



Introduction
Age-related macular degeneration (AMD) is an eye disorder affecting the elderly which can induce an irreversible loss of central visual function [1]. Its limited treatment possibilities and the rising number of senior citizens in developed countries are reasons for concern; indeed, it has been estimated that 200 million individuals will suffer from AMD in 2020 and almost 300 million will be affected in 2040 [2]. Aging plays a major role in AMD pathogenesis [3]; smoking, cataract surgery, a high body mass index (BMI), a history of cardiovascular disease, and a family history of AMD are additional risk factors [4,5].
The Age-Related Eye Disease Study (AREDS) has classified AMD into early, intermediate, and late stage [6], while the Clinical Age-Related Maculopathy Staging (CARMS) system divides patients into five mutually exclusive categories based on slit-lamp assessment of clinical features [7]. However, there is no consensus on the staging and progression terminology. In 2013, the Beckman Initiative for Macular Research Classification Committee proposed a new clinical classification based on the three AREDS stages: stage 1, normal, aging phenotype (small drusen < 63 μm without pigmentary changes); stage 2, early AMD with medium drusen (63 - 125 μm) and no pigmentary abnormalities; and stage 3, which is subdivided into intermediate – large drusen and/or pigmentary changes – and advanced – choroidal neovascularization (CNV, exudative or neovascular AMD) or geographic atrophy (GA; dry or non-exudative AMD) [8]. Anti-vascular endothelial growth factor (anti-VEGF) has long been the mainstay of treatment for neovascular AMD, whereas no effective treatment is available for the more common dry form [9–12].
AMD is the result of complex interactions among metabolic, functional, genetic, and environmental factors [13]. It is characterized by degeneration of the retinal pigment epithelium (RPE), death of photoreceptors and degradation of choriocapillaries, which together lead to impairment and loss of central vision. The RPE is a polarized non-proliferative cell monolayer lying between the neural retina and the vascularized choroid. It serves several functions that are essential for vision and for the survival of retinal neurons. Its dysfunction, due to oxidative stress, mitochondrial destabilization, and complement dysregulation, has been implicated in AMD pathogenesis [14,15]. During aging, RPE cells undergo a number of functional alterations that result in the development of age-related eye disorders, including AMD. RPE cell damage induced by proinflammatory factors – including tumor necrosis factor (TNF)-α – and cell death are able to promote activation of the alternative pathway (AP) of the complement system at the retina-choroid interface, a process that has been associated to RPE cell death; aging also induces a reduction in the number and an increase in the size and multinucleation of RPE cells [16]. The activation of different protein kinase C isoforms has also been implicated in the age-related formation of multinucleated RPE cells [17].
Neovascular AMD and GA are characterized by RPE dysfunction [18]; in GA, formation of large confluent drusen and hyperpigmentation (presumably related to the RPE dysfunction) seem to be the initial insult, while drusen resorption and RPE loss (hypopigmentation) are believed to predict its progression. Photoreceptor and choriocapillary dysfunction and death appear to be secondary to RPE loss; loss of choriocapillaries with an intact RPE has also been described in wet AMD [18]. According to a recent study, RPE cells from AMD patients show a different phenotype as well as functional changes such as altered autophagy, mitochondrial dysfunction, and susceptibility to oxidative stress compared to those from normal individuals [19].
The early and intermediate stages of AMD are characterized by changes in lipid metabolism, autophagy, and inflammation; molecular signaling pathways, such as inflammation, cellular senescence and cell death, play a key role in the progression of late-stage dry AMD, whereas angiogenesis predominates in the neovascular form [3]. A greater understanding of the molecular pathways that are involved in the various stages of AMD would contribute to the development of innovative therapies.
An imbalance of circulating inflammatory molecules seems to characterize most age-related diseases (ARDs). Aging is characterized by a state of chronic, low-grade inflammation, known as inflammaging [20], which also appears to be involved in all stages of AMD development and progression.
Senescent cells are non-proliferating cells capable of secreting proinflammatory cytokines, thus contributing to ARD development and ARD-related morbidity. Cellular senescence is characterized by cell growth arrest, altered DNA synthesis and repair, resistance to apoptosis, and increased cell size [21]. Telomere shortening, DNA damage, and oxidative stress are capable of activating senescence processes [22].
Mesenchymal stem cells (MSCs) have been isolated from different adult tissues, including the RPE [23]. Ease of isolation, high proliferation potential, and low immunogenicity make them ideal for cell-based therapies. MSC function declines with age; senescent MSCs acquire a senescence-associated secretory phenotype (SASP) that contributes to driving aging and the development and progression of ARDs, including AMD [24].
The human RPE contains a subpopulation of stem-like MSCs (RPESCs) [23]. In a previous work, we isolated and characterized human RPESCs, demonstrated their ability to differentiate into mesenchymal (adipogenic, osteogenic and chondrogenic) lineages, and analyzed their differentiation potential into neuronal and retinal lineages [25]. In a recent study of an AMD rat model, transplantation of RPESCs isolated from human RPE was able to prevent visual loss [26].
The mechanisms involved in the activation of differentiation of resident RPESCs into mature RPE cells and the role of RPESCs in AMD pathogenesis are still unclear. A growing number of studies have been addressing the role of persistent inflammation in AMD development and progression. The aim of this work is explore the molecular mechanisms that are involved in the acquisition of the senescent phenotype by RPESCs and the role of its proinflammatory factors in altering the function of aged RPE cells. To do so, we investigated the characteristics of replicative and stress-induced senescence of RPESCs, their morphological and genetic features, and their acquisition of the SASP, all of which seem to play a role in AMD pathogenesis.
Results
Replicative senescence of human RPESCs
RPESCs were isolated, characterized, and cultured as described in a previous work by our group [25]. They were isolated from a healthy eye from a 21-year-old donor. All experiments were conducted using 3 different batches of exponentially growing cells at the 3rd passage (confluence about 75%) in which replicative senescence was induced.
Replicative senescence was documented at the 16th passage (P16) by arrested proliferation, increased β-galactosidase (SA β-gal) activity, and telomere length reduction. In particular, RPESC proliferation rate increased steeply from P2 to P11 and plateaued at P13; growth stopped at P18 (Figure 1A). As illustrated in Figure 1B, the proportion of β-gal-positive cells increased significantly (P < 0.05) from 6 ± 0.04% in young cells (P3-P6) to 80 ± 12.1% in senescent cells (P15-P18). Telomere length declined from 14 kb at P3 to 4 kb at P16 (Figure 1C). TRIC-phalloidin staining demonstrated that senescent RPESCs had a flatter and enlarged morphology compared to young cells (Figure 1D).
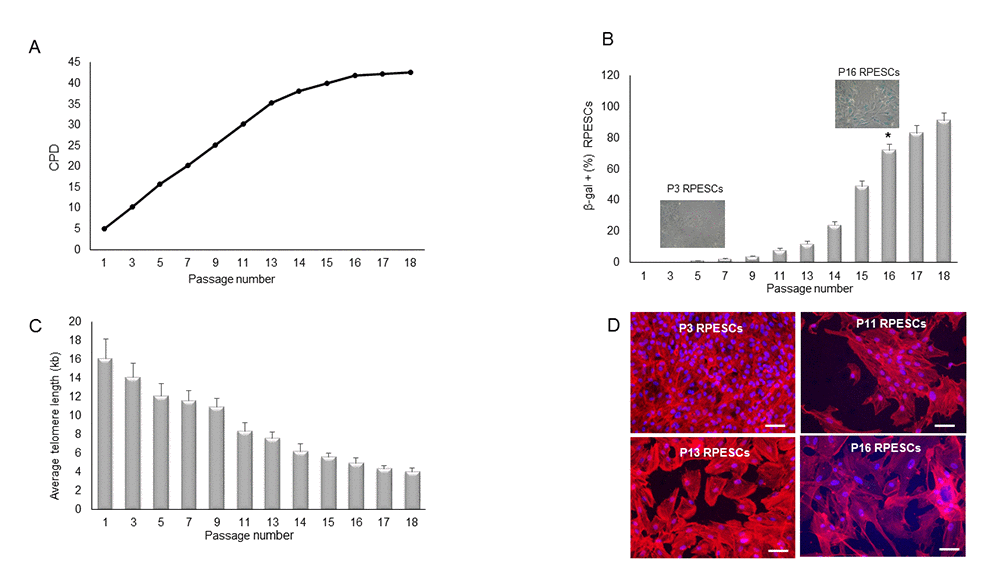
Figure 1. Proliferation rate, β-gal positivity, telomere length, and cell morphology during RPESC replicative senescence. RPESC replicative senescence. (A) Cumulative number of population doublings (CPD) in RPESCs grown to senescence. (B) Percentage of β-gal-positive cells detected during RPESC replicative senescence from P1 to P16. P11, number of culture passages. Data are reported as mean ± SD. *P =0.039. (C) RPESC telomere length during replicative senescence was analyzed from P1 to P18; data are reported as mean ± SD of 3 independent experiments. (D) Morphological analysis of young (P3) and senescent (P16) RPESCs by the TRIC-phalloidin immunofluorescence assay. Senescent RPESCs appear enlarged and flattened. Magnification 20X, scale bar 200 µm. Pictures are representative of 3 independent experiments.
Acquisition of the secretory phenotype by senescent RPESCs
SASP acquisition by senescent REPSCs was determined by analyzing a panel of proinflammatory molecules: interleukin (IL)-6, interferon (INF)-γ, TNF-α, IL-12, and transforming growth factor β (TGFβ)1 - in young (P3) and senescent (P16) RPESCs. The analysis was extended by determination of the levels of the anti-inflammatory molecules IL-4, IL-10, and IL-13.
The results of ELISA indicated that senescent RPESCs secreted higher levels of IL-6, IL-12, IL-17, TNF-α, TGFβ1, and INF-γ compared to young RPESCs (Figure 2A-F), reflecting a proinflammatory state and SASP acquisition. They also secreted lower levels of several anti-inflammatory molecules, including IL-4, IL-10, and IL-13, compared to young RPESCs (Figure 2G-I).
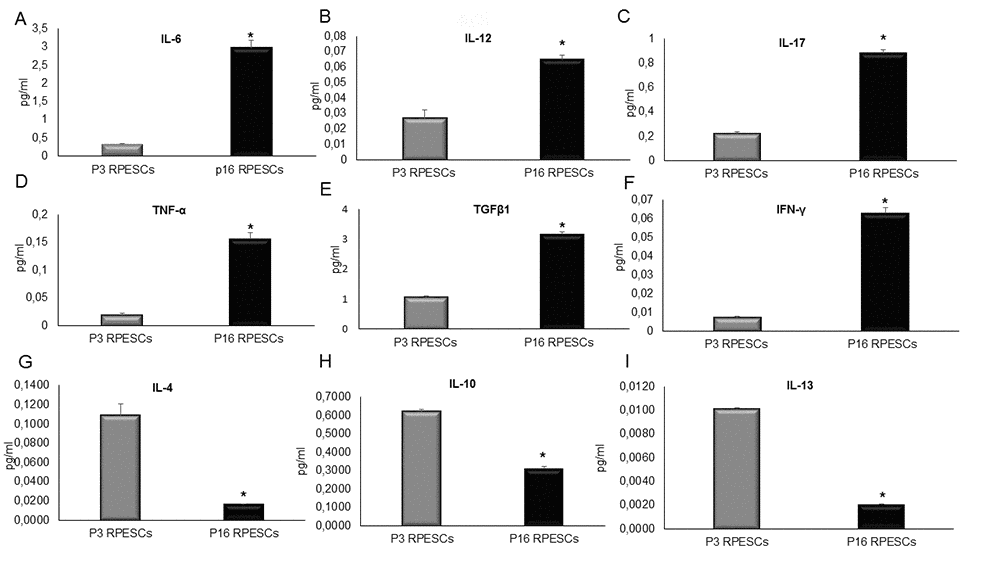
Figure 2. SASP induction in senescent RPESCs. Young (P3) and senescent (P16) RPESCs were maintained in culture for 48 h. The supernatant was analyzed for IL-6 (A), IL-12 (B), IL-17 (C), TNF-α (D), TGFβ1 (E), INF-γ (F), IL-4 (G), IL-10 (H), and IL-13 (I), by ELISA. Data are mean ± SD of 3 independent experiments. *P = from 0.021 to 0.041.
Expression of stemness, reprogramming, and RPE-specific genes in young and senescent RPESCs
The expression of RPE-specific (OTX2, PEDF, PAX6, RPE65, MITF, and CRALBP), stemness, and reprogramming (SOX2, KLF4 and c-MYC) genes was investigated in young and senescent RPECS by qRT-PCR.
As shown in Figure 3A, the two sets of cells showed similar expression levels of SOX2, KLF4 and c-MYC, whereas senescent RPESCs exhibited downregulation of OTX2, PEDF, PAX6, RPE65, MITF, and CRALBP (Figure 3B).
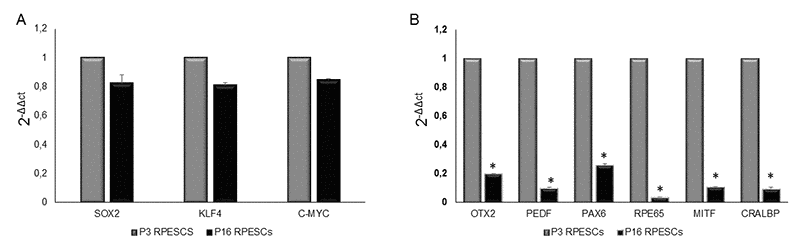
Figure 3. mRNA expression levels of stemness and RPE-specific genes in senescent and young RPESCs. (A) qRT-PCR analysis of the expression levels of stemness genes in senescent (P16) and young (P3) RPESCs. (B) qRT-PCR analysis of the mRNA levels of RPE-specific genes in senescent and young RPESCs. Data are mean ± SD of 3 independent experiments. *P = from 0.026 to 0.036.
Determination of the mRNA levels of senescence-associated genes in senescent and young RPESCs by PCR Array
The expression of senescence-associated genes in senescent and young RPESCs was measured by analyzing their mRNA levels using Cellular Senescence RT2 Profiler PCR Array (PAHS, Qiagen). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and ribosomal protein, large, P0 (RPLP0) were used as inner controls to normalize gene expression levels.
Fifteen genes related to human cellular senescence were differentially expressed and showed a 4-fold change in senescent compared to young RPESCs: 8 genes, ALDH1A3 (aldehyde dehydrogenase 1 family member A3), CDKN1A (cyclin-dependent kinase inhibitor 1A, p21), CDKN2A (cyclin-dependent kinase inhibitor 2A, P16INK4), IGFBP3 (insulin-like growth factor binding protein 3), IRF5 (interferon regulatory factor 5), SERPINB2 (serpin family B member 2, PAI2), SERPINE1 (serpin family E member 1, PAI1), and THBS1 (thrombospondin 1), were upregulated (Figure 4A), whereas 7 genes, CCNA2 (cyclin A2), CCNB1 (cyclin B1), CDC25C (cell division cycle 25C), CDK2 (cyclin-dependent kinase 2), EGR1 (early growth response 1), TXB2 (T-box 2), and PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha), were downregulated (Figure 4B).
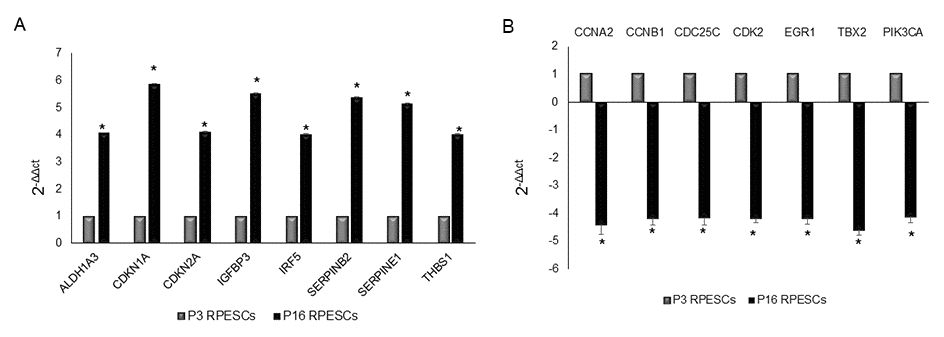
Figure 4. Senescence-associated gene expression profile in senescent and young RPESCs. PCR array performed to analyze the mRNA expression levels of senescence-associated genes in 3 senescent (P16) and 3 young (P3) RPESCs. Data are reported as fold change. A 4-fold difference was considered significant and only mRNAs with a ΔΔCt greater than 4 (A) or lower than -4 (B) are reported. *P = from 0.018 to 0.046.
p21 and p53 protein expression in young and senescent RPESCs
Western blot analysis demonstrated that p21 and p53 protein were significantly upregulated in senescent compared to young RPESCs (Figure 5A) and that intermediate-passage cells (P11) were downregulated compared to both young and senescent RPESCs. The results of densitometric analysis and normalization to GAPDH expression (Figure 5B) were confirmed by real time PCR, which demonstrated that p21 and p53 mRNA was upregulated in senescent (P16) compared to young RPESCs (P3) (Figure 5C and D). In contrast, P11 RPESCs showed higher p21 and p53 mRNA and lower protein levels compared to young RPESCs (P3).
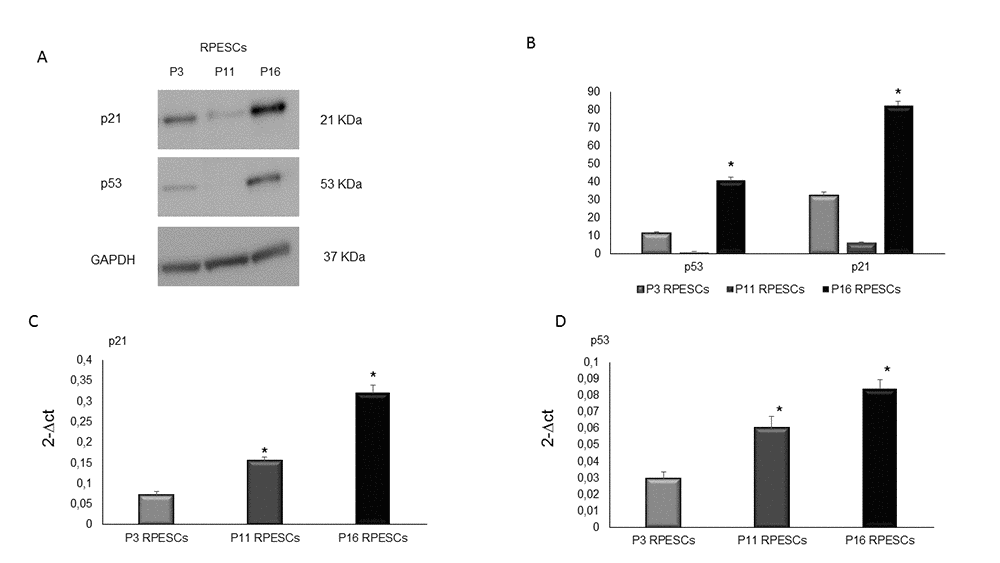
Figure 5. Human p53 and p21 protein expression levels in senescent and young RPESCs. p53 and p21 protein expression levels (A) Western blot analysis (B) and densitometric analysis of blots. Data are mean ± SD of 3 independent experiments. *P = from 0.022 to 0.046. (C) Relative expression levels of mRNA related to p21 and (D) p53 genes in young (P3), pre-senescent (P11) and senescent (P16) RPESCs cells. Data are mean ± SD of 3 independent experiments. *P = from 0.031 to 0.044.
Discussion
Age-related inflammation is a major risk factor in the aging process. AMD is a highly common, irreversible cause of severe loss of vision among the elderly in developed countries [2]. It has a multifactorial etiology, where advanced age and genetic predisposition are the strongest risk factors [27,28]. Although the mechanisms involved in its pathogenesis are unclear, inflammatory pathways have been reported to play a key role in its development and progression, as in the case of other ARDs [29]. Recent data have lent support to the hypothesis that senescent cells accumulate in the aging primate RPE [30]. Senescent cells accumulate with aging in human tissues and organs, promoting ARD development and progression; cell exposure to recurrent or chronic stress, including oxidative stress, may also result in their accumulation [31,32]. Our group has recently characterized a stem cell-like population derived from the human RPE [25] as RPESCs (RPE progenitor cells). Notably, MSCs enter replicative senescence after a limited number of cell divisions, a fact that needs to be considered in experiments involving cell cultures, especially in investigations of regenerative medicine approaches. The effects of aging, senescence, and oxidative stress can induce loss of differentiation and proliferative potential in adult MSCs, including RPESCs [33]. Senescent cells acquire the SASP and release various proteins, including proinflammatory molecules, thus contributing to ARDs and the associated morbidity [34]. Critically, although human RPESCs have been identified as a stem-cell like population, they are unable to differentiate into mature RPE cells, replacing those that are lost due to AMD. In our previous study we isolated, cultured, and characterized RPESCs from the human RPE and investigated their differentiation potential [25]. In the present work, induction of replicative senescence in RPESCs resulted in reduced proliferative and multilineage differentiation potential, SASP acquisition, and release of inflammatory proteins that may be involved in AMD development and progression. Senescent RPESCs exhibited telomere length shortening and a characteristic large and flattened morphology. They also stained for SA β-gal and showed upregulation of p21 and p53 protein compared to young RPESCs. p21 and p53 are key components of the senescence machinery, playing a critical role as regulators of stem cell functions [35]. Senescent RPESCs expressed higher mRNA levels of senescence-associated genes compared to their young counterparts. These genes, which include CDKN1A (p21), IGFBP3, SERPINE1, and SERPINB2, have been implicated in the maintenance of replicative and stress-induced cellular senescence mechanisms [36,37]. In addition, SERPINE1 (PAI1) is responsible for decreased extracellular matrix (ECM) degradation through inhibition of metalloprotease activation, mechanisms that may be involved in ECM accumulation in the RPE of AMD patients [38].
The irreversible growth arrest of senescent RPESCs was confirmed by the downregulation of the genes involved in cell cycle progression, including CCNA2, CCNB1, and CDK2 [39,40]. The transcription factors SOX2, KLF4, and c-MYC have been reported to play a regulatory role in stem cell self-renewal, i.e. reprogramming. Interestingly, these stem-cell specific genes showed a similar expression level in senescent and young RPESCs, whereas the RPE-specific genes (RPE65, MITF, OTX2, PAX6, CRALBP, and PEDF) were downregulated in senescent RPESCs. These data suggest that despite their loss of differentiation potential during senescence, the reprogramming ability of these cells is preserved.
Senescent cells are metabolically active and release high concentrations of proinflammatory cytokines, chemokines, growth factors, and proteases into the culture medium [24,41]. Senescent RPESCs (P16) secreted higher IL-6, IL-12, IL-17, INF-γ, TNF-α, and TGFβ1 concentrations compared to young RPESCs. IFN-γ, TNF-α, and IL-17 are involved in Th1 and Th17 inflammation response pathways [42]. A recent study of the CD4+ T cell compartment in AMD patients has found that these cells play a proinflammatory role in an IFN-γ- and IL-17-dependent fashion [43]. Such proinflammatory cytokines are also likely to play a key role in AMD pathogenesis, and their effect may well be reinforced by senescent RPESCs.
Interestingly, proinflammatory cytokines may induce activation of the anti-oxidative stress response in mature RPE cells, as reported in a study where protective anti-oxidant pathways were activated in mature RPE cells treated with oxidative agents and cultured with peripheral blood mononuclear cell-conditioned medium or with IFN-γ/TNF-α [44]. Moreover, T-cell–derived proinflammatory cytokines were able to induce in mature RPE cells the production and secretion of multiple chemokines, which can affect the immune homeostasis in the retina [45]. Indeed, there is growing evidence for a role of the adaptive immune system in the pathogenesis of neovascular AMD. Several studies have addressed the crucial role of macrophages in the development of choroidal neovascularization [46–48] and of atrophic changes in the AMD retina [49], while an association has been described between AMD and systemic leukocyte activity [50].
Singh and co-workers have demonstrated that the age-related decrement in Th1 frequency seen in healthy controls is absent in AMD patients, since the percentage of CD4+ T-cells expressing CCR6 was significantly reduced in patients with non-exudative as well as exudative AMD [51]. There is also evidence that CCR2 expression in circulating monocytes may play a role in the development of neovascular AMD [52,46]. Furthermore, significantly accelerated T-cell differentiation and aging have been described in the CD8+ T-cell compartment of patients with neovascular AMD [53].
As regards the pro/anti-inflammatory phenotype of senescent RPESCs, the present study found a significant downregulation of the anti-inflammatory cytokines IL-4, IL-10, and IL-13 in these cells. An increased or similar expression of these cytokines has been reported in the serum and aqueous humor of AMD patients compared with controls [54,55], probably due to the acquisition by senescent RPESCs of a specific proinflammatory senescence-associated phenotype and to the downregulation of anti-inflammatory cytokines in vitro.
This study is preliminary and as such suffers from some limitations. Replicative senescence was induced in vitro in RPESCs isolated from the eye of a single healthy young donor. The examination of samples from aged AMD patients and healthy subjects is expected to provide insight into the senescent proinflammatory status of RPESCs in the elderly and into their role in disease pathogenesis. It would also be interesting to investigate the effects of oxidative agents on young and senescent RPESCs from the mature RPE in terms of induction of apoptosis or activation of anti-inflammatory anti-oxidant pathways.
Altogether, the present findings indicate that RPESCs can undergo replicative senescence, which affects their proliferation and differentiation ability. In addition, senescent RPESCs acquired the SASP, which probably compounds the inflammatory RPE microenvironment during AMD development and progression. A greater understanding of the role of RPESCs in AMD pathogenesis is needed to find means to control the disease and modulate its progression.
Materials and Methods
RPESC culture
RPESCs were isolated, characterized, and cultured as described previously [25]. Cells were maintained in RPE medium [MEM-α modified medium containing 2 mM L-glutamine, penicillin/streptomycin (1:100), 1% Na-pyruvate, 10% fetal bovine serum], supplemented with taurine, hydrocortisone, triiodothyronine (THT), and N1 (all from Sigma-Aldrich, Milano, Italy). RPESCs were incubated at 37 °C in a 5% humidified CO2 incubator. The medium was replaced every 3 days.
Replicative senescence was induced by culturing cells up to the 18th passage (P18). Viable cells were counted at each passage by trypan blue staining using an automated cell counter (Thermo Fisher, Milano, Italy). Population doublings (PDs) were determined as current PDs = last PDs+log2 (collected cell number / seeded cell number); cumulative PD (CPD) was calculated as the sum of PD changes, as described previously [56].
Telomere length measurement
Telomere length was measured using the Relative Human Telomere Length quantification qPCR assay kit (ScienCell Research Laboratories, San Diego, CA, USA) according to the manufacturer’s protocol and a quantitative RT-PCR technique (Cawthon’s method [57]), as described previously [56].
Senescence-associated β-galactosidase (SA β-gal) staining
β-gal staining was performed using the Senescence β-Galactosidase Staining kit (Cell Signaling Technology, Leiden, The Netherlands) according to the manufacturer’s protocol.
Phalloidin staining
For phalloidin staining, young (P3), intermediate passage (P11) and senescent (P16) cells were seeded on chamber slides and cultured for 3 days in RPE medium. They were then fixed in 4% PFA for 30 min, blocked with bovine serum albumin (BSA), 2.5% in PBS for 30 min, and permeabilized with Triton X-100, 0.2% in BSA/PBS for 10 min at room temperature. Subsequently, cells were stained with TRITC-labeled phalloidin (Sigma Aldrich) according to the manufacturer’s protocol. Nuclear staining was obtained by applying Hoechst solution (Molecular Probes, Thermo Fisher, Milano, Italy) for 10 min. Slides were mounted with Vectashield (Vector Laboratories, Burlingame, CA, USA).
Analysis of the secretory phenotype
The concentration of IL-4, IL-6, IL-10, IL-12, IL-13, INF-γ, TNF-α, and TGFβ1 was measured in the supernatant by an ELISA method (Multi-Analyte ELISArray kit, Qiagen; Affymetrix Ebiosciences, Vienna, Austria). Briefly, the supernatants were collected at the end of each passage before trypsinization, centrifuged, and stored at -20 °C. Optical density at 450 nm was determined using a microtiter plate reader (Multiskan Go, Thermo Scientific, Monza, Italy). Cytokine concentration was determined as pg/ml by comparing their absorbance to the antigen standards. Each experiment was performed three times.
RNA isolation
Total RNA from P3 and P16 RPESCs was isolated using PerfectPure RNA Cell and Tissue Kit (5 PRIME, Hamburg, Germany). The concentration and purity of total RNA samples were determined using a NanoDrop One Spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE, USA). RNA was stored at -80 °C until use. About 300 ng of total RNA extracted from both RPESC sets was reverse transcribed using the GoScript™ Reverse Transcription System (Promega, Milano Italy) according to the manufacturer’s protocol.
RT-PCR analysis
Real-time PCR was performed with a Master Cycle (Eppendorf, Hamburg, Germany) apparatus using EVA Green PCR Master Mix (Bio-Rad, Milano, Italy) according to the manufacturer's instructions. Conditions were as follows: denaturation 98 °C for 2 min and 40 cycles of 98 °C for 60 s and 60 °C for 60 s. A melting stage was added at the end of amplification. There was no non-specific amplification as determined by the melting curve. All samples were tested in triplicate with the reference genes β-actin and RPL30 for data normalization. Genes and related primer sequences (SOX2, KLF4, c-MYC, RPE65, CRALBP, PEDF, OTX2, MITF, p21 and p53) were as described previously [58]. The mRNA expression level of all tested genes was analyzed in young and senescent RPESCs with the 2-ΔΔCt method: Δ (ΔCt) = ΔCt (senescent) − ΔCt (young). The relative expression values of the genes of interest are reported as mean ± standard deviation (SD) of three independent experiments.
mRNA profiling
Total RNA from young (P3) and senescent (P16) RPESCs was isolated with the PerfectPure RNA Cell and Tissue Kit. cDNA synthesis was performed with RT2 First Strand Kit (Qiagen, Milano, Italy) and Cellular Senescence RT Profiler PCR Array (PAHS-050ZA; Qiagen). The average of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and ribosomal protein, large, P0 (RPLP0) was used for normalization. Only mRNAs with reads < 35 Ct in all three biological replicates were included in the analysis. After amplification, melting curves were acquired. mRNA expression was quantified with the 2-ΔΔCt method. Relative gene expression values are reported as mean ± SD of three independent experiments.
Protein extraction and immunoblotting
Total protein was extracted from young (P3) and senescent (P16) RPESCs (1 x 106 cells/sample) as described previously [16], with some modifications. Membranes were incubated overnight with the primary p21 and p53 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) using GAPDH as the endogenous control, followed by incubation with the secondary antibody conjugated to horseradish peroxidase (Santa Cruz Biotechnology). Protein detection on the membrane was performed using the Clarity Western ECL Substrate Kit (Bio-Rad). The signals were captured with an Alliance Mini (UVITEC Cambridge, Cambridge, UK) system; the p21 and p53 bands were quantified with UVITEC software and their intensity was normalized by comparison to the housekeeping protein β-actin, used as a loading control. The intensity of each band was compared to the negative controls and any change was expressed as a percentage.
Statistical analysis
Results are expressed as mean ± SD. Comparisons between groups were analyzed by paired-sample t test comparisons using SPSS 20.0 software. Significance was analyzed in data from at least three independent experiments. P values ≤ 0.05 were considered significant.
Author Contributions
Raffaella Lazzarini, principal author, performed analysis on all samples, interpreted data and wrote manuscript. Michele Nicolai, principal co-author, helped in data interpretation and manuscript evaluation. Vittorio Pirani helped to evaluate and edit the manuscript. Cesare Mariotti, wrote manuscript and acted as corresponding author. Roberto Di Primio, supervised the findings of this work. All authors discussed the results and contributed to the final manuscript.
Acknowledgments
The authors are grateful to Word Designs (www.silviamodena.com) for the language revision.
Conflicts of Interest
The authors have declared that no competing interests exist.
References
- 1. Hyttinen JM, Błasiak J, Niittykoski M, Kinnunen K, Kauppinen A, Salminen A, Kaarniranta K. DNA damage response and autophagy in the degeneration of retinal pigment epithelial cells-Implications for age-related macular degeneration (AMD). Ageing Res Rev. 2017; 36:64–77. https://doi.org/10.1016/j.arr.2017.03.006 [PubMed]
- 2. Reibaldi M, Longo A, Pulvirenti A, Avitabile T, Russo A, Cillino S, Mariotti C, Casuccio A. Geo-Epidemiology of Age-Related Macular Degeneration: New Clues Into the Pathogenesis. Am J Ophthalmol. 2016; 161:78–93.e1, 2. https://doi.org/10.1016/j.ajo.2015.09.031 [PubMed]
- 3. Blasiak J, Piechota M, Pawlowska E, Szatkowska M, Sikora E, Kaarniranta K. Cellular Senescence in Age-Related Macular Degeneration: Can Autophagy and DNA Damage Response Play a Role? Oxid Med Cell Longev. 2017; 2017:5293258. https://doi.org/10.1155/2017/5293258 [PubMed]
- 4. Chakravarthy U, Wong TY, Fletcher A, Piault E, Evans C, Zlateva G, Buggage R, Pleil A, Mitchell P. Clinical risk factors for age-related macular degeneration: a systematic review and meta-analysis. BMC Ophthalmol. 2010; 10:31. https://doi.org/10.1186/1471-2415-10-31 [PubMed]
- 5. Lim LS, Mitchell P, Seddon JM, Holz FG, Wong TY. Age-related macular degeneration. Lancet. 2012; 379:1728–38. https://doi.org/10.1016/S0140-6736(12)60282-7 [PubMed]
- 6. Age-Related Eye Disease Study Research Group. Risk factors associated with age-related macular degeneration. A case-control study in the age-related eye disease study: Age-Related Eye Disease Study Report Number 3. Ophthalmology. 2000; 107:2224–32. [PubMed]
- 7. Seddon JM, Sharma S, Adelman RA. Evaluation of the clinical age-related maculopathy staging system. Ophthalmology. 2006; 113:260–66. https://doi.org/10.1016/j.ophtha.2005.11.001 [PubMed]
- 8. Miller JW, Bagheri S, Vavvas DG. Advances in Age-related Macular Degeneration Understanding and Therapy. US Ophthalmic Rev. 2017; 10:119–30. https://doi.org/10.17925/USOR.2017.10.02.119 [PubMed]
- 9. Abd AJ, Kanwar RK, Kanwar JR. Aged macular degeneration: current therapeutics for management and promising new drug candidates. Drug Discov Today. 2017; 22:1671–79. https://doi.org/10.1016/j.drudis.2017.07.010 [PubMed]
- 10. Gillies MC, Campain A, Barthelmes D, Simpson JM, Arnold JJ, Guymer RH, McAllister IL, Essex RW, Morlet N, Hunyor AP, and Fight Retinal Blindness Study Group. Long-Term Outcomes of Treatment of Neovascular Age-Related Macular Degeneration: Data from an Observational Study. Ophthalmology. 2015; 122:1837–45. https://doi.org/10.1016/j.ophtha.2015.05.010 [PubMed]
- 11. Yaspan BL, Williams DF, Holz FG, Regillo CD, Li Z, Dressen A, van Lookeren Campagne M, Le KN, Graham RR, Beres T, Bhangale TR, Honigberg LA, Smith A, et al, and MAHALO Study Investigators. Targeting factor D of the alternative complement pathway reduces geographic atrophy progression secondary to age-related macular degeneration. Sci Transl Med. 2017; 9:eaaf1443. https://doi.org/10.1126/scitranslmed.aaf1443 [PubMed]
- 12. Holz FG, Sadda SR, Busbee B, Chew EY, Mitchell P, Tufail A, Brittain C, Ferrara D, Gray S, Honigberg L, Martin J, Tong B, Ehrlich JS, Bressler NM, and Chroma and Spectri Study Investigators. Efficacy and Safety of Lampalizumab for Geographic Atrophy Due to Age-Related Macular Degeneration: Chroma and Spectri Phase 3 Randomized Clinical Trials. JAMA Ophthalmol. 2018; 136:666–77. https://doi.org/10.1001/jamaophthalmol.2018.1544 [PubMed]
- 13. Nowak JZ. Age-related macular degeneration (AMD): pathogenesis and therapy. Pharmacol Rep. 2006; 58:353–63. [PubMed]
- 14. Mitter SK, Song C, Qi X, Mao H, Rao H, Akin D, Lewin A, Grant M, Dunn W
Jr , Ding J, Bowes Rickman C, Boulton M. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy. 2014; 10:1989–2005. https://doi.org/10.4161/auto.36184 [PubMed] - 15. Luo C, Zhao J, Chen M, Xu H. The expression of C1 inhibitor (C1INH) in macrophages is upregulated by retinal pigment epithelial cells - implication in subretinal immune privilege in the aging eye. Aging (Albany NY). 2018; 10:1380–89. https://doi.org/10.18632/aging.101474 [PubMed]
- 16. Chen M, Rajapakse D, Fraczek M, Luo C, Forrester JV, Xu H. Retinal pigment epithelial cell multinucleation in the aging eye - a mechanism to repair damage and maintain homoeostasis. Aging Cell. 2016; 15:436–45. https://doi.org/10.1111/acel.12447 [PubMed]
- 17. Rajapakse D, Chen M, Curtis TM, Xu H. PKCζ-dependent upregulation of p27kip1 contributes to oxidative stress induced retinal pigment epithelial cell multinucleation. Aging (Albany NY). 2017; 9:2052–68. https://doi.org/10.18632/aging.101299 [PubMed]
- 18. Bhutto I, Lutty G. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol Aspects Med. 2012; 33:295–317. https://doi.org/10.1016/j.mam.2012.04.005 [PubMed]
- 19. Golestaneh N, Chu Y, Xiao YY, Stoleru GL, Theos AC. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2017; 8:e2537. https://doi.org/10.1038/cddis.2016.453 [PubMed]
- 20. Cevenini E, Monti D, Franceschi C. Inflamm-ageing. Curr Opin Clin Nutr Metab Care. 2013; 16:14–20. https://doi.org/10.1097/MCO.0b013e32835ada13 [PubMed]
- 21. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011; 192:547–56. https://doi.org/10.1083/jcb.201009094 [PubMed]
- 22. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014; 15:482–96. https://doi.org/10.1038/nrm3823 [PubMed]
- 23. Salero E, Blenkinsop TA, Corneo B, Harris A, Rabin D, Stern JH, Temple S. Adult human RPE can be activated into a multipotent stem cell that produces mesenchymal derivatives. Cell Stem Cell. 2012; 10:88–95. https://doi.org/10.1016/j.stem.2011.11.018 [PubMed]
- 24. Lunyak VV, Amaro-Ortiz A, Gaur M. Mesenchymal Stem Cells Secretory Responses: Senescence Messaging Secretome and Immunomodulation Perspective. Front Genet. 2017; 8:220. https://doi.org/10.3389/fgene.2017.00220 [PubMed]
- 25. Mariotti C, Lazzarini R, Nicolai M, Saitta A, Orsini E, Orciani M, Di Primio R. Comparative study between amniotic-fluid mesenchymal stem cells and retinal pigmented epithelium (RPE) stem cells ability to differentiate towards RPE cells. Cell Tissue Res. 2015; 362:21–31. https://doi.org/10.1007/s00441-015-2185-9 [PubMed]
- 26. Davis RJ, Alam NM, Zhao C, Müller C, Saini JS, Blenkinsop TA, Mazzoni F, Campbell M, Borden SM, Charniga CJ, Lederman PL, Aguilar V, Naimark M, et al. The Developmental Stage of Adult Human Stem Cell-Derived Retinal Pigment Epithelium Cells Influences Transplant Efficacy for Vision Rescue. Stem Cell Reports. 2017; 9:42–49. https://doi.org/10.1016/j.stemcr.2017.05.016 [PubMed]
- 27. DeAngelis MM, Owen LA, Morrison MA, Morgan DJ, Li M, Shakoor A, Vitale A, Iyengar S, Stambolian D, Kim IK, Farrer LA. Genetics of age-related macular degeneration (AMD). Hum Mol Genet. 2017; 26:R45–50. https://doi.org/10.1093/hmg/ddx228 [PubMed]
- 28. Al-Zamil WM, Yassin SA. Recent developments in age-related macular degeneration: a review. Clin Interv Aging. 2017; 12:1313–30. https://doi.org/10.2147/CIA.S143508 [PubMed]
- 29. Nowak JZ. AMD--the retinal disease with an unprecised etiopathogenesis: in search of effective therapeutics. Acta Pol Pharm. 2014; 71:900–16. [PubMed]
- 30. Mishima K, Handa JT, Aotaki-Keen A, Lutty GA, Morse LS, Hjelmeland LM. Senescence-associated beta-galactosidase histochemistry for the primate eye. Invest Ophthalmol Vis Sci. 1999; 40:1590–93. [PubMed]
- 31. Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015; 21:1424–35. https://doi.org/10.1038/nm.4000 [PubMed]
- 32. Naylor RM, Baker DJ, van Deursen JM. Senescent cells: a novel therapeutic target for aging and age-related diseases. Clin Pharmacol Ther. 2013; 93:105–16. https://doi.org/10.1038/clpt.2012.193 [PubMed]
- 33. Denu RA, Hematti P. Effects of Oxidative Stress on Mesenchymal Stem Cell Biology. Oxid Med Cell Longev. 2016; 2016:2989076. https://doi.org/10.1155/2016/2989076 [PubMed]
- 34. He S, Sharpless NE. Senescence in Health and Disease. Cell. 2017; 169:1000–11. https://doi.org/10.1016/j.cell.2017.05.015 [PubMed]
- 35. Zon LI. Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature. 2008; 453:306–13. https://doi.org/10.1038/nature07038 [PubMed]
- 36. Elzi DJ, Lai Y, Song M, Hakala K, Weintraub ST, Shiio Y. Plasminogen activator inhibitor 1--insulin-like growth factor binding protein 3 cascade regulates stress-induced senescence. Proc Natl Acad Sci USA. 2012; 109:12052–57. https://doi.org/10.1073/pnas.1120437109 [PubMed]
- 37. Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M, O’Hara SP, LaRusso NF, Miller JD, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015; 14:644–58. https://doi.org/10.1111/acel.12344 [PubMed]
- 38. Fuchshofer R, Yu AL, Teng HH, Strauss R, Kampik A, Welge-Lussen U. Hypoxia/reoxygenation induces CTGF and PAI-1 in cultured human retinal pigment epithelium cells. Exp Eye Res. 2009; 88:889–99. https://doi.org/10.1016/j.exer.2008.11.036 [PubMed]
- 39. Kim J, Kim Y, Choi H, Kwon A, Jekarl DW, Lee S, Jang W, Chae H, Kim JR, Kim JM, Kim M. Ubiquitin C decrement plays a pivotal role in replicative senescence of bone marrow mesenchymal stromal cells. Cell Death Dis. 2018; 9:139. https://doi.org/10.1038/s41419-017-0032-5 [PubMed]
- 40. Krenning L, Feringa FM, Shaltiel IA, van den Berg J, Medema RH. Transient activation of p53 in G2 phase is sufficient to induce senescence. Mol Cell. 2014; 55:59–72. https://doi.org/10.1016/j.molcel.2014.05.007 [PubMed]
- 41. Rodier F. Detection of the senescence-associated secretory phenotype (SASP). Methods Mol Biol. 2013; 965:165–73. https://doi.org/10.1007/978-1-62703-239-1_10 [PubMed]
- 42. Leung S, Liu X, Fang L, Chen X, Guo T, Zhang J. The cytokine milieu in the interplay of pathogenic Th1/Th17 cells and regulatory T cells in autoimmune disease. Cell Mol Immunol. 2010; 7:182–89. https://doi.org/10.1038/cmi.2010.22 [PubMed]
- 43. Chen J, Wang W, Li Q. Increased Th1/Th17 Responses Contribute to Low-Grade Inflammation in Age-Related Macular Degeneration. Cell Physiol Biochem. 2017; 44:357–67. https://doi.org/10.1159/000484907 [PubMed]
- 44. Juel HB, Faber C, Svendsen SG, Vallejo AN, Nissen MH. Inflammatory cytokines protect retinal pigment epithelial cells from oxidative stress-induced death. PLoS One. 2013; 8:e64619. https://doi.org/10.1371/journal.pone.0064619 [PubMed]
- 45. Juel HB, Faber C, Udsen MS, Folkersen L, Nissen MH. Chemokine expression in retinal pigment epithelial ARPE-19 cells in response to coculture with activated T cells. Invest Ophthalmol Vis Sci. 2012; 53:8472–80. https://doi.org/10.1167/iovs.12-9963 [PubMed]
- 46. Tsutsumi C, Sonoda KH, Egashira K, Qiao H, Hisatomi T, Nakao S, Ishibashi M, Charo IF, Sakamoto T, Murata T, Ishibashi T. The critical role of ocular-infiltrating macrophages in the development of choroidal neovascularization. J Leukoc Biol. 2003; 74:25–32. https://doi.org/10.1189/jlb.0902436 [PubMed]
- 47. McLeod DS, Bhutto I, Edwards MM, Silver RE, Seddon JM, Lutty GA. Distribution and Quantification of Choroidal Macrophages in Human Eyes With Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci. 2016; 57:5843–55. https://doi.org/10.1167/iovs.16-20049 [PubMed]
- 48. Camelo S, Calippe B, Lavalette S, Dominguez E, Hur J, Devevre E, Guillonneau X, Raoul W, Sennlaub F. Thinning of the RPE and choroid associated with T lymphocyte recruitment in aged and light-challenged mice. Mol Vis. 2015; 21:1051–59. [PubMed]
- 49. Sennlaub F, Auvynet C, Calippe B, Lavalette S, Poupel L, Hu SJ, Dominguez E, Camelo S, Levy O, Guyon E, Saederup N, Charo IF, Rooijen NV, et al. CCR2(+) monocytes infiltrate atrophic lesions in age-related macular disease and mediate photoreceptor degeneration in experimental subretinal inflammation in Cx3cr1 deficient mice. EMBO Mol Med. 2013; 5:1775–93. https://doi.org/10.1002/emmm.201302692 [PubMed]
- 50. Subhi Y, Lykke Sørensen T. New neovascular age-related macular degeneration is associated with systemic leucocyte activity. Acta Ophthalmol. 2017; 95:472–80. https://doi.org/10.1111/aos.13330 [PubMed]
- 51. Singh A, Subhi Y, Krogh Nielsen M, Falk MK, Matzen SM, Sellebjerg F, Sørensen TL. Systemic frequencies of T helper 1 and T helper 17 cells in patients with age-related macular degeneration: A case-control study. Sci Rep. 2017; 7:605. https://doi.org/10.1038/s41598-017-00741-4 [PubMed]
- 52. Subhi Y, Krogh Nielsen M, Molbech CR, Sørensen TL. Altered proportion of CCR2+ and CX3CR1+ circulating monocytes in neovascular age-related macular degeneration and polypoidal choroidal vasculopathy. Clin Experiment Ophthalmol. 2018; 46:661–69. https://doi.org/10.1111/ceo.13152 [PubMed]
- 53. Subhi Y, Nielsen MK, Molbech CR, Oishi A, Singh A, Nissen MH, Sørensen TL. T-cell differentiation and CD56+ levels in polypoidal choroidal vasculopathy and neovascular age-related macular degeneration. Aging (Albany NY). 2017; 9:2436–52. https://doi.org/10.18632/aging.101329 [PubMed]
- 54. Nassar K, Grisanti S, Elfar E, Lüke J, Lüke M, Grisanti S. Serum cytokines as biomarkers for age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 2015; 253:699–704. https://doi.org/10.1007/s00417-014-2738-8 [PubMed]
- 55. Spindler J, Zandi S, Pfister IB, Gerhardt C, Garweg JG. Cytokine profiles in the aqueous humor and serum of patients with dry and treated wet age-related macular degeneration. PLoS One. 2018; 13:e0203337. https://doi.org/10.1371/journal.pone.0203337 [PubMed]
- 56. Olivieri F, Lazzarini R, Recchioni R, Marcheselli F, Rippo MR, Di Nuzzo S, Albertini MC, Graciotti L, Babini L, Mariotti S, Spada G, Abbatecola AM, Antonicelli R, et al. MiR-146a as marker of senescence-associated pro-inflammatory status in cells involved in vascular remodelling. Age (Dordr). 2013; 35:1157–72. https://doi.org/10.1007/s11357-012-9440-8 [PubMed]
- 57. Cawthon RM. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002; 30:e47. https://doi.org/10.1093/nar/30.10.e47 [PubMed]
- 58. Amatori S, Bagaloni I, Macedi E, Formica M, Giorgi L, Fusi V, Fanelli M. Malten, a new synthetic molecule showing in vitro antiproliferative activity against tumour cells and induction of complex DNA structural alterations. Br J Cancer. 2010; 103:239–48. https://doi.org/10.1038/sj.bjc.6605745 [PubMed]