




Introduction
Circular RNAs (circRNAs) are a class of closed circular RNA molecules that rarely encode proteins and have no 5 ‘caps and 3’ poly(A) tails. Their unique characteristics, such as better stability and resistance to nucleases make circRNAs potential candidates for clinical diagnostic/prognostic biomarkers [1]. The molecular mechanisms of circRNAs are diverse. In addition to acting as miRNA sponges, they may also play a role by binding protein or DNA sequences [2–4]. Some of them can also regulate gene expression by affecting RNA splicing or translating into short peptides [5]. They are conserved among different species, and also exhibit tissue-specific and developmental-stage specific traits [6]. Emerging evidences suggest that circRNAs are likely to play important function in different physiological or pathological processes. A multitude of studies have demonstrated that circRNAs are associated with various diseases, including cancer, Alzheimer’s disease, ovarian endometriosis, preeclampsia [7–11]. Moreover, a previous study has reported that there are 15,996 circRNAs in normal human testis and more than 20 testis-derived circRNAs are stably expressed in seminal plasma [5]. All the above evidences indicate that circRNAs might play a role in spermatogenesis and maturation. However, the global expression and function of circRNAs in non-obstructive azoospermia (NOA) are largely unknown.
NOA is considered a major cause of male infertility [12, 13], which can’t be completely cured by assisted reproductive technology (ART). Several genetic and epigenetic factors, especially Y chromosome microdeletion, are associated with the pathogenesis NOA [14–18]. In recent years, genome-wide association analysis (GWAS), has led to the identification of a few key NOA risk loci including PRMT6, PEX10, HLA-DRA [14, 16]. In addition to these genetic factors, epigenetic factors also play an important role in the development of NOA. For instance, Chencheng Yao et al. found that there were many differentially expressed miRNAs in testicular spermatogonia, pachytene spermatocytes, and round spermatids between NOA and OA patients [13]. Further, another group also found that miRNA-188-3p could regulate spermatogenic cell apoptosis by targeting MLH1 [19]. However, the epigenetic regulatory mechanisms, especially the role of circRNAs, in NOA are yet unknown.
Hence, the purpose of this study was to elucidate the expression profile of circRNAs and unravel the possible functions and interactions of circRNAs in NOA.
Results
Identification of the dysregulated circRNAs between NOA and OA groups
We performed microarray-based profiling analyses to screen the differentially expressed circRNAs in 6 NOA and 3 OA tissues. The box plot depicts similar distributions of the intensities from all the normalized datasets (Figure 1A). Raw variation of circRNA expression between the two groups is shown in the scatter plot (Figure 1B). Volcano plots and hierarchical clustering exhibit the differentially expressed circRNAs between the NOA and OA samples (Figure 2A and 2B). Using a threshold of fold change (FC) ≥ 1.5 and a p-value < 0.05, we identified 82 differentially expressed circRNAs, of which 16 were up-regulated and 66 were down-regulated in NOA tissues compared with OA tissues (Table 1 and Table 2). According to the annotation of human circRNAs, we found 81.71% of the dysregulated circRNAs were exonic circRNAs, 9.76% were intronic circRNAs and 8.54% were sense overlapping circRNAs (Figure 2C). Further analysis revealed that these dysregulated circRNAs were mostly distributed on chr1, chr2, chr3, chr4, chr5, chr6, chr7, chr8, chr9, chr10, chr11, chr12, chr13, chr14, chr16, chr17, chr19, chr20, chr21 and chr22, but not on chr15, chr18, chrX and chrY (Figure 2D).
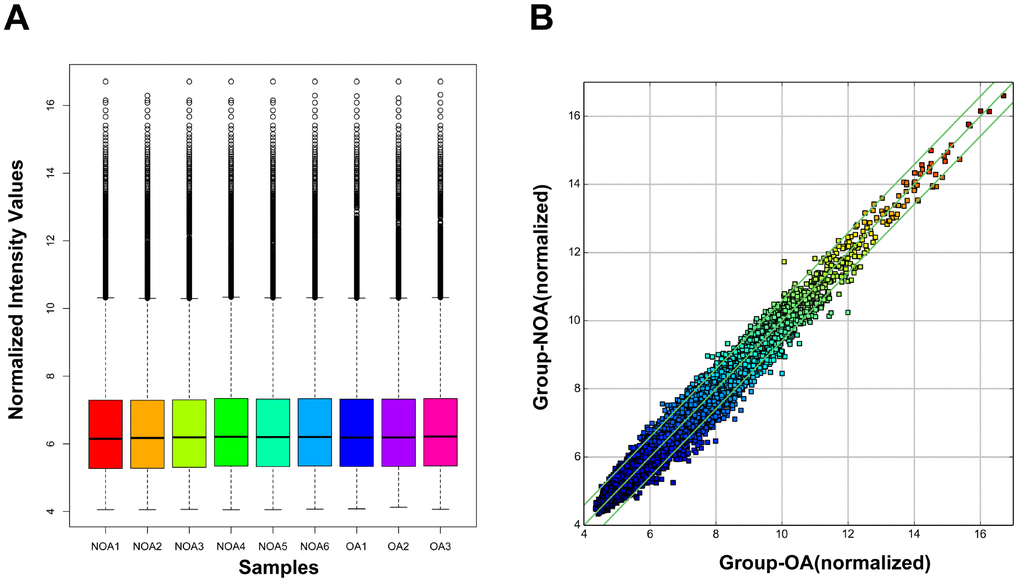
Figure 1. Box plot and scatter plot of circRNA signal value variation in OA and NOA samples. (A) The distribution of circRNAs for the nine samples. (OA: obstructive azoospermia; NOA: non-obstructive azoospermia). (B) Scatter plot showed variations in circRNA expression between OA and NOA samples. CircRNAs located above the top green line and below the bottom green line were more than 1.5-fold between OA and NOA samples.
Table 1. Significantly upregulated circRNAs in NOA.
| Significantly upregulated circRNAs | |||||
| probeID | P-value | FDR | FC (abs) | Regulation | circRNA |
| ASCRP3001555 | 0.0461916 | 0.957391589 | 1.52275 | up | hsa_circRNA_401590 |
| ASCRP3002427 | 0.016655094 | 0.957391589 | 1.5038532 | up | hsa_circRNA_002032 |
| ASCRP3002765 | 0.035853368 | 0.957391589 | 2.2698958 | up | hsa_circRNA_072697 |
| ASCRP3003870 | 0.010571567 | 0.957391589 | 1.7599646 | up | hsa_circRNA_101130 |
| ASCRP3004000 | 0.03573483 | 0.957391589 | 1.5243845 | up | hsa_circRNA_100452 |
| ASCRP3006656 | 0.019589109 | 0.957391589 | 1.7348182 | up | hsa_circRNA_103916 |
| ASCRP3007758 | 0.028997996 | 0.957391589 | 1.5351706 | up | hsa_circRNA_070294 |
| ASCRP3007963 | 0.019122487 | 0.957391589 | 1.7931198 | up | hsa_circRNA_001512 |
| ASCRP3009230 | 0.027775787 | 0.957391589 | 1.5701942 | up | hsa_circRNA_031757 |
| ASCRP3009252 | 0.004415576 | 0.957391589 | 1.5314721 | up | hsa_circRNA_104078 |
| ASCRP3009351 | 0.01558938 | 0.957391589 | 1.5958358 | up | hsa_circRNA_073942 |
| ASCRP3010355 | 0.013282587 | 0.957391589 | 1.6874562 | up | hsa_circRNA_030050 |
| ASCRP3010888 | 0.036692735 | 0.957391589 | 1.6901888 | up | hsa_circRNA_103075 |
| ASCRP3011370 | 0.042166787 | 0.957391589 | 1.5615877 | up | hsa_circRNA_406996 |
| ASCRP3011901 | 0.042668697 | 0.957391589 | 2.3337021 | up | hsa_circRNA_001506 |
| ASCRP3012974 | 0.037931142 | 0.957391589 | 1.5484401 | up | hsa_circRNA_002006 |
Table 2. Significantly downregulated circRNAs in NOA
| Significantly downregulated circRNAs | |||||
| probeID | P-value | FDR | FC (abs) | Regulation | circRNA |
| ASCRP3000334 | 0.016382909 | 0.957391589 | 1.7917313 | down | hsa_circRNA_403874 |
| ASCRP3000537 | 0.004831071 | 0.957391589 | 1.6995075 | down | hsa_circRNA_001937 |
| ASCRP3000663 | 0.021040239 | 0.957391589 | 1.6627497 | down | hsa_circRNA_406402 |
| ASCRP3000825 | 0.002705609 | 0.957391589 | 1.5361052 | down | hsa_circRNA_406267 |
| ASCRP3000954 | 0.005248263 | 0.957391589 | 1.746061 | down | hsa_circRNA_402130 |
| ASCRP3000991 | 0.037238178 | 0.957391589 | 1.730753 | down | hsa_circRNA_406822 |
| ASCRP3001155 | 0.045260941 | 0.957391589 | 1.536961 | down | hsa_circRNA_029998 |
| ASCRP3001768 | 0.025224767 | 0.957391589 | 2.7399933 | down | hsa_circRNA_406168 |
| ASCRP3001923 | 0.048642805 | 0.957391589 | 3.3645337 | down | hsa_circRNA_025349 |
| ASCRP3002888 | 0.049366594 | 0.957391589 | 1.6916213 | down | hsa_circRNA_402150 |
| ASCRP3002953 | 0.012038305 | 0.957391589 | 1.6022409 | down | hsa_circRNA_406503 |
| ASCRP3003279 | 0.036135037 | 0.957391589 | 1.9818485 | down | hsa_circRNA_406775 |
| ASCRP3003518 | 0.035892518 | 0.957391589 | 2.4505456 | down | hsa_circRNA_101053 |
| ASCRP3003843 | 0.018020351 | 0.957391589 | 1.9115564 | down | hsa_circRNA_406194 |
| ASCRP3003942 | 0.00052197 | 0.957391589 | 1.5953273 | down | hsa_circRNA_406419 |
| ASCRP3003975 | 0.009862311 | 0.957391589 | 1.5153189 | down | hsa_circRNA_103161 |
| ASCRP3004118 | 0.036664644 | 0.957391589 | 1.5911543 | down | hsa_circRNA_103141 |
| ASCRP3004393 | 0.026087538 | 0.957391589 | 1.5282233 | down | hsa_circRNA_067007 |
| ASCRP3004478 | 0.010656101 | 0.957391589 | 1.5616605 | down | hsa_circRNA_102558 |
| ASCRP3004662 | 0.033021538 | 0.957391589 | 1.7230277 | down | hsa_circRNA_000780 |
| ASCRP3004702 | 0.022526843 | 0.957391589 | 1.5259744 | down | hsa_circRNA_015279 |
| ASCRP3004757 | 0.012437973 | 0.957391589 | 2.2150382 | down | hsa_circRNA_104935 |
| ASCRP3005255 | 0.047336724 | 0.957391589 | 1.5978168 | down | hsa_circRNA_007328 |
| ASCRP3005451 | 0.001349981 | 0.957391589 | 1.5599409 | down | hsa_circRNA_103867 |
| ASCRP3005694 | 0.04141577 | 0.957391589 | 1.6261871 | down | hsa_circRNA_101894 |
| ASCRP3005793 | 0.031346564 | 0.957391589 | 1.6079474 | down | hsa_circRNA_102966 |
| ASCRP3006067 | 0.009529304 | 0.957391589 | 1.5492371 | down | hsa_circRNA_103348 |
| ASCRP3006096 | 0.038650961 | 0.957391589 | 1.5329346 | down | hsa_circRNA_023576 |
| ASCRP3006334 | 0.010459305 | 0.957391589 | 1.5086231 | down | hsa_circRNA_400185 |
| ASCRP3006335 | 0.023372267 | 0.957391589 | 2.2086086 | down | hsa_circRNA_404655 |
| ASCRP3006336 | 0.046143308 | 0.957391589 | 1.8350186 | down | hsa_circRNA_104940 |
| ASCRP3006936 | 0.025160274 | 0.957391589 | 1.9992925 | down | hsa_circRNA_102166 |
| ASCRP3007419 | 0.025860647 | 0.957391589 | 1.5221658 | down | hsa_circRNA_006604 |
| ASCRP3007490 | 4.4193E-05 | 0.587369562 | 1.5977581 | down | hsa_circRNA_406768 |
| ASCRP3007702 | 0.047580725 | 0.957391589 | 1.5580697 | down | hsa_circRNA_000883 |
| ASCRP3007956 | 0.034250275 | 0.957391589 | 2.0042991 | down | hsa_circRNA_006710 |
| ASCRP3008513 | 0.032785099 | 0.957391589 | 2.0605802 | down | hsa_circRNA_403457 |
| ASCRP3008758 | 0.022383408 | 0.957391589 | 2.1321055 | down | hsa_circRNA_400696 |
| ASCRP3008931 | 0.035547528 | 0.957391589 | 1.7530169 | down | hsa_circRNA_001695 |
| ASCRP3009069 | 0.014371347 | 0.957391589 | 1.5358115 | down | hsa_circRNA_103601 |
| ASCRP3009323 | 0.046773984 | 0.957391589 | 1.6990883 | down | hsa_circRNA_100166 |
| ASCRP3009406 | 0.027542185 | 0.957391589 | 2.1979834 | down | hsa_circRNA_102232 |
| ASCRP3009440 | 0.031767553 | 0.957391589 | 1.7600919 | down | hsa_circRNA_007624 |
| ASCRP3009894 | 0.021930582 | 0.957391589 | 1.6907468 | down | hsa_circRNA_002141 |
| ASCRP3009982 | 0.009261095 | 0.957391589 | 2.1517926 | down | hsa_circRNA_002796 |
| ASCRP3010075 | 0.006608806 | 0.957391589 | 1.7673139 | down | hsa_circRNA_051123 |
| ASCRP3010266 | 0.042141617 | 0.957391589 | 1.6013384 | down | hsa_circRNA_103181 |
| ASCRP3010752 | 0.049588744 | 0.957391589 | 1.531321 | down | hsa_circRNA_405511 |
| ASCRP3011101 | 0.031160151 | 0.957391589 | 1.9640111 | down | hsa_circRNA_059085 |
| ASCRP3011253 | 0.020745727 | 0.957391589 | 1.6280942 | down | hsa_circRNA_003145 |
| ASCRP3011607 | 0.027895917 | 0.957391589 | 1.6782159 | down | hsa_circRNA_092478 |
| ASCRP3011736 | 0.039612091 | 0.957391589 | 1.7179174 | down | hsa_circRNA_407124 |
| ASCRP3011751 | 0.023198686 | 0.957391589 | 1.6108817 | down | hsa_circRNA_100812 |
| ASCRP3011979 | 0.048109527 | 0.957391589 | 1.5443097 | down | hsa_circRNA_008142 |
| ASCRP3012222 | 0.047250017 | 0.957391589 | 1.6431699 | down | hsa_circRNA_001752 |
| ASCRP3012384 | 0.032114065 | 0.957391589 | 1.9400004 | down | hsa_circRNA_403560 |
| ASCRP3013001 | 0.021611589 | 0.957391589 | 2.4807294 | down | hsa_circRNA_003671 |
| ASCRP3013007 | 0.002843475 | 0.957391589 | 1.5463175 | down | hsa_circRNA_102552 |
| ASCRP3013008 | 0.001979569 | 0.957391589 | 1.6561938 | down | hsa_circRNA_102551 |
| ASCRP3013058 | 0.033675352 | 0.957391589 | 1.6553273 | down | hsa_circRNA_406350 |
| ASCRP3013126 | 0.043940509 | 0.957391589 | 2.3814219 | down | hsa_circRNA_402731 |
| ASCRP3013133 | 0.035888866 | 0.957391589 | 1.5808639 | down | hsa_circRNA_000328 |
| ASCRP3013332 | 0.004952971 | 0.957391589 | 1.5817435 | down | hsa_circRNA_104168 |
| ASCRP3013372 | 0.042368459 | 0.957391589 | 1.8241931 | down | hsa_circRNA_068259 |
| ASCRP3013501 | 0.029805804 | 0.957391589 | 1.6140515 | down | hsa_circRNA_100900 |
| ASCRP3013569 | 0.038372041 | 0.957391589 | 2.7556443 | down | hsa_circRNA_100981 |
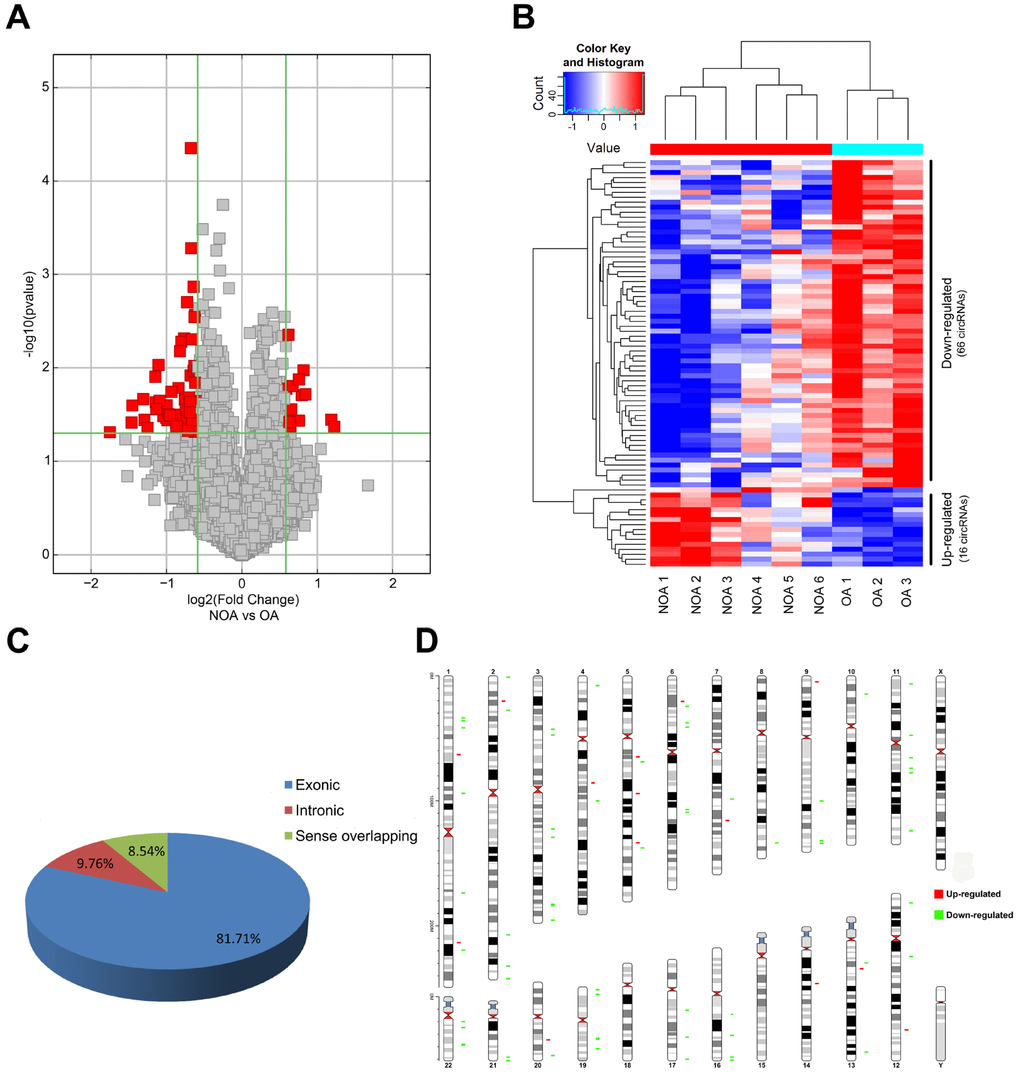
Figure 2. Characterization of differentially expressed circRNA in OA and NOA samples. (A) The volcano plots display the differential circRNAs expression between the two groups. Red points denote the differentially expressed circRNAs with statistical significance. (fold change ≥1.5 and p <0.05). (B) Heatmap shows the 16 up-regulated circRNAs and 66 down-regulated circRNAs in NOA. (C) The pie charts show the origin of transcription of differentially expressed circRNAs. (D) The chromosomal location of differentially expressed circRNAs is shown in the last figure. Red: up-regulated circRNAs in NOA; green: down-regulated circRNAs in NOA.
Validation of circRNAs using qRT-PCR
To further confirm the results of circRNA microarray, we chose six dysregulated circRNAs at random, including three down-regulated (hsa_circRNA_402130, hsa_circRNA_100812 and hsa_circRNA_406168) and three up-regulated (hsa_circRNA_072697, hsa_circ RNA_104078 and hsa_circRNA_030050). qRT-PCR in NOA and OA samples showed that the expression of hsa_circRNA_072697, hsa_circRNA_402130, hsa_ circRNA_100812, hsa_circRNA_030050 and hsa_circ RNA_406168 was consistent with the microarray data. And the difference in expression of hsa_circ RNA_072697, hsa_circRNA_030050, hsa_circRNA_ 100812, hsa_circRNA_402130 and hsa_circRNA_406168 was statistically significant (Figure 3A, 3C, 3D, 3E, 3F, p=0.0332, p=0.0156, p=0.0482, p=0.0139, p=0.0164). The expression of hsa_circRNA_104078, however, did not follow the microarray and was not statistically significant (Figure 3B, p=0.2795).
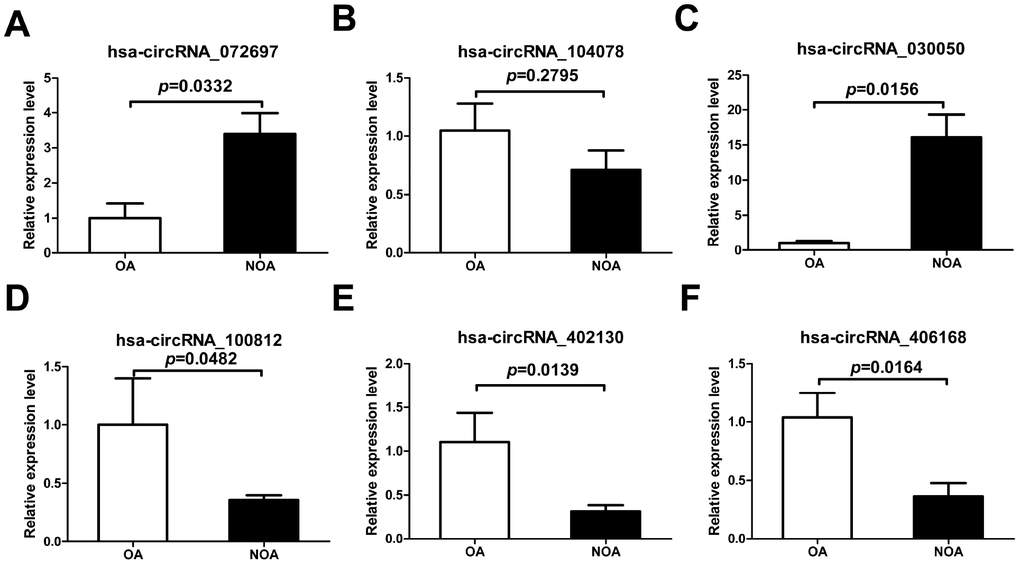
Figure 3. Confirmation of the six candidate circRNAs by qRT-PCR. All RNAs of 6 NOA and 3 OA patients were included. The experiments were repeated three times. 2-ΔΔCt method was used to measure gene expression. The values are presented as the mean ± SE.
MicroRNA response elements analysis of validated circRNAs
Several studies have demonstrated that circRNAs regulate the expression of their target genes by competing with their target miRNAs for binding [8, 20, 21]. The Arraystar’s homemade miRNA target prediction software based on TargetScan and miRanda was utilized to predict the circRNA-microRNA interactions of two confirmed circRNAs (hsa_circRNA_402130 and hsa_circRNA_ 072697). The top 5 targeted miRNAs were then identified based on the miRNA support vector regression (mirSVR) scores. Studies have shown that some NOA-related miRNAs are dysregulated in the testicular tissues of patients with NOA [22, 23]. So we selected the the potential miRNAs shared by the software and the reports to construct circRNA–miRNA interactions. The 2D structure was created from the sequence analysis of microRNA response elements (MREs) (Figure 4A and 4B). For hsa_circRNA_402130, the potential miRNA targets included hsa-let-7b-5p, hsa-let-7c-5p and hsa-let-7i-5p, and for hsa_circRNA_072697 the miRNA included hsa-miR-23c and hsa-miR-182-5p.
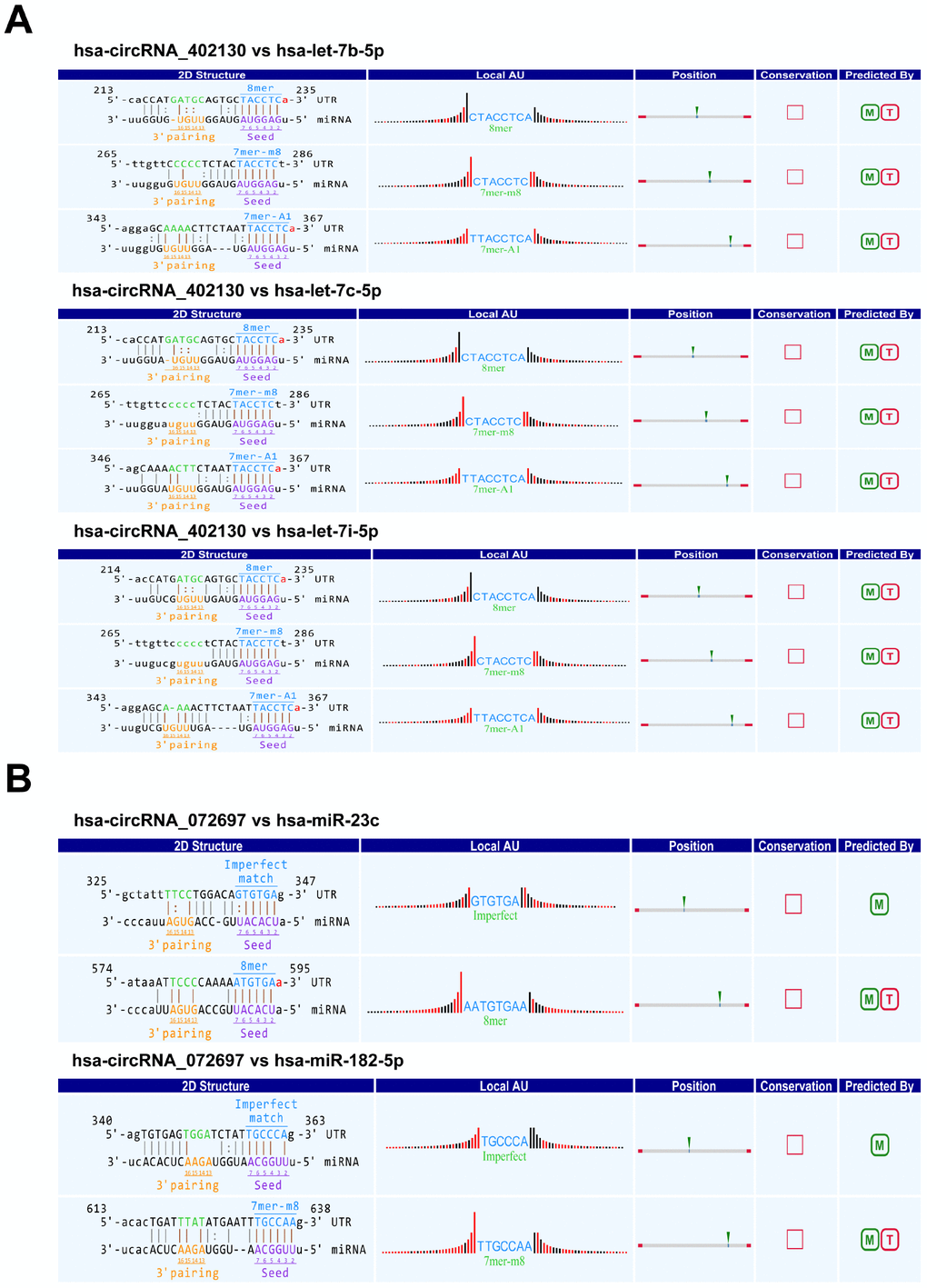
Figure 4. Prediction of circRNA/miRNA interactions by Arraystar’s homemade miRNA target prediction software based on TargetScan and miRanda. (A) The potential miRNA targets of hsa_circRNA_402130. (B) The potential miRNA targets of hsa_circRNA_072697. Local AU: Schematic diagram of the AU weights on both sides of the seed site. The empty square under “Conservation” indicates the lack of conserved information of circRNA between the species. M: miRanda; T: TargetScan.
CircRNA-miRNA-mRNA interaction analysis
To dissect out the functions of the two validated circRNAs, starBase v2.0 and miRDB identified down-stream target genes of the abovementioned candidate miRNAs. The two circRNA-miRNA-mRNA regulatory networks (built by OmicShare tools) included 3 miRNAs and 212 mRNAs in the ceRNA network of hsa_circRNA_402130 (Figure 5A, Supplementary Table 1), and 2 miRNAs and 411 mRNAs in the ceRNA network of hsa_circRNA_072697 (Figure 5B, Supplementary Table 2). As shown in Figure 5A, we found that this network contains multiple transcription factors such as HMGA2, SOX13, E2F5, LIN28B and so on. Similarly, multiple transcription factors such as FOXO3, FOXP2 and SP3 are included in the regulatory network of Figure 5B. These regulatory networks revealed that circRNAs may play a crucial part in the process of NOA.
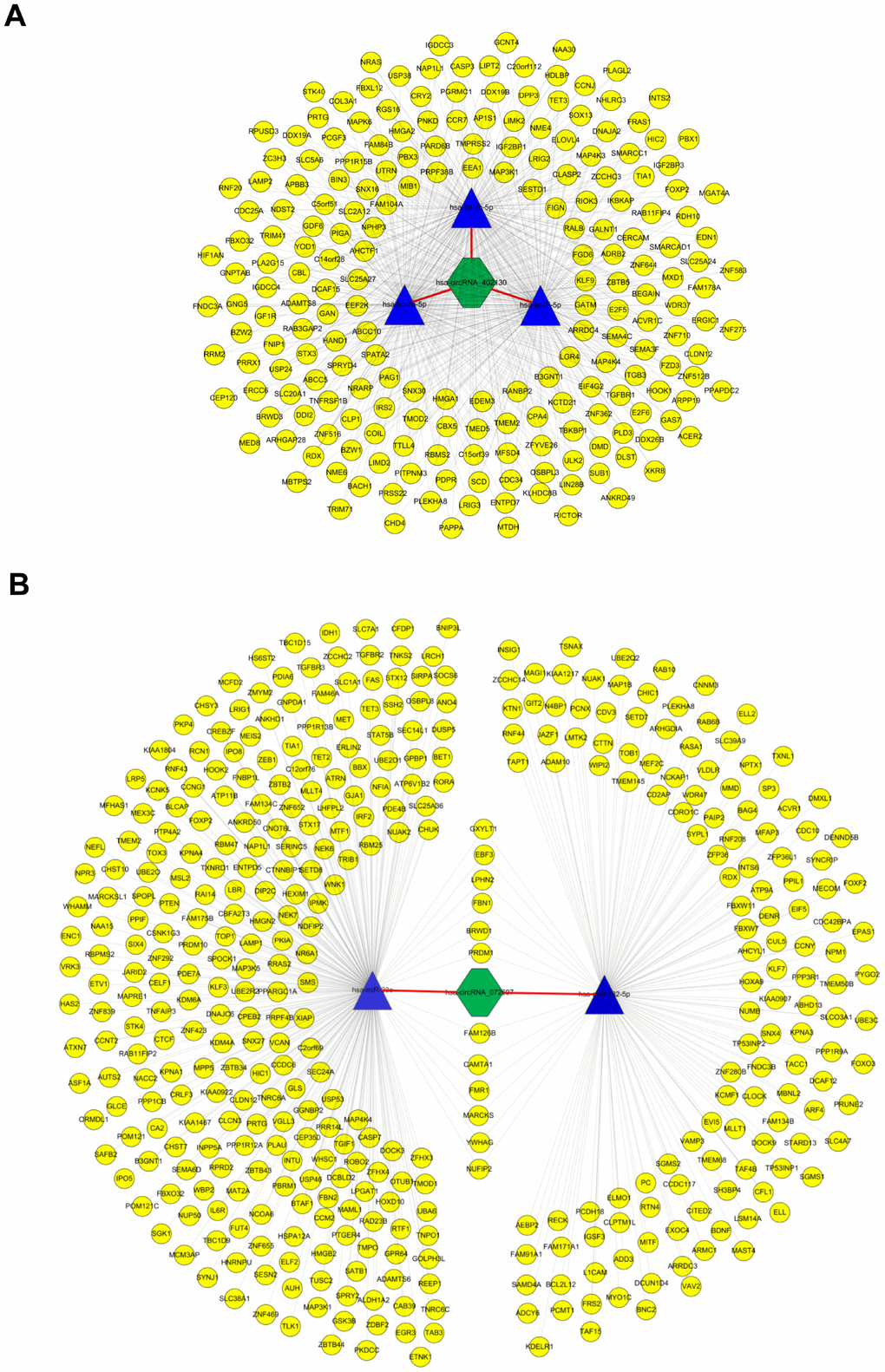
Figure 5. CircRNA-miRNA-mRNA network analysis. (A) The ceRNA network of hsa_circRNA_402130. The network consists of 3 miRNAs and 212 genes with 631 relationships. (B) The ceRNA network of hsa_circRNA_072697. The network consists of 2 miRNAs and 411 genes with 425 relationships. The yellow round node represents a protein-coding gene, blue triangle node represents a miRNA and a green hexagon represents a circRNA.
GO and KEGG enrichment analysis of the predicted network genes for hsa_circRNA_402130 and hsa_circRNA_072697
The functions of all the target genes were analyzed by online biological classification tool DAVID. The top 10 GO terms and KEGG terms are displayed as bubble diagram. The GO biological process (BP) analysis (Figure 6A) showed that the target genes were significantly enriched in positive regulations including transcription from RNA polymerase II promoter, transcription, cell migration, protein phosphorylation, palate development, pathway-restricted SMAD protein phosphorylation, circadian regulation of gene expression. KEGG pathway analysis (Figure 6B), showed target genes were enriched for microRNAs in cancer, MAPK signaling pathway, signaling pathways regulating pluripotency of stem cells, FoxO signaling pathway, ubiquitin mediated proteolysis, axon guidance, transcriptional misregulation in cancer, proteoglycans in cancer and TNF signaling pathway. These findings imply that circRNAs might participate in the development of NOA through multiple signaling pathways.
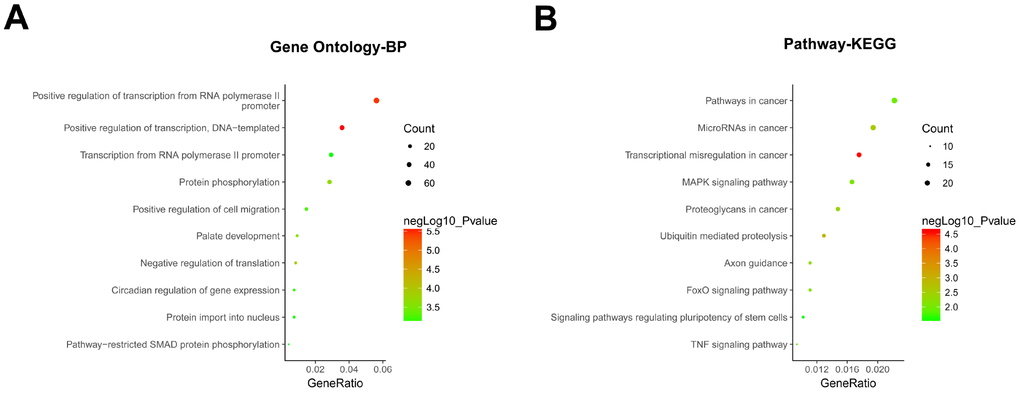
Figure 6. GO and KEGG enrichment analysis of hsa_circRNA_402130 and hsa_circRNA_072697. (A) Gene ontology (GO) enrichment analysis of the two circRNAs predicted target genes in terms of biological processes (BP). (B) KEGG enrichment analysis of the two circRNAs predicted target genes. The top 10 significantly enriched activities were shown above. The size of each circle indicates the counting number on each part, while the color represents the negative logarithm of p value of the enrichment analysis.
Discussion
Results from the Human Genome Project and the DNA Elements Encyclopedia Project indicated that protein-coding gene sequences account for only 1-3% of the human genome sequence. Also, most of the transcribed sequences in the human genome are non-coding RNA sequences [24]. Studies demonstrated that non-coding RNAs including miRNAs and lncRNAs participate in the occurrence and development of NOA [13, 18, 22, 23, 25–27]. However, the role of circRNAs, a class of non-coding RNA, has not been reported in NOA.
Here, we constructed the expression profile of circRNAs in NOA by high-throughput circRNA microarray for the first time. 16 up-regulated and 66 down-regulated circRNAs in NOA were identified. As most of these circRNAs originate from exons or introns, these results imply that dysregulated expression of circRNAs may be involved in the process of NOA. Subsequently, differential expression of circRNAs was confirmed by qRT-PCR. As circRNAs usually function as RNA sponge to regulate gene expression [4, 20, 21], bioinformatics was used to predict target miRNAs of hsa_circRNA_402130 and hsa_circRNA_072697. The results showed that hsa-let-7b-5p, hsa-let-7c-5p and hsa-let-7i-5p had a binding site for hsa_circRNA_402130, and hsa-miR-23c and hsa-miR-182-5p had a binding site for hsa_circRNA_ 072697. These indicated that hsa-let-7c-5p, hsa-let-7b-5p and hsa-let-7i-5p were the targets for hsa_ circRNA_402130, and hsa-miR-23c and hsa-miR-182-5p were the targets for hsa_circRNA_072697. In addition, these predicted miRNAs were previously reported that hsa-let-7b-5p, hsa-let-7c-5p and hsa-let-7i-5p were up-regulated and hsa-miR-23c and hsa-miR-182-5p were down-regulated in NOA [22, 23]. Among these target miRNAs, let-7 family is an important regulator of stemness by regulating LIN28A, a gene that plays pivotal roles in organogenesis, embryonic development and tumorigenesis [28–30]. Jixin Tang et al. [31] indicated that let-7 miRNA family (let-7c-5p and let-7i-5p) regulate spermatogenesis in mice through LIN28A. Another member of the let-7 miRNA family let-7g could significantly reduce the germ cell pool in mice [32]. Based on these findings, we speculate that hsa_circRNA_402130 may cause NOA by regulating the expression of let-7 miRNA family.
Moreover, based on the above two circRNAs, we constructed “circRNA-miRNA-mRNA” ceRNA regulatory networks. From these regulatory networks, we found many transcription factors that regulate spermatogenesis. Studies have shown that SP3 presents a unique expression pattern during spermatogenesis in mice and other mammalian [33, 34]. It has been confirmed that HMGA2 was expressed during the process from spermatocyte to spermatid in mice, and the phenotype of HMGA2-null mice was few spermatids and complete absence of spermatozoa [35]. We also found some protein coding genes that are affected by let-7 miRNAs involved in spermatogenesis. TET3 is expressed in round spermatids and sperm and its effect on sperm methylome might be essential in human [36]. As early as 2006, a group found that CDC25A was down-regulated in the testis tissue of male infertile patients [37]. Subsequent studies suggested that CDC25A might be regulated by BOLL and participate in human spermatogenesis [38]. These evidences all indirectly indicated that circRNAs were involved in spermatogenesis. However, experiments are required to confirm these interactions and, to elucidate the localization of circRNAs and target miRNAs.
Additionally, we also performed GO and KEGG enrichment analysis of miRNA target genes for the first time. GO enrichment analysis revealed miRNA target genes that positively regulate transcription from RNA polymerase II promoter, transcription, cell migration, protein phosphorylation, palate development, pathway-restricted SMAD protein phosphorylation, circadian regulation of gene expression and so on. Given that phosphorylation of SMAD promotes differentiation of mouse SSC [39], and proliferation of germ cells in mice [40], it is possible that circRNAs may regulate spermatogenesis by affecting SMAD activity in NOA patients. Among the KEGG pathways associated with the two circRNAs, FoxO signaling pathway and MAPK signaling pathway are strongly associated with spermatogenesis in mice [41–43]. Based on our and previous data, we can easily say that humans and mice share many common ways in the regulation of spermatogenesis. More studies on NOA patients should be conducted to improve our understanding of the mechanisms of spermatogenesis and NOA to validate the results of animal experiments. In addition, we identified a signaling pathway involved in regulation pluripotency of stem cells within the network of the dysregulated circRNAs. This is significant as self-renewal and mitosis of spermatogonial stem cells are important steps in human spermatogenesis. Based on the abovementioned ceRNA networks and enrichment analysis results, we have discovered many molecules and pathways related to stemness. Thus, we suspect that these two circRNAs may be involved in the development of NOA by regulating the stemness of spermatogonial stem cells. In a word, our study provides new ideas and strategies for NOA research. But whether circRNAs are involved in these processes deserves to be confirmed by subsequent in vitro and in vivo experiments.
Our study also has two limitations. First, more testicular tissue samples could be included in this study. This might reduce the error caused by individual differences of patients. In addition, due to the difficulties in acquiring testicular samples, we were unable to use the testicular samples of volunteers who had already been known fertility and normal spermatogenesis as normal controls.
In summary, this is the first report that reveals the expression profile of differentially expressed circRNAs and, in conjunction with bioinformatics analysis, provides the first assessment of ceRNA networks and circRNAs associated pathways in NOA. Results indicated that circRNAs may play important functions and have potential to become therapeutic targets for NOA.
Materials and Methods
Testicular tissue samples collection
The study was approved by the Ethics Committee of the Institute of Human Reproduction and Stem Cell Engineering of Central South University. Testicular biopsies were collected from 6 NOA and 3 OA patients. All participants signed informed consent, and routine semen analysis based on the World Health Organization criteria showed no sperm. Most patients have been tested for follicle-stimulating hormone (FSH), luteinizing hormone (LH) and testosterone (T). All the patients underwent testicular fine needle aspiration for histological examination at Reproductive and Genetic Hospital of CITIC-Xiangya. Those patients without testicular sperm were defined as NOA patients. OA patients had normal spermatogenic function accompanied by blockage of the vas deferens, but no other congenital diseases. The results of the pathological examinations are in the (Supplementary Figure 1). None of the patients were exposed to radiation, high temperature or toxic substances prior to their hospitalization. NOA patients with chromosomal abnormalities, Y chromosome microdeletion, and varicocele were excluded from the study.
Total RNA extraction
Nine RNA samples of testicular biopsies from NOA and OA patients were extracted using TRIzol (Invitrogen, USA) following the manufacturer’s protocol. The quantity and purity of total RNA was measured on the NanoDrop® ND-1000 spectrophotometer (Thermo Fisher, USA). RNA integrity was measured by denaturing agarose gel electrophoresis.
Microarray detection
All RNA samples were analyzed by Arraystar circRNA Microarray at Kangchen Bio-tech (Shanghai, China). Sample labeling and array hybridization were performed according to the manufacturer’s protocol (Arraystar Inc.). The hybridized arrays were washed, fixed and scanned on the Agilent Scanner G2505C. Agilent Feature Extraction software was used for raw data extraction. The data was then processed using the limma package of R software. Low intensity was filtered after quantile normalization of the raw data. Student’s t-test was used to estimate the statistical significance between groups. CircRNAs with a fold change ≥ 1.5 and p < 0.05 were considered significant.
Quantitative real-time reverse transcription PCR
The cDNA was prepared from 1ug of total RNA using Super Script TMIII Reverse Transcriptase kit (Invitrogen) according to the manufacturer’s instructions. Quantitative real-time reverse transcription PCR (qRT-PCR) was performed using a 2×PCR master mix (Arraystar) on the ViiA 7 Real-time PCR System (Applied Biosystems) to detect the relative expression levels of target circRNAs. The primer sequences for the target circRNAs are as follows: hsa_ circRNA_402130 forward: 5′-GTGGCCGAGGACTTTGATTG-3′, reverse: 5′-CCTGTAACAACGCATCTCATATT-3′; hsa_circRNA_100812 forward: 5′-TATTCTCAAGCTGTCACAGGACATT-3′, reverse: 5′-TGAGGGTAGCAGCAGAACGAG-3′; hsa_circRNA_072697 forward: 5′-TGATAGAAAAGTTAGAATTTTCAGA-3′, reverse: 5′-ACTCTTTCAAACTCTAAGAGCTTAG-3′; hsa_circRNA_104078 forward: 5′-GCTTATGGCTATAAAATTACAGAGA-3′, reverse: 5′-CGGGACAACATCCTTTCTTAC-3′; hsa_circRNA_030050 forward: 5′-GGGAGAAGCAGCTAGAACCA-3′; reverse: 5′-TT TGCCAGAATACCCCTTTG-3′; hsa_circRNA_406168 forward: 5′-ATTGGGTTCTTTGCCTGTTG-3′; reverse: 5′-GGGGCAGACAGATGAGAAAG-3′; β-actin forward: 5′-GTGGCCGAGGACTTTGATTG-3′, reverse: 5′-CCTGTAACAACGCATCTCATATT-3′. Transcript level of the housekeeping gene β-actin was used to normalize the relative expression of circRNAs. Data is represented as mean ± SE of three independent experiments.
Annotation and functional prediction of hsa_circRNA_402130 and hsa_circRNA_072697
The circRNA-microRNA interactions of hsa_circRNA_402130 and hsa_circRNA_072697 were predicted with Arraystar’s home-made microRNA target prediction software based on TargetScan [44] and miRanda [45]. The analysis process of the software is as follows: First, it performs target prediction by using the miRNA information in the latest miRBase on the target sequence. Then, candidate miRNAs are screened by Context+<=-0.05 and Context<=-0.05. Finally, according to the miRNA coverage of MREs (MicroRNA Recoginition Elements) on the target sequence, the required circRNA-microRNA interactions are obtained. The microRNA-mRNA interactions were predicted by starBase v2.0 (http://starbase.sysu.edu.cn/browser.php) [46, 47] and miRDB (http://www.mirdb.org) [48, 49]. GO and KEGG enrichment analysis of circRNA-targeting genes were performed by DAVID https://david.ncifcrf.gov/. Then OmicShare tools (http://www.omicshare.com/tools) was used to create potential maps of the circRNA/miRNA/mRNA interaction networks of hsa_circ_402130 and hsa_circ_072697.
Statistical analysis
Student’s t-test (two-tailed) was used to estimate statistical significance between groups. Data analysis was performed using GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA). A p-value < 0.05 was considered significant.
Abbreviations
ART: assisted reproductive technology; BP: biological processce; ceRNA: competing endogenous RNA; circRNAs: Circular RNAs; FSH: follicle-stimulating hormone; GWAS: genome-wide association analysis; LH: luteinizing hormone; MREs: microRNA response elements; NOA: non-obstructive azoospermia; OA: obstructive azoospermia; qRT-PCR: quantitative real-time reverse transcription PCR; T: testosterone.
Author Contributions
Liqing Fan designed and guided the research. Hao Bo and Fang Zhu performed the experiments. Hao Bo, Han Zhang, Zhizhong Liu and Xingming Wang analyzed the data. Ruiling Tang, Fang Zhu, Dai Zhou and Guanghui Gong collected and prepared the samples. Hao Bo and Zhizhong Liu wrote the paper. Wenbin Zhu, Yueqiu Tan and Liqing Fan revised the manuscript, and all authors had approved the final manuscript.
Acknowledgments
We thank Kangchen Bio-tech (Shanghai, China) for the excellent technical assistance.
Conflicts of Interest
The authors declare there are no conflicts of interest in this work.
Funding
This work was supported by the National Natural Science Foundation of China (No. 31472054), the National Key Research and Development Program of China (2016YFC1000600), the Fundamental Research Funds for the Central Universities of Central South University (2019zzts715) and the Special Fund of Clinical Medicine of Chinese Medical Association (17020280697).
References
- 1. Suzuki H, Zuo Y, Wang J, Zhang MQ, Malhotra A, Mayeda A. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res. 2006; 34:e63. https://doi.org/10.1093/nar/gkl151 [PubMed]
- 2. Li Z, Huang C, Bao C, Chen L, Lin M, Wang X, Zhong G, Yu B, Hu W, Dai L, Zhu P, Chang Z, Wu Q, et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol. 2015; 22:256–64. https://doi.org/10.1038/nsmb.2959 [PubMed]
- 3. Holdt LM, Stahringer A, Sass K, Pichler G, Kulak NA, Wilfert W, Kohlmaier A, Herbst A, Northoff BH, Nicolaou A, Gäbel G, Beutner F, Scholz M, et al. Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat Commun. 2016; 7:12429. https://doi.org/10.1038/ncomms12429 [PubMed]
- 4. Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B, Luo Y, Lyu D, Li Y, Shi G, Liang L, Gu J, He X, Huang S. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 2016; 7:11215. https://doi.org/10.1038/ncomms11215 [PubMed]
- 5. Dong WW, Li HM, Qing XR, Huang DH, Li HG. Identification and characterization of human testis derived circular RNAs and their existence in seminal plasma. Sci Rep. 2016; 6:39080. https://doi.org/10.1038/srep39080 [PubMed]
- 6. Szabo L, Morey R, Palpant NJ, Wang PL, Afari N, Jiang C, Parast MM, Murry CE, Laurent LC, Salzman J. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 2015; 16:126. https://doi.org/10.1186/s13059-015-0690-5 [PubMed]
- 7. Qian Y, Lu Y, Rui C, Qian Y, Cai M, Jia R. Potential Significance of Circular RNA in Human Placental Tissue for Patients with Preeclampsia. Cell Physiol Biochem. 2016; 39:1380–90. https://doi.org/10.1159/000447842 [PubMed]
- 8. Qiu M, Xia W, Chen R, Wang S, Xu Y, Ma Z, Xu W, Zhang E, Wang J, Fang T, Hu J, Dong G, Yin R, et al. The circular RNA circPRKCI promotes tumor growth in lung adenocarcinoma. Cancer Res. 2018; 78:2839–51. https://doi.org/10.1158/0008-5472.CAN-17-2808 [PubMed]
- 9. Sand M, Bechara FG, Sand D, Gambichler T, Hahn SA, Bromba M, Stockfleth E, Hessam S. Circular RNA expression in basal cell carcinoma. Epigenomics. 2016; 8:619–32. https://doi.org/10.2217/epi-2015-0019 [PubMed]
- 10. Shen L, Zhang Y, Zhou W, Peng Z, Hong X, Zhang Y. Circular RNA expression in ovarian endometriosis. Epigenomics. 2018; 10:559–72. https://doi.org/10.2217/epi-2017-0079 [PubMed]
- 11. Wang Z, Xu P, Chen B, Zhang Z, Zhang C, Zhan Q, Huang S, Xia ZA, Peng W. Identifying circRNA-associated-ceRNA networks in the hippocampus of Aβ1-42-induced Alzheimer’s disease-like rats using microarray analysis. Aging (Albany NY). 2018; 10:775–88. https://doi.org/10.18632/aging.101427 [PubMed]
- 12. Wosnitzer M, Goldstein M, Hardy MP. Review of Azoospermia. Spermatogenesis. 2014; 4:e28218. https://doi.org/10.4161/spmg.28218 [PubMed]
- 13. Yao C, Yuan Q, Niu M, Fu H, Zhou F, Zhang W, Wang H, Wen L, Wu L, Li Z, He Z. Distinct Expression Profiles and Novel Targets of MicroRNAs in Human Spermatogonia, Pachytene Spermatocytes, and Round Spermatids between OA Patients and NOA Patients. Mol Ther Nucleic Acids. 2017; 9:182–94. https://doi.org/10.1016/j.omtn.2017.09.007 [PubMed]
- 14. Hu Z, Xia Y, Guo X, Dai J, Li H, Hu H, Jiang Y, Lu F, Wu Y, Yang X, Li H, Yao B, Lu C, et al. A genome-wide association study in Chinese men identifies three risk loci for non-obstructive azoospermia. Nat Genet. 2011; 44:183–86. https://doi.org/10.1038/ng.1040 [PubMed]
- 15. Yatsenko AN, Georgiadis AP, Röpke A, Berman AJ, Jaffe T, Olszewska M, Westernströer B, Sanfilippo J, Kurpisz M, Rajkovic A, Yatsenko SA, Kliesch S, Schlatt S, Tüttelmann F. X-linked TEX11 mutations, meiotic arrest, and azoospermia in infertile men. N Engl J Med. 2015; 372:2097–107. https://doi.org/10.1056/NEJMoa1406192 [PubMed]
- 16. Zhao H, Xu J, Zhang H, Sun J, Sun Y, Wang Z, Liu J, Ding Q, Lu S, Shi R, You L, Qin Y, Zhao X, et al. A genome-wide association study reveals that variants within the HLA region are associated with risk for nonobstructive azoospermia. Am J Hum Genet. 2012; 90:900–06. https://doi.org/10.1016/j.ajhg.2012.04.001 [PubMed]
- 17. Wu W, Qin Y, Li Z, Dong J, Dai J, Lu C, Guo X, Zhao Y, Zhu Y, Zhang W, Hang B, Sha J, Shen H, et al. Genome-wide microRNA expression profiling in idiopathic non-obstructive azoospermia: significant up-regulation of miR-141, miR-429 and miR-7-1-3p. Hum Reprod. 2013; 28:1827–36. https://doi.org/10.1093/humrep/det099 [PubMed]
- 18. Lü M, Tian H, Cao YX, He X, Chen L, Song X, Ping P, Huang H, Sun F. Downregulation of miR-320a/383-sponge-like long non-coding RNA NLC1-C (narcolepsy candidate-region 1 genes) is associated with male infertility and promotes testicular embryonal carcinoma cell proliferation. Cell Death Dis. 2015; 6:e1960. https://doi.org/10.1038/cddis.2015.267 [PubMed]
- 19. Song WY, Meng H, Wang XG, Jin HX, Yao GD, Shi SL, Wu L, Zhang XY, Sun YP. Reduced microRNA-188-3p expression contributes to apoptosis of spermatogenic cells in patients with azoospermia. Cell Prolif. 2017; 50:50. https://doi.org/10.1111/cpr.12297 [PubMed]
- 20. Han D, Li J, Wang H, Su X, Hou J, Gu Y, Qian C, Lin Y, Liu X, Huang M, Li N, Zhou W, Yu Y, Cao X. Circular RNA circMTO1 acts as the sponge of microRNA-9 to suppress hepatocellular carcinoma progression. Hepatology. 2017; 66:1151–64. https://doi.org/10.1002/hep.29270 [PubMed]
- 21. Wu J, Jiang Z, Chen C, Hu Q, Fu Z, Chen J, Wang Z, Wang Q, Li A, Marks JR, Guo C, Chen Y, Zhou J, et al. CircIRAK3 sponges miR-3607 to facilitate breast cancer metastasis. Cancer Lett. 2018; 430:179–92. https://doi.org/10.1016/j.canlet.2018.05.033 [PubMed]
- 22. Lian J, Zhang X, Tian H, Liang N, Wang Y, Liang C, Li X, Sun F. Altered microRNA expression in patients with non-obstructive azoospermia. Reprod Biol Endocrinol. 2009; 7:13. https://doi.org/10.1186/1477-7827-7-13 [PubMed]
- 23. Zhuang X, Li Z, Lin H, Gu L, Lin Q, Lu Z, Tzeng CM. Integrated miRNA and mRNA expression profiling to identify mRNA targets of dysregulated miRNAs in non-obstructive azoospermia. Sci Rep. 2015; 5:7922. https://doi.org/10.1038/srep07922 [PubMed]
- 24. Maher B. ENCODE: the human encyclopaedia. Nature. 2012; 489:46–48. https://doi.org/10.1038/489046a [PubMed]
- 25. Ji J, Qin Y, Zhou R, Zang R, Huang Z, Zhang Y, Chen M, Wu W, Song L, Ling X, Shen H, Hu Z, Xia Y, et al. X chromosome-wide identification of SNVs in microRNA genes and non-obstructive azoospermia risk in Han Chinese population. Oncotarget. 2016; 7:49122–29. https://doi.org/10.18632/oncotarget.8759 [PubMed]
- 26. Tang D, Huang Y, Liu W, Zhang X. Up-Regulation of microRNA-210 is Associated with Spermatogenesis by Targeting IGF2 in Male Infertility. Med Sci Monit. 2016; 22:2905–10. https://doi.org/10.12659/MSM.897340 [PubMed]
- 27. Luk AC, Chan WY, Rennert OM, Lee TL. Long noncoding RNAs in spermatogenesis: insights from recent high-throughput transcriptome studies. Reproduction. 2014; 147:R131–41. https://doi.org/10.1530/REP-13-0594 [PubMed]
- 28. Repetto E, Briata P, Kuziner N, Harfe BD, McManus MT, Gherzi R, Rosenfeld MG, Trabucchi M. Let-7b/c enhance the stability of a tissue-specific mRNA during mammalian organogenesis as part of a feedback loop involving KSRP. PLoS Genet. 2012; 8:e1002823. https://doi.org/10.1371/journal.pgen.1002823 [PubMed]
- 29. Madison BB, Jeganathan AN, Mizuno R, Winslow MM, Castells A, Cuatrecasas M, Rustgi AK. Let-7 Represses Carcinogenesis and a Stem Cell Phenotype in the Intestine via Regulation of Hmga2. PLoS Genet. 2015; 11:e1005408. https://doi.org/10.1371/journal.pgen.1005408 [PubMed]
- 30. Kloosterman WP, Wienholds E, Ketting RF, Plasterk RH. Substrate requirements for let-7 function in the developing zebrafish embryo. Nucleic Acids Res. 2004; 32:6284–91. https://doi.org/10.1093/nar/gkh968 [PubMed]
- 31. Tang JX, Li J, Cheng JM, Hu B, Sun TC, Li XY, Batool A, Wang ZP, Wang XX, Deng SL, Zhang Y, Chen SR, Huang X, Liu YX. Requirement for CCNB1 in mouse spermatogenesis. Cell Death Dis. 2017; 8:e3142. https://doi.org/10.1038/cddis.2017.555 [PubMed]
- 32. Shinoda G, De Soysa TY, Seligson MT, Yabuuchi A, Fujiwara Y, Huang PY, Hagan JP, Gregory RI, Moss EG, Daley GQ. Lin28a regulates germ cell pool size and fertility. Stem Cells. 2013; 31:1001–09. https://doi.org/10.1002/stem.1343 [PubMed]
- 33. Yoshioka H, Geyer CB, Hornecker JL, Patel KT, McCarrey JR. In vivo analysis of developmentally and evolutionarily dynamic protein-DNA interactions regulating transcription of the Pgk2 gene during mammalian spermatogenesis. Mol Cell Biol. 2007; 27:7871–85. https://doi.org/10.1128/MCB.00990-07 [PubMed]
- 34. Ma W, Horvath GC, Kistler MK, Kistler WS. Expression patterns of SP1 and SP3 during mouse spermatogenesis: SP1 down-regulation correlates with two successive promoter changes and translationally compromised transcripts. Biol Reprod. 2008; 79:289–300. https://doi.org/10.1095/biolreprod.107.067082 [PubMed]
- 35. Chieffi P, Battista S, Barchi M, Di Agostino S, Pierantoni GM, Fedele M, Chiariotti L, Tramontano D, Fusco A. HMGA1 and HMGA2 protein expression in mouse spermatogenesis. Oncogene. 2002; 21:3644–50. https://doi.org/10.1038/sj.onc.1205501 [PubMed]
- 36. Ni K, Dansranjavin T, Rogenhofer N, Oeztuerk N, Deuker J, Bergmann M, Schuppe HC, Wagenlehner F, Weidner W, Steger K, Schagdarsurengin U. TET enzymes are successively expressed during human spermatogenesis and their expression level is pivotal for male fertility. Hum Reprod. 2016; 31:1411–24. https://doi.org/10.1093/humrep/dew096 [PubMed]
- 37. Cheng YS, Kuo PL, Teng YN, Kuo TY, Chung CL, Lin YH, Liao RW, Lin JS, Lin YM. Association of spermatogenic failure with decreased CDC25A expression in infertile men. Hum Reprod. 2006; 21:2346–52. https://doi.org/10.1093/humrep/del163 [PubMed]
- 38. Lin YM, Chung CL, Cheng YS. Posttranscriptional regulation of CDC25A by BOLL is a conserved fertility mechanism essential for human spermatogenesis. J Clin Endocrinol Metab. 2009; 94:2650–57. https://doi.org/10.1210/jc.2009-0108 [PubMed]
- 39. Li Y, Zhang Y, Zhang X, Sun J, Hao J. BMP4/Smad signaling pathway induces the differentiation of mouse spermatogonial stem cells via upregulation of Sohlh2. Anat Rec (Hoboken). 2014; 297:749–57. https://doi.org/10.1002/ar.22891 [PubMed]
- 40. Wu FJ, Lin TY, Sung LY, Chang WF, Wu PC, Luo CW. BMP8A sustains spermatogenesis by activating both SMAD1/5/8 and SMAD2/3 in spermatogonia. Sci Signal. 2017; 10:10. https://doi.org/10.1126/scisignal.aal1910 [PubMed]
- 41. Goertz MJ, Wu Z, Gallardo TD, Hamra FK, Castrillon DH. Foxo1 is required in mouse spermatogonial stem cells for their maintenance and the initiation of spermatogenesis. J Clin Invest. 2011; 121:3456–66. https://doi.org/10.1172/JCI57984 [PubMed]
- 42. Huang P, Zhou Z, Shi F, Shao G, Wang R, Wang J, Wang K, Ding W. Effects of the IGF-1/PTEN/Akt/FoxO signaling pathway on male reproduction in rats subjected to water immersion and restraint stress. Mol Med Rep. 2016; 14:5116–24. https://doi.org/10.3892/mmr.2016.5880 [PubMed]
- 43. Almog T, Naor Z. Mitogen activated protein kinases (MAPKs) as regulators of spermatogenesis and spermatozoa functions. Mol Cell Endocrinol. 2008; 282:39–44. https://doi.org/10.1016/j.mce.2007.11.011 [PubMed]
- 44. Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003; 5:R1. https://doi.org/10.1186/gb-2003-5-1-r1 [PubMed]
- 45. Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet. 2012; 13:271–82. https://doi.org/10.1038/nrg3162 [PubMed]
- 46. Yang JH, Li JH, Shao P, Zhou H, Chen YQ, Qu LH. starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 2011 (Data Issue); 39:D202–09. https://doi.org/10.1093/nar/gkq1056 [PubMed]
- 47. Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014; 42:D92–97. https://doi.org/10.1093/nar/gkt1248 [PubMed]
- 48. Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015; 43:D146–52. https://doi.org/10.1093/nar/gku1104 [PubMed]
- 49. Wang X. Improving microRNA target prediction by modeling with unambiguously identified microRNA-target pairs from CLIP-ligation studies. Bioinformatics. 2016; 32:1316–22. https://doi.org/10.1093/bioinformatics/btw002 [PubMed]