



Introduction
Hepatocellular carcinoma (HCC) is the most common type of the liver cancer. The incidence and death rate of the liver cancer increase rapidly worldwide [1]. In China, the liver cancer remains the third cause of cancer-related deaths and the third commonly diagnosed cancer [2]. Some therapeutic strategies such as curative resection, liver transplantation, radiofrequency ablation (RFA) and transarterial embolization (TAE) may be promising for the treatment of HCC, whereas its prognosis remains unsatisfactory due to high recurrence and metastasis rates [3–6]. The underlying mechanisms may involve epigenetic alterations and genetic mutations, as well as lack of reliable gene signatures [7–10]. It is hence of clinical importance to construct a robust molecular model that can reliably predict the prognosis of HCC and has the potential to guide personalized therapeutic first-line treatment strategies.
Tremendous improvement has been made over the past decade in seeking prognostic genetic biomarkers for HCC [11]. In particular, the development of high-throughput platforms including DNA microarrays and RNA sequencing contributes greatly to the identification of potential biomarkers and therapeutic targets [12]. A large panel of genes have been reported to be associated with clinical outcomes of HCC, and some genetic prediction models have developed accordingly [13–16]. Besides, other types of genetic forms, such as long non-coding RNA (lncRNA) have been demonstrated to play a role in the pathogenesis of HCC [17]. However, few of these genetic biomarkers have been applied to clinical practice owing to lack of general extrapolation and experimental or clinical validation. Thus, identification of more reliable genetic biomarkers that can accurately predict HCC prognosis is still in the process of exploration and perfection.
Considering that HCC is an extremely heterogeneous disease and targeted therapies and personalized management become promising these days, the specific genomic sub-classification of HCC patients is of great significance for the improvement of prognosis assessment [18]. Here, we attempted to identify specific genes that are significantly dysregulated only in HCC rather than in the other subtypes of the liver cancer like intrahepatic cholangiocarcinoma (ICC) and the secondary liver cancer. Although therapeutic approaches for ICC are similar to HCC, the clinical outcome is relatively poorer than HCC, partly due to different molecular pathologic mechanisms [19,20]. As for the metastatic liver cancer (MLC), colorectal cancer liver metastasis (CRCLM) was adopted as an example here, which was a common type of secondary liver cancer [21]. Like other MLC, the treatment for CRCLM needs multidisciplinary approaches because of complex complications of primary cancer and secondary liver cancer [22–24]. Though hepatectomy is an optimal approach, the prognosis of CRCLM largely depended on primary cancer that differed from HCC [25–28]. Therefore, the specificity of the prognostic model is fetal to the precise prognostic prediction and therapeutic decision-making of HCC. In this study, we aimed to develop a specific, accurate and robust genetic prognostic risk score system for HCC.
Results
Identification of HCC-specific gene list
The overall workflow of this study is presented in Figure 1A. After background correcting, normalization and quality control of the selected raw profiles of four datasets (GSE5851, GSE28691, GSE15765 and GSE14323), 70 HCC samples, 75 CRCLM samples, 12 ICC samples and 19 normal control samples were obtained and considered eligible for further analysis. The relative expression of all samples pre- and post-normalization is shown in Figure 2B. Next, we compared three subtypes of the liver cancer samples with control samples, respectively. Three lists of dysregulated genes were identified and subjected to Venn selection for cancer-specific genes. A total of 1103 HCC-specific genes were identified including 816 up-regulated and 287 down-regulated (Figure 1C). The list of HCC-specific genes and their comparisons between HCC and non-tumor samples are shown in Supplementary Table S1. Moreover, gene ontology analysis of these genes revealed that “positive regulation of transferase activity”, “NGF signaling via TRKA from the plasma membrane”, and “transmembrane receptor protein tyrosine kinase signaling pathway” were significantly enriched biological processes and pathways which might be associated with HCC progression (Figure 2B). The protein-protein interaction (PPI) network of HCC-specific genes and top 20 significant enriched terms are presented in Figure 2A and Figure 2C.
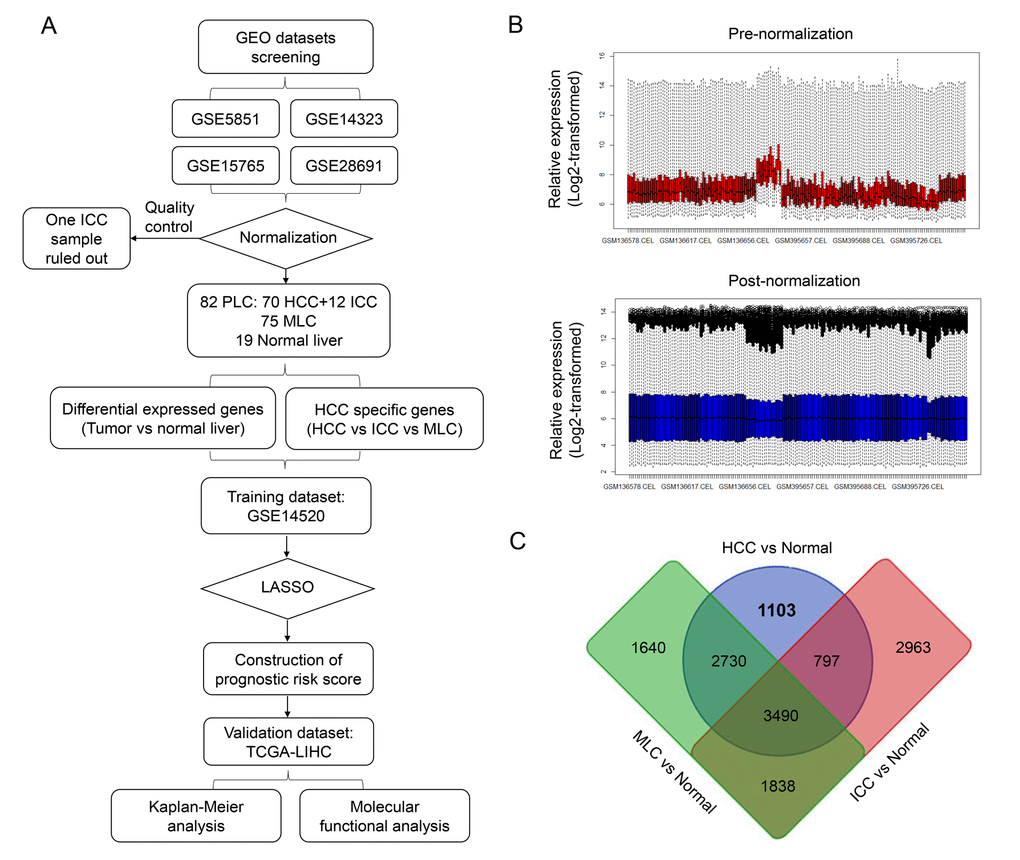
Figure 1. Identification of HCC-specific gene list. (A) Overview of the overall design and analytic procedure of the study. (B) Relative expression of all the included sample before (Upper & Red) and after (Lower & blue) RMA normalization. All the expression value was transformed by “log2()” algorism. (C) Venn diagram among three lists of dysregulated genes between three different subtypes of liver cancer (HCC, ICC and MLC) and normal liver. 1103 HCC-specific genes, 2963 ICC-specific genes and 1640 MLC-specific genes were generated through Venn selection.
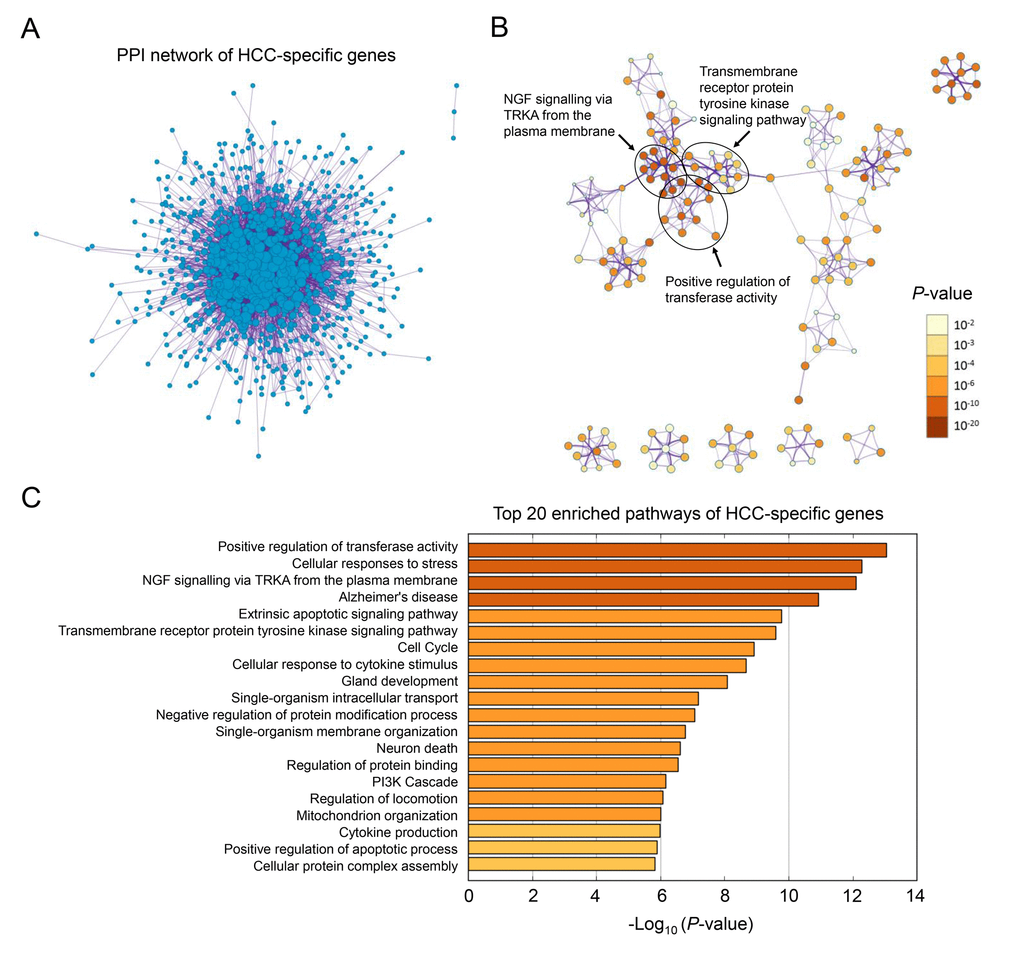
Figure 2. Gene ontology analysis of HCC-specific genes. (A) The PPI network of all the HCC-specific genes illustrated in Cytoscape. Each node represented a protein translated by an HCC-specific gene. (B) Network of 20 top-score modules (clusters) visualized in Cytoscape. Each cluster was made up of 10 best enriched GO terms within the threshold of Kappa-statistical similarity (0.3). Each node represented one enriched term and was colored by P value. In the figure, 3 representative pathways and the clusters they belonged to were marked. (C) The bar chart of 20 most enriched terms of HCC-specific genes arranged by -Log10P value.
Construction of HCC-specific gene risk score system
To explore the prognostic value of HCC-specific genes, univariate survival analysis by Cox proportional hazards models of each gene from the training data (GSE14520) was conducted. All the genes with significant P values were screened for next-step model construction. Eventually, the score formula comprised of 12 optimal genes was developed by LASSO: Risk score = 0.66 × (expression value of RNF24) + (-0.61) × (expression value of COPS8) + 0.40 × (expression value of EWSR1) + (-0.40) × (expression value of SUGCT) + 0.38 × (expression value of PCSK5) + 0.35 × (expression value of POLR3C) + 0.31 × (expression value of NRBP1) + 0.27 × (expression value of MNAT1) + 0.18 × (expression value of EIF5B) + (-0.15) × (expression value of DUSP10) + 0.08 × (expression value of WASF1) + 0.07 × (expression value of CCDC88A). In this risk score system, three genes (COPS8, SUGCT and DUSP10) were proved to be positively associated with OS, while nine of them (EIF5B, MNAT1, WASF1, EWSR1, POLR3C, RNF24, PCSK5, NRBP1 and CCDC88A) were negatively related to OS according to the negativity or positivity of their coefficients. The contribution of each gene made to this risk score model was weighted by absolute value of coefficients. Every patient would get a risk score according to the expressions of the 12 HCC-specific genes of themselves (Figure 3A). This risk score was considered to correlate with the individual overall survival (OS). The median of all patients’ scores used as the cut-off value divided the whole group into the high-risk and the low-risk groups (Figure 3A). The OS and DFS status of each patient in the training dataset was shown in Figure 3B and Figure 3C. From Kaplan-Meier analysis of GSE14520, high-risk group was thought to be associated with poor prognosis while low-risk group was predicted to have the opposite outcome (Figure 3D & 3E).
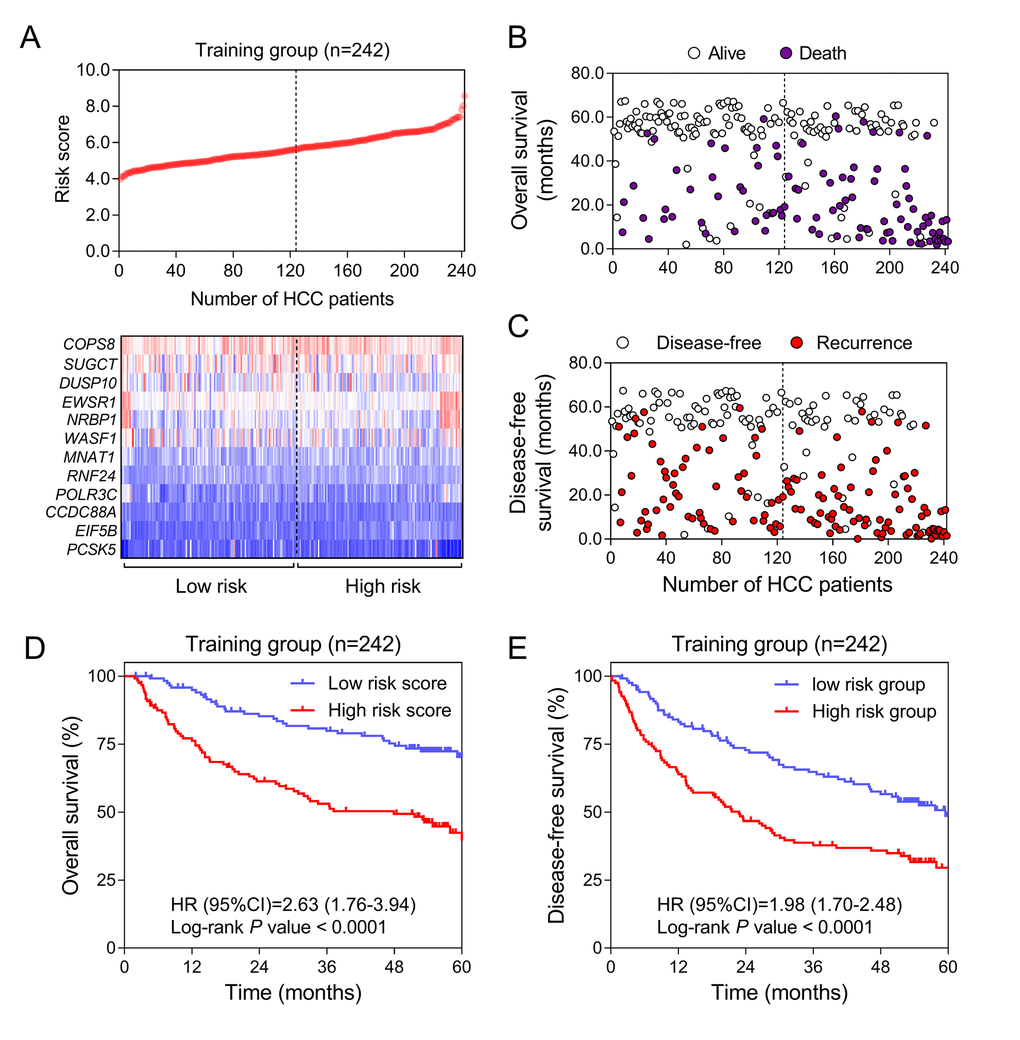
Figure 3. Construction of HCC-specific gene risk score system using GSE14520. (A) HCC-specific risk score analysis in GSE14520. (Upper) The distribution of the risk score of 242 included samples. (Lower) Heatmap of the expression value of each gene in HCC-specific gene signature corresponding to each patient above. Red: high expression; Blue: low expression. (B and C) Survival (B) and recurrence (C) status of every patient in the training dataset (N=242). (D and E) Kaplan-Meier curves to compare OS (D) and DFS (E) of high-risk and low-risk groups in GSE14520.
Validation and development of 12 HCC-specific gene signature for prognosis
To confirm the potentiality of the 12-HCC-specific gene prognostic model, Kaplan-Meier curve was performed to evaluate the association between the OS and DFS and our gene signature in validation dataset (TCGA) (Figure 4A). The cut-off values of TCGA cohort was 8.9. From the results, those high-risk patients had significantly shorter survival and earlier recurrence (P < 0.01).
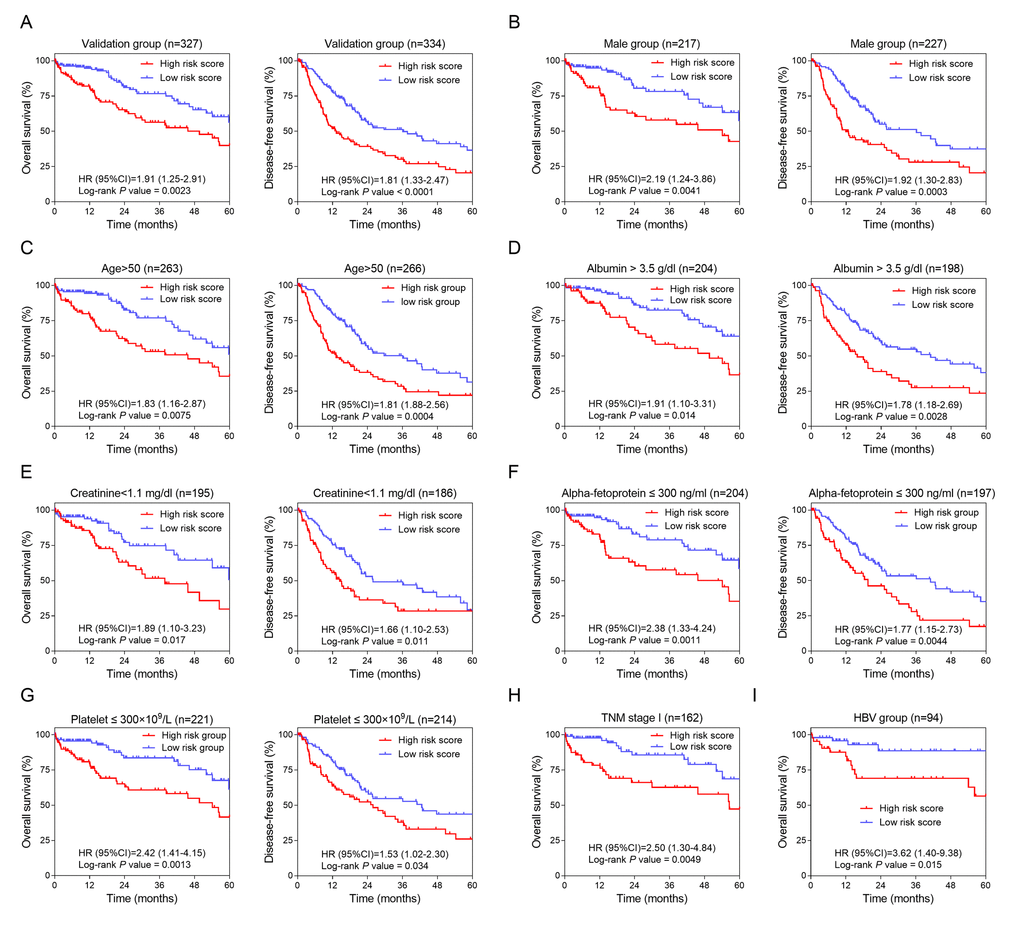
Figure 4. Validation and development of HCC-specific risk score system. (A) Kaplan-Meier curves of OS (Left) and DFS (Right) in the validation dataset. (B, C, D, E, F and G) Kaplan-Meier curves of OS (Left) and DFS (Right) in the subgroups stratified by gender (Male) (B), age (>50) (C), ALB (> 3.5 g/dl) (D), CRE (< 1.1 mg/dl) (E), AFP (≤ 300 ng/ml) (F) and PLT (≤ 300×109/L) (G). (H and I) Kaplan-Meier curves of OS in the subgroups stratified by TNM stage (stage I) (H) and HBV infection (I).
In order to investigate the prognostic value of the risk score system in different patient groups with different characteristics, we firstly performed univariate and multivariate Cox regression analyses to confirm the relevance between different characteristics including the risk score and OS or DFS. From the Cox regression results, the risk score (> 8.9) and the race (THE White) were independent risk factors of OS (Table 1). And of DFS, the risk score (> 8.9), TNM stage (higher grade) and ECOG score (higher score) were independent risk factors (Table 2). Secondly, stratified analyses based on these clinical characteristics were carried out to identify the suitable patient groups of the risk score system (Table 3). The cut-off value (8.9) of each subgroup for survival analysis was consistent with the mother group. The stratified results of the validation dataset showed our HCC-specific gene signature was more applicable to the elderly male patients with preoperative relatively lower serum level of CRE (creatinine), AFP (alpha-fetoprotein) and PLT (platelet) and higher serum level of ALB (albumin) (Figure 4B, 4C, 4D, 4E, 4F and 4G). Besides, this risk score system exhibited better performance in predicting OS particularly in patients with HBV infection and in early stage (Figure 4H & 4I).
Table 1. Univariate/multivariate Cox regression analysis of clinicopathologic factors associated with OS in TCGA cohort.
| Variables | Univariate analysis | Multivariate analysis | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HR (95% CI) | P value | HR (95% CI) | P value | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Risk score (> 8.9/≤ 8.9) | 1.910(1.250-2.919) | 0.003* | 1.617(1.021-2.560) | 0.040* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TNM stage (I/II/III/IV) | 1.282(0.993-1.655) | 0.057 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Hepatitis (HBV/HCV/neither) | 0.751(0.527-1.069) | 0.112 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Alcohol consumption (yes/no) | 0.835(0.514-1.359) | 0.469 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Gender (female/male) | 0.773(0.504-1.185) | 0.237 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age (>50/≤50) | 1.967(1.043-3.710) | 0.037* | 1.454(0.711-2.971) | 0.305 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cirrhosis (yes/no) | 0.865(0.471-1.587) | 0.639 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Albumin (≤3.5/>3.5 g/dl) | 1.378(0.834-2.277) | 0.211 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Creatinine (<1.1/≥1.1 mg/dl) | 0.739(0.455-1.199) | 0.221 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AFP a (≤300/>300 ng/ml) | 0.900(0.513-1.582) | 0.715 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Platelet (≤300/>300×109/L) | 0.753(0.466-1.216) | 0.246 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Race (Asian/White) | 0.760(0.638-0.904) | 0.002* | 0.766(0.630-0.930) | 0.007* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BMI b (≥25<25) | 1.028(0.650-1.626) | 0.905 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Family history (yes/no) | 1.800(1.152-2.812) | 0.010* | 1.271(0.778-2.077) | 0.339 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ECOG c | 1.406(0.956-2.066) | 0.083 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Histological grade (G3-4/G1-2) | 1.247(0.802-1.938) | 0.327 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviations: OS, overall survival; HR, hazard ratio; 95% CI, 95% confidence interval. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| *: Statistically significant; | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a: Alpha-fetoprotein; | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| b: body mass index; | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| c: Eastern Cooperative Oncology Group. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 2. Univariate/multivariate Cox regression analysis of clinicopathologic factors associated with DFS in TCGA cohort.
| Variables | Univariate analysis | Multivariate analysis | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HR (95% CI) | P value | HR (95% CI) | P value | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Risk score (> 8.9/≤ 8.9) | 1.841(1.358-2.494) | <0.001* | 1.483(1.038-2.117) | 0.030* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TNM stage (I/II/III/IV) | 1.727(1.441-2.070) | <0.001* | 1.568(1.274-1.929) | <0.001* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Hepatitis (HBV/HCV/neither) | 0.943(0.760-1.170) | 0.592 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Alcohol consumption (yes/no) | 1.061(0.767-1.468) | 0.720 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Gender (female/male) | 0.982(0.711-1.355) | 0.911 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age (> 50/≤ 50) | 1.015(0.693-1.487) | 0.940 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cirrhosis (yes/no) | 1.271(0.861-1.877) | 0.228 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Albumin (≤ 3.5/> 3.5 g/dl) | 1.033(0.702-1.519) | 0.870 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Creatinine (< 1.1/≥ 1.1 mg/dl) | 0.739(0.511-1.069) | 0.109 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AFP a (≤ 300/> 300 ng/ml) | 1.035(0.681-1.573) | 0.873 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Platelet (≤ 300/> 300×109/L) | 1.415(0.976-2.052) | 0.067 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Race (Asian/White) | 0.787(0.575-1.078) | 0.136 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BMI b (≥ 25/< 25 kg/m2) | 0.882(0.643-1.211) | 0.437 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Family history (yes/no) | 0.920(0.655-1.292) | 0.630 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ECOG c | 1.697(1.406-2.049) | <0.001* | 1.389(1.138-1.695) | 0.001* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Histological grade (G3-4/G1-2) | 1.186(0.867-1.621) | 0.286 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviations: DFS, disease-free survival; HR, hazard ratio; 95% CI, 95% confidence interval. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| *: Statistically significant; | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a: Alpha-fetoprotein; | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| b: body mass index; | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| c: Eastern Cooperative Oncology Group. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 3. Stratified analysis of overall and disease-free survival in TCGA samples.
| Characteristics | Overall survival | Disease-free survival | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| High-risk / low-risk | HR (95% CI) | P value | High-risk / low-risk | HR (95% CI) | P value | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Overall | 154/173 | 1.905 (1.248-2.910) | 0.0023* | 164/170 | 1.811 (1.329-2.466) | <0.0001* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TNM stage | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Stage I | 68/94 | 2.502 (1.295-4.835) | 0.0049* | 63/95 | 1.643 (0.9616-2.806) | 0.0509 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Stage II | 36/38 | 1.383 (0.5167-3.700) | 0.5161 | 39/37 | 1.098 (0.5908-2.041) | 0.7632 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Stage III | 38/26 | 2.263 (0.9245-5.539) | 0.0524 | 49/25 | 1.714 (0.9881-2.972) | 0.0592 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Hepatitis | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HBV | 44/50 | 3.622 (1.398-9.384) | 0.015* | 44/48 | 1.736 (0.9153-3.294) | 0.0873 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HCV | 25/29 | 2.661 (0.8091-8.750) | 0.0644 | 23/28 | 1.263 (0.5953-2.682) | 0.5234 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Non-hepatitis | 81/84 | 1.643 (0.9681-2.790) | 0.0539 | 90/84 | 2.157 (1.421-3.276) | 0.0002* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Alcohol consumption | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Yes | 40/54 | 2.235 (0.8735-5.719) | 0.0464* | 49/58 | 3.069 (1.736-5.426) | <0.0001* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| No | 110/109 | 0.807 (1.107-2.951) | 0.0188* | 108/102 | 1.429 (0.9693-2.105) | 0.0691 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Gender | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Male | 94/123 | 2.192 (1.243-3.863) | 0.0041* | 103/124 | 1.920 (1.304-2.826) | 0.0003* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Female | 60/50 | 1.420 (0.7451-2.704) | 0.2847 | 61/46 | 1.711 (1.008-2.903) | 0.048* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ≤ 50 | 32/32 | 1.553 (0.4715-5.112) | 0.4636 | 38/30 | 1.765 (0.8905-2.497) | 0.0972 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > 50 | 122/141 | 1.828 (1.165-2.868) | 0.0075* | 126/140 | 1.808 (1.278-2.558) | 0.0004* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cirrhosis | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Yes | 34/42 | 3.445 (1.244-9.539) | 0.0237* | 33/42 | 0.9595 (0.522-1.764) | 0.8938 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| No | 51/77 | 1.323 (0.7002-2.500) | 0.3701 | 52/74 | 1.866 (1.114-3.125) | 0.0114* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Albumin (g/dl) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ≤ 3.5 | 44/38 | 2.379 (1.030-5.495) | 0.061 | 41/36 | 1.203 (0.6193-2.337) | 0.5811 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > 3.5 | 87/117 | 1.911 (1.102-3.312) | 0.0139* | 82/116 | 1.784 (1.183-2.691) | 0.0028* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Creatinine(mg/dl) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| < 1.1 | 87/108 | 1.888 (1.102-3.234) | 0.0171* | 81/105 | 1.665 (1.097-2.529) | 0.0113* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ≥ 1.1 | 47/48 | 2.148 (0.9684-4.765) | 0.0507 | 44/45 | 1.428 (0.7646-2.668) | 0.2451 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Alpha-fetoprotein(ng/ml) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ≤ 300 | 79/125 | 2.379 (1.334-4.243) | 0.0011* | 76/121 | 1.770 (1.147-2.731) | 0.0044* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > 300 | 45/18 | 2.148 (0.7583-6.082) | 0.2203 | 41/17 | 1.425 (0.665-3.054) | 0.3896 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Platelet(×109/L) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ≤ 300 | 101/120 | 2.421 (1.413-4.147) | 0.0013* | 95/119 | 1.527 (1.016-2.295) | 0.0337* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > 300 | 34/38 | 1.338 (0.5988-2.988) | 0.4481 | 31/34 | 1.772 (0.9258-3.391) | 0.0671 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Race | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Asian | 65/63 | 4.354 (1.719-11.03) | 0.0041* | 83/65 | 2.046 (1.270-3.297) | 0.0034* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| White | 76/95 | 1.612 (0.9556-2.720) | 0.0623 | 69/91 | 1.901 (1.234-2.928) | 0.0015* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BMIa | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| < 25 | 78/71 | 2.331 (1.201-4.523) | 0.0175* | 93/71 | 1.472 (0.9494-2.281) | 0.0855 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ≥ 25 | 62/91 | 1.858 (0.9573-3.608) | 0.0454* | 56/87 | 2.549 (1.546-4.204) | <0.0001* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Family history | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Yes | 52/57 | 1.723 (0.9518-3.118) | 0.0669 | 85/91 | 1.543 (0.8052-2.957) | 0.1744 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| No | 48/52 | 1.882 (1.073-3.301) | 0.0214* | 101/90 | 1.639 (1.099-2.446) | 0.0135* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ECOGb | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| =0 | 63/95 | 2.958 (1.476-5.931) | 0.002* | 59/93 | 1.463 (0.889-2.408) | 0.1159 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| >0 | 47/52 | 1.454 (0.6708-3.150) | 0.3143 | 62/52 | 2.165 (1.353-3.463) | 0.001* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Histological grade | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| G1/2 | 73/127 | 1.445 (0.805-2.596) | 0.1908 | 82/126 | 1.936 (1.256-2.983) | 0.0008* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| G3/4 | 77/45 | 3.357 (1.698-6.636) | 0.0042* | 78/43 | 1.684 (1.033-2.747) | 0.0457* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviations: HR, hazard ratio; 95% CI, 95% confidence interval. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| *: Statistically significant; | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a: body mass index; | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| b: Eastern Cooperative Oncology Group. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Functional annotation of the established 12-gene signature
Gene set enrichment analysis (GSEA) was carried out in the high-risk group of the TCGA cohort to investigate key biological and cellular processes linked with poor prognosis. 16 in all significantly enriched BioCarta pathways are listed in Figure 5A. There were notable enriched KEGG pathways in high-risk patients included pathways connected with cell cycle like MCM pathway (Figure 5B), NF-κB signaling like MAL and TNFR1 pathways (Figure 5C) and classic MAPK-associated pathways such as FAS, Rho and PYK2 pathways (Figure 5D). These pathways mainly involved in cell proliferation and migration that might contributed to HCC metastasis and recurrence.
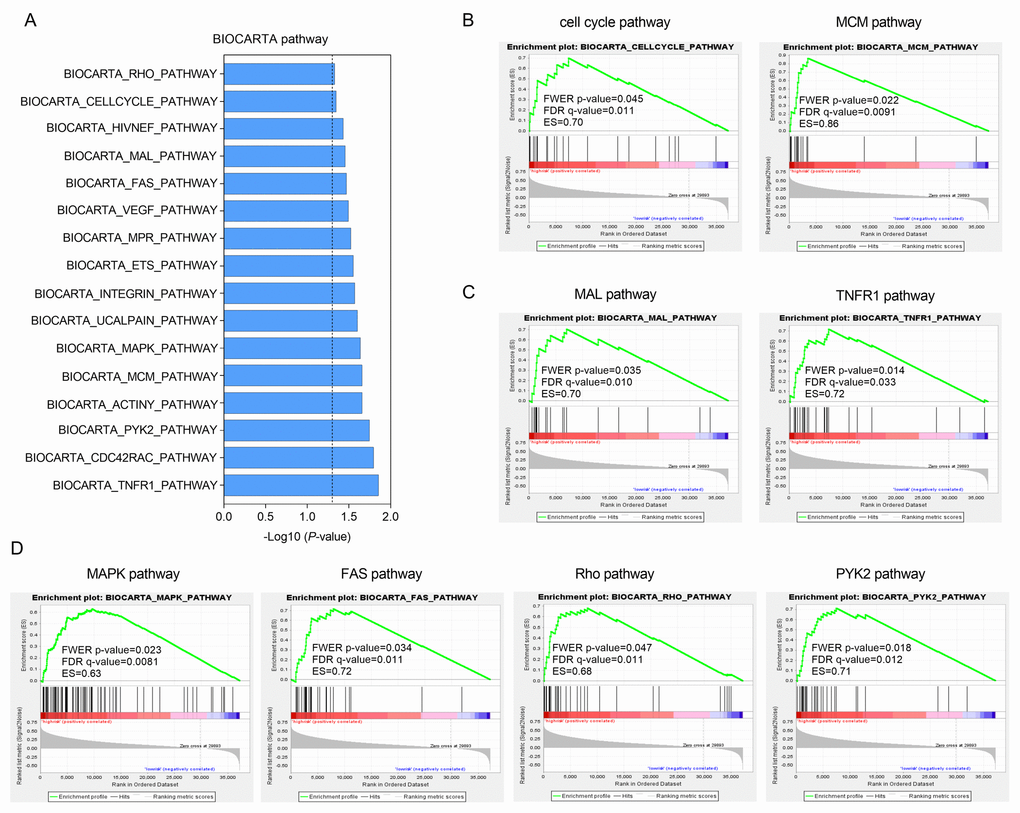
Figure 5. Functional enrichment of HCC-specific gene signature in high-risk patients of TCGA series. (A) The bar chart of 16 significantly enriched BIOCARTA pathways through GSEA. (B, C and D) Significantly enriched pathways associated with cell cycle (B), TNF-κB signaling (C) and MAPK pathway (D).
Discussion
In this study, we identified and validated a twelve-HCC-specific gene risk score system for predicting OS and DFS of HCC by multistep comparisons and screening. Firstly, to maintain the coincidence of all the datasets, we employed four mRNA expression profiles (GSE5851, GSE28691, GSE15765 and GSE14323) conducted by the same platform and integrated all datasets for normalization. After a comprehensive analysis, 1103 genes were selected as HCC-specific genes, among which 816 up-regulated genes were identified as risk factors, while 287 down-regulated as protective ones. In addition, pathway enrichment analysis revealed that these genes might influence cell cycle, migration and inflammation. Finally, 12-prognostic gene signature was constituted by LSAAO algorism and successfully validated in another independent dataset.
Of 12 identified genes in this prognostic model, three (COPS8, SUGCT and DUSP10) played a protective role in HCC patients, and by contrast the other nine (EIF5B, MNAT1, WASF1, EWSR1, POLR3C, RNF24, PCSK5, NRBP1 and CCDC88A) acted as risk factors for HCC. COPS8 (COP9 signalosome subunit 8) encodes a highly conserved protein complex that has been reported to be associated with gastric cancer and kidney cancer [29,30]. It has been proved in murine models that COPS8 deficiency can impair ubiquitin-proteasome system (UPS) in the liver and the heart, respectively [31,32]. UPS is critical to protein degradation and homeostasis that is important to normal liver function [33]. SUGCT (succinyl-CoA: glutarate-CoA transferase) was demonstrated to be related to mitochondrial diseases and glutaric aciduria type III that might play a role in hepatic energetic metabolism [34–36]. DUSP10 (dual specificity phosphatase 10) negatively regulate mitogen-activated protein kinase (MAPK) family, novel members of cellular proliferation and differentiation [37]. A recent study showed downregulation of DUSP10 was associated with HCC metastasis [38]. EIF5B, one of eukaryotic translation initiation factors, was demonstrated to be involved in cell-cycle arrest in case of up-regulation [39,40]. MNAT1, a factor of the CDK-activating kinase (CAK) enzymatic complex, is vital to transcription. MNAT1 was correlated with poor prognosis of several types of cancer including breast cancer, gastric cancer and colorectal cancer [41–43]. WASF1, also known as WAVE1, is a member of the Wiskott-Aldrich syndrome protein (WASP)-family highly expressed in brain and testis [44]. EWSR1 (Ewing sarcoma breakpoint region 1 gene) has relation with gene expression, cell signaling and translocation, which is momentous to tumorigenesis [45]. There is evidence that POLR3C (RNA polymerase III subunit C) was be associated with virus infection [46]. RNF24 (ring finger protein 24) encode a membrane protein that can interact with transient receptor potential cation channel subfamily C (TRPC) proteins [47]. The study by Wang et al. showed that RNF24 correlated with the occurrence of esophageal adenocarcinoma [48]. PCSK5 (proprotein convertase subtilisin/kexin type 5) was found to be dysregulated in different subtypes of triple-negative breast cancer (TNBC) [49]. A recent study by Bajikar et al. demonstrated PCSK5 can inhibit TNBC metastasis by mediating retention of growth-differentiation factor 11 (GDF11) [50]. NRBP1 (nuclear receptor binding protein 1) acts as a tumor suppressor and is commonly downregulated in a series of cancers such as breast cancer [51,52]. However, in prostate cancer, NRBP1 was highly expressed and correlated with poor survival [53]. CCDC88A (coiled-coil domain containing 88A) regulates cytoskeleton remodeling and cell motility. Recent studies have suggested that CCDC88A played a role in metastasis and radio-resistance of HCC [54,55]. Of the 12 genes, 7 genes (EIF5B, MNAT1, WASF1, POLR3C, RNF24, PCSK5 and NRBP1) were first reported to be associated with HCC prognosis in this study, which might give a few hints for future research into molecular mechanisms of HCC.
Gene enrichment analysis in the high-risk patients showed pathways involved with cell cycle, inflammation and migration were significantly enriched. Minichromosome maintenance (MCM) proteins are a group of ATPase, fundamental to the replication of DNA and the process of cell cycle [56]. Tumor necrosis factor receptor-1 (TNFR1) is the receptor of tumor necrosis factor (TNF), if activated, can cause proliferation or death of cells in different cellular context [57]. One of key roles of TNFR1 is to trigger NF-κB signaling by activating IκB kinase (IKK) complex [58]. MAL is an adaptor protein in the activation of Toll-like receptor 4 (TLR4)/ NF-κB pathway [59]. NF-κB is a nuclear transcription factor that acts as a regulator in various biological and pathological processes including inflammation, cell apoptosis, immune responses and tumorigenesis [60–62]. The mitogen-activated protein kinase/ extracellular signal-regulated (MAPK/ERK) pathway was a novel oncogenic pathway in most cancers [63]. MAPK mediates cellular apoptosis by Fas/FasL signaling pathways [64]. Rho is a kind of small GTP-binding protein triggering transduction of signaling cascades of MAPK pathways [65]. Proline-rich kinase-2 (Pyk2) is a non-receptor protein tyrosine kinase which participates in several pathways including MAPK and regulates cell proliferation, differentiation, adhesion and migration [66]. In a word, the findings of GSEA indicated that the HCC-specific gene signature might have potentials in the regulation of cell apoptosis, inflammatory responses, invasion and metastasis of HCC.
However, there are some limitations for this present study. First, the combination of samples for screening HCC-specific genes was small. Second, we constructed risk score system merely based on the gene expression levels, without considering the mutation, methylation, or other genetic events of genes that probably have an effect on the initiation and progression of cancer. Third, nearly 90% of patients in the discovery dataset had HBV, so the risk score system was established based on an HBV background. And further stratified analysis in validation cohort also demonstrated that this prognostic model was more applicable to HBV patients. Last but not the least, our HCC prognostic signature still needs to be validated in a larger population of patients from various backgrounds.
In conclusion, we constructed and confirmed an HCC-specific prognostic risk score system comprised of 12 genes. This risk score system could serve as a potential predictor for OS particularly in elderly male patients with HBV infection but in relatively better general conditions by risk-dependent stratification. From the results of functional annotations, pathways involved in cell cycle, NF-κB- and MAPK-associated pathways were significantly enriched, which might help better understand the molecular mechanisms underlying the initiation and progression of HCC. Moreover, our data provide new promising evidence on prediction biomarkers and targeted therapy for HCC.
Materials and Methods
Microarray data collection and pre-processing
All microarray datasets were retrieved from the GEO database (https://www.ncbi.nlm.nih.gov/geo/) and the Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov/) [67]. GSE5851, GSE28691, GSE15765 and GSE14323 from GEO were conducted through GPL571 (Affymetrix Human Genome U133A 2.0 Array). GPL571 platform was comprised of 22277 unique probes and tested more than 13500 genes (http://www.affymetrix.com/support/technical/byproduct.affx?product= hgu133-20). We selected 75 CRCLM samples from GSE5851 (61) and GSE28691 (14), 70 HCC samples and 13 ICC samples from GSE15765 and 19 normal liver tissue samples from GSE14323 for integrated normalization and analysis. One of ICC samples, GSM395714, was ruled out through quality control. GSE14520, as the training series downloaded from GEO database, included 247 HCC samples, 239 non-tumor tissue samples and 2 normal liver samples from healthy donors and was conducted by GPL571 and GPL3921 (Affymetrix HT Human Genome U133A Array), respectively. All the tumor samples (n=247) from GSE14520 were used as the training dataset. The raw fluorescence intensity profiles (*.CEL) of all the selected data from GEO were downloaded and normalized and further transformed to expression values through RMA algorism in the R environment (v3.4.3) [68]. 357 HCC samples from TCGA cohort were included as the validation group. The mRNA-seq data were preprocessed and submitted into analysis as the upper quantile normalized FPKM values.
Screening of HCC-specific genes and gene ontology analysis
Differentially expressed genes (DEGs) between tumor groups (HCC, ICC and CRCLM) and normal group were obtained from GSE5851, GSE28691, GSE15765 and GSE14323. Only fold change (FC) ≥ 1.5 and P value for t-test < 0.05 were considered statistically significant. We then carried out Venn selection of cancer-specific genes among three DEG lists of the three types of hepatic neoplasms by Venny 2.1.0 (http://bioinfogp.cnb.csic.es/tools/venny/). Univariate survival analysis based on Cox proportional hazards of the HCC-specific genes was performed and genes with significant P values (< 0.05) from Log-rank tests were selected. Gene ontology (GO) analysis of HCC-specific genes was performed by Metascape, a user-friendly web tool for gene annotation and also a plugin of Cytoscape.
LASSO statistical modeling
The 12-HCC-specific gene signature was derived from the least absolute shrinkage and selection operator (LASSO). LASSO is a linear regression algorism capable of variable selection and regularization simultaneously [69]. We carried out LASSO fitting method based on a series of λ using ‘glmnet’ package in the R environment (v3.4.3) [70]. The coefficients of each gene in risk score system were generated based on the expressions of each tissue sample in R studio at the same time.
Confirmation and evaluation of risk score system
The HCC risk score model was validated and evaluated in TCGA cohort and the primary dataset (GSE14520). Univariate and multivariate Cox regression analyses were carried out with the validating series to estimate the association between various clinical characteristics containing the risk score and OS or DFS. Then stratified analysis based on clinical information was conducted in TCGA series. The median of TCGA cohort was accepted as optimal cut-off values to divide each group into the high-risk and the low-risk subgroups for survival analysis. All Kaplan-Meier curves were plotted and P values and hazard ratio (HR) with 95% confidence interval (CI) from log-rank tests were generated in GraphPad Prism 7.0.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) was one of computational methods to identify significantly enriched biological processes and pathways. GSEA was carried out by the JAVA program (http://www.broadinstitute.org/gsea) based on Molecular Signature Database (MSigDB) [71]. Here, BioCarta (http://cgap.nci.nih.gov/Pathways/BioCarta_Pathways) pathway was enriched through GSEA in both high-risk and low-risk groups [72]. Each gene set would get an enrichment score (ES) that represented the number of overexpressed genes in this gene set. The false discovery rate (FDR) and the FWER P value of the gene sets < 0.05 were considered statistically significant.
Statistical methods
Statistical analyses were performed using STATA/SE software (v12.0). False discovery rate (FDR) was applied to compare the expression of genes between tumor and non-tumor samples. FDR or P value < 0.05 was considered statistically different.
Supplementary Materials
Author Contributions
K.Q., J.G. and J.S. designed the study and performed data analysis; W.N. and C.L. revised the paper; X.Z., X.M., and R.M. performed literature search and data collection; X.X. and Y.F. constructed figures and improved the language; K.Q. and J.S. directed the overall project. All authors reviewed the manuscript.
Conflicts of Interest
The authors have declared no conflicts of interest in this study.
Funding
This work was supported by the National Science Foundation of China (No. 81871998), the Natural Science Basic Research Plan in Shaanxi Province of China (Nos. 2018JM7013 and 2017JM8039), the Fundamental Research Fund for the Central Universities (No. 2016qngz05) and the Clinical Research Award of the First Affiliated Hospital of Xi'an Jiaotong University (No. XJTU1AF-CRF-2015-011).
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017; 67:7–30. https://doi.org/10.3322/caac.21387 [PubMed]
- 2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016; 66:115–32. https://doi.org/10.3322/caac.21338 [PubMed]
- 3. Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut. 2014; 63:844–55. https://doi.org/10.1136/gutjnl-2013-306627 [PubMed]
- 4. Dutkowski P, Linecker M, DeOliveira ML, Müllhaupt B, Clavien PA. Challenges to liver transplantation and strategies to improve outcomes. Gastroenterology. 2015; 148:307–23. https://doi.org/10.1053/j.gastro.2014.08.045 [PubMed]
- 5. Fong ZV, Tanabe KK. The clinical management of hepatocellular carcinoma in the United States, Europe, and Asia: a comprehensive and evidence-based comparison and review. Cancer. 2014; 120:2824–38. https://doi.org/10.1002/cncr.28730 [PubMed]
- 6. Villanueva A, Hernandez-Gea V, Llovet JM. Medical therapies for hepatocellular carcinoma: a critical view of the evidence. Nat Rev Gastroenterol Hepatol. 2013; 10:34–42. https://doi.org/10.1038/nrgastro.2012.199 [PubMed]
- 7. Ma L, Chua MS, Andrisani O, So S. Epigenetics in hepatocellular carcinoma: an update and future therapy perspectives. World J Gastroenterol. 2014; 20:333–45. https://doi.org/10.3748/wjg.v20.i2.333 [PubMed]
- 8. Liu M, Jiang L, Guan XY. The genetic and epigenetic alterations in human hepatocellular carcinoma: a recent update. Protein Cell. 2014; 5:673–91. https://doi.org/10.1007/s13238-014-0065-9 [PubMed]
- 9. Marrero JA, Fontana RJ, Barrat A, Askari F, Conjeevaram HS, Su GL, Lok AS. Prognosis of hepatocellular carcinoma: comparison of 7 staging systems in an American cohort. Hepatology. 2005; 41:707–16. https://doi.org/10.1002/hep.20636 [PubMed]
- 10. Maida M, Orlando E, Cammà C, Cabibbo G. Staging systems of hepatocellular carcinoma: a review of literature. World J Gastroenterol. 2014; 20:4141–50. https://doi.org/10.3748/wjg.v20.i15.4141 [PubMed]
- 11. Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology. 2015; 149:1226–1239.e4. https://doi.org/10.1053/j.gastro.2015.05.061 [PubMed]
- 12. Murakami Y, Tanahashi T, Okada R, Toyoda H, Kumada T, Enomoto M, Tamori A, Kawada N, Taguchi YH, Azuma T. Comparison of hepatocellular carcinoma miRNA expression profiling as evaluated by next generation sequencing and microarray. PLoS One. 2014; 9:e106314. https://doi.org/10.1371/journal.pone.0106314 [PubMed]
- 13. Yuan S, Wang J, Yang Y, Zhang J, Liu H, Xiao J, Xu Q, Huang X, Xiang B, Zhu S, Li L, Liu J, Liu L, Zhou W. The Prediction of Clinical Outcome in Hepatocellular Carcinoma Based on a Six-Gene Metastasis Signature. Clin Cancer Res. 2017; 23:289–97. https://doi.org/10.1158/1078-0432.CCR-16-0395 [PubMed]
- 14. Roessler S, Jia HL, Budhu A, Forgues M, Ye QH, Lee JS, Thorgeirsson SS, Sun Z, Tang ZY, Qin LX, Wang XW. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 2010; 70:10202–12. https://doi.org/10.1158/0008-5472.CAN-10-2607 [PubMed]
- 15. Wu DH, Wang TT, Ruan DY, Li X, Chen ZH, Wen JY, Lin Q, Ma XK, Wu XY, Jia CC. Combination of ULK1 and LC3B improve prognosis assessment of hepatocellular carcinoma. Biomed Pharmacother. 2018; 97:195–202. https://doi.org/10.1016/j.biopha.2017.10.025 [PubMed]
- 16. Nault JC, De Reyniès A, Villanueva A, Calderaro J, Rebouissou S, Couchy G, Decaens T, Franco D, Imbeaud S, Rousseau F, Azoulay D, Saric J, Blanc JF, et al. A hepatocellular carcinoma 5-gene score associated with survival of patients after liver resection. Gastroenterology. 2013; 145:176–87. https://doi.org/10.1053/j.gastro.2013.03.051 [PubMed]
- 17. Klingenberg M, Matsuda A, Diederichs S, Patel T. Non-coding RNA in hepatocellular carcinoma: Mechanisms, biomarkers and therapeutic targets. J Hepatol. 2017; 67:603–18. https://doi.org/10.1016/j.jhep.2017.04.009 [PubMed]
- 18. Llovet JM, Villanueva A, Lachenmayer A, Finn RS. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol. 2015; 12:408–24. https://doi.org/10.1038/nrclinonc.2015.103 [PubMed]
- 19. Bridgewater J, Galle PR, Khan SA, Llovet JM, Park JW, Patel T, Pawlik TM, Gores GJ. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J Hepatol. 2014; 60:1268–89. https://doi.org/10.1016/j.jhep.2014.01.021 [PubMed]
- 20. Yazici C, Niemeyer DJ, Iannitti DA, Russo MW. Hepatocellular carcinoma and cholangiocarcinoma: an update. Expert Rev Gastroenterol Hepatol. 2014; 8:63–82. https://doi.org/10.1586/17474124.2014.852468 [PubMed]
- 21. Ribero D, Viganò L, Amisano M, Capussotti L. Prognostic factors after resection of colorectal liver metastases: from morphology to biology. Future Oncol. 2013; 9:45–57. https://doi.org/10.2217/fon.12.159 [PubMed]
- 22. Giuliani J, Marzola M, Pansini G. Liver metastases from primary rectal cancer: a multidisciplinary reverse approach. J Gastrointest Cancer. 2013; 44:368–69. https://doi.org/10.1007/s12029-012-9444-8 [PubMed]
- 23. Bester L, Meteling B, Boshell D, Chua TC, Morris DL. Transarterial chemoembolisation and radioembolisation for the treatment of primary liver cancer and secondary liver cancer: a review of the literature. J Med Imaging Radiat Oncol. 2014; 58:341–52. https://doi.org/10.1111/1754-9485.12163 [PubMed]
- 24. Page AJ, Weiss MJ, Pawlik TM. Surgical management of noncolorectal cancer liver metastases. Cancer. 2014; 120:3111–21. https://doi.org/10.1002/cncr.28743 [PubMed]
- 25. Ikoma N, You YN, Bednarski BK, Rodriguez-Bigas MA, Eng C, Das P, Kopetz S, Messick C, Skibber JM, Chang GJ. Impact of Recurrence and Salvage Surgery on Survival After Multidisciplinary Treatment of Rectal Cancer. J Clin Oncol. 2017; 35:2631–38. https://doi.org/10.1200/JCO.2016.72.1464 [PubMed]
- 26. Ayez N, Burger JW, van der Pool AE, Eggermont AM, Grunhagen DJ, de Wilt JH, Verhoef C. Long-term results of the “liver first” approach in patients with locally advanced rectal cancer and synchronous liver metastases. Dis Colon Rectum. 2013; 56:281–87. https://doi.org/10.1097/DCR.0b013e318279b743 [PubMed]
- 27. McNally SJ, Parks RW. Surgery for colorectal liver metastases. Dig Surg. 2013; 30:337–47. https://doi.org/10.1159/000351442 [PubMed]
- 28. Choti MA, Sitzmann JV, Tiburi MF, Sumetchotimetha W, Rangsin R, Schulick RD, Lillemoe KD, Yeo CJ, Cameron JL. Trends in long-term survival following liver resection for hepatic colorectal metastases. Ann Surg. 2002; 235:759–66. https://doi.org/10.1097/00000658-200206000-00002 [PubMed]
- 29. Mahmoodi M, Nguyen-Dumont T, Hammet F, Pope BJ, Park DJ, Southey MC, Darlow JM, Bruinsma F, Winship I. Mutation screening of ACKR3 and COPS8 in kidney cancer cases from the CONFIRM study. Fam Cancer. 2017; 16:411–16. https://doi.org/10.1007/s10689-016-9961-x [PubMed]
- 30. Crone SG, Jacobsen A, Federspiel B, Bardram L, Krogh A, Lund AH, Friis-Hansen L. microRNA-146a inhibits G protein-coupled receptor-mediated activation of NF-κB by targeting CARD10 and COPS8 in gastric cancer. Mol Cancer. 2012; 11:71. https://doi.org/10.1186/1476-4598-11-71 [PubMed]
- 31. Lei D, Li F, Su H, Liu J, Wei N, Wang X. Hepatic deficiency of COP9 signalosome subunit 8 induces ubiquitin-proteasome system impairment and Bim-mediated apoptosis in murine livers. PLoS One. 2013; 8:e67793. https://doi.org/10.1371/journal.pone.0067793 [PubMed]
- 32. Liu J, Su H, Wang X. The COP9 signalosome coerces autophagy and the ubiquitin-proteasome system to police the heart. Autophagy. 2016; 12:601–02. https://doi.org/10.1080/15548627.2015.1136773 [PubMed]
- 33. Chen YJ, Wu H, Shen XZ. The ubiquitin-proteasome system and its potential application in hepatocellular carcinoma therapy. Cancer Lett. 2016; 379:245–52. https://doi.org/10.1016/j.canlet.2015.06.023 [PubMed]
- 34. Calvo SE, Compton AG, Hershman SG, Lim SC, Lieber DS, Tucker EJ, Laskowski A, Garone C, Liu S, Jaffe DB, Christodoulou J, Fletcher JM, Bruno DL, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med. 2012; 4:118ra10. https://doi.org/10.1126/scitranslmed.3003310 [PubMed]
- 35. Hendrickson SL, Lautenberger JA, Chinn LW, Malasky M, Sezgin E, Kingsley LA, Goedert JJ, Kirk GD, Gomperts ED, Buchbinder SP, Troyer JL, O’Brien SJ. Genetic variants in nuclear-encoded mitochondrial genes influence AIDS progression. PLoS One. 2010; 5:e12862. https://doi.org/10.1371/journal.pone.0012862 [PubMed]
- 36. Marlaire S, Van Schaftingen E, Veiga-da-Cunha M. C7orf10 encodes succinate-hydroxymethylglutarate CoA-transferase, the enzyme that converts glutarate to glutaryl-CoA. J Inherit Metab Dis. 2014; 37:13–19. https://doi.org/10.1007/s10545-013-9632-0 [PubMed]
- 37. Low HB, Zhang Y. Regulatory Roles of MAPK Phosphatases in Cancer. Immune Netw. 2016; 16:85–98. https://doi.org/10.4110/in.2016.16.2.85 [PubMed]
- 38. Ng KY, Chan LH, Chai S, Tong M, Guan XY, Lee NP, Yuan Y, Xie D, Lee TK, Dusetti NJ, Carrier A, Ma S. TP53INP1 Downregulation Activates a p73-Dependent DUSP10/ERK Signaling Pathway to Promote Metastasis of Hepatocellular Carcinoma. Cancer Res. 2017; 77:4602–12. https://doi.org/10.1158/0008-5472.CAN-16-3456 [PubMed]
- 39. Lee S, Truesdell SS, Bukhari SI, Lee JH, LeTonqueze O, Vasudevan S. Upregulation of eIF5B controls cell-cycle arrest and specific developmental stages. Proc Natl Acad Sci USA. 2014; 111:E4315–22. https://doi.org/10.1073/pnas.1320477111 [PubMed]
- 40. Nag N, Lin KY, Edmonds KA, Yu J, Nadkarni D, Marintcheva B, Marintchev A. eIF1A/eIF5B interaction network and its functions in translation initiation complex assembly and remodeling. Nucleic Acids Res. 2016; 44:7441–56. https://doi.org/10.1093/nar/gkw552 [PubMed]
- 41. Santarpia L, Iwamoto T, Di Leo A, Hayashi N, Bottai G, Stampfer M, André F, Turner NC, Symmans WF, Hortobágyi GN, Pusztai L, Bianchini G. DNA repair gene patterns as prognostic and predictive factors in molecular breast cancer subtypes. Oncologist. 2013; 18:1063–73. https://doi.org/10.1634/theoncologist.2013-0163 [PubMed]
- 42. Fejzo MS, Anderson L, Chen HW, Anghel A, Zhuo J, Anchoori R, Roden R, Slamon DJ. ADRM1-amplified metastasis gene in gastric cancer. Genes Chromosomes Cancer. 2015; 54:506–15. https://doi.org/10.1002/gcc.22262 [PubMed]
- 43. Kap EJ, Seibold P, Richter S, Scherer D, Habermann N, Balavarca Y, Jansen L, Becker N, Pfütze K, Popanda O, Hoffmeister M, Ulrich A, Benner A, et al. Genetic variants in DNA repair genes as potential predictive markers for oxaliplatin chemotherapy in colorectal cancer. Pharmacogenomics J. 2015; 15:505–12. https://doi.org/10.1038/tpj.2015.8 [PubMed]
- 44. Ceglia I, Reitz C, Gresack J, Ahn JH, Bustos V, Bleck M, Zhang X, Martin G, Simon SM, Nairn AC, Greengard P, Kim Y. APP intracellular domain-WAVE1 pathway reduces amyloid-β production. Nat Med. 2015; 21:1054–59. https://doi.org/10.1038/nm.3924 [PubMed]
- 45. Skálová A, Weinreb I, Hyrcza M, Simpson RH, Laco J, Agaimy A, Vazmitel M, Majewska H, Vanecek T, Talarčik P, Manajlovic S, Losito SN, Šteiner P, et al. Clear cell myoepithelial carcinoma of salivary glands showing EWSR1 rearrangement: molecular analysis of 94 salivary gland carcinomas with prominent clear cell component. Am J Surg Pathol. 2015; 39:338–48. https://doi.org/10.1097/PAS.0000000000000364 [PubMed]
- 46. Ogunjimi B, Zhang SY, Sørensen KB, Skipper KA, Carter-Timofte M, Kerner G, Luecke S, Prabakaran T, Cai Y, Meester J, Bartholomeus E, Bolar NA, Vandeweyer G, et al. Inborn errors in RNA polymerase III underlie severe varicella zoster virus infections. J Clin Invest. 2017; 127:3543–56. https://doi.org/10.1172/JCI92280 [PubMed]
- 47. Lussier MP, Lepage PK, Bousquet SM, Boulay G. RNF24, a new TRPC interacting protein, causes the intracellular retention of TRPC. Cell Calcium. 2008; 43:432–43. https://doi.org/10.1016/j.ceca.2007.07.009 [PubMed]
- 48. Wang XW, Wei W, Wang WQ, Zhao XY, Guo H, Fang DC. RING finger proteins are involved in the progression of barrett esophagus to esophageal adenocarcinoma: a preliminary study. Gut Liver. 2014; 8:487–94. https://doi.org/10.5009/gnl13133 [PubMed]
- 49. Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, et al. Molecular portraits of human breast tumours. Nature. 2000; 406:747–52. https://doi.org/10.1038/35021093 [PubMed]
- 50. Bajikar SS, Wang CC, Borten MA, Pereira EJ, Atkins KA, Janes KA. Tumor-Suppressor Inactivation of GDF11 Occurs by Precursor Sequestration in Triple-Negative Breast Cancer. Dev Cell. 2017; 43:418–435.e13. https://doi.org/10.1016/j.devcel.2017.10.027 [PubMed]
- 51. Wilson CH, Crombie C, van der Weyden L, Poulogiannis G, Rust AG, Pardo M, Gracia T, Yu L, Choudhary J, Poulin GB, McIntyre RE, Winton DJ, March HN, et al. Nuclear receptor binding protein 1 regulates intestinal progenitor cell homeostasis and tumour formation. EMBO J. 2012; 31:2486–97. https://doi.org/10.1038/emboj.2012.91 [PubMed]
- 52. Wei H, Wang H, Ji Q, Sun J, Tao L, Zhou X. NRBP1 is downregulated in breast cancer and NRBP1 overexpression inhibits cancer cell proliferation through Wnt/β-catenin signaling pathway. OncoTargets Ther. 2015; 8:3721–30. https://doi.org/10.2147/OTT.S89779 [PubMed]
- 53. Ruiz C, Oeggerli M, Germann M, Gluderer S, Stocker H, Andreozzi M, Thalmann GN, Cecchini MG, Zellweger T, Stürm S, Koivisto PA, Helin HJ, Gelmann EP, et al. High NRBP1 expression in prostate cancer is linked with poor clinical outcomes and increased cancer cell growth. Prostate. 2012; 72:1678–87. https://doi.org/10.1002/pros.22521 [PubMed]
- 54. Ke Y, Bao T, Zhou Q, Wang Y, Ge J, Fu B, Wu X, Tang H, Shi Z, Lei X, Zhang C, Tan Y, Chen H, et al. Discs large homolog 5 decreases formation and function of invadopodia in human hepatocellular carcinoma via Girdin and Tks5. Int J Cancer. 2017; 141:364–76. https://doi.org/10.1002/ijc.30730 [PubMed]
- 55. Yu L, Sun Y, Li J, Wang Y, Zhu Y, Shi Y, Fan X, Zhou J, Bao Y, Xiao J, Cao K, Cao P. Silencing the Girdin gene enhances radio-sensitivity of hepatocellular carcinoma via suppression of glycolytic metabolism. J Exp Clin Cancer Res. 2017; 36:110. https://doi.org/10.1186/s13046-017-0580-7 [PubMed]
- 56. Noseda M, Niessen K, McLean G, Chang L, Karsan A. Notch-dependent cell cycle arrest is associated with downregulation of minichromosome maintenance proteins. Circ Res. 2005; 97:102–04. [PubMed]
- 57. Cabal-Hierro L, Lazo PS. Signal transduction by tumor necrosis factor receptors. Cell Signal. 2012; 24:1297–305. https://doi.org/10.1016/j.cellsig.2012.02.006 [PubMed]
- 58. Wajant H, Scheurich P. TNFR1-induced activation of the classical NF-κB pathway. FEBS J. 2011; 278:862–76. https://doi.org/10.1111/j.1742-4658.2011.08015.x [PubMed]
- 59. Ghosh S, Dass JF. Study of pathway cross-talk interactions with NF-κB leading to its activation via ubiquitination or phosphorylation: A brief review. Gene. 2016; 584:97–109. https://doi.org/10.1016/j.gene.2016.03.008 [PubMed]
- 60. DiDonato JA, Mercurio F, Karin M. NF-κB and the link between inflammation and cancer. Immunol Rev. 2012; 246:379–400. https://doi.org/10.1111/j.1600-065X.2012.01099.x [PubMed]
- 61. Zhuang Z, Li H, Lee H, Aguilar M, Gocho T, Ju H, Iida T, Ling J, Fu J, Wu M, Sun Y, Lu Y, Chiao PJ. NEMO peptide inhibits the growth of pancreatic ductal adenocarcinoma by blocking NF-κB activation. Cancer Lett. 2017; 411:44–56. https://doi.org/10.1016/j.canlet.2017.09.018 [PubMed]
- 62. Banoth B, Chatterjee B, Vijayaragavan B, Prasad MV, Roy P, Basak S. Stimulus-selective crosstalk via the NF-κB signaling system reinforces innate immune response to alleviate gut infection. eLife. 2015; 4:4. https://doi.org/10.7554/eLife.05648 [PubMed]
- 63. De Luca A, Maiello MR, D’Alessio A, Pergameno M, Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets. 2012 (Suppl 2); 16:S17–27. https://doi.org/10.1517/14728222.2011.639361 [PubMed]
- 64. Liu WH, Cheng YC, Chang LS. ROS-mediated p38alpha MAPK activation and ERK inactivation responsible for upregulation of Fas and FasL and autocrine Fas-mediated cell death in Taiwan cobra phospholipase A(2)-treated U937 cells. J Cell Physiol. 2009; 219:642–51. https://doi.org/10.1002/jcp.21713 [PubMed]
- 65. Okada T, Sinha S, Esposito I, Schiavon G, López-Lago MA, Su W, Pratilas CA, Abele C, Hernandez JM, Ohara M, Okada M, Viale A, Heguy A, et al. The Rho GTPase Rnd1 suppresses mammary tumorigenesis and EMT by restraining Ras-MAPK signalling. Nat Cell Biol. 2015; 17:81–94. https://doi.org/10.1038/ncb3082 [PubMed]
- 66. Anand AR, Bradley R, Ganju RK. LPS-induced MCP-1 expression in human microvascular endothelial cells is mediated by the tyrosine kinase, Pyk2 via the p38 MAPK/NF-kappaB-dependent pathway. Mol Immunol. 2009; 46:962–68. https://doi.org/10.1016/j.molimm.2008.09.022 [PubMed]
- 67. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell. 2017; 169:1327–1341.e1323. https://doi.org/10.1016/j.cell.2017.05.046 [PubMed]
- 68. Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003; 4:249–64. https://doi.org/10.1093/biostatistics/4.2.249 [PubMed]
- 69. Tibshirani R. Regression Shrinkage and Selection via the Lasso. J R Stat Soc Ser A Stat Soc. 1996; 58:267–88.
- 70. Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw. 2010; 33:1–22. https://doi.org/10.18637/jss.v033.i01 [PubMed]
- 71. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005; 102:15545–50. https://doi.org/10.1073/pnas.0506580102 [PubMed]
- 72. Nishimura D. BioCarta. Biotech Softw Internet Rep. 2001; 2:117–20. https://doi.org/10.1089/152791601750294344