



Introduction
Nickel (Ni) has been recognized as a ubiquitous environmental contaminant, which is commonly used in electronic processing and medical industries, such as electroplating and the manufacturing processes of battery, electronic device and stainless steel [1]. Several epidemiological reports have indicated that occupational Ni exposure is associated with a higher prevalence of human nasal and lung cancers [2, 3]. Many other studies have suggested that Ni or Ni compounds can induce carcinogenicity, cytotoxicity, genotoxicity, immunotoxicity and mutagenicity both in vivo and in vitro [4, 5]. The broad application of Ni has resulted in its elevated levels in biogeochemical cycles, and increased its environmental exposure in humans [6].
At present, Ni-containing alloys are commonly used as biomaterials for cardiovascular, dental and orthopedic applications [7, 8]. It has been reported that Ni2+ is released from Ni-alloy during corrosion process [9, 10]. Ni2+ is released not only in medical devices such as dental restorations, surgical instruments, orthopedic implants and vascular stents, but also from coins, jewelry, mobile phones, piercing materials and synthetic nanoparticles [11]. Ni2+ is among the most frequent causes of allergic contact dermatitis in humans [12], which can affect the local and systemic immunity by suppressing the immune system or activating different inflammatory mediators such as intracellular adhesion molecule 1 and pro-inflammatory cytokines [13].
Inflammation represents cellular responses to infection, stress or injury [14]. Ni2+ can directly activate pro-inflammatory intracellular signal transduction cascades that stimulate mitogen-activated protein kinase (MAPK) p38 and nuclear factor-κB (NF-κB) [15]. It has been also shown that Ni and Ni compounds can induce the up-regulation of interleukin-1β (IL-1β), -6, -8, -18, tumor necrosis factor-α (TNF-α) and cyclooxygenase-2 (COX-2) [16, 17]. The maturation and release of IL-1β and IL-18 in macrophages are regulated by an inflammatory signaling platform, namely, “inflammasome” [18]. Inflammasome triggers the activation of caspase-1 in responses to cellular stresses and pathogenic infections [19]. The most studied inflammasome is Nod-like receptor 3 (NLRP3) inflammasome, which can be activated by numerous stimuli including infection and metabolic disorders [20]. NLRP3 inflammasome contains three subunits: NLRP3; caspase-1, the effector subunit; and apoptosis-associated speck-like protein containing a CARD (ASC) [20]. Although it is not completely clear, mitochondrial ROS generation and mtDNA release are the plausible stimulators for the production of NLRP3 inflammasome [18, 21]. Recruitment of caspase-1 into the inflammasome complexes can result in its full activation, auto-processing and substrate cleavage. Additionally, few studies have shown that Ni2+ can activate inflammasome pathway [22, 23].
In recent years, substantial attention has been paid to Ni uptake by macrophages [24–26]. Besides, direct T cell interaction or soluble mediator release can modulate the responses of macrophages to metal-containing alloys [27]. Here, we investigated the mechanism of nickel chloride (NiCl2)-induced inflammatory response, such as NF-κB, MAPKs and interferon regulatory factor 3 (IRF3) signaling pathways as well as NLRP3 inflammasome pathway in bone marrow-derived macrophages (BMDMs). Our findings would reveal a novel molecular mechanism underlying NI-induced inflammatory responses, which can improve future therapies against Ni-induced allergic reactions.
Results
NiCl2 induces cytotoxicity in BMDMs
Our previous work has shown that NiCl2 can inhibit the immune response in chicken. To evaluate the cytotoxicity of NiCl2, BMDMs were exposed to various doses of NiCl2 (0, 0.1, 0.5 and 1.0 mM) for 24 h. It was found that NiCl2 suppressed BMDMs viability in a dose-dependent manner (Figure 1A and 1B). Notably, the viabilities of BMDMs were significantly (p < 0.01) decreased in 0.5 and 1.0 mM NiCl2 exposure groups compared to control group.
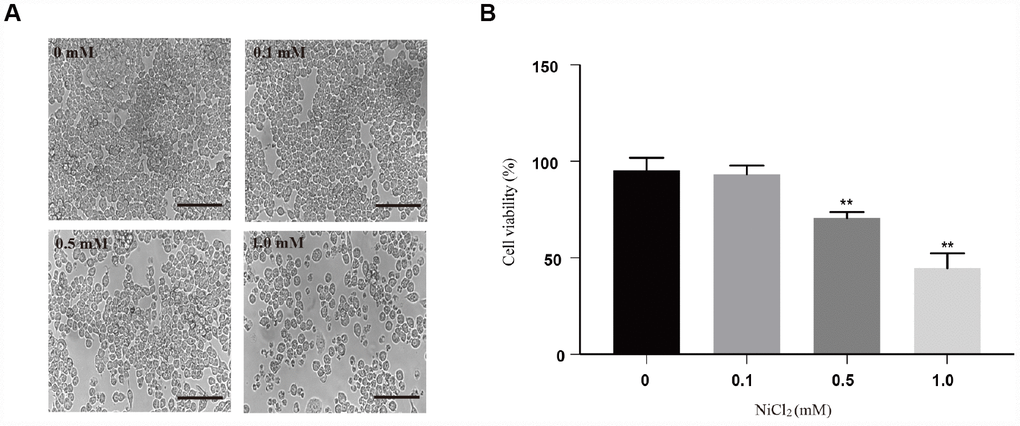
Figure 1. Cytotoxicity of NiCl2 in BMDMs. (A) BMDMs are treated with NiCl2 (0, 0.1, 0.5 and 1.0 mM) for 24h, and changes of cell numbers were observed by microscopy. Scale bar 50 μm. (B) Cell viability is analyzed by MTT assay. Data are presented with the means ± standard deviation (n=5). *p < 0.05 and **p < 0.01, compared with the control group.
NiCl2 activates NF-κB, MAPKs and IRF3 pathways in BMDMs
To investigate the inflammatory potential of NiCl2, NF-κB pathway was tested. The activation of NF-κB transcription factor may trigger an inflammatory response [15]. NF-κB proteins are bound and inhibited by IκBα proteins. NiCl2 treatment increased the phosphorylation levels of IκBα protein expression, and decreased the total IκBα protein expression levels (Figure 2A and 2B). Phosphorylation of IκBα caused its ubiquitination and proteasomal degradation, freeing NF-κB complexes. In Figure 2C and 2E, the results showed that NF-κB was translocated to the nucleus, and thus inducing target gene expression.
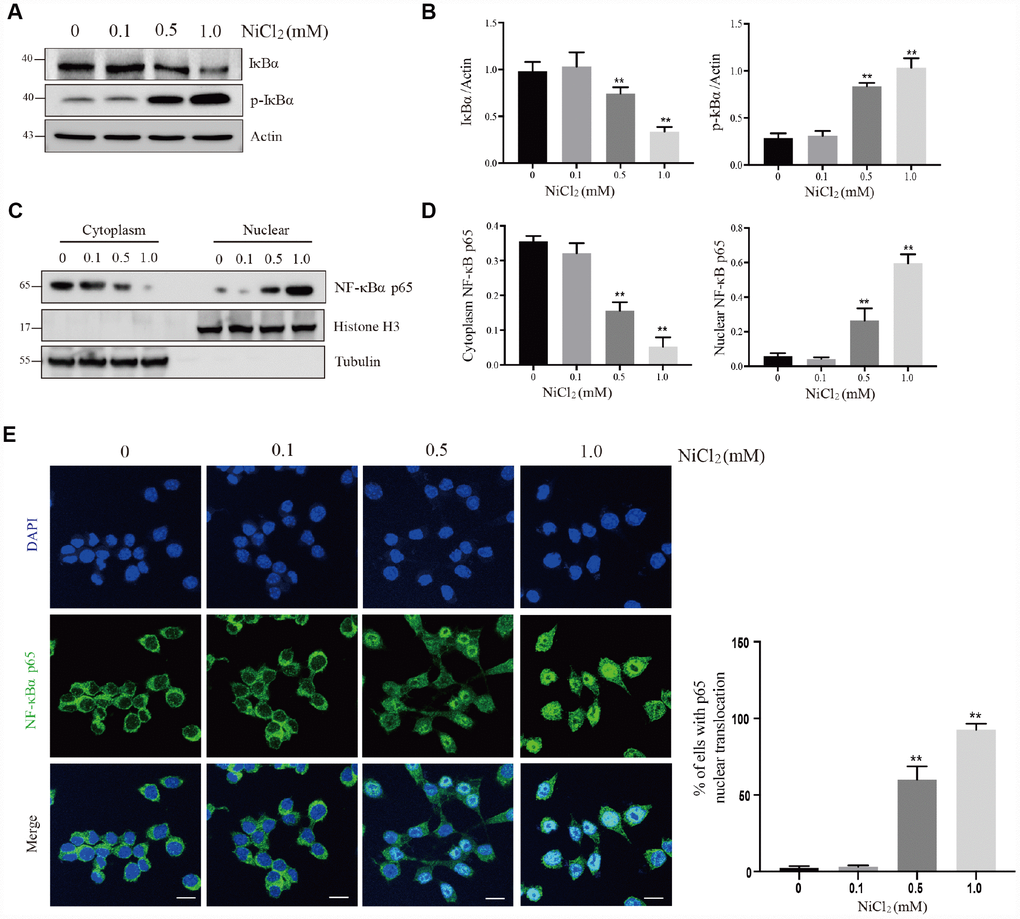
Figure 2. NiCl2 activates NF-κB pathway in BMDMs. (A and B) BMDMs are treated with NiCl2 (0, 0.1, 0.5 and 1.0 mM) for 24h, and immunoblotted for the whole cell lysis IκBα and p-IκBα protein expression. (C and D) cells are treated with NiCl2 (0, 0.1, 0.5 and 1.0 mM) for 24h, and immunoblotted for the cytoplasm and nuclear cell lysis NF-κB p65 protein expression. (E) The translocation of NF-κB p65 protein determined by NF-κB p65 staining of NiCl2-primed BMDMs. Scale bar 50 μm. Data are presented with the means ± standard deviation (n=5). *p < 0.05 and **p < 0.01, compared with the control group.
ERK1/2, Jun amino-terminal kinases (JNK) and p38 play crucial roles in cell growth and differentiation as well as inflammatory response and cellular stress [28]. In Figure 3A and 3B, our results showed that the protein expression levels of p-ERK, p-JNK and p-p38 were remarkably (p < 0.01) increased in 0.5 and 1.0 mM NiCl2 exposure groups.
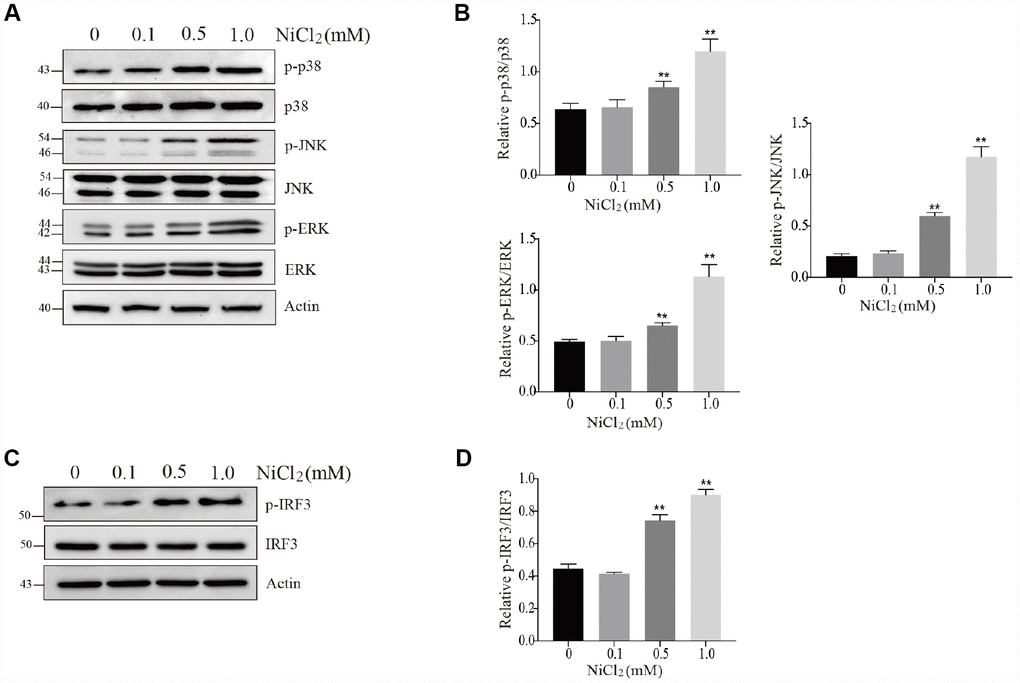
Figure 3. NiCl2 activates MAPKs and IRF3 pathway in BMDMs. (A and B) BMDMs are treated with NiCl2 (0, 0.1, 0.5 and 1.0 mM) for 24h, and immunoblotted for the whole cell lysis p-p38, p38, p-JNK, JNK, p-ERK and ERK protein expression. (C and D) BMDMs are treated with NiCl2 (0, 0.1, 0.5 and 1.0 mM) for 24h, and immunoblotted for the p-IRF3 and IRF3 protein expression. Data are presented with the means ± standard deviation (n=5). *p < 0.05 and **p < 0.01, compared with the control group.
Interferon regulatory factor 3 (IRF3) can suppress cell proliferation and regulate the expression levels of genes related to innate immunity [29]. As shown in Figure 3C and 3D, NiCl2 exposure markedly (p < 0.01) increased the protein expression levels of p-IRF3.
NiCl2 induces pro-inflammatory cytokine production in BMDMs
The above results showed that NiCl2 activated NF-κB, MAPKs and IRF3 pathways, by upregulating the expression levels of pro-inflammatory cytokines. In Figure 4A, the mRNA expression levels of IL-1β, -6, -8, -18, TNF-α and IFN-β were remarkably (p < 0.01) increased in 0.5 and 1.0 mM NiCl2 exposure groups compared to those in control group. The ELISA data revealed that NiCl2 treatment markedly (p < 0.01) increased the production of IL-1β, -6, -8, -18, TNF-α and IFN-β (Figure 4B).
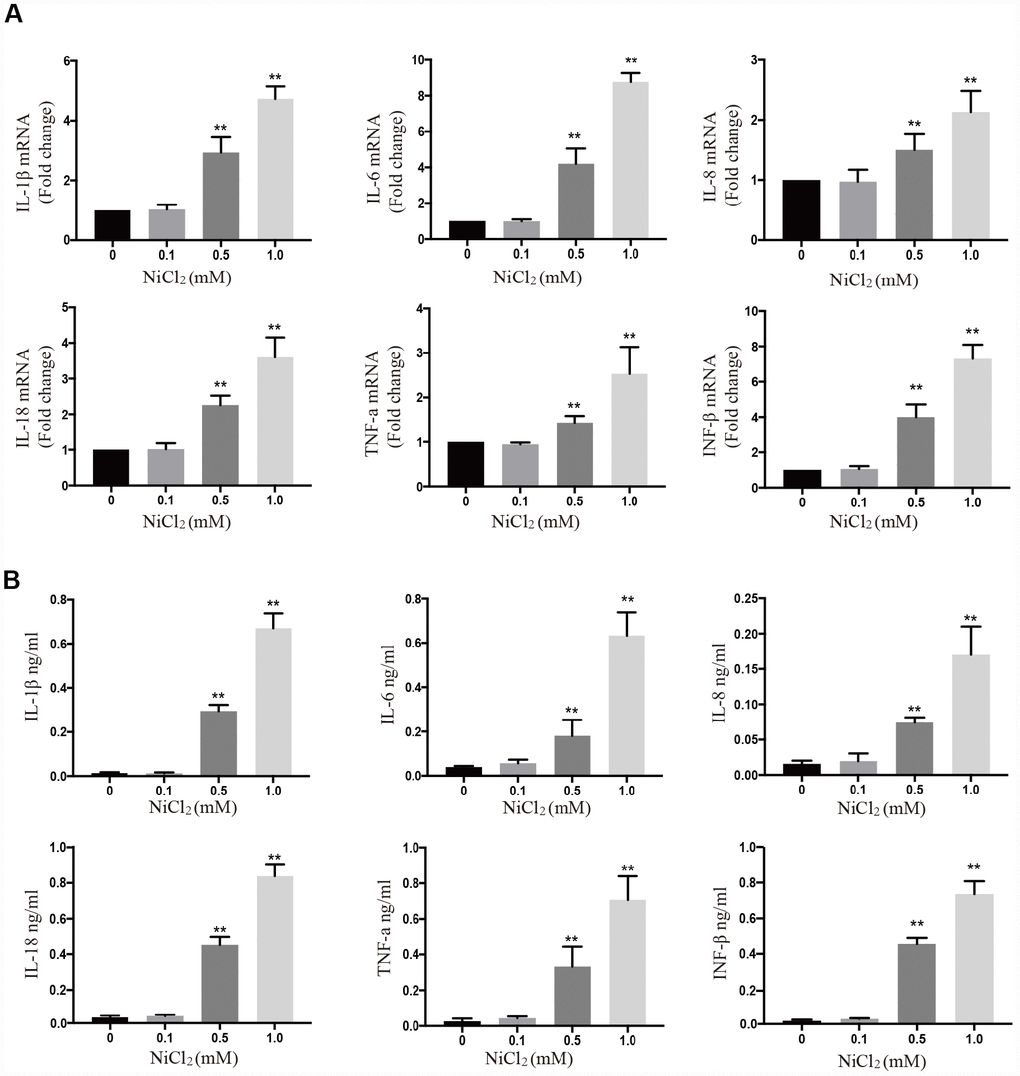
Figure 4. NiCl2 induces pro-inflammatory cytokine production in BMDMs. (A) The mRNA expression levels of inflammatory cytokines after NiCl2 (0, 0.1, 0.5 and 1.0 mM) treatment for 24h. (B) Inflammatory cytokine expression is quantified in BMDMs cultural medium by ELISA after NiCl2 (0, 0.1, 0.5 and 1.0 mM) treatment for 24h. Data are presented with the means ± standard deviation (n=5). *p < 0.05 and **p < 0.01, compared with the control group.
NiCl2 activates NLRP3 inflammasome pathway in BMDMs
NiCl2 activated caspase-1-mediated inflammatory cytokine production in BMDMs. Inflammasomes are multi-protein signaling complexes that promote inflammatory caspases activation and IL-1β maturation. We tested whether NLRP3-ASC-caspase-1 inflammasome is activated after NiCl2 treatment. It was found that NiCl2 induced the cleavage of IL-1β and caspase-1 (Figure 5E).
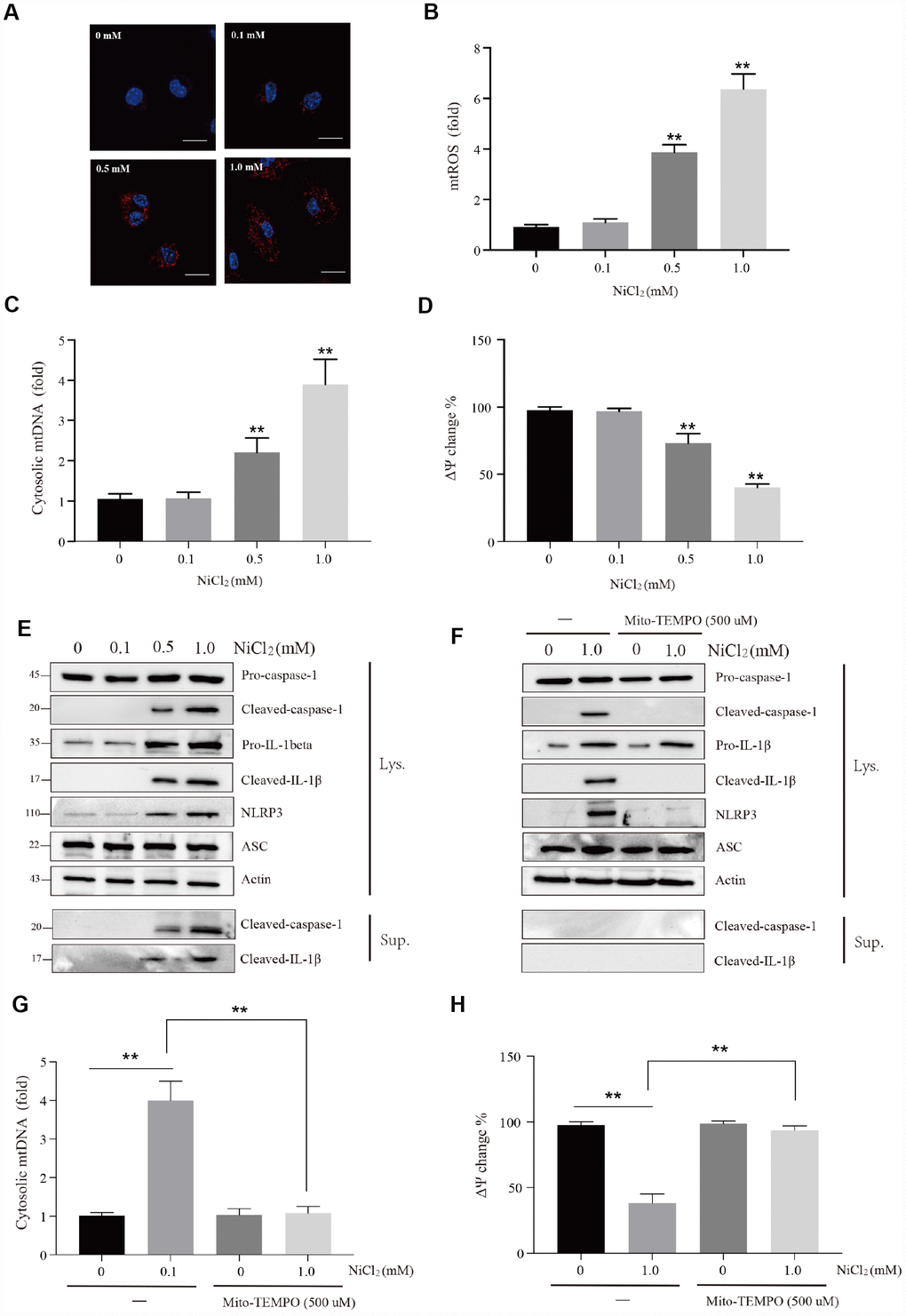
Figure 5. NiCl2 activates NLRP3 inflammasome pathway in BMDMs. (A and B) Relative mtROS amounts determined by MitoSOX-red staining of NiCl2-primed BMDMs. Scale bar 50 μm. (C) Relative cytosolic mtDNA expression in NiCl2-primed BMDMs. (D) NiCl2-induced changes in mitochondrial membrane potential (Ψm) in BMDMs measured by TMRM fluorescence. (E) Immunoblot analysis of pro-caspase-1, cleaved-caspase-1, pro-IL-1β, cleaved- IL-1β, NLRP3 and ASC in lysates of NiCl2-treated BMDMs, and cleaved-caspase-1and cleaved- IL-1β in the supernatant. (F) Immunoblot analysis of pro-caspase-1, cleaved-caspase-1, pro-IL-1β, cleaved- IL-1β, NLRP3 and ASC in lysates of Mito-TEMPO (500 μM)-pre-treated 1h before 24h of NiCl2 stimulation. (G) Relative cytosolic mtDNA expression in NiCl2-treated (24h) BMDMs in the presence/absence of Mito-TEMPO (500 μM, 1h) pre-treatment. (H) Changes of mitochondrial membrane potential (Ψm) in NiCl2-treated (24h) BMDMs in the presence/absence of Mito-TEMPO (500 μM, 1h) pre-treatment. Data are presented with the means ± standard deviation (n=5). *p < 0.05 and **p < 0.01, compared with the control group.
The mechanism underlying NiCl2-induced caspase-1 activation was explored in this study. The results showed that NiCl2 induced mitochondria damage such as increase of mitochondrial ROS (mtROS) production (Figure 5A and 5B), elevation of mitochondrial DNA (mtDNA) release (Figure 5C) and decrease of mitochondrial membrane potential (Figure 5D). We further assessed whether mitochondria-specific ROS production is associated with NiCl2-induced activation of caspase-1. Mito-TEMPO, a mitochondria-specific ROS scavenger, suppressed the formation of cleaved-IL-1β and cleaved-caspase-1 in BMDMs in response to NiCl2 (Figure 5F). Additionally, Mito-TEMPO attenuated the elevation of mtDNA release and loss of mitochondrial membrane potential (Figure 5G and 5H). These data demonstrate that NiCl2-induced caspase-1 activation largely relies on the mitochondrial generation of ROS.
NiCl2 induces apoptosis in BMDMs
In Figure 6A and 6B, we observed that the protein expression levels of cleaved-caspase-3, -8, -9 and cleaved-poly (ADP-ribose) polymerase (PARP) were significantly (p < 0.01) increased in 0.5 and 1.0 mM NiCl2 exposure groups (Figure 6A and 6B).
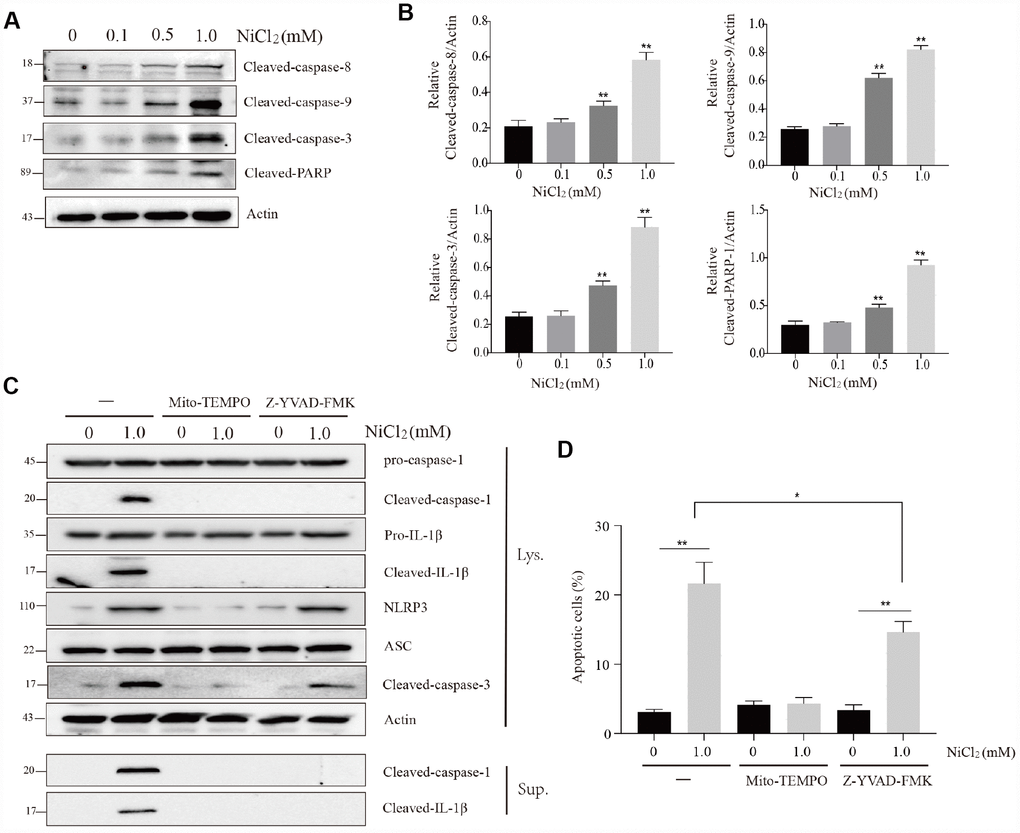
Figure 6. NiCl2 induces apoptosis in BMDMs. (A and B) Immunoblot analysis of cleaved-caspase-8, cleaved-caspase-9, cleaved-caspase-3 and cleaved-PARP in lysates of the NiCl2-treated BMDMs. (C) Immunoblot analysis of pro-caspase-1, cleaved-caspase-1, pro-IL-1β, cleaved- IL-1β, NLRP3, ASC and cleaved-caspase-3 in the NiCl2-treated (24h) BMDMs in the presence/absence of Mito-TEMPO (500 μM, 1h) or Z-YVAD-FMK (100 μM, 1h) pre-treatment, and cleaved-caspase-1and cleaved- IL-1β in the supernatant. (D) Flow cytometry analysis apoptosis in the NiCl2-treated (24h) BMDMs in the presence/absence of Mito-TEMPO (500 μM, 1h) or Z-YVAD-FMK (100 μM, 1h) pre-treatment. Data are presented with the means ± standard deviation (n=5). *p < 0.05 and **p < 0.01, compared with the control group.
MtROS generation diminishes mitochondrial membrane potential, and subsequently induces mitochondria-mediated apoptosis. Our above-mentioned results showed that NiCl2 treatment resulted in excessive mitochondrial ROS production and decreased mitochondrial membrane potential. Next, we examined whether mitochondrial ROS and caspase-1 activation are involved in NiCl2-induced apoptosis.
Cotreatment with NiCl2 and Mito-TEMPO suppressed the cleavage of caspase-1, -3 and IL-1β (Figure 6C). Cotreatment with NiCl2 and Z-YVAD-FMK, a potent inhibitor of caspase-1, suppressed the cleavage of IL-1β and caspase-1 (Figure 6C). However, the caspase-1 inhibitor only partly inhibited the cleaved-caspase-3 protein expression (Figure 6C). The percentage of apoptosis was significantly (p < 0.01) higher in NiCl2 treatment groups and NiCl2+Z-YVAD-FMK treatment group than that in control group (Figure 6D).
Taken altogether, these findings indicate that NiCl2-induced apoptosis is dependent on mitochondrial ROS production, and caspase-1 activation may also partly contribute to the apoptotic process.
Discussion
In the present study, our findings indicated that NiCl2 was extremely toxic to macrophages invitro. In addition, our results demonstrated that the excessive inflammatory responses and apoptosis were both associated with NiCl2 toxicity in BMDMs.
In our previous studies, we have found that NiCl2 induces inflammatory destruction in the kidney and liver of broiler chickens, by activating NF-κB pathway activation and upregulating the mRNA levels of IL-1β, -6, -8 and TNF-α [30, 31]. Here, we showed that NiCl2 activated NF-κB pathway. Typically, NF-κB sequestration is initiated by IκB in the cytoplasm. IκB degradation may translocate NF-κB into the nucleus from the cytoplasm (Figure 2C), where it regulates the transcription of specific genes encoding inducible enzymes, pro-inflammatory cytokines and chemokines. After NiCl2 treatment, the mRNA and protein levels of IL-1β, -6, -8, -18, TNF-α and INF-β were remarkably increased (Figure 3). Further, we investigated whether MAPKs and IRF3 pathways are also activated in BMDMs following NiCl2 treatment. Indeed, NiCl2 treatment results in the phosphorylation of p38, JNK, ERK and IRF3, which translocates to the nucleus in order to activate its binding site-containing promoter regions, and then upregulates the transcription of pro-inflammatory cytokines. Besides, it has been demonstrated that nickel oxide nanoparticles (NiONPs) activate MAPKs (p38 and JNK) and NF-κB pathways in lung-derived A549 and BEAS-2B cell lines [16].
In macrophages, inflammasome controls the maturation and release of IL-1β [20, 32]. In this study, NiCl2 upregulated the mRNA and protein levels of IL-1β via NF-κB, MAPKs and IRF3 signaling pathways, and enhanced the maturation and production of IL-1β through NLRP3 inflammasome induction. Li and colleagues [22] have demonstrated that Ni induces the secretion of IL-1β via NLRP3-ASC-caspase-1 pathway. In addition, a previous study has shown that the induction of NLRP3 inflammasome largely depends on mitochondrial damage [18]. In this study, we observed increased mtROS generation and reduced mitochondrial membrane potential (Ψm). As a consequence of mtROS accumulation and mitochondrial membrane potential (Ψm), we found that NiCl2 treatment released mtDNA into the cytosolic compartment. Next, we tested whether mtROS generation is a relatively upstream step during NiCl2-induced NLRP3 inflammasome. Treatment with Mito-TEMPO (mitochondrial ROS scavenger) abolished the activation of NLRP3 inflammasome, such as the inhibition of cleaved-caspase-1 and IL-1β. Mito-TEMPO also suppressed mtDNA release and loss of mitochondrial membrane potential (Ψm). It has been demonstrated that mtROS generation is critical for the stimulation of NLRP3 inflammasome [33, 34]. Nonetheless, there is a report that mtDNA plays a pivotal role during inflammasome activation [35]. Collectively, our results indicate that NiCl2 induces NLRP3 inflammasome activation through mtROS production.
In this study, NiCl2 also induces the apoptosis of BMDMs, and macrophages are critical components of both innate and adaptive immunity, suggesting that NiCl2-induced BMDM apoptosis inhibits the immune function. In the three NiCl2 treatment groups, the transcription levels cleaved-caspase-3, -8, -9 and cleaved-PARP were upregulated. Furthermore, Mito-TEMPO could abolish the enhance levels of cleaved-caspase-3 and BMDM apoptosis. The above-mentioned results show that NiCl2 triggers mtROS generation and loss of mitochondrial membrane potential (Ψm). Taken together, NiCl2 aggravates BMDM apoptosis via mitochondrial ROS-associated pathway. These findings are consistent with those of Zuo et al. [36], in which nickel sulfate induces apoptosis in rat Leydig cells through activation of ROS-dependent mitochondria pathway. Likewise, the data of this study are compatible with our previous work that NiCl2 induces apoptosis in the kidney of broiler chickens via mitochondria-dependent apoptotic pathway [37]. Interestingly, Z-YVAD-FMK (caspase-1 inhibitor) can decrease at least part of the apoptotic process. Thus, NiCl2-induced NLRP3 inflammasome activation may contribute to cell apoptosis.
In summary, this study reveals that NiCl2 triggers the transcription levels of pro-inflammatory cytokines, including IL-1β, -6, -8, -18, TNF-α and INF-β in BMDMs through NF-κB, MAPKs and IRF3 signaling pathways. Moreover, NiCl2 stimulates caspase-1 activation via NLRP3 inflammasome activation. Furthermore, NiCl2 induces apoptosis through mitochondrial ROS-mediated pathway, and the activation of NLRP3 inflammasome can contribute to apoptotic process (as shown in the Figure 7).
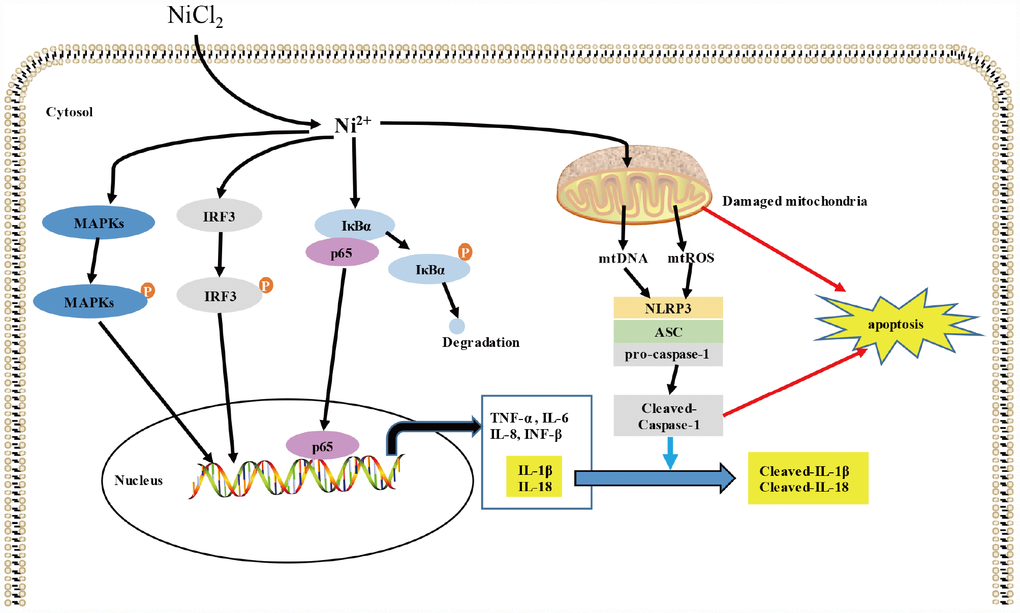
Figure 7. Schematic diagram of the possible inflammatory response induced by NiCl2. NiCl2 triggers the transcription of pro-inflammatory cytokines, including IL-1β, IL-6, IL-8, IL-18, TNF-α and INF-β through the NF-κB, MAPKs, IRF3 signaling pathways in the BMDM. And NiCl2 also can induce caspase-1 activation via NRLP3 inflammasome activation. Moreover, NiCl2 induces apoptosis through mitochondrial ROS-mediated pathway, and also, NLRP3 inflammasome activation contributes to the apoptosis.
Materials and Methods
Materials and reagents
NiCl2 (451193) was purchased from Sigma Aldrich Corporation. The mitochondria-targeted antioxidant Mito-TEMPO (CAS 1569257-94-8) was bought from Santa Cruz Biotechnology. Tetramethylrhodamine, methyl ester (TMRM) was obtained from AnaSpec Inc. (#CA94555). Z-YVAD-FMK (ALX-260-154-R100) was purchased from Enzo Life Sciences. MitoSOX (M36008) was obtained from Invitrogen.
The antibodies such as mouse anti-IκBα (#4814, 1:1000 for WB), rabbit anti-phospho (p)-IκBα (#2859, 1:1000 for WB), rabbit anti-NF-κB p65 (#8242, 1:1000 for WB, 1:200 for IF), mouse anti-β-Actin (#3700, 1:1000 for WB), rabbit anti-p38 (#8690, 1:1000 for WB), rabbit anti-p-p38 (#4511, 1:1000 for WB), rabbit anti-JNK (#9252, 1:1000 for WB), rabbit anti-p-JNK (#4668, 1:1000 for WB), rabbit anti-ERK (#4695, 1:1000 for WB), rabbit anti-p-ERK (#9101, 1:1000 for WB), rabbit anti-IRF3 (#11904, 1:1000 for WB), rabbit anti-p-IRF3 (#83611, 1:1000 for WB), rabbit anti-NLRP3 (#15101, 1:1000 for WB), rabbit anti-IL-1β (#12426S, 1:1000 for WB), rabbit anti-cleaved-caspase-3 (#9664, 1:1000 for WB), rabbit anti-cleaved-caspase-8 (#8592, 1:1000 for WB), rabbit anti-cleaved-caspase-9 (#9509, 1:1000 for WB), rabbit anti-PARP (#9548, 1:1000 for WB) were supplied by Cell Signaling Technology. Mouse anti-Histone H3 (sc-517576, 1:1000 for WB) and mouse anti-Tubulin (sc-73242, 1:1000 for WB) were obtained from Santa Cruz Biotechnology, while mouse anti-caspase-1 p20 (#AG-20B-0042-C100, 1:1000 for WB) and mouse anti-ASC (#AG-25B-0006-C100, 1:1000 for WB) were purchased from Adipogen.
IL-1β (MLB00C), IL-6 (M6000B), IL-18 (7625), TNF-α (MTA00B) and IFN-β (42400-1) ELISA kits were purchased from R&D system, while IL-8 (MBS261967) ELISA kit was obtained from MyBioSource.
Cell isolation and culture
Preparation of primary BMDM was carried out as proposed by Nakahira et al. [35]. Briefly, BMDMs were collected from the femur and tibia of each mouse, and seeded on a sterile petri dish. The BMDMs were cultured for 7 days in DMEM supplemented with 10% heat-inactivated fetal bovine serum, penicillin, streptomycin and 25% conditioned medium from L929 mouse fibroblasts. Upon reaching confluency, the BMDMs were exposed to different concentrations (0, 0.1, 0.5 and 1.0 mM) of NiCl2 for 24 h. In the cotreatment with NiCl2 and Mito-TEMPO experiment, the Mito-TEMPO (500 μM) or Z-YVAD-FMK (100 μM) were treated 1h before 24h of NiCl2 treatment. In the cotreatment with NiCl2 and Z-YVAD-FMK experiment, the Z-YVAD-FMK (100 μM) were treated 1h before 24h of NiCl2 treatment.
Cytotoxicity assessment
The viability of BMDMs was assessed using MTT assay. Briefly, BMDMs (1 × 106 cells/ml) were suspended in complete culture media, and 150 μL of the cell suspension was cultured 48-well plates for 24 h. After treatment with NiCl2, 0.5 mg/ml MTT solution was added onto the BMDMs, followed by a 4 h incubation period. The obtained formazan crystals were dissolved in dimethyl sulfoxide, and the absorbance was recorded at 540 nm using a microplate reader (PerkinElmer). Data were the mean ± SD of 5 independent experiments conducted in triplicate.
Western blotting
After NiCl2 treatment, cell lysis was performed with ice-cold RIPA buffer. After centrifuging at 15000 g for 15 min at 4 °C, the protein lysates were separated by 12% SDS-PAGE and then transferred onto PVDF membranes. Subsequently, the membranes were blocked with 5% non-fat milk in TBST, and then incubated with primary antibodies, followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies and enhanced chemiluminescent (ECL) detection (GE Healthcare, Piscataway, NJ, USA). The protein bands were visualized using Bio-Rad ChemiDoc XRS+ System (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The statistical differences in protein expression levels were computed by an ImageJ2x software.
The supernatant protein is extracted by the method of trichloroacetic acid (TCA) precipitation of proteins according to the reference [38], followed by the detection of protein content using BCA protein assay. The expression levels of cleaved-caspase-1 and IL-1β are detected by western blotting.
Determination of NF-κB p65 protein expression
After treatment with NiCl2 for 24 h, BMDMs were collected and total protein was isolated from cytoplasm and nucleus by using Cytoplasmic and Nuclear Protein Extraction Kit (SINP001 Viagene Biotech). The protein expression was determined by Western blotting.
BMDMs were seeded and allowed to be adhered on a coverslip for 24 h. After exposure to NiCl2 for 24 h, the BMDMs were fixed and permeabilized with 4% paraformaldehyde and 0.05% Triton X-100, respectively. After blocking with PBS containing 5% BSA, the BMDMs were stained with rabbit anti-rabbit NF-κB p65, followed by Alexa Fluor 488-conjugated anti-rabbit IgG secondary antibody (Jackson ImmunoResearch). The nuclei of BMDMs were stained with propidium iodide (PI; Thermo Fisher). All images were acquired with a confocal microscope. For image quantification, 100 cells randomly chosen from 10 high-power fields and pooled from five independent experiments, were evaluated for the distribution pattern of the indicated molecules. Then, we quantified the percentage of cells with NF-kB nuclear translocation.
Determination of mitochondrial ROS
MitoSOX was used to measure mtROS production in BMDMs. After NiCl2 treatment, the BMDMs were rinsed in PBS and then stained with 5 μM of MitoSOX-red at 37 °C, 5% CO2 for 15 min. After MitoSOX staining, the BMDMs were washed with PBS, followed by fixing and staining with DAPI. All samples were visualized and recorded using Zeiss Axio Imager A2 fluorescence microscope. For image quantification, 100 cells randomly chosen from 10 high-power fields and pooled from five independent experiments, were evaluated for the distribution pattern of the indicated molecules. The red fluorescence of each cell is detected by image J.
Measurements of mitochondrial membrane potential (Ψm)
Mitochondrial membrane potential (Ψm) was measured using TMRM as proposed by Zhong et al. [39]. Briefly, after NiCl2 exposure, the BMDMs were rinsed twice in PBS, incubated with 200 nM of TMRM for 30 min, and then rinsed twice in 50 nM of TMRM. The BMDMs were resuspended in PBS, and the fluorescence intensities recorded using FilterMax F5 multimode plate reader (Molecular Devices).
Cellular fractionation and determination of cytosolic mtDNA
MtDNA in cytosol was performed as described previously [35]. Total DNA was extracted from the cytosolic fraction (200 μL) using DNeasy Blood and Tissue kit (Cat. 69504 Qiagen). Quantitative PCR was adopted for the measurement of mtDNA by using the established mitochondrial and nuclear gene primers and SYBR® Premix Ex TaqTMII (RR820A, Takara, China). The copy numbers of mtDNA were normalized to those of nuclear DNA, and the data were expressed as the ratio of mtDNA (cytochrome c oxidase I) to nuclear DNA (18S rRNA). The accession number, sequences, product size and annealing temperature of each primer are presented in Table 1.
Table 1. Primer sequences of genes selected for analysis mtDNA.
| Target gene | Accession number | Primer | Primer sequence (5′-3′) | Product size | Tm (°C) |
| 18S | EU120032 | Forward | TAGAGGGACAAGTGGCGTTC | 104bp | 60 |
| Reverse | CGCTGAGCCAGTCAGTGT | ||||
| mouse cytochrome c oxidase I | XM_006980540 | Forward | GCCCCAGATATAGCATTCCC | 221bp | 59 |
| Reverse | GTTCATCCTGTTCCTGCTCC |
RNA isolation and quantitative Real-Time PCR (qRT-PCR)
Total RNA was isolated from the treated BMDM and was reverse transcribed. QRT-PCR reactions were carried out as proposed by Guo et al. [30]. QRT-PCR data were analyzed by the 2−ΔΔCT method [40]. The accession number, sequences, product size and annealing temperature of each primer are listed in Table 2.
Table 2. Primer sequences of genes selected for analysis pro-inflammatory cytokines.
| Target gene | Accession number | Primer | Primer sequence (5′-3′) | Product size | Tm (°C) |
| IL-1β | NM_008361 | Forward | AATGCCACCTTTTGACAGTGAT | 132bp | 60 |
| Reverse | TGCTGCGAGATTTGAAGCTG | ||||
| IL-6 | NM_001314054 | Forward | AGGATACCACTCCCAACAGACC | 140bp | 60 |
| Reverse | AAGTGCATCATCGTTGTTCATACA | ||||
| IL-8 | NM_ 011339 | Forward | TTTCCACCGGCAATGAAG | 118bp | 59 |
| Reverse | TAGAGGTCTCCCGAATTGGA | ||||
| IL-18 | NM_008360 | Forward | GGCTGCCATGTCAGAAGACT | 239bp | 60 |
| Reverse | GTCTGGTCTGGGGTTCACTG | ||||
| TNF-a | NM_013693 | Forward | CACGTCGTAGCAAACCACC | 88bp | 59 |
| Reverse | TGAGATCCATGCCGTTGGC | ||||
| INF-β | NM_010510 | Forward | GGCTTCCATCATGAACAACAGGT | 167bp | 61 |
| Reverse | AGGTGAGGTTGATCTTTCCATTCAG | ||||
| β-actin | NM_007393 | Forward | GCTGTGCTATGTTGCTCTAG | 117bp | 59 |
| Reverse | CGCTCGTTGCCAATAGTG |
Enzyme-linked immunosorbent assay
After NiCl2 treatment, ELISA kit was used to examine the concentration of cytokines in cell culture supernatants as per the manufacturer's protocols.
Apoptosis analysis by flow cytometry
After NiCl2 treatment, the BMDMs were rinsed twice with ice-cold phosphate buffer saline (PBS, pH 7.2-7.4), and then suspended in PBS to the desired concentration of 1×106 cells/ml. The staining protocol was consistent with the description of Guo et al. [37]. Briefly, 100 μL of the cell suspension was placed into a 5 mL tube. The cell suspension was stained with 5 μL of Annexin V-FITC (Cat: 51-65874X, BD, USA) followed by 5 μL of PI (Cat: 51-66211E, BD, USA) at 25°C in darkness. After 15-min incubation, 400 μl of 1× binding buffer were added, and the stained cells were evaluated by a flow cytometry (BD FACS Calibur) within 40 min of preparation. Flow cytometry data analysis was performed using the ModFit LT v3.0 application software.
Statistical analysis
All data were expressed as mean ± standard deviation. The statistical differences between the three experimental groups and control group were compared using one-way analysis of variance (SPSS 17.0 statistical software). P-value below 0.05 was considered statistically significant.
Ethics statement
The animal protocols and all procedures of the experiment were performed in compliance with the laws and guidelines of Animal Care and Use Committee, Sichuan Agricultural University (Approval No: 2012- 024).
Author Contributions
H. Guo, H. Liu and H. Cui designed and performed experiments, collected and analyzed data, and wrote the paper. J. Fang, Z. Zuo, J. Deng, Y. Li, X. Wang, L. Zhao, Y. Geng, P. Ouyang, W. Lai, Z. Chen and C. Huang performed experiments, collected and analyzed data. All authors contributed discussions and interpretations.
Conflicts of Interest
The authors declare that there are no conflicts of interest.
Funding
This research was supported by the program for changjiang scholars and the university innovative research team (IRT 0848), and the Shuangzhi Project of Sichuan Agricultural University (03571800; 03572437).
References
- 1. Nestle FO, Speidel H, Speidel MO. Metallurgy: high nickel release from 1- and 2-euro coins. Nature. 2002; 419:132–132. https://doi.org/10.1038/419132a [PubMed]
- 2. Canaz E, Kilinc M, Sayar H, Kiran G, Ozyurek E. Lead, selenium and nickel concentrations in epithelial ovarian cancer, borderline ovarian tumor and healthy ovarian tissues. J Trace Elem Med Biol. 2017; 43:217–23. https://doi.org/10.1016/j.jtemb.2017.05.003 [PubMed]
- 3. Lee YW, Klein CB, Kargacin B, Salnikow K, Kitahara J, Dowjat K, Zhitkovich A, Christie NT, Costa M. Carcinogenic nickel silences gene expression by chromatin condensation and DNA methylation: a new model for epigenetic carcinogens. Mol Cell Biol. 1995; 15:2547–57. https://doi.org/10.1128/mcb.15.5.2547 [PubMed]
- 4. Cameron KS, Buchner V, Tchounwou PB. Exploring the molecular mechanisms of nickel-induced genotoxicity and carcinogenicity: a literature review. Rev Environ Health. 2011; 26:81–92. https://doi.org/10.1515/reveh.2011.012 [PubMed]
- 5. Das KK, Das SN, Dhundasi SA. Nickel, its adverse health effects and oxidative stress. Indian J Med Res. 2008; 128:412–25. [PubMed]
- 6. Guo H, Chen L, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Wu B. Research Advances on Pathways of Nickel-Induced Apoptosis. Int J Mol Sci. 2015; 17:10. https://doi.org/10.3390/ijms17010010 [PubMed]
- 7. Fujii N, Asano R, Nagayama M, Tobaru T, Misu K, Hasumi E, Hosoya Y, Iguchi N, Aikawa M, Watanabe H, Umemura J, Sumiyoshi T. Long-term outcome of first-generation metallic coronary stent implantation in patients with coronary artery disease: observational study over a decade. Circ J. 2007; 71:1360–65. https://doi.org/10.1253/circj.71.1360 [PubMed]
- 8. Cobb AG, Schmalzreid TP. The clinical significance of metal ion release from cobalt-chromium metal-on-metal hip joint arthroplasty. Proc Inst Mech Eng H. 2006; 220:385–98. https://doi.org/10.1243/09544119JEIM78 [PubMed]
- 9. Wylie CM, Shelton RM, Fleming GJ, Davenport AJ. Corrosion of nickel-based dental casting alloys. Dent Mater. 2007; 23:714–23. https://doi.org/10.1016/j.dental.2006.06.011 [PubMed]
- 10. Ries MW, Kampmann C, Rupprecht HJ, Hintereder G, Hafner G, Meyer J. Nickel release after implantation of the Amplatzer occluder. Am Heart J. 2003; 145:737–41. https://doi.org/10.1067/mhj.2003.7 [PubMed]
- 11. Lidén C, Skare L, Vahter M. Release of nickel from coins and deposition onto skin from coin handling—comparing euro coins and SEK. Contact Dermatitis. 2008; 59:31–37. https://doi.org/10.1111/j.1600-0536.2008.01363.x [PubMed]
- 12. Schäfer T, Böhler E, Ruhdorfer S, Weigl L, Wessner D, Filipiak B, Wichmann HE, Ring J. Epidemiology of contact allergy in adults. Allergy. 2001; 56:1192–96. https://doi.org/10.1034/j.1398-9995.2001.00086.x [PubMed]
- 13. Wataha IC, Sun ZL, Hanks CT, Fang DN. Effect of Ni ions on expression of intercellular adhesion molecule 1 by endothelial cells. J Biomed Mater Res. 1997; 36:145–51. https://doi.org/10.1002/(SICI)1097-4636(199708)36:2<145::AID-JBM2>3.0.CO;2-K [PubMed]
- 14. Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006; 444:860–67. https://doi.org/10.1038/nature05485 [PubMed]
- 15. Goebeler M, Roth J, Bröcker EB, Sorg C, Schulze-Osthoff K. Activation of nuclear factor-kappa B and gene expression in human endothelial cells by the common haptens nickel and cobalt. J Immunol. 1995; 155:2459–67. [PubMed]
- 16. Capasso L, Camatini M, Gualtieri M. Nickel oxide nanoparticles induce inflammation and genotoxic effect in lung epithelial cells. Toxicol Lett. 2014; 226:28–34. https://doi.org/10.1016/j.toxlet.2014.01.040 [PubMed]
- 17. Cai T, Li X, Ding J, Luo W, Li J, Huang C. A cross-talk between NFAT and NF-κB pathways is crucial for nickel-induced COX-2 expression in Beas-2B cells. Curr Cancer Drug Targets. 2011; 11:548–59. https://doi.org/10.2174/156800911795656001 [PubMed]
- 18. Sorbara MT, Girardin SE. Mitochondrial ROS fuel the inflammasome. Cell Res. 2011; 21:558–60. https://doi.org/10.1038/cr.2011.20 [PubMed]
- 19. Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. 2012; 28:137–61. https://doi.org/10.1146/annurev-cellbio-101011-155745 [PubMed]
- 20. Guarda G, So A. Regulation of inflammasome activity. Immunology. 2010; 130:329–36. https://doi.org/10.1111/j.1365-2567.2010.03283.x [PubMed]
- 21. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012; 36:401–14. https://doi.org/10.1016/j.immuni.2012.01.009 [PubMed]
- 22. Li X, Zhong F. Nickel induces interleukin-1β secretion via the NLRP3-ASC-caspase-1 pathway. Inflammation. 2014; 37:457–66. https://doi.org/10.1007/s10753-013-9759-z [PubMed]
- 23. Hamilton RF
Jr , Buford M, Xiang C, Wu N, Holian A. NLRP3 inflammasome activation in murine alveolar macrophages and related lung pathology is associated with MWCNT nickel contamination. Inhal Toxicol. 2012; 24:995–1008. https://doi.org/10.3109/08958378.2012.745633 [PubMed] - 24. Zhang Y, Zhang ZW, Xie YM, Wang SS, Qiu QH, Zhou YL, Zeng GH. Toxicity of nickel ions and comprehensive analysis of nickel ion-associated gene expression profiles in THP-1 cells. Mol Med Rep. 2015; 12:3273–78. https://doi.org/10.3892/mmr.2015.3878 [PubMed]
- 25. Moitra S, Ghosh J, Firdous J, Bandyopadhyay A, Mondal M, Biswas JK, Sahu S, Bhattacharyya S, Moitra S. Exposure to heavy metals alters the surface topology of alveolar macrophages and induces respiratory dysfunction among Indian metal arc-welders. Toxicol Ind Health. 2018; 34:748233718804426. https://doi.org/10.1177/0748233718804426 [PubMed]
- 26. Edwards DL, Wataha JC, Hanks CT. Uptake and reversibility of uptake of nickel by human macrophages. J Oral Rehabil. 1998; 25:2–7. https://doi.org/10.1046/j.1365-2842.1998.00197.x [PubMed]
- 27. Trindade MC, Lind M, Sun D, Schurman DJ, Goodman SB, Smith RL. In vitro reaction to orthopaedic biomaterials by macrophages and lymphocytes isolated from patients undergoing revision surgery. Biomaterials. 2001; 22:253–59. https://doi.org/10.1016/S0142-9612(00)00181-2 [PubMed]
- 28. Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002; 298:1911–12. https://doi.org/10.1126/science.1072682 [PubMed]
- 29. Kumari M, Wang X, Lantier L, Lyubetskaya A, Eguchi J, Kang S, Tenen D, Roh HC, Kong X, Kazak L, Ahmad R, Rosen ED. IRF3 promotes adipose inflammation and insulin resistance and represses browning. J Clin Invest. 2016; 126:2839–54. https://doi.org/10.1172/JCI86080 [PubMed]
- 30. Guo H, Cui H, Fang J, Zuo Z, Deng J, Wang X, Zhao L, Chen K, Deng J. Nickel chloride (NiCl2) in hepatic toxicity: apoptosis, G2/M cell cycle arrest and inflammatory response. Aging (Albany NY). 2016; 8:3009–27. https://doi.org/10.18632/aging.101108 [PubMed]
- 31. Guo H, Deng H, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Wu B, Chen K. Nickel chloride (NiCl2)-caused inflammatory responses via activation of NF-κB pathway and reduction of anti-inflammatory mediator expression in the kidney. Oncotarget. 2015; 6:28607–20. https://doi.org/10.18632/oncotarget.5759 [PubMed]
- 32. Martinon F. Signaling by ROS drives inflammasome activation. Eur J Immunol. 2010; 40:616–19. https://doi.org/10.1002/eji.200940168 [PubMed]
- 33. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011; 469:221–25. https://doi.org/10.1038/nature09663 [PubMed]
- 34. Alfonso-Loeches S, Ureña-Peralta JR, Morillo-Bargues MJ, Oliver-De La Cruz J, Guerri C. Role of mitochondria ROS generation in ethanol-induced NLRP3 inflammasome activation and cell death in astroglial cells. Front Cell Neurosci. 2014; 8:216. https://doi.org/10.3389/fncel.2014.00216 [PubMed]
- 35. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011; 12:222–30. https://doi.org/10.1038/ni.1980 [PubMed]
- 36. Zou L, Su L, Sun Y, Han A, Chang X, Zhu A, Liu F, Li J, Sun Y. Nickel sulfate induced apoptosis via activating ROS-dependent mitochondria and endoplasmic reticulum stress pathways in rat Leydig cells. Environ Toxicol. 2017; 32:1918–26. https://doi.org/10.1002/tox.22414 [PubMed]
- 37. Guo H, Cui H, Fang J, Zuo Z, Deng J, Wang X, Zhao L, Wu B, Chen K, Deng J. Nickel chloride-induced apoptosis via mitochondria- and Fas-mediated caspase-dependent pathways in broiler chickens. Oncotarget. 2016; 7:79747–60. https://doi.org/10.18632/oncotarget.12946 [PubMed]
- 38. Link AJ, LaBaer J. Trichloroacetic acid (TCA) precipitation of proteins. Cold Spring Harb Protoc. 2011; 2011:993–94. https://doi.org/10.1101/pdb.prot5651 [PubMed]
- 39. Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR, McGeough MD, Ellisman MH, Seki E, et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell. 2016; 164:896–910. https://doi.org/10.1016/j.cell.2015.12.057 [PubMed]
- 40. Nolan T, Hands RE, Bustin SA. Quantification of mRNA using real-time RT-PCR. Nat Protoc. 2006; 1:1559–82. https://doi.org/10.1038/nprot.2006.236 [PubMed]