



Cellular Senescence and Senotherapies
Causes of senescence
Aging is accompanied by the accumulation of senescent cells in tissues. Cellular senescence was first discovered in cultured fibroblast cells, where prolonged passaging and replicative exhaustion led to growth arrest due to critically short telomeres [1,2]. Senescence was eventually also observed in cells subjected to a variety of stressors including oncogene induction, oxidative stress, acute DNA damage or mitochondrial dysfunction [3,4]. Senescent cells are characterized by (a) a cell-autonomous proliferative arrest [1], (b) expression of lysosomal beta-galactosidase [5], (c) resistance to apoptosis [6,7] and mitogenic signals, (d) release of inflammatory cytokines and chemokines (also known as senescence-associated secretory phenotype or SASP) [8], (e) persistent DNA damage [9] and (f) formation of senescence-associated heterochromatic foci (SAHF) [10] and changes in DNA methylation [11]. At the molecular level, growth arrest is initiated by DNA damage that signals the upregulation of CDKN1A/p21CIP1/WAF1 by p53 stabilization and binding to promoter of the p21 gene. An independent upregulation of CDKN2A/p16INK4A, an inhibitor of cyclin dependent kinases 4 and 6, cooperates with p21 to reduce the phosphorylation of retinoblastoma (Rb) protein and arrest cells at the G1 phase [4,12]. These upstream signals also maintain the active metabolic state of senescent cells while promoting marked changes in the gene expression program such as downregulation of cell cycle genes and upregulation of SASP genes [13,14].
A number of factors including DNA damage and dysfunctional mitochondria are causal to the senescent phenotype. For example, telomere shortening is the primary cause of replicative senescence (RS) resulting from exposure of telomeric ends due to removal of the Shelterin complex, and their recognition as double strand breaks [15]. Activation of oncogenes such as RAS or BRAF results in oncogene-induced senescence (OIS) by engaging three machineries; repression of pro-proliferative E2F target genes, replication stress-activated DNA damage response and SASP [16]. Mitochondria can modulate the senescence phenotype by two related mechanisms: hyperactivation of the TCA cycle and upregulation of SASP. Changes in the levels of TCA cycle metabolite malate in senescence is influenced by p53-mediated repression of malic enzyme 2 [17]. Senescence can be directly influenced by overexpressing or inhibiting specific enzymes in the TCA cycle [18]. In OIS, increased pyruvate oxidation due to elevated pyruvate dehydrogenase levels produces reactive oxygen species (ROS) that activates the DNA damage response. The DNA damage response triggers mitochondrial biogenesis through the ATM, Akt, mTORC1, PGC1ß pathway thus further increasing ROS production and genome damage. Selective ablation of mitochondria in senescent cells can reduce the pro-oxidative (ROS) and pro-inflammatory (SASP) arms of senescence [19].
Despite the causal triggers of senescence, their direct link to epigenomic changes is less clear. Studies from lower organisms provide key mechanistic insights that could influence the senescent cell epigenome. For example, mid-life flies show enhanced acetyl-CoA (an acetyl group donor to TCA cycle) levels, increased histone acetylation and longevity-altering transcriptome changes [20]. Along these lines, elevated pyruvate dehydrogenase levels in OIS cells also increase acetyl-CoA production and can potentially influence histone acetylation [21]. These findings provoke a theory that chronic DNA damage and altered metabolism in midlife can trigger the onset of senescence and ultimately tissue aging via chromatin [22]. However, the exact mechanisms remain to be tested.
Taken together, the anti-proliferative nature of senescent cells serves a potent tumor suppressive mechanism. However, the chronic DNA damage, ROS and SASP promotes both local and systemic dysfunction at the tissue and organ level [23].
Consequences of senescence
Senescent cells accumulate in aged tissues due to exhaustion of proliferation-competent cells and adult stem cells [24]. In age-related diseases, senescent cells are often found at sites of pathology [25–27] in mice and humans. This accumulation in turn, disrupts tissue homeostasis, reduces regenerative capacity and remodels the tissue micro-environment mimicking a chronic low-grade inflammation status with positive beta-galactosidase staining in multiple tissues, ultimately resulting in an irreversible structural and functional decline [13].
It is important to note that the total percentage of senescent cells in aged tissues is usually <20%. Quantitative estimation of beta galactosidase positive cells in aged murine tissues ranged from ~14% in subcutaneous stromal cells, 6.9% in lung epithelial cells, 3.3-3.5% in small intestinal and spleen cells respectively and 1.48% in lymph node cells [28]. In the skin of old baboons, senescent dermal fibroblasts were quantified to be ~15% [29]. In aged human skin, beta galactosidase positive keratinocytes and fibroblasts are notably higher in aged donors [5]. Although, the overall percentage of senescent cells in aged tissues seem low, it is important to remember that (a) their number correlates positively with age [5,28–30], (b) they release inflammatory cytokines capable of amplifying damage in surrounding tissue via a paracrine action [31] and (c) if senescence occurs in a particularly important fraction of cells, they can have deleterious consequences on tissue health and function [32].
Challenges and alternative approaches to developing new senotherapeutics
Despite monumental advancements in developing or repurposing drugs to target and kill senescent cells, the scientific community faces major challenges in designing therapies that are highly specific to the rare senescent cell population. Alternative approaches to senolytics will be to delay the onset of senescence altogether or restore senescent cells to their youthful state [33]. Senescent cells share similarities with terminally differentiated cells and one strategy to revert the bad effects of senescence is to induce dedifferentiation by overexpressing Yamanaka factors [42]. This method has achieved remarkable success both in vitro [42] and in vivo [43]. However, as an important note, these studies aim to only partially reprogram cells without re-entry into cell cycle. Since senescence is a potent tumor suppressor, mechanisms that provoke cell cycle re-entry can have deleterious pro-cancer outcomes [44]. An alternative safer strategy is to develop therapies that target epigenetic enzymes acting on the chromatin in senescent cells [45]. Although challenging, this strategy may be able to switch gene expression programs in senescent cells restoring youthful morphology, shutting down SASP and achieving metabolic balance.
The following sections discuss the accumulating evidence of chromatin changes in senescent cells both in vitro and in vivo. The goal of this review is to encourage the readers to identify emerging trends and devise novel epigenetic senotherapies to ameliorate age-related functional decline and disease. The final section of this review discusses senolytic alternatives and novel epigenetic approaches to prevent senescence-related damage in aged tissues.
Chromatin Changes in Senescence
Breaking the histone code in senescent cells
The histone code refers to the combinatorial patterns of posttranslational histone modifications primarily on the tails (but also on the globular domains) of basic histone proteins that form an integral part of chromatin [46]. There are four canonical histone proteins that constitute the histone octamer with two copies each of H2A, H2B, H3 and H4 forming a spool around which DNA is wound [47]. Additional diversity is provided by variant histones [48] (see below). A catalog of histone modifications on canonical and variant histones have now been identified using molecular biology, genomics and proteomic approaches including acetylation (ac), methylation (me), phosphorylation (p), ubiquitination (ub) among others [49]. Specific histone modifications are correlated with open/closed or active/repressed chromatin states [50]. For example, histone 3 lysine 4 trimethylation (H3K4me3) and H3K27me3 are common epigenetic modifications with opposing controls on transcription and which have been directly linked to longevity regulation in many systems [14,51]. H3K4me3 is an activating modification found at gene promoters and drive active transcription by RNA polymerase II. In contrast, H3K27me3 is a repressive modification that marks facultative heterochromatin. Senescence and aging in diverse organisms entails an imbalance of these activating and repressive histone marks as evidenced by global assessments such as western blotting, immunofluorescence as well as genomics-based profiling with consequences at the transcription level. However, there is no consensus on a working model that fully explains the effects of these activating and repressive changes on lifespan in all organisms [52].
Heterochromatic alterations: formation of SAHFs
The quantitative assessment of the extent of imbalance of histone marks in senescent cells is confounded by declining levels of histone proteins triggered by DNA damage [53,54]. Despite this decline in total histone levels, distinct chromatin changes are visible by microscopy. OIS in human diploid fibroblasts is accompanied by a breakdown of the nuclear lamina (see below), loss of heterochromatin domains and large scale spatial rearrangements of chromatin, forming nuclear structures known as SAHFs [10]. SAHFs are formed by condensation of individual chromosomes into a single SAHF focus as identified by chromosome painting [55]. Confocal microscopy further elucidated a multilayer concentric structure of SAHFs with the repressive mark H3K9me3 enriched in the core and H3K9me2 covering the whole area of SAHF. In marked contrast to H3K9me3, H3K27me3 exhibited a “ring” structure, surrounding the H3K9me3 “core” separating it from H3K36me3-enriched transcriptionally active regions. In the genomic space, SAHFs coincide with late replicating regions. Furthermore, SAHFs are enriched in heterochromatic proteins HP1, chromatin architectural proteins of the HMGA family and histone variant macroH2A while excluding euchromatic marks such as H3K9ac, H3K4me3 and linker histone H1 [56]. SAHFs have been widely used as a marker of senescence with their mechanism of formation carefully mapped out. Prior to formation of SAHFs, HP1, HIRA and ASF1a (a chaperone complex for H3.3, see section on histone variants) transiently enter promyelocytic leukaemia (PML) bodies in a rate-limiting step. Subsequently, macroH2A is deposited onto chromatin and helps to stabilize SAHFs. These concerted events then promote cell cycle exit and durable growth arrest in senescence [57].
In addition to the microscopic studies that identified SAHFs, genomics analysis of a panel of histone modifications in growing and OIS cells, showed altered occupancy of active (H3K4me3 and H3K36me3) and repressive (H3K9me3, H3K9me2, and H3K27me3) histone marks with all except H3K9me3 being correlated with alterations in transcription. In addition, cluster analysis of ChIP signal of H3K9me3, H3K27me3 and H3K9me2 recapitatulated SAHF structure. However, although the marks were redistributed in some genic regions, the global pattern was highly static, suggesting that 3D repositioning rather than spreading of pre-existing H3K9me3 and H3K27me3 is involved in SAHF formation [56].
Genome-wide profiling of activating and repressive histone modifications: canyons, mesas and new enhancers
A complementary study analyzing H3K4me3 and H3K27me3 distributions by ChIP-seq in RS, OIS and cells from Hutchinson Gilford Progeria Syndrome (HGPS) patients showed large-scale changes such as domains of H3K4me3 and H3K27me3-enriched “mesas” and H3K27me3-depleted “canyons”. Cells from HGPS patients therefore represent a population of “aged” but not necessarily true senescent cells unless passaged a few times ex vivo. Hereon, we refer to cells isolated from aged tissues as aged cells.
Mesas form at lamin B1-associated domains (LADs) while canyons mostly form between LADs and are enriched in genes and enhancers. Forced reduction of lamin B1 results in mesas and canyons. Localized H3K27me3 loss in canyons strongly correlates with upregulation of key senescence and anti-proliferation genes, including the canonical SASP genes [58]. In support, a decrease in the expression of the H3K27 methylase EZH2, a polycomb group protein, during RS and OIS directs a decrease of repression-associated H3K27me3 and rapid senescence in primary human cells, in part through upregulation of p16 [59]. On the contrary, inhibition of the H3K4 methylase MLL1, decreased the levels of H3K4me3 modestly and Υ-H2AX dramatically over SASP gene bodies, with a concomitant inactivation of their gene expression. Υ-H2AX is a variant histone deposited on DNA damage in SASP genes suggesting that MLL1 is a key regulator of the secretory phenotype through a DNA damage response pathway [45].
Other histone modifications implicated in senescence include H4K20me3 (repressive), H4K16ac and H3K27ac (activating). H4K20me3 is enriched in senescent cells and aged cells and especially at SAHFs with H3K9me3 which recruits the cognate H4K20 methylase SUV420H2, as well as at specific non-genic and genic repeats [60–63]. In contrast, RS cells show strong enrichment of H4K16ac at promoter elements of all expressed genes and its retention is dependent of histone chaperone HIRA [64]. Like H4K20me3, global levels of H4K16ac do not strongly correlate with gene expression changes in senescent cells. A similar pattern of H4K16ac enrichment was also observed with aged neurons in human brain samples [65]. A systematic profiling of H3K27ac in proliferating, quiescent and OIS cells revealed the dynamic remodeling of the regulatory enhancer landscape in senescence. Loss of narrow typical enhancers adjacent to the promoters of proliferation genes correlated with the shut-down of proliferation in senescent cells. In contrast, the formation of new super enhancers in senescence occurs close to genes related to the SASP program. A subset of these super enhancers are bound by BRD4, a bromodomain containing protein. BRD4 inhibition by BET inhibitors did not bypass senescence but specifically modulated SASP levels and reduced immune-surveillance both in vitro and in vivo [66].
Taken together, the chromatin landscape in senescent cells presents a unique environment that promotes formation of features such as SAHFs which reinforce a tumor suppressive phenotype, as well as large regulatory elements that activate SASP programs. Interestingly, the breadth of H3K4me3 domains and enhancer score are important predictors of aging in murine tissues as identified using machine-learning models [67]. Overall, the balance in activating and repressive marks is tipped towards an “opening” of chromatin structure that likely promotes genome instability while maintaining the senescent transcriptome. A summary of histone modification changes is shown in Figure 1.
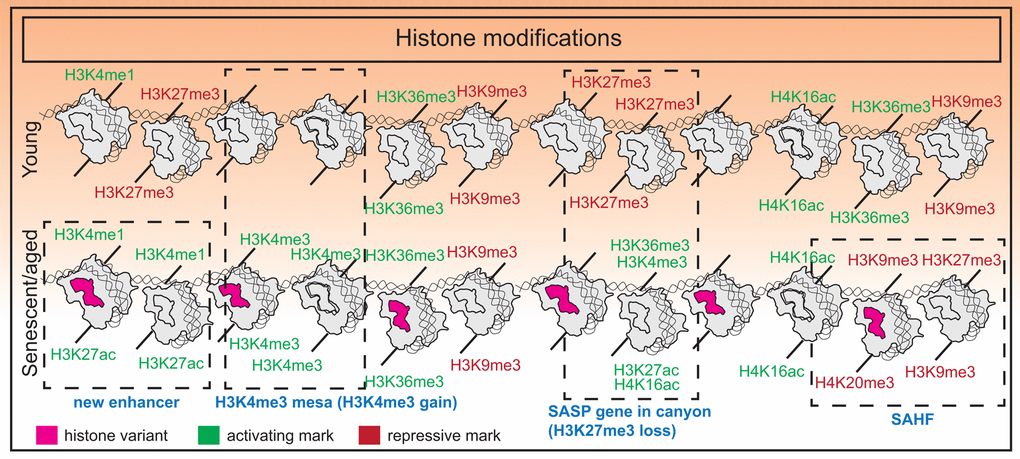
Figure 1. Histone modification changes in senescence. Senescence is associated with an imbalance of histone modifications with a tendency towards accumulating euchromatin marks. Additional features include formation of new super-enhancers near SASP genes in OIS, H3K27me3 “canyons” where SASP genes reside, H3K4me3 “mesas”, and formation of SAHFs.
Variant histones and altered nucleosomal composition in senescence
Histone variants are non-allelic counterparts of the core canonical histone proteins varying in one or few amino acids located primarily in the C terminus. Generally, genes encoding the canonical forms of H3 (H3.1 and H3.2) contain one exon, lack introns, have a stem loop terminator and are not polyadenylated. Hence they undergo rapid turnover in contrast to variant histones. Additionally, canonical histones are synthesized only in the S phase whereas variant histones are made constitutively throughout the cell cycle in a replication-independent manner [68,69]. Thus, in non-cycling senescent cells, canonical histone production declines (in part due to reduced synthesis and in part due to high turnover) and variant histones tend to accumulate [53,64]. However, expression of a small subset of two-exon histone genes encoding of H2A, H2B and H4 are upregulated in senescence, allowing the formation of nucleosomes with altered composition [64].
One variant of histone H3 is H3.3, which differs from H3.1 and H3.2 (the canonical counterparts) at five and four amino acid positions respectively [69]. RS cells show strong expression of H3.3 and incorporation at promoter elements of active genes by ChIP-seq. H3.3 peaks overlap with HIRA, a chaperone responsible for H3.3 and H4 deposition on chromatin. HIRA is also required for the steady state maintenance of the histone modification H4K16ac, which largely overlaps with H3.3 peaks in senescent cells. These events then maintain senescence-related gene expression and enforce tumor suppression [64]. In RS and OIS cells, the deposited H3.3 can undergo N-terminal tail cleavage at two sites by the lysosomal protease Cathepsin L1. This processing removes histone N-terminal posttranslational modifications such as H3K14ac, H3K18ac, H3K4me3 etc. that critically control the expression of cell cycle regulators. HIRA-mediated loading of cleaved H3.3 is sufficient to promote cellular senescence [70].
MacroH2A isoforms (macroH2A1.1, macroH2A1.2 and macroH2A.2) are variants of H2A that have a 30KDa “macro” domain at their C-terminus. MacroH2A1 is found predominantly in heterochromatic regions such as the inactive X chromosome [71]. In senescent cells, macroH2A1 is localized to SAHFs. Although not critical for SAHF formation, macroH2A1 serves a maintenance role to stabilize SAHFs [57]. In RS cells, the poly-ADP-ribose (PAR) binding isoform macroH2A1.1 (but not PAR-binding deficient isoform macroH2A1.2) becomes enriched as cells exit the cell cycle [72]. Another study in OIS cells, showed an intricate model of macroH2A1 regulation of SASP genes. In proliferating cells, macroH2A1 binds to transcriptionally inactive SASP genes where it likely poises SASP genes for activation. Upon senescence induction by oncogenes and the initial burst of SASP activity, macroH2A1 is required for autocrine and paracrine effects of SASP. Sustained SASP however, allows for activation of the ER stress signaling and DNA damage response, elevating ATM activity, which in a negative feedback loop removes macroH2A.1 and repression of SASP genes [73,74].
The histone variant H2A.J differs from canonical H2A protein by the presence of an SQK motif near the C-terminus and an A11V substitution in the N terminal tail. Mass spectrometric studies showed that H2A.J accumulates in RS and DNA-damage induced senescence increasing in amount by almost ten-fold. Additionally, elevated H2A.J was found in senescent keratinocytes upon carcinogen treatment as well as in hair follicle stem cells and interfollicular epidermal cells of old mice, irradiated mice and aged human epidermal cells. Depletion of H2A.J by RNAi was found to reduce the expression of genes encoding proteins bound to the cell surface and those involved in the SASP response. However, this response could not be explained by differential incorporation of H2A.J in SASP gene promoters as measured by ChIP-seq, begging further analysis [75].
Overall, the abundance of variant histones (over canonical histones) during senescence promotes a permissive chromatin for establishment and maintenance of the senescent state.
Profound alterations in the aging DNA methylome
DNA methylation is a second type of epigenetic control that involves either cytosine-5 methylation (5mC) within CpG dinucleotides or adenine-6 methylation (6mA). 5mC is a prominent and extensively studied modification in mammalian systems whereas other commonly studied aging models such as worms and flies either lack or have limited DNA methylation. DNA methylation is established during development through the action of de novo methylases DNMT3A and 3B, while DNMT1 plays a maintenance role [76]. Conversely, the ten-eleven-translocation (TET) proteins (TET1-3) mediate the removal of the methyl group in a three step iterative oxidation process, an intermediate of which is 5-hydroxymethyl cytosine (5hmC) [77]. 5mC and 5hmC together make up the bulk of methylated DNA with other oxidative products such as 5-formyl cytosine (5fC) and 5-carboxyl cytosine (5caC) being an order of magnitude less abundant [78].
5mC is recognized by a host of methyl CpG-binding domain (MBD) proteins including MBD1-4 and MeCP2 and together with co-repressors, prevent transcription from genes [79]. 5hmC on the other hand has been shown to be enriched in promoters, gene bodies and enhancers surrounding transcription factor binding sites and correlates with active transcription [80]. The genome-wide profiles of 6mA or 5hmC have not been characterized in senescent cells.
Alterations in the global distribution of 5mC
Senescence and aging markedly alter the DNA methylation (5mC) landscape with global DNA hypomethylation co-occuring with focal hypermethylation [11,14]. Hypomethylation occurs primarily in repetitive regions (LINEs and SINEs) or late-replicating pericentromeric satellites and lamin-associated domains in the genome that normally correlate with constitutive heterochromatin. In senescent cells, one consequence of hypomethylated DNA at repeat regions of the genome is distension (senescence-associated distension of satellites or SADS) and derepression [81]. For example, in both RS and OIS cells, SADS can be visualized via 3D DNA fluorescent in situ hybridization (FISH) experiments on pericentric satellite II and centromeric alpha satellites [13,82,83]. FAIRE (formaldehyde assisted isolation of regulatory elements) data from RS cells in addition showed that chromatin from major retrotransposon classes, Alu, SVA and L1 become more open, ultimately resulting in more transcription and transposition during deep senescence [84]. Increased retroviral repeat element transcription is also evident in aged mouse heart, liver, cerebellum and olfactory bulb [67]. Hypermethylation occurs at promoter CpGs, and mostly correlates with gene repression. In RS, whole genome bisulfite sequencing revealed hypermethylation at promoters of gene related to cell cycle and tumor suppressors suggesting that the senescent DNA methylome may sensitize aged cells to malignancy. Genome-wide, differentially methylated regions (DMRs) in senescence, senescence-bypass and cancer showed partial but significant overlap. Importantly, methylation changes retained in bypass cells (compared to senescent cells) were enriched for methylation changes in cancer [11]. In RS, expression of DNMT1 and DNMT3B is downregulated [11], as well as TET1 and TET3 [85] and therefore cannot predict the direction of DNA methylation change. However, hypomethylation was shown to occur even in proliferation competent near-senescent cells suggesting a failure of DNMT1 to maintain methylation. This observation, together with the lack of DNMT1 nuclear puncta in senescent cells and premature senescence induced by DNMT1 knockdown strongly supports a dominant role of the maintenance methylase in driving the senescent phenotype [11]. DNA methylation changes are also a feature of tissue aging. Comparative analysis of the DNA methylome in young and old mice livers revealed that hypomethylated DMRs were enriched at intragenic enhancers in highly expressed liver-specific genes and hypermethylated DMRs were enriched at bivalent CpG islands [86,87]. Taken together, senescence and aging involves a bidirectional epigenetic drift in the DNA methylome that contributes to cellular dysfunction and likely cancer progression.
Development of a DNA methylation-based epigenetic clock
Regression modeling of the methylation status of a set of CpGs has inspired the development of an “epigenetic clock”, serving as a robust biomarker of biological aging [88–90]. In the original study from UCLA, Horvath developed a multi-tissue predictor of age using 8000 samples from 82 Illumina DNA methylation array datasets, encompassing 51 healthy tissues and cell types [89,90]. He discovered that DNA methylation at 353 CpGs and no other epigenetic modifications provided an accurate age estimate. In a parallel study, Hannum et al performed genome-wide methylomic profiling of whole blood taken from a large cohort of individuals spanning a wide age range and variety of races. The study identified 71 CpGs as highly predictive of age [91]. Although the “clock” varies markedly across species and tissue types, several studies have refined CpG signatures to include a smaller subset that predict age independently of sex, tissue type, disease state and array platform [88]. Additionally, the "clock" has now been accurately defined in specific cell types and tissues for researchers working with those models. For example, mice liver “clocks” have shown that longevity altering interventions such as rapamycin and dietary restriction affects the biological age [86]. The “skin and blood clock” accurately predicts age for a variety of skin and blood cells and shows rapid age acceleration when cultured ex vivo [92]. Interestingly, the “clock” has now been commercialized as a direct-to-customer product by Zymo Research (sold as My Dnage) for predicting biological age in humans.
It is important to note that although the “epigenetic clock” correlates with cell passage, it is not a marker of cellular senescence. In one study that investigated RS, OIS and irradiation-induced senescence (IR), it was interestingly noted that RS and OIS cells were aged (as measured by the epigenetic clock) but not IR cells. This observation implies that DNA damage does not cause cellular aging and that cellular aging and senescence are uncoupled [93].
Despite the accuracy of the “clock”, the mechanisms accelerating or slowing down the "clock" are not clear and in conjunction with other epigenetic predictors, would be a fascinating area of study. Additionally, the underlying biological meaning of the "clock" is also unknown, and genes associated with the clock CpGs show no differences in age-related gene expression with two exceptions. A genome-wide association study (GWAS) revealed specific variants inside a putative RNA helicase DHX57 and near mTOR complex 2 gene MLST8, had cis-effects on gene expression in the cerebellum [94]. However, the apparent general lack of correlation with gene expression suggests that the "clock" is perhaps a highly sensitive readout of upstream signals such as hormonal changes or immune signaling resulting in 3D spatial or phase changes inside the nucleus. A summary of DNA methylome changes in senescence is summarized in Figure 2.
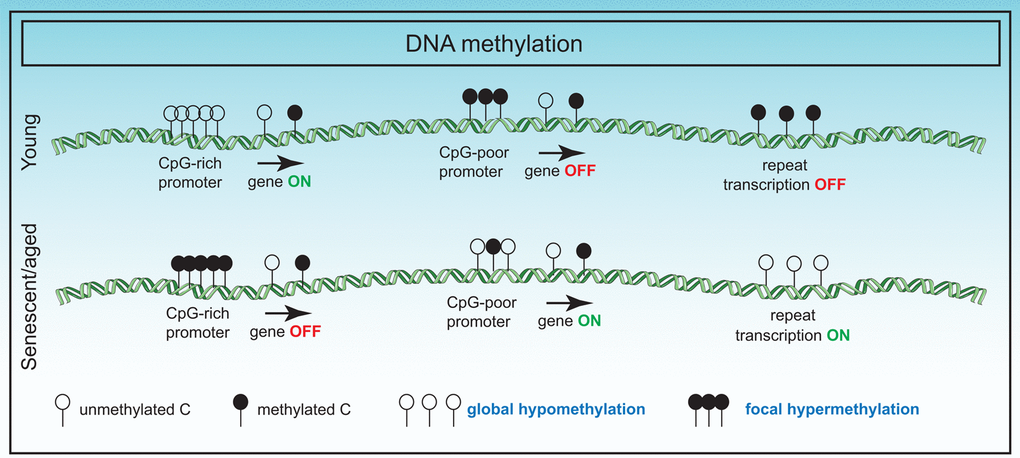
Figure 2. Changes in the DNA methylome in senescence. DNA methylation changes in senescence primarily involve global hypomethylation particularly at repeat regions and focal hypermethylation at CpG rich promoter sequences. These changes have adverse effects on gene expression.
The nuclear lamina structure and its disintegration in senescence
The nuclear envelope separates nuclear functions from cytoplasmic functions and at its inner surface, provides a docking site for chromatin. The inner nuclear membrane is lined by the nuclear lamina which is composed of a complex meshwork of proteins viz., the type V intermediate filament proteins and the nuclear lamins. Lamins are either A or B type based on sequence homology. In mammals, two major A-type lamins (lamin A and C) and two major B-type lamins (lamin B1 and B2) have been characterized. A-type lamins are derived from one gene (LMNA) by alternative splicing whereas B-type lamins are encoded by different genes [95].
The lamins are composed of a long central α-helical rod domain containing heptad repeats, flanked by globular N-terminal (head) and C-terminal (tail) domains [96]. A CaaX box (where C is cysteine, a is an aliphatic amino acid and X is any amino acid) located at the C terminal of lamins B1 and B2 and lamin A (but not lamin C), undergoes extensive post-translational processing. First, the cysteine residue of the CaaX box is farnesylated and subsequently the aaX is removed by endopeptidases such as RCE1 or ZMPSTE24/FACE1, following which, the cysteine is carboxymethylated. Lamin A undergoes a further maturation step with the removal of 15 amino acids from the C terminus by the enzyme ZMPSTE24 to form mature lamin A [97].
Perturbations in nuclear lamina-chromatin interactions
Loss of lamin B1 is a notable biomarker of senescence in primary human and murine cells [98]. In senescent cells, lamin B1 expression is downregulated at two levels: at the mRNA level [98] as cells exit the cell cycle, and at the protein level, by autophagic degradation [99]. Consequently, lamin B1 is found in conjunction with associated DNA and histones in cytoplasmic chromatin fragments (CCFs). The cytoplasmic chromatin activates the innate immunity arm via cytosolic DNA sensing cGAS-STING and NF-κB pathway leading to both short-term and chronic inflammation by activation of SASP [54,100,101]. A consequence of lamin B1 downregulation is a detachment of chromatin domains normally attached to the nuclear lamina [102] leading to the redistribution of heterochromatin from the nuclear periphery to the interior. This is evidenced by epigenetic profiling of senescent cells which show formation of large-scale domains of H3K4me3- and H3K27me3-enriched mesas and H3K27me3-depleted canyons. Mesas form at LADs in RS and OIS and overlap DNA hypomethylation regions in cancer providing further support that the senescent epigenome precedes cancer progression [58]. Lamin B1 reduction in proliferating cells triggers senescence and the formation of mesas and canyons. The anti-proliferative effect of lamin B1 silencing requires the activation of p53, but not Rb, whereas full induction of premature senescence requires both proteins [103]. Importantly, immunofluorescence studies in liver sections derived from irradiated mice showed lower staining of lamin B1 (but not lamin C) suggesting that it is also a feature of aged cells [98].
Laminopathies and link to premature senescence
Lamins are key proteins linked to premature human aging [104]. The group of heterogeneous diseases caused by lamin dysfunction is called “laminopathies”. Laminopathies among other diseases include HGPS or childhood progeria and Werner’s syndrome or adult progeria, both rare sporadic disorders characterized by accelerated aging [95]. A majority of HGPS patients carry the G608G (GGC>GGT) mutation within exon 11 of lamin A, activating a cryptic splice donor site that results in production of a dominant negative form of a truncated lamin A protein, called progerin that remains farnesylated [105]. With this mutation, HGPS cells exhibit severe abnormalities in nuclear morphology including nuclear blebbing, compromised DNA damage repair, alterations in chromosome organization, abnormal heterochromatin, and accelerated rates of cellular senescence [106]. At the chromatin level, HGPS fibroblasts (made senescent ex vivo) show evidence of H3K4me3 mesa formation [58], suggesting a link between abnormal nuclear morphology, premature chromatin changes and accelerated cellular senescence. Additionally, late passage senescent HGPS cells exhibit reduction in H3K9me3 and H3K27me3 levels, and H3K27me3 methylase EZH2 but an increase in another heterochromatic mark, H4K20me3 [62,104]. A recent genome-wide analysis of H3K27me3 in HGPS cells suggested a redistribution of the remaining amount of this modification across the genome that correlates with gene expression changes [107]. Interestingly, progerin is found to accumulate in RS cells [108], and skin cells in the elderly [105] suggesting that it may drive alterations in nuclear morphology, disruption of heterochromatin and downstream gene expression changes in senescence and aging. Werner syndrome patients commonly carry mutations in the WRN helicase. However, a subset of patients may not exhibit WRN mutations but rather carry mutations in the heptad repeats of lamin A (atypical Werner’s). Altered nuclear morphology and mislocalized lamina are also characteristic of these patient-derived aged cells and likely contribute to the accelerated aging phenotype also seen in HGPS [95].
The exact cause of lamin B1 downregulation during senescence is unknown although it is speculated that sustained DNA damage may play a role. Alternatively, loss of lamin B1 may be due to the exit from cell cycle coupled to selective autophagy mechanisms as discussed above. Nevertheless, the consequence of this loss is dramatic, affecting lamina structure, nuclear morphology and genome organization. In the case of HGPS patients, some of these detrimental changes can be ameliorated by farnesyltransferase inhibitors that prevent the farnesylation and incorporation of progerin into the nuclear membrane [109,110]. An overall summary of lamina changes in senescence is shown in Figure 3.
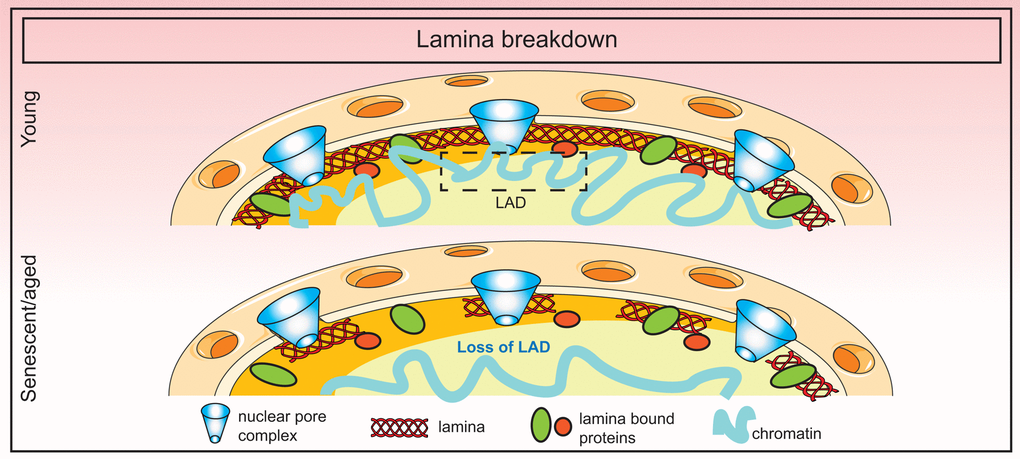
Figure 3. Breakdown of the nuclear lamina in senescence. Loss of lamin B1 in senescence triggers the detachment of constitutive heterochromatic regions (lamin-associated domains or LADs) which disorganizes the spatial arrangement of the genome in the nucleus.
3D genome (dis)organization in senescence
The genome within the nucleus is hierarchically organized in three dimensions; the first dimension (1D genome) being the linear genome sequence, the second dimension (2D genome) being the arrangement of genes and regulatory elements forming a functional network of the genome, and the third dimension (3D genome) being the spatial organization of the genome in the nuclear space. It has been observed that 3D organization is non-random and that active regions of the genome are segregated from inactive domains. At its highest resolution, the 3D genome is able to define how gene expression is controlled by “looping” of regulatory elements to gene promoters despite being separated in the first dimension [111]. A fourth dimension (4D nucleome), “time”, has been added which integrates the 1D, 2D and 3D information and traces changes in genome organization during processes such as development, adaptation to stress, aging, and disease [112,113].
Higher-order chromatin organization has attracted great attention in recent years due to progress in chromosome conformation capture (3C) methods and high-resolution nuclear microscopy. Using these methodologies researchers elegantly illustrated a top-down view of the 3D genome in which chromosomes are spatially segregated into territories (50-250 Mb), compartments (~5 Mb), topologically associated domains (TADs; ~1 Mb), sub-TADs (0.1-1 Mb) and loops (5-300Kb). Within each territory are clusters of compartments which may be either active (A) or repressive (B). Within each compartment, there are TADs which in turn contain sub-TADs. Sub-TADs contain loops (usually between regulatory enhancers and gene promoters). Each individual structure is physically separated from one another by boundary regions. Three ubiquitously expressed proteins, cohesin, CTCF and YY1, control the organization of loops, sub-TADs and TADs using a loop extrusion mechanism but are dispensable for compartments and territories [114–117]. Condensin II, on the other hand, controls intermixing of territories [118]. Loops and sub-TAD structures are dynamic whereas TADs are largely stable units of transcription and replication but are cell-type specific [119,120].
Compartment switching and TAD behavior
During senescence and aging, there are dramatic changes in the chromatin landscape including changes in histone modification, DNA methylation and nucleosome organization [52], and therefore logically these changes transpire into changes in 3D genome organization [121]. Hi-C experiments in human diploid fibroblast (LF1) cells in deep RS showed (as expected) that compartments and TADs were largely unaltered compared to proliferating or quiescent cells. However, there was a significant decrease in long-range interactions and an increase in short-range interactions. Additionally, a subset of TADs showed compartment switching from B to A (higher frequency) or A to B (lower frequency; cell cycle genes) with gene expression changes in the expected direction. The popular senescence-associated genes such as SASP were in stable A compartments [82]. A higher-resolution Hi-C map in HUVEC, IMR90 and MSC (mesenchymal stromal cells) afforded a finer examination of TAD behavior in senescence whereby ~50% of the TADs were unchanged but the rest showed either a shifting, separating or fusing behavior with the latter being most frequent. HMGB2 was discovered as a novel looping factor found at TAD boundaries that insulates CTCF sites and prevents them from forming long-range interactions. A down-regulation of HMGB2 in senescence prompted the formation of large senescence-induced CTCF clusters (SICCs) [122]. However, this study contradicts the previous study in that the authors found changes in both short- and long-range interactions and limited compartment switching.
In contrast to RS, OIS in WI38 cells showed that long-range cross-boundary interactions were significantly gained but short-range local intra-TAD interactions were lost. This local loss in short-range interactions appeared to occur in regions that correspond to constitutive heterochromatin enriched in H3K9me3, are late replicating, have low GC% and are lamina-associated. The authors thus concluded that they represent regions of heterochromatin disruption. Interestingly, similar changes were also observed in aged fibroblast cells from Progeria patients which are known to undergo nuclear lamina destabilization concomitant with a near complete loss of long-range interactions. In OIS cells uniquely, there was a spatial clustering of the decondensing regions that likely represent SAHFs [123]. These observations while reconciling the observed differences in SAHF formation in OIS (more SAHF) and RS/HGPS (less SAHF) cells, do not fully explain the mechanism of SAHF formation. It is speculated that structures such as SAHFs and SICCs may represent outcomes of liquid phase separation in senescent cells although that remains to be tested.
Orthologous approaches such as chromosome painting, FISH distance measurements, microscopy, FAIRE and DNaseI sensitivity further point to the compaction of chromosomes and increase in overall nuclear volume in senescent cells [124]. Together with a general increase in short-range interactions (in RS and HGPS cells), a collapsing coil model has been proposed [82]. This model envisions that the changes in the 3D genome in senescence and aging manifests in the shrinking of chromosome arms non-homogeneously at different levels of organization. A summary of the frequent alterations in 3D genome organization is presented in Figure 4.
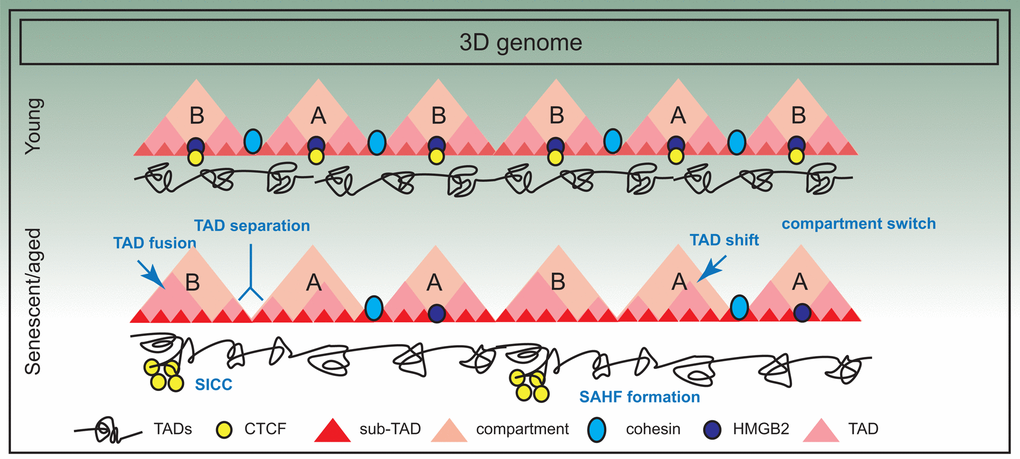
Figure 4. Three dimensional spatial changes in the genome during senescence. The 3D arrangement of the genome suffers significant changes in senescence; for example there is evidence of compartment switching, TAD fusing, TAD separation and TAD shifting. Some of these changes are also triggered by decline in chromatin architectural proteins such as HMGB2, a consequence of which is SICC formation. However, in general, TAD structure is maintained.
Future Directions and Translational Perspectives
The “coming of age” of the senescence field
When cellular senescence was first characterized in in vitro cell culture, links to tissue and organismal aging were proposed [1,125]. Critics of cellular senescence questioned its relevance to in vivo aging, their possibility of being an artefact and the inevitable lack of senescence despite normal aging in lower organisms. In fact, it was recently discovered that the extremely long lived rodent, naked mole rat, can undergo various forms of cellular senescence that apparently does not affect its longevity [126]. On the contrary, senescence as a pro-aging phenomenon gained popularity with the discovery of biomarkers such as p16 and beta-galactosidase in multiple aged tissues [127]. Mechanistically, the idea of senescent cells being causal in chronic inflammation characteristic of aging, also gained momentum with the discovery of SASP [8].
The senescence field came of age with four major milestones, (a) two proof-of-concept studies showed major improvement in healthspan and lifespan in mice by the targeted ablation of senescent cells [26,27], (b) development of small molecule senolytics as a therapeutic strategy for clearing senescent cells [33], (c) demonstration that senolytics improve physiological function and lifespan in aged mice [34,35,37] and (d) the success of senolytics in pre-clinical studies of a range of age-related conditions [128]. Below, we discuss and illustrate (Figure 5) potential alternatives to senolytics that can deploy epigenetic proteins as “switches” to turn on/off specific pathways in senescent cells for their effective elimination.
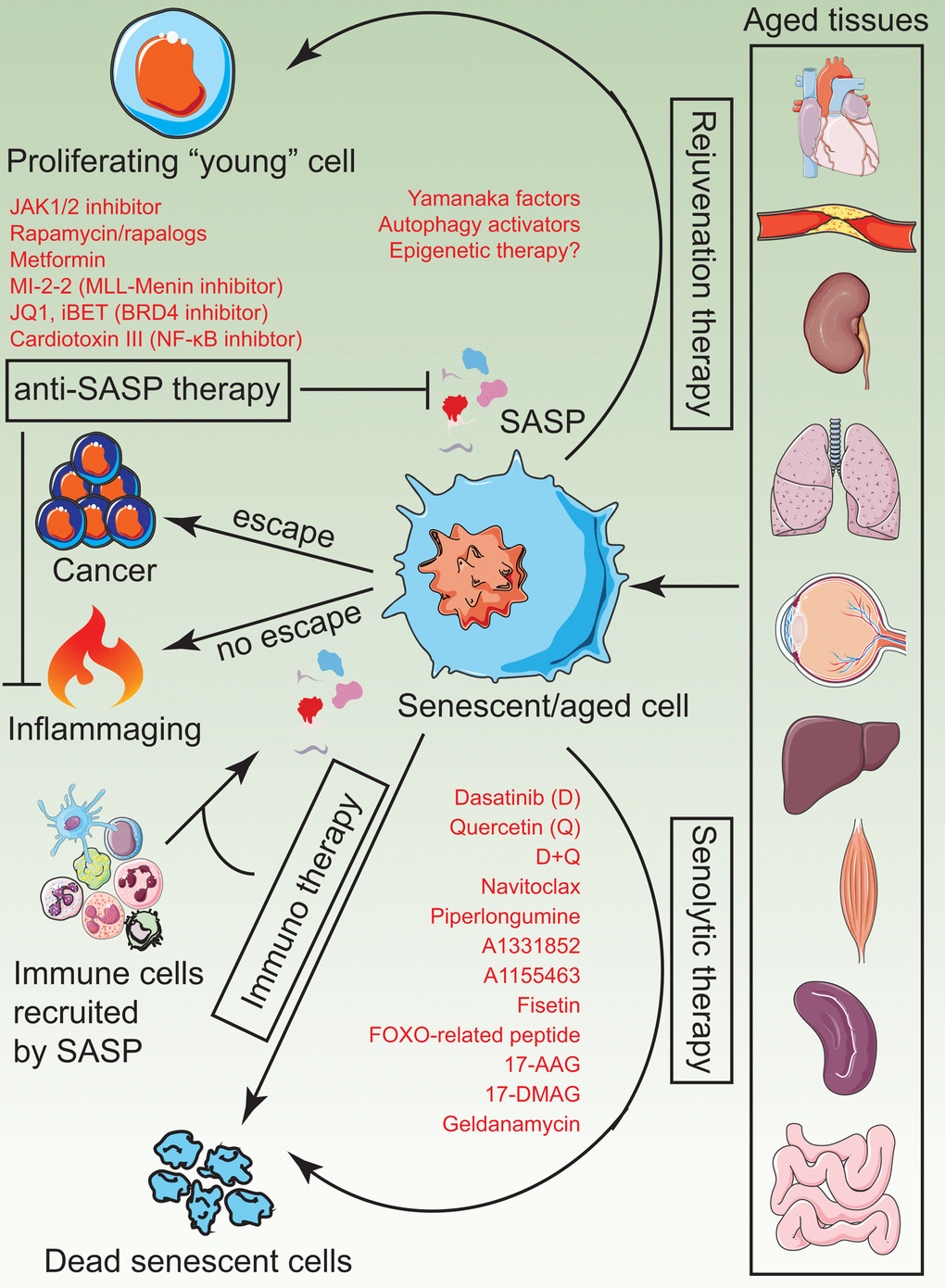
Figure 5. A visual summary of current senotherapies. Aged tissues tend to accumulate senescent cells which impose detrimental changes to tissue structure, regenerative ability and physiological function due to chronic inflammation. Current and plausible strategies to treat these adverse effects include administration of senolytics, rejuvenation therapy by induce partial reprogramming to a “youthful” state, anti-SASP therapy to prevent the generation and release of inflammatory cytokines and immunotherapy to activate innate immune mechanisms of the body, which in turn, clear senescent cells naturally.
SASP inhibitors
Despite the overwhelming success of senolytics, fundamental concerns about specificity and safety prevail. Additionally, the potential benefit of senolytics in treating age-related disease remains to be tested. A second class of molecules that have shown promise in anti-aging rejuvenation therapies is SASP inhibitors. The concept of annihilating the pro-aging arm of senescent cells while preserving the anti-tumor arm is a very attractive treatment option in the elderly who have a high incidence of cancer. Both rapamycin [39] and metformin [129] have shown anti-SASP effects and are on the road to clinical trial for aging. Alternatively, epigenetic enzymes that play a key role in turning on SASP genes (MLL1 [45] and BRD4 [66]) can be inhibited by small molecules to prevent its deleterious effects.
Autophagy activation
Autophagy is a self-degenerative process that clears and recycles damaged cellular components. In a seminal publication, it was reported that basal autophagy is essential to maintain the stem-cell quiescent state while preventing senescence of muscle satellite cells in mice [130]. Furthermore, autophagy declines during aging, calorie restriction activates autophagy, and dysfunctional autophagy is evident in Alzheimer’s disease pathology [131]. Thus, boosting general macroautophagy (non-selective) is a viable anti-aging avenue. The challenge of autophagy-promoting strategies however comes from observations that autophagy of “nuclear” substrates [99,100] might in fact contribute to senescence, aging and inflammation. Selective substrate-specific activation methods (in this case, activation of non-nuclear substrates), need to be developed for use as anti-aging therapy. As an example, selective activation of autophagy directed against damaged mitochondria (mitophagy) that accumulates in senescence and aging has been accomplished [132]. For further development, it is important to understand upstream triggers of macroautophagy and key epigenetic factors that may play in its activation while suppressing nuclear autophagy.
Immune-mediated clearance
Senescent cells are naturally cleared by innate immune mechanisms with the macrophage playing a central role. However, immune cells themselves undergo progressive decline in function (termed immunosenescence) that actively contributes to senescent cell accumulation [14,133]. Furthermore, it has been proposed that subsets of senescent cells become resistant to immune-mediated clearance. Therefore, epigenetic interventions that boost immune surveillance in aged tissues or antibody-based therapies that revert the immune-resistance of senescent cells may also be future rejuvenation strategies [134].
Rejuvenation therapy
Regenerative medicine is a field that provides strategies to repair and restore organ function due to injury, disease and congenital defects. The principles of regenerative medicine can also be applied in aging and age-related disease. Expression of pluripotency factors in senescent cells have been shown to allow cell cycle entry with reset telomere size, gene expression profiles, oxidative stress, and mitochondrial metabolism [42]. Additionally, their expression in mice has also shown amelioration of a panel of age-related phenotypes [43]. Epigenetic factors that can potentiate reprogramming can be used to rejuvenate senescent/aged cells. However, it is important to be cautious with regenerative therapy in the elderly because of its potential to be pro-tumorigenic.
Other potential epigenetic therapies
This review summarizes the major findings of chromatin and nuclear changes in senescent cells (listed in Table 1). The emerging conceptual themes that arise from the observations are (a) a gradual euchromatinization of the genome, (b) loss or disorganization of constitutive heterochromatin due to (c) breakdown of the nuclear lamina and changes in nuclear morphology and (d) loss of spatial organization of the genome. These large-scale changes manifest in profound transcriptional alterations that ultimately activate programs such as SASP and contribute to transcriptional noise. Systematic screens for epigenetic factors will likely yield potential candidates that can be targeted to prevent or reverse the detrimental effects of senescence. Two exemplary discoveries in this field are (a) the discovery of MLL1 (and potentially inhibition of the MLL/Menin interaction) [45] and (b) BRD4 (and potentially BET inhibitors) [66] as direct SASP ameliorating targets. Inhibitors of other epigenetic enzymes, some of which are already in the market for chemotherapy, can be repurposed to provide anti-aging benefits. With the first RNAi therapy being approved by the FDA, the possibilities of epigenetic therapy are limitless.
Table 1. Epigenetic themes from studies in senescence in vitro and in vivo.
| Theme | in vitro senescence type | in vivo aging | Possible therapy | Reference |
| SAHF with repressive chromatin marks, HP1 and macroH2A | RS, OIS, HGPS | [10,55–57] | ||
| SASP | RS, OIS | Multiple aged tissues | Anti-SASP therapy (Fig. 5) | [8,23,44] |
| Decline in total histone | RS | Boost expression of canonical histones | [53,54] | |
| Canyons and mesas | RS, OIS and HGPS | Histone methylase/demethylase inhibitors | [58] | |
| Increase of H4K20me3 | RS, OIS, HGPS | Aged rat liver and kidney | [60–63] | |
| Increase of H4K16ac | RS | Aged human brains | Sirtuin activators | [64,65] |
| Enhancer formation and score | OIS | Aged mouse heart, liver, cerebellum and olfactory bulb | BRD4 and BET inhibitors | [66,67] |
| Increase in expression and deposition of histone variants, histone clipping | RS, OIS | Boost expression of canonical histones, Inhibition of cathepsin | [45,57,64,69–71,75] | |
| Global DNA hypomethylation (5mC), focal hypermethylation | RS | Aged mouse liver | TET inhibition | [11,86,87] |
| SADS | RS, OIS | [82,83] | ||
| Derepression of repeat elements | RS | Aged mouse heart, liver, cerebellum and olfactory bulb | [67,84] | |
| Epigenetic clock | RS, OIS (clock predicts cellular age but not senescence) | Multiple human cells and tissues from aged and diseased donors including skin and blood, mouse liver etc. | [87–93] | |
| Loss of lamin B1, nuclear blebs, progerin accumulation | RS, OIS, HGPS | Skin cells from HGPS patients and old humans | Farnesyltransferase inhibitors | [58,98–100,102,103,108–110] |
| CCF | RS, OIS, DNA-damage induced | Inhibition of unknown endonuclease | [54,99,100] | |
| Compartment switching | RS, OIS, HGPS | [82,122,123] | ||
| TAD fusion, separation, shift | RS, OIS, HGPS | [82,122,123] | ||
| SICC | RS | Increase expression of HMGB2 | [122] |
Taken together, anti-aging and senescence-clearing therapies can be devised around many well founded principles and what will likely benefit in the end are combinatorial approaches that rejuvenate senescent cells while preserving its anti-proliferative state and blocking its pro-inflammatory properties. Epigenetic approaches provide tractable solutions in this direction.
Acknowledgements
All figures were made using images available through Servier Medical Art by Servier, licensed under a Creative Commons Attribution 3.0 Unported License.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding
We wish to acknowledge the National Institute on Aging Intramural Research Program, National Institutes of Health, for financial and resource support. This work was supported in part by AFAR Irene Diamond Transition Award DIAMOND 17113 to P.S.
References
- 1. Hayflick L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp Cell Res. 1965; 37:614–36. https://doi.org/10.1016/0014-4827(65)90211-9 [PubMed]
- 2. Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990; 345:458–60. https://doi.org/10.1038/345458a0 [PubMed]
- 3. Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007; 8:729–40. https://doi.org/10.1038/nrm2233 [PubMed]
- 4. Adams PD. Healing and hurting: molecular mechanisms, functions, and pathologies of cellular senescence. Mol Cell. 2009; 36:2–14. https://doi.org/10.1016/j.molcel.2009.09.021 [PubMed]
- 5. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995; 92:9363–67. https://doi.org/10.1073/pnas.92.20.9363 [PubMed]
- 6. Hampel B, Malisan F, Niederegger H, Testi R, Jansen-Dürr P. Differential regulation of apoptotic cell death in senescent human cells. Exp Gerontol. 2004; 39:1713–21. https://doi.org/10.1016/j.exger.2004.05.010 [PubMed]
- 7. Chen QM, Liu J, Merrett JB. Apoptosis or senescence-like growth arrest: influence of cell-cycle position, p53, p21 and bax in H2O2 response of normal human fibroblasts. Biochem J. 2000; 347:543–51. https://doi.org/10.1042/bj3470543 [PubMed]
- 8. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010; 5:99–118. https://doi.org/10.1146/annurev-pathol-121808-102144 [PubMed]
- 9. Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009; 11:973–79. https://doi.org/10.1038/ncb1909 [PubMed]
- 10. Narita M, Nũnez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003; 113:703–16. https://doi.org/10.1016/S0092-8674(03)00401-X [PubMed]
- 11. Cruickshanks HA, McBryan T, Nelson DM, Vanderkraats ND, Shah PP, van Tuyn J, Singh Rai T, Brock C, Donahue G, Dunican DS, Drotar ME, Meehan RR, Edwards JR, et al. Senescent cells harbour features of the cancer epigenome. Nat Cell Biol. 2013; 15:1495–506. https://doi.org/10.1038/ncb2879 [PubMed]
- 12. Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003; 22:4212–22. https://doi.org/10.1093/emboj/cdg417 [PubMed]
- 13. Nacarelli T, Liu P, Zhang R. Epigenetic Basis of Cellular Senescence and Its Implications in Aging. Genes (Basel). 2017; 8. https://doi.org/10.3390/genes8120343 [PubMed]
- 14. Sidler C, Kovalchuk O, Kovalchuk I. Epigenetic Regulation of Cellular Senescence and Aging. Front Genet. 2017; 8:138. https://doi.org/10.3389/fgene.2017.00138 [PubMed]
- 15. Bernadotte A, Mikhelson VM, Spivak IM. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging (Albany NY). 2016; 8:3–11. https://doi.org/10.18632/aging.100871 [PubMed]
- 16. Dimauro T, David G. Ras-induced senescence and its physiological relevance in cancer. Curr Cancer Drug Targets. 2010; 10:869–76. https://doi.org/10.2174/156800910793357998 [PubMed]
- 17. Jiang P, Du W, Mancuso A, Wellen KE, Yang X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature. 2013; 493:689–93. https://doi.org/10.1038/nature11776 [PubMed]
- 18. Sun N, Youle RJ, Finkel T. The Mitochondrial Basis of Aging. Mol Cell. 2016; 61:654–66. https://doi.org/10.1016/j.molcel.2016.01.028 [PubMed]
- 19. Correia-Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A, Rushton MD, Charles M, Jurk D, et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016; 35:724–42. https://doi.org/10.15252/embj.201592862 [PubMed]
- 20. Peleg S, Feller C, Forne I, Schiller E, Sévin DC, Schauer T, Regnard C, Straub T, Prestel M, Klima C, Schmitt Nogueira M, Becker L, Klopstock T, et al. Life span extension by targeting a link between metabolism and histone acetylation in Drosophila. EMBO Rep. 2016; 17:455–69. https://doi.org/10.15252/embr.201541132 [PubMed]
- 21. Aird KM, Zhang R. Metabolic alterations accompanying oncogene-induced senescence. Mol Cell Oncol. 2014; 1:e963481. https://doi.org/10.4161/23723548.2014.963481 [PubMed]
- 22. Baker DJ, Peleg S. Biphasic Modeling of Mitochondrial Metabolism Dysregulation during Aging. Trends Biochem Sci. 2017; 42:702–11. https://doi.org/10.1016/j.tibs.2017.06.005 [PubMed]
- 23. Campisi J. Aging and cancer: the double-edged sword of replicative senescence. J Am Geriatr Soc. 1997; 45:482–88. https://doi.org/10.1111/j.1532-5415.1997.tb05175.x [PubMed]
- 24. van Deursen JM. The role of senescent cells in ageing. Nature. 2014; 509:439–46. https://doi.org/10.1038/nature13193 [PubMed]
- 25. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016; 354:472–77. https://doi.org/10.1126/science.aaf6659 [PubMed]
- 26. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, Khazaie K, Miller JD, van Deursen JM. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016; 530:184–89. https://doi.org/10.1038/nature16932 [PubMed]
- 27. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011; 479:232–36. https://doi.org/10.1038/nature10600 [PubMed]
- 28. Biran A, Zada L, Abou Karam P, Vadai E, Roitman L, Ovadya Y, Porat Z, Krizhanovsky V. Quantitative identification of senescent cells in aging and disease. Aging Cell. 2017; 16:661–71. https://doi.org/10.1111/acel.12592 [PubMed]
- 29. Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006; 311:1257. https://doi.org/10.1126/science.1122446 [PubMed]
- 30. Mishima K, Handa JT, Aotaki-Keen A, Lutty GA, Morse LS, Hjelmeland LM. Senescence-associated beta-galactosidase histochemistry for the primate eye. Invest Ophthalmol Vis Sci. 1999; 40:1590–93. [PubMed]
- 31. Zhu Y, Armstrong JL, Tchkonia T, Kirkland JL. Cellular senescence and the senescent secretory phenotype in age-related chronic diseases. Curr Opin Clin Nutr Metab Care. 2014; 17:324–28. https://doi.org/10.1097/MCO.0000000000000065 [PubMed]
- 32. He S, Sharpless NE. Senescence in Health and Disease. Cell. 2017; 169:1000–11. https://doi.org/10.1016/j.cell.2017.05.015 [PubMed]
- 33. Kirkland JL, Tchkonia T. Cellular Senescence: A Translational Perspective. EBioMedicine. 2017; 21:21–28. https://doi.org/10.1016/j.ebiom.2017.04.013 [PubMed]
- 34. Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature. 2018; 562:578–82. https://doi.org/10.1038/s41586-018-0543-y [PubMed]
- 35. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG, Onken JL, Johnson KO, Verzosa GC, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. 2018; 24:1246–56. https://doi.org/10.1038/s41591-018-0092-9 [PubMed]
- 36. Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, Chung JW, Kim DH, Poon Y, David N, Baker DJ, van Deursen JM, Campisi J, Elisseeff JH. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. 2017; 23:775–81. https://doi.org/10.1038/nm.4324 [PubMed]
- 37. Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, Ling YY, Melos KI, Pirtskhalava T, Inman CL, McGuckian C, Wade EA, Kato JI, et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine. 2018; 36:18–28. https://doi.org/10.1016/j.ebiom.2018.09.015 [PubMed]
- 38. Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, White TA, Johnson KO, Stout MB, Mezera V, Giorgadze N, Jensen MD, LeBrasseur NK, Kirkland JL. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci USA. 2015; 112:E6301–10. https://doi.org/10.1073/pnas.1515386112 [PubMed]
- 39. Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, Limbad C, Demaria M, Li P, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015; 17:1049–61. https://doi.org/10.1038/ncb3195 [PubMed]
- 40. Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, Lonetto MA, Maecker HT, Kovarik J, Carson S, Glass DJ, Klickstein LB. mTOR inhibition improves immune function in the elderly. Sci Transl Med. 2014; 6:268ra179. https://doi.org/10.1126/scitranslmed.3009892 [PubMed]
- 41. Mannick JB, Morris M, Hockey HP, Roma G, Beibel M, Kulmatycki K, Watkins M, Shavlakadze T, Zhou W, Quinn D, Glass DJ, Klickstein LB. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci Transl Med. 2018; 10:10. https://doi.org/10.1126/scitranslmed.aaq1564 [PubMed]
- 42. Lapasset L, Milhavet O, Prieur A, Besnard E, Babled A, Aït-Hamou N, Leschik J, Pellestor F, Ramirez JM, De Vos J, Lehmann S, Lemaitre JM. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011; 25:2248–53. https://doi.org/10.1101/gad.173922.111 [PubMed]
- 43. Ocampo A, Reddy P, Martinez-Redondo P, Platero-Luengo A, Hatanaka F, Hishida T, Li M, Lam D, Kurita M, Beyret E, Araoka T, Vazquez-Ferrer E, Donoso D, et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell. 2016; 167:1719–1733.e12. https://doi.org/10.1016/j.cell.2016.11.052 [PubMed]
- 44. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013; 75:685–705. https://doi.org/10.1146/annurev-physiol-030212-183653 [PubMed]
- 45. Capell BC, Drake AM, Zhu J, Shah PP, Dou Z, Dorsey J, Simola DF, Donahue G, Sammons M, Rai TS, Natale C, Ridky TW, Adams PD, Berger SL. MLL1 is essential for the senescence-associated secretory phenotype. Genes Dev. 2016; 30:321–36. https://doi.org/10.1101/gad.271882.115 [PubMed]
- 46. Jenuwein T, Allis CD. Translating the histone code. Science. 2001; 293:1074–80. https://doi.org/10.1126/science.1063127 [PubMed]
- 47. Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997; 389:251–60. https://doi.org/10.1038/38444 [PubMed]
- 48. Henikoff S, Smith MM. Histone variants and epigenetics. Cold Spring Harb Perspect Biol. 2015; 7:a019364. https://doi.org/10.1101/cshperspect.a019364 [PubMed]
- 49. Zhao Y, Garcia BA. Comprehensive Catalog of Currently Documented Histone Modifications. Cold Spring Harb Perspect Biol. 2015; 7:a025064. https://doi.org/10.1101/cshperspect.a025064 [PubMed]
- 50. Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007; 447:407–12. https://doi.org/10.1038/nature05915 [PubMed]
- 51. Han S, Brunet A. Histone methylation makes its mark on longevity. Trends Cell Biol. 2012; 22:42–49. https://doi.org/10.1016/j.tcb.2011.11.001 [PubMed]
- 52. Sen P, Shah PP, Nativio R, Berger SL. Epigenetic Mechanisms of Longevity and Aging. Cell. 2016; 166:822–39. https://doi.org/10.1016/j.cell.2016.07.050 [PubMed]
- 53. O’Sullivan RJ, Kubicek S, Schreiber SL, Karlseder J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol. 2010; 17:1218–25. https://doi.org/10.1038/nsmb.1897 [PubMed]
- 54. Ivanov A, Pawlikowski J, Manoharan I, van Tuyn J, Nelson DM, Rai TS, Shah PP, Hewitt G, Korolchuk VI, Passos JF, Wu H, Berger SL, Adams PD. Lysosome-mediated processing of chromatin in senescence. J Cell Biol. 2013; 202:129–43. https://doi.org/10.1083/jcb.201212110 [PubMed]
- 55. Zhang R, Chen W, Adams PD. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol Cell Biol. 2007; 27:2343–58. https://doi.org/10.1128/MCB.02019-06 [PubMed]
- 56. Chandra T, Kirschner K, Thuret JY, Pope BD, Ryba T, Newman S, Ahmed K, Samarajiwa SA, Salama R, Carroll T, Stark R, Janky R, Narita M, et al. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol Cell. 2012; 47:203–14. https://doi.org/10.1016/j.molcel.2012.06.010 [PubMed]
- 57. Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, Erzberger JP, Serebriiskii IG, Canutescu AA, Dunbrack RL, Pehrson JR, Berger JM, Kaufman PD, Adams PD. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell. 2005; 8:19–30. https://doi.org/10.1016/j.devcel.2004.10.019 [PubMed]
- 58. Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, Aggarwala V, Cruickshanks HA, Rai TS, McBryan T, Gregory BD, Adams PD, Berger SL. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013; 27:1787–99. https://doi.org/10.1101/gad.223834.113 [PubMed]
- 59. Ito T, Teo YV, Evans SA, Neretti N, Sedivy JM. Regulation of Cellular Senescence by Polycomb Chromatin Modifiers through Distinct DNA Damage- and Histone Methylation-Dependent Pathways. Cell Reports. 2018; 22:3480–92. https://doi.org/10.1016/j.celrep.2018.03.002 [PubMed]
- 60. Nelson DM, Jaber-Hijazi F, Cole JJ, Robertson NA, Pawlikowski JS, Norris KT, Criscione SW, Pchelintsev NA, Piscitello D, Stong N, Rai TS, McBryan T, Otte GL, et al. Mapping H4K20me3 onto the chromatin landscape of senescent cells indicates a function in control of cell senescence and tumor suppression through preservation of genetic and epigenetic stability. Genome Biol. 2016; 17:158. https://doi.org/10.1186/s13059-016-1017-x [PubMed]
- 61. Chicas A, Kapoor A, Wang X, Aksoy O, Evertts AG, Zhang MQ, Garcia BA, Bernstein E, Lowe SW. H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proc Natl Acad Sci USA. 2012; 109:8971–76. https://doi.org/10.1073/pnas.1119836109 [PubMed]
- 62. Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, Jenuwein T, Goldman RD. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci USA. 2006; 103:8703–08. https://doi.org/10.1073/pnas.0602569103 [PubMed]
- 63. Sarg B, Koutzamani E, Helliger W, Rundquist I, Lindner HH. Postsynthetic trimethylation of histone H4 at lysine 20 in mammalian tissues is associated with aging. J Biol Chem. 2002; 277:39195–201. https://doi.org/10.1074/jbc.M205166200 [PubMed]
- 64. Rai TS, Cole JJ, Nelson DM, Dikovskaya D, Faller WJ, Vizioli MG, Hewitt RN, Anannya O, McBryan T, Manoharan I, van Tuyn J, Morrice N, Pchelintsev NA, et al. HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev. 2014; 28:2712–25. https://doi.org/10.1101/gad.247528.114 [PubMed]
- 65. Nativio R, Donahue G, Berson A, Lan Y, Amlie-Wolf A, Tuzer F, Toledo JB, Gosai SJ, Gregory BD, Torres C, Trojanowski JQ, Wang LS, Johnson FB, et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat Neurosci. 2018; 21:497–505. https://doi.org/10.1038/s41593-018-0101-9 [PubMed]
- 66. Tasdemir N, Banito A, Roe JS, Alonso-Curbelo D, Camiolo M, Tschaharganeh DF, Huang CH, Aksoy O, Bolden JE, Chen CC, Fennell M, Thapar V, Chicas A, et al. BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer Discov. 2016; 6:612–29. https://doi.org/10.1158/2159-8290.CD-16-0217 [PubMed]
- 67. Benayoun BA, Pollina E, Singh PP, Mahmoudi S, Harel I, Casey K, Dulken B, Kundaje A, Brunet A. Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. bioRxiv. https://www.biorxiv.org/content/early/2018/05/31/3361722018. .
- 68. Kamakaka RT, Biggins S. Histone variants: deviants? Genes Dev. 2005; 19:295–310. https://doi.org/10.1101/gad.1272805 [PubMed]
- 69. Szenker E, Ray-Gallet D, Almouzni G. The double face of the histone variant H3.3. Cell Res. 2011; 21:421–34. https://doi.org/10.1038/cr.2011.14 [PubMed]
- 70. Duarte LF, Young AR, Wang Z, Wu HA, Panda T, Kou Y, Kapoor A, Hasson D, Mills NR, Ma’ayan A, Narita M, Bernstein E. Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nat Commun. 2014; 5:5210. https://doi.org/10.1038/ncomms6210 [PubMed]
- 71. Chadwick BP, Willard HF. Cell cycle-dependent localization of macroH2A in chromatin of the inactive X chromosome. J Cell Biol. 2002; 157:1113–23. https://doi.org/10.1083/jcb.200112074 [PubMed]
- 72. Sporn JC, Kustatscher G, Hothorn T, Collado M, Serrano M, Muley T, Schnabel P, Ladurner AG. Histone macroH2A isoforms predict the risk of lung cancer recurrence. Oncogene. 2009; 28:3423–28. https://doi.org/10.1038/onc.2009.26 [PubMed]
- 73. Kozlowski M, Ladurner AG. ATM, MacroH2A.1, and SASP: The Checks and Balances of Cellular Senescence. Mol Cell. 2015; 59:713–15. https://doi.org/10.1016/j.molcel.2015.08.010 [PubMed]
- 74. Chen H, Ruiz PD, McKimpson WM, Novikov L, Kitsis RN, Gamble MJ. MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype. Mol Cell. 2015; 59:719–31. https://doi.org/10.1016/j.molcel.2015.07.011 [PubMed]
- 75. Contrepois K, Coudereau C, Benayoun BA, Schuler N, Roux PF, Bischof O, Courbeyrette R, Carvalho C, Thuret JY, Ma Z, Derbois C, Nevers MC, Volland H, et al. Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression. Nat Commun. 2017; 8:14995. https://doi.org/10.1038/ncomms14995 [PubMed]
- 76. Li E, Zhang Y. DNA methylation in mammals. Cold Spring Harb Perspect Biol. 2014; 6:a019133. https://doi.org/10.1101/cshperspect.a019133 [PubMed]
- 77. Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013; 502:472–79. https://doi.org/10.1038/nature12750 [PubMed]
- 78. Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016; 30:733–50. https://doi.org/10.1101/gad.276568.115 [PubMed]
- 79. Mellén M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012; 151:1417–30. https://doi.org/10.1016/j.cell.2012.11.022 [PubMed]
- 80. Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A, Li X, Dai Q, Shen Y, Park B, Min JH, Jin P, Ren B, He C. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012; 149:1368–80. https://doi.org/10.1016/j.cell.2012.04.027 [PubMed]
- 81. Criscione SW, Teo YV, Neretti N. The Chromatin Landscape of Cellular Senescence. Trends Genet. 2016; 32:751–61. https://doi.org/10.1016/j.tig.2016.09.005 [PubMed]
- 82. Criscione SW, De Cecco M, Siranosian B, Zhang Y, Kreiling JA, Sedivy JM, Neretti N. Reorganization of chromosome architecture in replicative cellular senescence. Sci Adv. 2016; 2:e1500882. https://doi.org/10.1126/sciadv.1500882 [PubMed]
- 83. Swanson EC, Manning B, Zhang H, Lawrence JB. Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J Cell Biol. 2013; 203:929–42. https://doi.org/10.1083/jcb.201306073 [PubMed]
- 84. De Cecco M, Criscione SW, Peckham EJ, Hillenmeyer S, Hamm EA, Manivannan J, Peterson AL, Kreiling JA, Neretti N, Sedivy JM. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell. 2013; 12:247–56. https://doi.org/10.1111/acel.12047 [PubMed]
- 85. Sakaki M, Ebihara Y, Okamura K, Nakabayashi K, Igarashi A, Matsumoto K, Hata K, Kobayashi Y, Maehara K. Potential roles of DNA methylation in the initiation and establishment of replicative senescence revealed by array-based methylome and transcriptome analyses. PLoS One. 2017; 12:e0171431. https://doi.org/10.1371/journal.pone.0171431 [PubMed]
- 86. Cole JJ, Robertson NA, Rather MI, Thomson JP, McBryan T, Sproul D, Wang T, Brock C, Clark W, Ideker T, Meehan RR, Miller RA, Brown-Borg HM, Adams PD. Diverse interventions that extend mouse lifespan suppress shared age-associated epigenetic changes at critical gene regulatory regions. Genome Biol. 2017; 18:58. https://doi.org/10.1186/s13059-017-1185-3 [PubMed]
- 87. Wang T, Tsui B, Kreisberg JF, Robertson NA, Gross AM, Yu MK, Carter H, Brown-Borg HM, Adams PD, Ideker T. Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol. 2017; 18:57. https://doi.org/10.1186/s13059-017-1186-2 [PubMed]
- 88. Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018; 19:371–84. https://doi.org/10.1038/s41576-018-0004-3 [PubMed]
- 89. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013; 14:R115. https://doi.org/10.1186/gb-2013-14-10-r115 [PubMed]
- 90. Horvath S. Erratum to: DNA methylation age of human tissues and cell types. Genome Biol. 2015; 16:96. https://doi.org/10.1186/s13059-015-0649-6 [PubMed]
- 91. Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, Deconde R, Chen M, Rajapakse I, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013; 49:359–67. https://doi.org/10.1016/j.molcel.2012.10.016 [PubMed]
- 92. Horvath S, Oshima J, Martin GM, Lu AT, Quach A, Cohen H, Felton S, Matsuyama M, Lowe D, Kabacik S, Wilson JG, Reiner AP, Maierhofer A, et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging (Albany NY). 2018; 10:1758–75. https://doi.org/10.18632/aging.101508 [PubMed]
- 93. Lowe D, Horvath S, Raj K. Epigenetic clock analyses of cellular senescence and ageing. Oncotarget. 2016; 7:8524–31. https://doi.org/10.18632/oncotarget.7383 [PubMed]
- 94. Lu AT, Hannon E, Levine ME, Hao K, Crimmins EM, Lunnon K, Kozlenkov A, Mill J, Dracheva S, Horvath S. Genetic variants near MLST8 and DHX57 affect the epigenetic age of the cerebellum. Nat Commun. 2016; 7:10561. https://doi.org/10.1038/ncomms10561 [PubMed]
- 95. Capell BC, Collins FS. Human laminopathies: nuclei gone genetically awry. Nat Rev Genet. 2006; 7:940–52. https://doi.org/10.1038/nrg1906 [PubMed]
- 96. Gruenbaum Y, Margalit A, Goldman RD, Shumaker DK, Wilson KL. The nuclear lamina comes of age. Nat Rev Mol Cell Biol. 2005; 6:21–31. https://doi.org/10.1038/nrm1550 [PubMed]
- 97. Davies BS, Fong LG, Yang SH, Coffinier C, Young SG. The posttranslational processing of prelamin A and disease. Annu Rev Genomics Hum Genet. 2009; 10:153–74. https://doi.org/10.1146/annurev-genom-082908-150150 [PubMed]
- 98. Freund A, Laberge RM, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell. 2012; 23:2066–75. https://doi.org/10.1091/mbc.e11-10-0884 [PubMed]
- 99. Dou Z, Xu C, Donahue G, Shimi T, Pan JA, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP, Catanzaro JM, Ricketts MD, Lamark T, et al. Autophagy mediates degradation of nuclear lamina. Nature. 2015; 527:105–09. https://doi.org/10.1038/nature15548 [PubMed]
- 100. Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, Capell BC, Xu C, Xu M, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017; 550:402–06. https://doi.org/10.1038/nature24050 [PubMed]
- 101. Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, Kogan SC, Lowe SW. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011; 25:2125–36. https://doi.org/10.1101/gad.17276711 [PubMed]
- 102. van Steensel B, Belmont AS. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell. 2017; 169:780–91. https://doi.org/10.1016/j.cell.2017.04.022 [PubMed]
- 103. Shimi T, Butin-Israeli V, Adam SA, Hamanaka RB, Goldman AE, Lucas CA, Shumaker DK, Kosak ST, Chandel NS, Goldman RD. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 2011; 25:2579–93. https://doi.org/10.1101/gad.179515.111 [PubMed]
- 104. Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006; 312:1059–63. https://doi.org/10.1126/science.1127168 [PubMed]
- 105. McClintock D, Ratner D, Lokuge M, Owens DM, Gordon LB, Collins FS, Djabali K. The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS One. 2007; 2:e1269. https://doi.org/10.1371/journal.pone.0001269 [PubMed]
- 106. Taimen P, Pfleghaar K, Shimi T, Möller D, Ben-Harush K, Erdos MR, Adam SA, Herrmann H, Medalia O, Collins FS, Goldman AE, Goldman RD. A progeria mutation reveals functions for lamin A in nuclear assembly, architecture, and chromosome organization. Proc Natl Acad Sci USA. 2009; 106:20788–93. https://doi.org/10.1073/pnas.0911895106 [PubMed]
- 107. McCord RP, Nazario-Toole A, Zhang H, Chines PS, Zhan Y, Erdos MR, Collins FS, Dekker J, Cao K. Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 2013; 23:260–69. https://doi.org/10.1101/gr.138032.112 [PubMed]
- 108. Cao K, Blair CD, Faddah DA, Kieckhaefer JE, Olive M, Erdos MR, Nabel EG, Collins FS. Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. J Clin Invest. 2011; 121:2833–44. https://doi.org/10.1172/JCI43578 [PubMed]
- 109. Gordon LB, Kleinman ME, Miller DT, Neuberg DS, Giobbie-Hurder A, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland R, Snyder BD, Fligor B, Bishop WR, Statkevich P, et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci USA. 2012; 109:16666–71. https://doi.org/10.1073/pnas.1202529109 [PubMed]
- 110. Capell BC, Olive M, Erdos MR, Cao K, Faddah DA, Tavarez UL, Conneely KN, Qu X, San H, Ganesh SK, Chen X, Avallone H, Kolodgie FD, et al. A farnesyltransferase inhibitor prevents both the onset and late progression of cardiovascular disease in a progeria mouse model. Proc Natl Acad Sci USA. 2008; 105:15902–07. https://doi.org/10.1073/pnas.0807840105 [PubMed]
- 111. Spielmann M, Lupiáñez DG, Mundlos S. Structural variation in the 3D genome. Nat Rev Genet. 2018; 19:453–67. https://doi.org/10.1038/s41576-018-0007-0 [PubMed]
- 112. Dekker J, Belmont AS, Guttman M, Leshyk VO, Lis JT, Lomvardas S, Mirny LA, O’Shea CC, Park PJ, Ren B, Politz JC, Shendure J, Zhong S, and 4D Nucleome Network. Corrigendum: The 4D nucleome project. Nature. 2017; 552:278. https://doi.org/10.1038/nature24667 [PubMed]
- 113. Dekker J, Belmont AS, Guttman M, Leshyk VO, Lis JT, Lomvardas S, Mirny LA, O’Shea CC, Park PJ, Ren B, Politz JC, Shendure J, Zhong S, and 4D Nucleome Network. The 4D nucleome project. Nature. 2017; 549:219–26. https://doi.org/10.1038/nature23884 [PubMed]
- 114. Gómez-Díaz E, Corces VG. Architectural proteins: regulators of 3D genome organization in cell fate. Trends Cell Biol. 2014; 24:703–11. https://doi.org/10.1016/j.tcb.2014.08.003 [PubMed]
- 115. Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day DS, Abraham BJ, Cohen MA, Nabet B, Buckley DL, Guo YE, Hnisz D, Jaenisch R, et al. YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell. 2017; 171:1573–1588.e28. https://doi.org/10.1016/j.cell.2017.11.008 [PubMed]
- 116. Merkenschlager M, Nora EP. CTCF and Cohesin in Genome Folding and Transcriptional Gene Regulation. Annu Rev Genomics Hum Genet. 2016; 17:17–43. https://doi.org/10.1146/annurev-genom-083115-022339 [PubMed]
- 117. Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Mirny LA. Formation of Chromosomal Domains by Loop Extrusion. Cell Reports. 2016; 15:2038–49. https://doi.org/10.1016/j.celrep.2016.04.085 [PubMed]
- 118. Rosin LF, Nguyen SC, Joyce EF. Condensin II drives large-scale folding and spatial partitioning of interphase chromosomes in Drosophila nuclei. PLoS Genet. 2018; 14:e1007393. https://doi.org/10.1371/journal.pgen.1007393 [PubMed]
- 119. Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, Vera DL, Wang Y, Hansen RS, Canfield TK, Thurman RE, Cheng Y, Gülsoy G, et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014; 515:402–05. https://doi.org/10.1038/nature13986 [PubMed]
- 120. Dixon JR, Gorkin DU, Ren B. Chromatin Domains: The Unit of Chromosome Organization. Mol Cell. 2016; 62:668–80. https://doi.org/10.1016/j.molcel.2016.05.018 [PubMed]
- 121. Sun L, Yu R, Dang W. Chromatin Architectural Changes during Cellular Senescence and Aging. Genes (Basel). 2018; 9. https://doi.org/10.3390/genes9040211 [PubMed]
- 122. Zirkel A, Nikolic M, Sofiadis K, Mallm JP, Brackley CA, Gothe H, Drechsel O, Becker C, Altmüller J, Josipovic N, Georgomanolis T, Brant L, Franzen J, et al. HMGB2 Loss upon Senescence Entry Disrupts Genomic Organization and Induces CTCF Clustering across Cell Types. Mol Cell. 2018; 70:730–744.e6. https://doi.org/10.1016/j.molcel.2018.03.030 [PubMed]
- 123. Chandra T, Ewels PA, Schoenfelder S, Furlan-Magaril M, Wingett SW, Kirschner K, Thuret JY, Andrews S, Fraser P, Reik W. Global reorganization of the nuclear landscape in senescent cells. Cell Reports. 2015; 10:471–83. https://doi.org/10.1016/j.celrep.2014.12.055 [PubMed]
- 124. Risca VI, Greenleaf WJ. Unraveling the 3D genome: genomics tools for multiscale exploration. Trends Genet. 2015; 31:357–72. https://doi.org/10.1016/j.tig.2015.03.010 [PubMed]
- 125. Hayflick L. The cell biology of human aging. N Engl J Med. 1976; 295:1302–08. https://doi.org/10.1056/NEJM197612022952308 [PubMed]
- 126. Zhao Y, Tyshkovskiy A, Muñoz-Espín D, Tian X, Serrano M, de Magalhaes JP, Nevo E, Gladyshev VN, Seluanov A, Gorbunova V. Naked mole rats can undergo developmental, oncogene-induced and DNA damage-induced cellular senescence. Proc Natl Acad Sci USA. 2018; 115:1801–06. https://doi.org/10.1073/pnas.1721160115 [PubMed]
- 127. Itahana K, Campisi J, Dimri GP. Methods to detect biomarkers of cellular senescence: the senescence-associated beta-galactosidase assay. Methods Mol Biol. 2007; 371:21–31. https://doi.org/10.1007/978-1-59745-361-5_3 [PubMed]
- 128. Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, Robbins PD. The Clinical Potential of Senolytic Drugs. J Am Geriatr Soc. 2017; 65:2297–301. https://doi.org/10.1111/jgs.14969 [PubMed]
- 129. Moiseeva O, Deschênes-Simard X, St-Germain E, Igelmann S, Huot G, Cadar AE, Bourdeau V, Pollak MN, Ferbeyre G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell. 2013; 12:489–98. https://doi.org/10.1111/acel.12075 [PubMed]
- 130. García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, Sandri M, Muñoz-Cánoves P. Autophagy maintains stemness by preventing senescence. Nature. 2016; 529:37–42. https://doi.org/10.1038/nature16187 [PubMed]
- 131. Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell. 2011; 146:682–95. https://doi.org/10.1016/j.cell.2011.07.030 [PubMed]
- 132. Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012; 393:547–64. https://doi.org/10.1515/hsz-2012-0119 [PubMed]
- 133. Aw D, Silva AB, Palmer DB. Immunosenescence: emerging challenges for an ageing population. Immunology. 2007; 120:435–46. https://doi.org/10.1111/j.1365-2567.2007.02555.x [PubMed]
- 134. Burton DG, Stolzing A. Cellular senescence: immunosurveillance and future immunotherapy. Ageing Res Rev. 2018; 43:17–25. https://doi.org/10.1016/j.arr.2018.02.001 [PubMed]