 and breast MFC-7 cancer cells. Chemosphere. 2015; 135:278–88. https://doi.org/10.1016/j.chemosphere.2015.03.079 [PubMed]
- 33. Zhao J, Bowman L, Zhang X, Shi X, Jiang B, Castranova V, Ding M. Metallic nickel nano- and fine particles induce JB6 cell apoptosis through a caspase-8/AIF mediated cytochrome c-independent pathway. J Nanobiotechnology. 2009; 7:2. https://doi.org/10.1186/1477-3155-7-2 [PubMed]
- 34. Urra H, Dufey E, Lisbona F, Rojas-Rivera D, Hetz C. When ER stress reaches a dead end. Biochim Biophys Acta. 2013; 1833:3507–17. https://doi.org/10.1016/j.bbamcr.2013.07.024 [PubMed]
- 35. Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013; 1833:3460–70. https://doi.org/10.1016/j.bbamcr.2013.06.028 [PubMed]
- 36. Hiramatsu N, Kasai A, Du S, Takeda M, Hayakawa K, Okamura M, Yao J, Kitamura M. Rapid, transient induction of ER stress in the liver and kidney after acute exposure to heavy metal: evidence from transgenic sensor mice. FEBS Lett. 2007; 581:2055–59. https://doi.org/10.1016/j.febslet.2007.04.040 [PubMed]
- 37. Dunnick JK, Elwell MR, Benson JM, Hobbs CH, Hahn FF, Haly PJ, Cheng YS, Eidson AF. Lung toxicity after 13-week inhalation exposure to nickel oxide, nickel subsulfide, or nickel sulfate hexahydrate in F344/N rats and B6C3F1 mice. Fundam Appl Toxicol. 1989; 12:584–94. https://doi.org/10.1016/0272-0590(89)90031-6 [PubMed]
- 38. Capasso L, Camatini M, Gualtieri M. Nickel oxide nanoparticles induce inflammation and genotoxic effect in lung epithelial cells. Toxicol Lett. 2014; 226:28–34. https://doi.org/10.1016/j.toxlet.2014.01.040 [PubMed]
- 39. Li X, Zhong F. Nickel induces interleukin-1β secretion via the NLRP3-ASC-caspase-1 pathway. Inflammation. 2014; 37:457–66. https://doi.org/10.1007/s10753-013-9759-z [PubMed]
- 40. Cortijo J, Milara J, Mata M, Donet E, Gavara N, Peel SE, Hall IP, Morcillo EJ. Nickel induces intracellular calcium mobilization and pathophysiological responses in human cultured airway epithelial cells. Chem Biol Interact. 2010; 183:25–33. https://doi.org/10.1016/j.cbi.2009.09.011 [PubMed]
- 41. Green DR, Llambi F. Cell Death Signaling. Cold Spring Harb Perspect Biol. 2015; 7:a006080. https://doi.org/10.1101/cshperspect.a006080 [PubMed]
- 42. Hartwell LH, Kastan MB. Cell cycle control and cancer. Science. 1994; 266:1821–28. https://doi.org/10.1126/science.7997877 [PubMed]
- 43. Gathwan KH, Al-Karkhi IH, Jaffar Al-Mulla EA. Hepatic toxicity of nickel chloride in mice. Res Chem Intermed. 2012; 39:2537–42. https://doi.org/10.1007/s11164-012-0780-x
- 44. Huang J, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wu B. The association between splenocyte apoptosis and alterations of Bax, Bcl-2 and caspase-3 mRNA expression, and oxidative stress induced by dietary nickel chloride in broilers. Int J Environ Res Public Health. 2013; 10:7310–26. https://doi.org/10.3390/ijerph10127310 [PubMed]
- 45. Siddiqui WA, Ahad A, Ahsan H. The mystery of BCL2 family: Bcl-2 proteins and apoptosis: an update. Arch Toxicol. 2015; 89:289–317. https://doi.org/10.1007/s00204-014-1448-7 [PubMed]
- 46. Steinbach JP, Weller M. Apoptosis in Gliomas: Molecular Mechanisms and Therapeutic Implications. J Neurooncol. 2004; 70:247–56. https://doi.org/10.1007/s11060-004-2753-4 [PubMed]
- 47. Wang YF, Shyu HW, Chang YC, Tseng WC, Huang YL, Lin KH, Chou MC, Liu HL, Chen CY. Nickel (II)-induced cytotoxicity and apoptosis in human proximal tubule cells through a ROS- and mitochondria-mediated pathway. Toxicol Appl Pharmacol. 2012; 259:177–86. https://doi.org/10.1016/j.taap.2011.12.022 [PubMed]
- 48. Duan WX, He MD, Mao L, Qian FH, Li YM, Pi HF, Liu C, Chen CH, Lu YH, Cao ZW, Zhang L, Yu ZP, Zhou Z. NiO nanoparticles induce apoptosis through repressing SIRT1 in human bronchial epithelial cells. Toxicol Appl Pharmacol. 2015; 286:80–91. https://doi.org/10.1016/j.taap.2015.03.024 [PubMed]
- 49. Pan J, Chang Q, Wang X, Son Y, Zhang Z, Chen G, Luo J, Bi Y, Chen F, Shi X. Reactive oxygen species-activated Akt/ASK1/p38 signaling pathway in nickel compound-induced apoptosis in BEAS 2B cells. Chem Res Toxicol. 2010; 23:568–77. https://doi.org/10.1021/tx9003193 [PubMed]
- 50. Zhang N, Chen Y, Jiang R, Li E, Chen X, Xi Z, Guo Y, Liu X, Zhou Y, Che Y, Jiang X. PARP and RIP 1 are required for autophagy induced by 11′-deoxyverticillin A, which precedes caspase-dependent apoptosis. Autophagy. 2011; 7:598–612. https://doi.org/10.4161/auto.7.6.15103 [PubMed]
- 51. Lee Y-J, Lim S-S, Baek BJ, An J-M, Nam H-S, Woo K-M, Cho M-K, Kim S-H, Lee S-H. Nickel(II)-induced nasal epithelial toxicity and oxidative mitochondrial damage. Environ Toxicol Pharmacol. 2016; 42:76–84. https://doi.org/10.1016/j.etap.2016.01.005 [PubMed]
- 52. Scott FL, Stec B, Pop C, Dobaczewska MK, Lee JJ, Monosov E, Robinson H, Salvesen GS, Schwarzenbacher R, Riedl SJ. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature. 2009; 457:1019–22. https://doi.org/10.1038/nature07606 [PubMed]
- 53. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010; 140:900–17. https://doi.org/10.1016/j.cell.2010.02.034 [PubMed]
- 54. Guo H, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Wu B, Chen K, Deng J. Nickel chloride (NiCl2) induces endoplasmic reticulum (ER) stress by activating UPR pathways in the kidney of broiler chickens. Oncotarget. 2016; 7:17508–19. [PubMed]
- 55. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011; 334:1081–86. https://doi.org/10.1126/science.1209038 [PubMed]
- 56. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001; 107:881–91. https://doi.org/10.1016/S0092-8674(01)00611-0 [PubMed]
- 57. Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A, Agostinis P. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012; 19:1880–91. https://doi.org/10.1038/cdd.2012.74 [PubMed]
- 58. Hetz CA. ER stress signaling and the BCL-2 family of proteins: from adaptation to irreversible cellular damage. Antioxid Redox Signal. 2007; 9:2345–55. https://doi.org/10.1089/ars.2007.1793 [PubMed]
- 59. Huang HL, Wu JL, Chen MH, Hong JR. Aquatic birnavirus-induced ER stress-mediated death signaling contribute to downregulation of Bcl-2 family proteins in salmon embryo cells. PLoS One. 2011; 6:e22935. https://doi.org/10.1371/journal.pone.0022935 [PubMed]
- 60. Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, Backes BJ, Oakes SA, Papa FR. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009; 138:562–75. https://doi.org/10.1016/j.cell.2009.07.017 [PubMed]
- 61. Jurczak MJ, Lee AH, Jornayvaz FR, Lee HY, Birkenfeld AL, Guigni BA, Kahn M, Samuel VT, Glimcher LH, Shulman GI. Dissociation of inositol-requiring enzyme (IRE1α)-mediated c-Jun N-terminal kinase activation from hepatic insulin resistance in conditional X-box-binding protein-1 (XBP1) knock-out mice. J Biol Chem. 2012; 287:2558–67. https://doi.org/10.1074/jbc.M111.316760 [PubMed]
- 62. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000; 287:664–66. https://doi.org/10.1126/science.287.5453.664 [PubMed]
- 63. Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996; 274:1664–72. https://doi.org/10.1126/science.274.5293.1664 [PubMed]
- 64. Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001; 20:1803–15. https://doi.org/10.1038/sj.onc.1204252 [PubMed]
- 65. Yin X, Zhang R, Feng C, Zhang J, Liu D, Xu K, Wang X, Zhang S, Li Z, Liu X, Ma H. Diallyl disulfide induces G2/M arrest and promotes apoptosis through the p53/p21 and MEK-ERK pathways in human esophageal squamous cell carcinoma. Oncol Rep. 2014; 32:1748–56. [PubMed]
- 66. Salnikow K, Davidson T, Kluz T, Chen H, Zhou D, Costa M. GeneChip analysis of signaling pathways effected by nickel. J Environ Monit. 2003; 5:206–09. https://doi.org/10.1039/b210262p [PubMed]
- 67. Salnikow K, An WG, Melillo G, Blagosklonny MV, Costa M. Nickel-induced transformation shifts the balance between HIF-1 and p53 transcription factors. Carcinogenesis. 1999; 20:1819–23. https://doi.org/10.1093/carcin/20.9.1819 [PubMed]
- 68. Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001; 107:7–11. https://doi.org/10.1172/JCI11830 [PubMed]
- 69. Freitas M, Fernandes E. Zinc, cadmium and nickel increase the activation of NF-κB and the release of cytokines from THP-1 monocytic cells. Metallomics. 2011; 3:1238–43. https://doi.org/10.1039/c1mt00050k [PubMed]
- 70. NRC. 1994. Nutrient Requirements of Poultry. 9th Revised Ed. National Academy Press Washington, DC.
- 71. Ling JR, Leach RM
Jr . Studies on nickel metabolism: interaction with other mineral elements. Poult Sci. 1979; 58:591–96. https://doi.org/10.3382/ps.0580591 [PubMed] - 72. Weber CW, Reid BL. Nickel toxicity in growing chicks. J Nutr. 1968; 95:612–16. [PubMed]
- 73. Szilagyi M, Szentmihalyi S, Anke M. Changes in some of the biochemical parameters in Ni and Mo deficient animals. Proceeding (Hungary). 1981; 1:257–60. goat, sheep, pig, chicken, rat.
- 74. Bersényi A, Fekete SG, Szilágyi M, Berta E, Zöldág L, Glávits R. Effects of nickel supply on the fattening performance and several biochemical parameters of broiler chickens and rabbits. Acta Vet Hung. 2004; 52:185–97. https://doi.org/10.1556/AVet.52.2004.2.7 [PubMed]
- 75. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402–08. https://doi.org/10.1006/meth.2001.1262 [PubMed]