



Introduction
The transcription factor SOX2, a master regulator of embryonic and induced pluripotent stem cells [1–4], is causally related to tumor initiation, aggressiveness, and metastasis likely due to its ability to induce and maintain the stemness of cancer stem cells (CSC) [5, 6]. Given that a key mechanism of drug resistance relates to the incapacity of most standard therapeutics to eradicate the minor subpopulation of CSC with self-renewal and seeding capacity, SOX2 has been suggested as an attractive anti-cancer target to prevent CSC-mediated clinical relapse [7, 8]. Unfortunately, the “undruggable” characteristics of transcription factors such as SOX2 has largely constrained the clinical potential of SOX2-centered therapeutic strategies in major cancer types.
Pre-clinical approaches such as SOX2-targeting siRNAs, shRNAs, or miRNAs offer little therapeutic value because of their poor efficacy and delivery. By the same token, zinc-finger-based artificial transcription factors, which can modify the epi-transcriptional state of endogenous promoters with single locus specificity [9, 10], have been employed to reduce SOX2 mRNA and protein via targeting of proximal SOX2 promoters in cultured cancer cells and xenografts [7, 11], but their poor in vivo delivery to solid tumor tissue limits their usefulness for stable SOX2 down-regulation in a clinical context. Targeting of SOX2-related upstream/downstream signaling pathways has become a more plausible approach, and pharmacological blockade of either the FBXW2-MSX2 axis with pevonedistat [12], the EGFR-STAT3 pathway with the cationic triphenylmethane pharmacophore gentian violet [13], or EGFR/SRC/AKT signaling with the EGFR inhibitors gefitinib and erlotinib and the Src inhibitor dasatinib [14], have been proposed as strategies to target human cancers with SOX2 overexpression. It is unknown, however, how much of the anti-cancer activity of these indirect approaches can be attributable to SOX2 depletion. Moreover, the aforementioned strategies mostly target the proximal promoters of the SOX2 gene driving SOX2 expression in the differentiated states of cancer cells, and epigenetic re-activation of stemness-specific enhancers that cause a subpopulation of tumor cells to shift towards a CSC state is unaffected. Mechanistically, such an approach can be achieved by inactivation of lysine-specific demethylase 1 (LSD1/KDM1A), a flavin adenine dinucleotide (FAD)-dependent homolog of the amine oxidase family that demethylates monomethyl or dimethyl lysine 4 (K4) of histone H3. LSD1 blockade with the small molecule inhibitor CBB1007 has been shown to enhance repressive H3K9 methylation at the stemness-specific enhancer of SOX2, thereby validating the notion that LSD1 might serve as a selective epigenetic target for therapeutic ablation of SOX2-driven cancer stemness [15]. Although CBB1007-like competitive LSD1 inhibitors, which have been developed based on the structure of LSD1 with a peptide inhibitor derived from the N-terminal tail of histone H3 [16], might be considered good candidates to selectively target CSC with SOX2-driven pluripotent stem cell properties [17], most of them are in a preclinical stage.
Iadademstat (formerly ORY-1001; Oryzon Genomics, Barcelona, Spain), a clinically proven, highly potent and selective covalent small-molecule inhibitor of LSD1 [18–22], is an emerging therapeutic in hematological malignancies. Iadademstat has been shown to induce blast cell differentiation and reduce the leukemia-propagating stem cell compartment in acute myeloid leukemia (AML). Initial results from a Phase I/IIa clinical trial of iadademstat demonstrated its safety and good tolerability together with preliminary signs of anti-leukemic activity in refractory and relapsed AML [20]. Based on these findings, the Phase IIa ALICE study is currently ongoing in elderly patients with AML not eligible for intensive chemotherapy to combine iadademstat with standard of care azacytidine (https://www.clinicaltrialsregister.eu/ctr-search/trial/2018-000482-36/ES). Beyond hematological cancers, blocking LSD1 with iadademstat has been proposed as a valid strategy in some solid tumors such as small-cell lung cancer (SCLC) and melanoma [21, 22]. Indeed, the Phase II CLEPSIDRA trial is recruiting relapsed SCLC patients to receive iadademstat in combination with platinum-etoposide chemotherapy (https://www.clinicaltrialsregister.eu/ctr-search/trial/2018-000469-35/ES). In addition, the capacity of iadademstat-driven inhibition of LSD1 activity to activate immune responses has recently been proposed as a new means to overcome resistance to immune checkpoint inhibitors in melanoma [22]. Iadademstat-driven reversion of tumor-driving undifferentiated cell states in genomically-diverse malignancies strongly supports the notion that LSD1 might serve as a highly selective epigenetic target for the elimination of cancer cells with pluripotent stem cell-like properties [15, 16, 23, 24]. To test this hypothesis, we here investigated the ability of iadademstat to target SOX2-driven CSC in breast cancer, an unexplored cancer type for iadademstat-based therapy.
Because the mechanism of action of iadademstat has been proposed to either impede the removal of the methyl group from mono-methylated and di-methylated K4 and K9 of histone 3 on LSD1-targeted genes via a catalytic/enzymatic mechanism [18], or to promote enhancer activation of subordinate genes through the displacement of LSD1 from chromatin via a scaffolding/structural mechanism [19], we first computationally investigated the capacity of iadademstat to target the LSD1-bound FAD cofactor and to disturb the anchorage of LSD1 and its co-repressor (RCOR1/CoREST) to chromatin. Second, because epigenetic re-activation of SOX2 expression via a pluripotency-specific enhancer can cause a subpopulation of tumor cells to dynamically acquire a CSC state, we evaluated the capacity of iadademstat to target the mammosphere-forming capacity -a well-accepted surrogate reporter of CSC activity- in established in vitro models bearing distinct mutational landscapes (i.e., BRCA1-mutated basal-like MDA-MB-436 and HER2 gene-amplified/luminal-B BT-474 cell lines) but sharing a common dependency on SOX2 expression to maintain their CSC phenotype. Third, because SOX2 confers sensitivity to LSD1 inhibition, we characterized the SOX2 expression pattern using the PAM50 classifier and the integrative clustering of transcriptional data available from the Molecular Taxonomy of Breast Cancer International consortium (METABRIC). Fourth, we finally evaluated the clinical relevance of iadademstat as a novel anti-SOX2 epigenetic breast cancer therapy by assessing its ability to impact both the expression of SOX2 and the tumorsphere-forming capacity of CSC-like cells derived from breast cancer patient-derived xenografts (PDX).
Results
Binding mode of iadademstat to the LSD1-CoREST-histone H3 complex
Two different models have been proposed to explain the mechanism of action of iadademstat. In the first, iadademstat rapidly and irreversibly binds the LSD1 cofactor FAD in a manner analogous to the monoamine oxidase inhibitor tranylcypromine [18]. In the second, iadademstat physically separates LSD1/RCOR1 from the SNAG-domain transcription repressor GFI1 and chromatin in a cell-type-specific manner [19]. Taking advantage of the solved three-dimensional structure of LSD1 in a ternary complex with its histone peptide substrate and RCOR1/CoREST, we aimed to computationally explore a working model whereby iadademstat might operate via both a catalytic/enzymatic mechanism involving highly potent, direct targeting of the FAD cofactor (at lower concentrations), and a scaffolding/structural mechanism involving inhibition of the chromatin binding activity of LSD1/RCOR1 (at higher concentrations). Docking simulations of iadademstat in a crystal structure of human LSD1 (chain A) including RCOR1/CoREST (chain B) – a co-repressor that collaborates to demethylate mono- and di-methylated H3-K9 in nucleosomes – and a histone H3 peptide (chain C), produced eight clusters of docking poses (Figure 1). When the docking results were ranked according to the ascent of the binding energies for iadademstat (up to -9.09 kcal/mol; Table 1), those clusters exhibiting the highest affinity (#1, #3, and #4, see inset on the top right of Figure 1) were in the nanomolar range and were predicted to occupy the same binding site as FAD in LSD1. Cluster #6 was predicted to interfere with the position of the histone H3 peptide, whereas clusters #7 and #8, with affinities in the low micromolar range, were predicted to interact with both the LSD1 enzyme and RCOR1/CoREST.
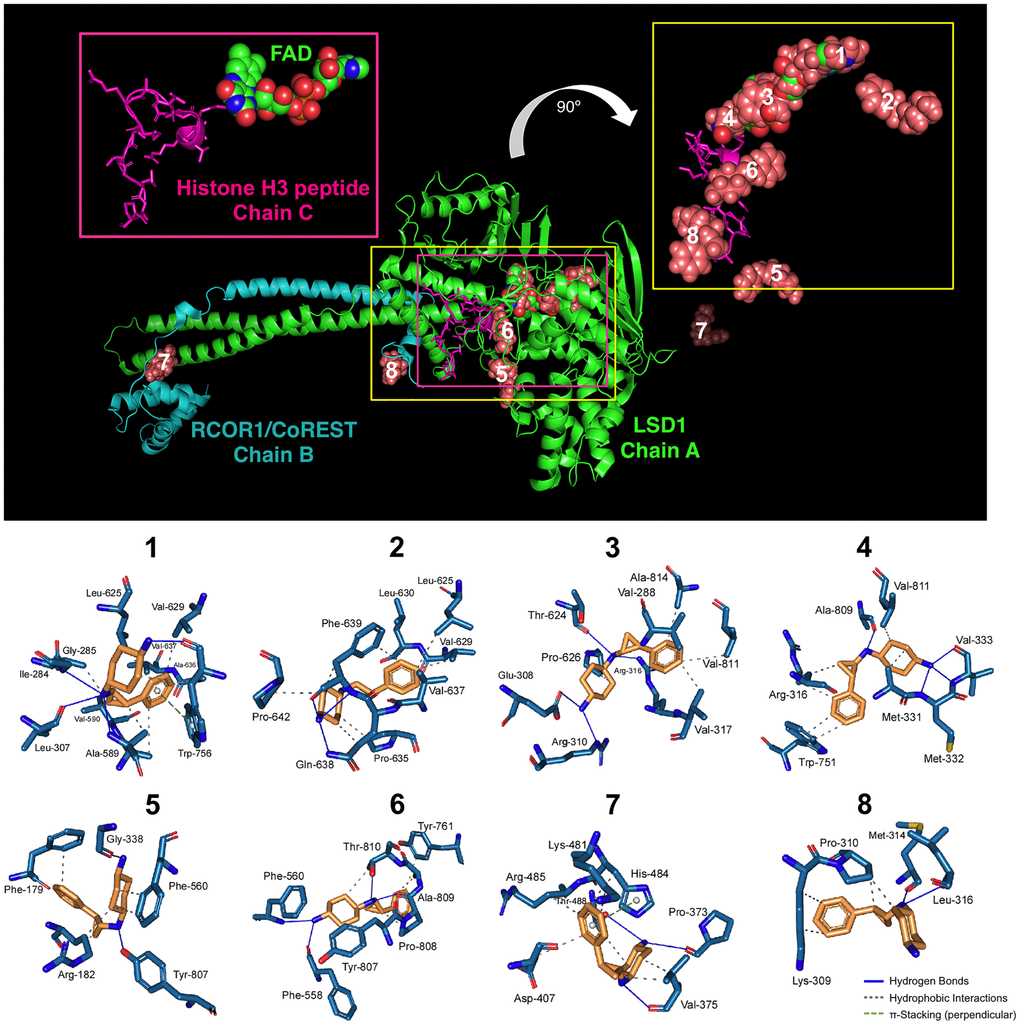
Figure 1. Binding mode of iadademstat to LSD1.Top. The backbone of LSD1 (chain A)/REST corepressor 1 (chain B)/histone H3 peptide (chain C) heterotrimeric complex is shown. For each cluster of the docked iadademstat (salmon color), only the molecule (spheres) with better binding energy is shown. The molecular docking was performed using the A and B chains in the absence of FAD and histone H3 peptide; however, the clusters of docked iadademstat are shown superimposed on the position that would occupy both the FAD and the histone H3 peptide. The cluster number is also indicated. The insert on the left shows the peptide histone H3 (chain C, backbone as cartoon and side chains as sticks) and the FAD (represented as spheres and with the green carbons). The insert on the right shows only the best pose of iadademstat docked in each cluster and the situation of the histone H3 peptide. The clusters #1, #3 and #4 of iadademstat would occupy the same position of the FAD and are shown superimposed. Bottom. The detailed map of the molecular interactions of iadademstat in each cluster is detailed (see also Table 1). Each inset shows the detailed interactions of each compound docked to the LSD1 heterodimer, indicating the participating amino acids involved in the interaction and the type of interaction (hydrogen bonds, hydrophilic interactions, salt bridges, Π-stacking, etc).
Table 1. Details of the interaction of iadademstat docked to the LSD1 heterodimer (see Figure 1A).
| Cluster number | ΔG, [kcal/mol] | Dissoc. constant, [μM] | Members | MM/PBSA solvation binding energy [kcal/mol] | Residues of the receptor that contact iadademstat | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | -9.09 | 0.21811 | 5% | -10.018 | Ile-284, Gly-285, Ser-286, Gly-287, Leu-307, Glu-308, Ala-309, Arg-310, Gly-315, Thr-588, Ala-589, Val-590, Arg-591, Thr-624, Leu-625, Pro-626, Val-629, Gln-632, Ala-636, Val-637, Trp-756, Ala-757 (chain A) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | -8.84 | 0.32872 | 9% | -21.945 | Leu-625, Val-629, Leu-630, Lys-631, Gln-632, Gln-633, Pro-635, Ala-636, Val-637, Gln-638, Phe-639, Val-640, Pro-642, Leu-643, Thr-648 (chain A) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | -8.75 | 0.38632 | 5.5% | -103.909 | Gly-285, Ser-286, Gly-287, Val-288, Ser-289, Gly-290, Glu-308, Ala-309, Arg-310, Gly-314, Gly-315, Arg-316, Val-317, Thr-624, Leu-625, Pro-626, Trp-756, Gly-800, Glu-801, Thr-810, Val-811, Ala-814 (chain A) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | -8.74 | 0.39379 | 17% | -111.598 | Arg-316, Leu-329, Gly-330, Ala-331, Met-332, Val-333, Thr-335, Tyr-571, Leu-659, Asn-660, Lys-661, Trp-751, Ser-760, Tyr-761, Ala-809, Thr-810, Val-811 (chain A) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | -7.95 | 1.49000 | 4.5% | -68.647 | Ala-178, Phe-179, Arg-182, Leu-183, Pro-184, His-185, Gly-338, Gly-339, Asp-557, Phe-558, Glu-559, Phe-560, Thr-561, Tyr-807 (chain A) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | -7.92 | 1.57000 | 2.5% | -29.621 | Thr-335, Ala-539, Asn 540, Trp-552, Asp-555, Phe-558, Glu-559, Phe-560, Tyr-761, Ser-762, Tyr-763, Val-764, Asn 806, Tyr-807, Pro-808, Ala-809, Thr 810, His-812 (chain A) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | -7.09 | 6.40000 | 16.5% | -18.198 | Lys-481, Ser-482, His-484, Arg-485, Thr-488 (chain A) and Leu-372, Pro-373, Glu-374, Val-375, Ile-376, Gln-377, Asp-407, Val-408, Gly-410 (chain B) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | -6.33 | 22.98000 | 8% | -32.309 | Tyr-391 (chain A) and Lys-309, Pro-310, Pro-311, Lys-312, Gly-313, Met-314, Phe-315, Leu-316, Ser-317, Gln-318 (chain B) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| For the best-docked iadademstat molecule of each cluster, the Gibbs free energy, the dissociation constant, the number of molecules members (as %), and the MM/PBSA solvation binding energy are shown. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
To add protein flexibility and provide additional information about different intra- and inter-molecular movements, we performed short molecular dynamics (MD) simulations over the course of 10 ns together with binding free energy calculations under the Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) approximation. The results highlighted the extremely high affinity of iadademstat at the FAD-targeted clusters #3, and #4, which reached -103.909 and -111.598 kcal/mol, respectively (Table 1).
Using an AlphaScreen™ assay with a biotinylated histone H3 peptide methylated at lysine 4, purified LSD1, and a highly specific antibody that recognizes demethylated substrate, we confirmed the in vitro efficacy of iadademstat to dose-dependently suppress the demethylase activity of LSD1, with a mean IC50 of 12 nmol/L (Supplementary Figure 1).
Iadademstat specifically suppresses the mammosphere-formation potential of breast cancer stem cells
One of the gold standards for evaluating the presence of CSC is their ability to form in vitro mammospheres in low-density non-adherent serum-free medium supplemented with growth factors [25, 26–29]. We assessed the anti-CSC activity of iadademstat in triple-negative breast cancer (TNBC), a highly aggressive breast cancer subtype driven by highly enriched CSC, which are related to therapy resistance, tumor relapse, and metastasis [25, 30]. Accordingly, estrogen receptor (ER)-negative/progesterone receptor (PR)-negative/ HER2-negative BRCA1mut/PTENmut MDA-MB-436 cells, which can form smooth and round spheres in suspension culture [25] and are dependent on SOX2 expression to maintain their CSC phenotype [31], were used in the Cell2Sphere™ assay to evaluate the impact of iadademstat on the ability of CSC to survive and proliferate as floating microtumors. Specifically, we examined the effects of iadademstat on the total number, size, and aspect of MDA-MB-436 mammospheres growing under stem cell-selective conditions. Compared with the untreated controls, exposure to graded concentrations of iadademstat resulted in a dose-dependent decrease in the total number of mammospheres (IC50 = 3.98 μmol/L; Figure 2). Notably, the strong decrease in mammosphere formation by iadademstat was not due to non-specific toxicity, as MTT-based cell viability assays run in parallel in 10% serum-supplemented adherent conditions showed no significant cytotoxic activity of iadademstat, even when employing concentrations as high as 30 μmol/L – a dose that completely prevented mammosphere formation in MDA-MB-436 cells (Figure 2).
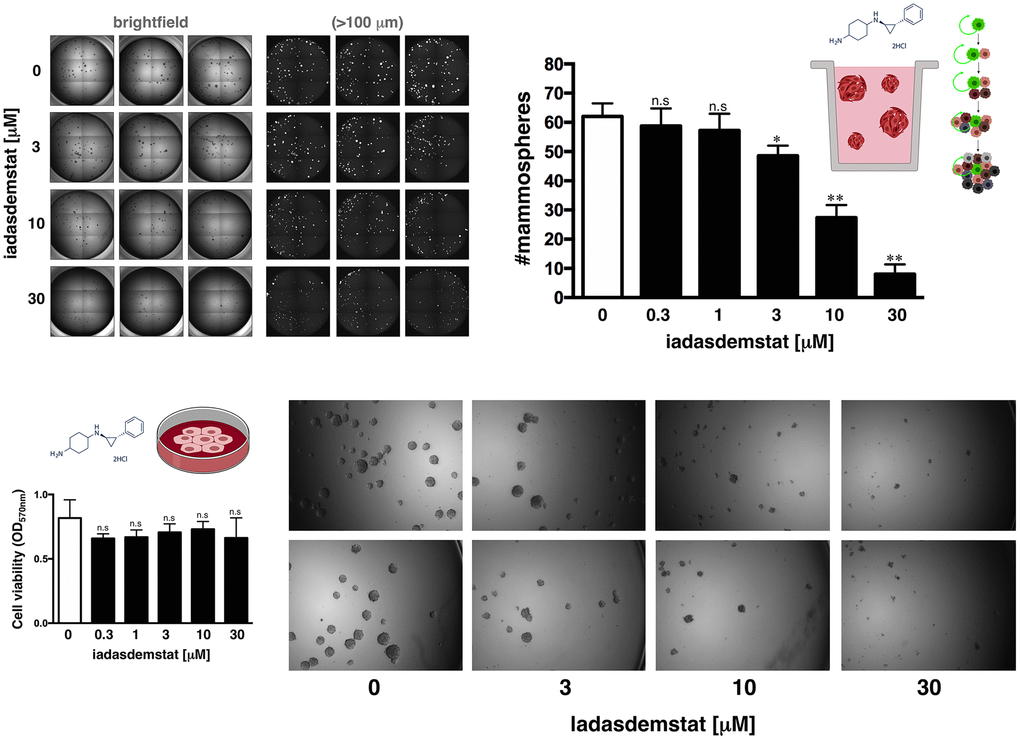
Figure 2. Iadademstat suppresses mammosphere formation in a basal-like established cell line. Figure shows representative microscope representations (×2.5 magnification) of mammospheres formed by MDA-MB-436 cells growing in sphere medium for 6 days in the absence or presence of graded concentrations of iadademstat. The number of mammospheres (>100 μm diameter) is expressed as means (columns) ± SD (bars). MTT uptake-based measurement of cell viability is expressed as percentages uptake (OD570) relative to untreated controls (=100% cell viability). The results are expressed as percentages means (columns) ± SD (bars). *P < 0.05 and **P < 0.005, statistically significant differences from the untreated (control) group.
Iadademstat suppresses pluripotency enhancer-driven activation of SOX2 in breast cancer stem cells
LSD1-blocking compounds are known to differentially target pluripotent cancer cells including teratocarcinoma, embryonic carcinoma, and seminoma, or embryonic stem cells that express SOX2, while having minimal growth-inhibitory effects on non-pluripotent cancer or normal somatic cells [15, 16, 23]. We therefore envisioned that iadademstat might suppress CSC function by repressing the re-activation of SOX2 in breast CSC, a transcriptional phenomenon that specifically occurs through activation of the distal enhancer of the SOX2 promoter that also controls SOX2 transcription in pluripotent stem cells [32, 33]. When we transfected tamoxifen-resistant, luminal-B/HER2+ BT-474 cells [34] with a luciferase reporter vector containing the SOX2 distal enhancer region, we observed a robust induction (9.2-fold on average) of reporter activity in mammosphere cultures when compared with the adherent culture control (Figure 3A). Of note, the enhancer-driven transcriptional activation of SOX2 differentially occurring in mammosphere cultures was dose-dependently suppressed by iadademstat (up to 80% reduction at 10 μmol/L; Figure 3A). Importantly, iadademstant-driven SOX2 silencing drastically reduced the number of BT-474 mammospheres (data not shown).
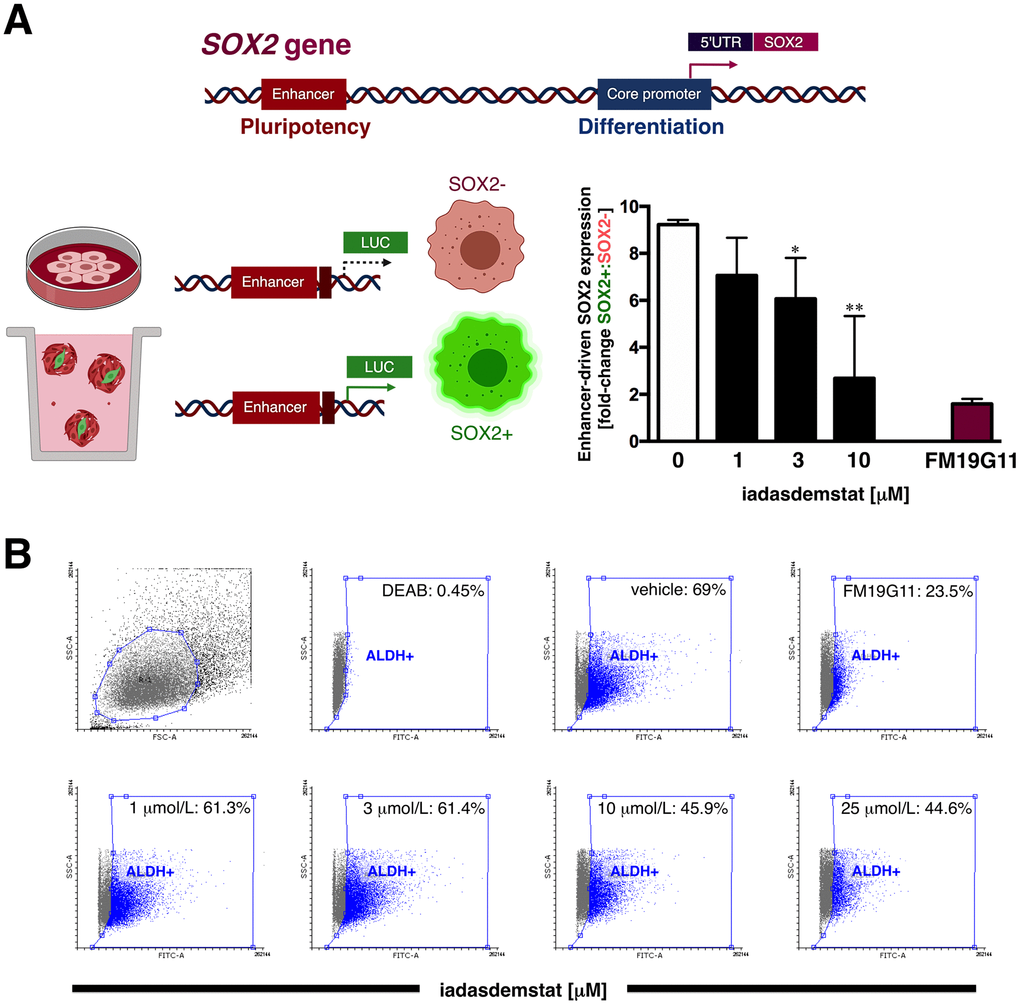
Figure 3. Iadademstat inhibits stemness-associated SOX2 expression in a luminal-B/HER2+ established cell line. (A) Schematic representation of SOX2 promoter structure indicating the proximal core promoter region and the location of the distal enhancer, which is induced exclusively upon CSC-driven mammosphere formation but not in cell-adherent differentiating conditions. Results are expressed as fold-induction of mammosphere culture-associated SOX2 reporter activity above adherent culture control in the absence or presence of graded concentrations of iadademstat. The results are expressed as percentages means (columns) ± SD (bars). *P < 0.05 and **P < 0.005, statistically significant differences from the untreated (control) group. (B) Representative Aldefluor® assay to identify BT-474 cells with high ALDH activity (ALDH+) in the absence or presence of graded concentrations of iadademstat for 3 days. The ALDH inhibitor diethylaminobenzaldehyde (DEAB) was used as negative control. Monolayer cultures were fed with iadademstat on day 1. (Note: 1 μmol/L FM19G11, an epigenetic repressor of key genes involved in stemness including SOX2 [98], was employed as a positive control).
Iadademstat fails to target ALDH+ breast cancer stem cells
We next tested whether the mechanism of action of iadademstat to specifically target mammosphere-forming CSC-like cells might be due to a more general phenomenon involving drivers of breast cancer stemness other than SOX2. Thus, we evaluated its capacity to target cells with high levels of aldehyde dehydrogenase-1 (ALDH1), a biomarker that has been suggested to label a tumorigenic cell fraction capable of self-renewal [35, 36]. ALDH1+ cell subpopulations enriched for cancer-initiating activity can be readily identified by flow cytometry using the Aldefluor® reagent, which quantifies ALDH activity by measuring the conversion of the ALDH substrate BODIPY aminoacetaldehyde to the fluorescent product BODIPY aminoacetate. Using HER2-overexpressing BT-474 cells as a breast cancer model naturally enriched with ALDH1+ cells (>50%), we detected a decrease in the proportion of ALDH1+ cells following 72 h treatment with increasing concentrations of iadademstat (up to 35% decrease at 25 μmol/L iadademstat) as compared with vehicle-treated controls (Figure 3B).
SOX2 expression status associates with luminal-B and HER2-positive breast carcinomas
Comprehensive comparison of molecular portraits between cell lines and breast cancer tumors confirmed the luminal-B intrinsic subtype classification of the ER+/HER2+ BT-474 cell line but revealed that the basal-like cell line MDA-MB-436 exhibits similar protein features to those of the luminal-A breast cancer subtype [37]. Because SOX2-overexpressing cells are particularly sensitive to LSD1 inhibitors [15, 17], we explored the possibility that SOX2 expression might associate with the luminal entity across breast tumor subtypes. When we examined the expression status of SOX2 in two sets of breast cancer cell lines organized by luminal, basal-A (i.e., basal-like breast cancer intrinsic subtype), and basal-B (i.e., claudin-low breast cancer intrinsic subtype) sub-classes [38, 39], most of the SOX2-overexpressing breast carcinoma cell lines were found to belong to the luminal subclass (Figure 4A).
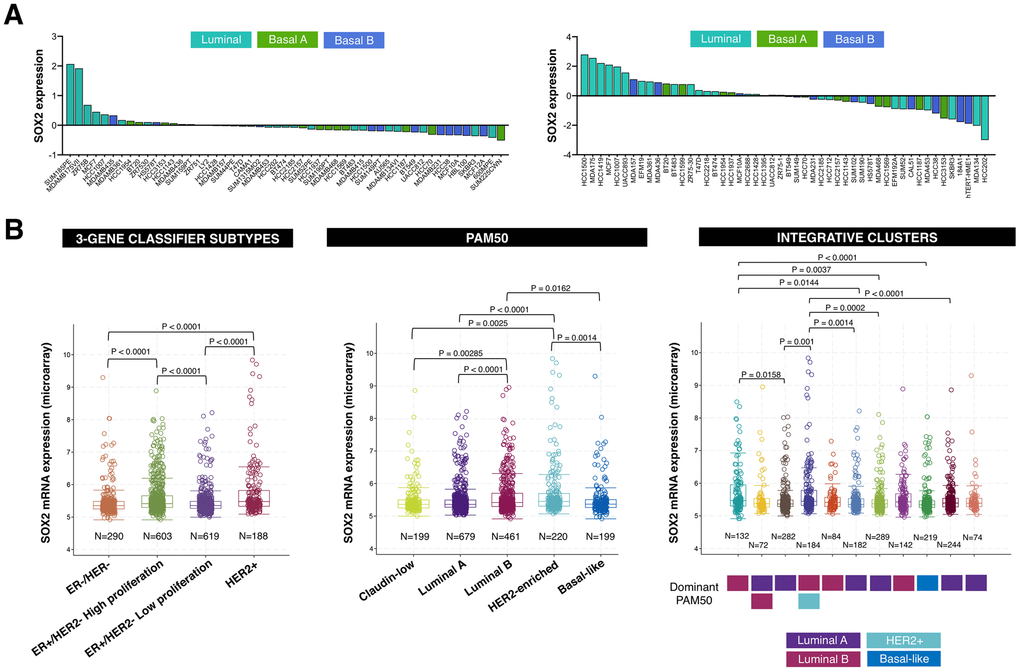
Figure 4. SOX2 expression is enriched in luminal-B and HER2-positive breast cancer subtypes. (A) Relative enrichment of SOX2 expression (213721_at) in breast cancer cell lines organized by luminal, basal A, and basal B sub-classes [38, 39]. (B) Box plots presents the SOX2 gene expression in primary breast tumors from the METABRIC project classified in distinct subtypes using 3-gene (left), PAM50 (middle), and integrative clusters (right) classifiers. The color line presents median, box shows interquartile region and whiskers – the highest (max) and the lowest (min) value.
We then extracted breast cancer date sets from the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) [40, 41] to explore the association between SOX2 expression and multiple breast cancer subtypes. When the METABRIC breast cancer data set was classified into each of the four Gene Expression prognostic Index Using Subtypes (GENIUS) subgroups using a 3-gene classifier (i.e., HER2+, ER-/HER-, ER+/HER2- high proliferation, ER+/HER2- low proliferation) [42, 43], SOX2 expression was found to be significantly higher in the HER2+ and ER+/HER2- high proliferation (luminal B-like) subgroups (Figure 4B, left panel). When the METABRIC breast cancer data set was classified into each of the five intrinsic subtypes (i.e., luminal-A, luminal-B, HER2-enriched, basal-like, and normal-like) using the research-based 50-gene prediction analysis of microarray (PAM50) classifier [44], SOX2 expression was found to be significantly higher in the luminal-B and HER2-enriched subtypes (Figure 4B, middle panel). When the METABRIC data set was classified into each of the 10 integrative clusters (IntClust1-10) each associated with distinct somatic aberrations (CNAs) and gene expression changes [45, 46], the transcript level of SOX2 was found to be significantly upregulated in the IntClust1, which is constituted by ER-positive tumors predominantly classified into the higher proliferation luminal-B intrinsic subtype, and in the IntClust5, which mostly encompasses HER2-amplified breast composed of both HER2-enriched and luminal-B intrinsic subtypes (Figure 4B, right panel).
Iadademstat suppresses the formation of CSC-enriched mammospheres derived from a multidrug-resistant luminal-B breast cancer patient in a SOX2-related manner
Because patient-derived xenograft (PDX) tumor models more faithful recapitulate human tumor biology and drug responsiveness than established human cancer cell lines [47, 48], we employed CSC-enriched mammospheres generated by growing single-cell suspensions from excised PDX tumors under adherent-free conditions [49] to test the anti-CSC activity of iadademstat in a clinically-relevant scenario (Figure 5). The BRE-0188 (ER+/PR+/HER2-) PDX model was generated from a clinical sample obtained from a 65-year-old female with luminal-A invasive ductal carcinoma with the presence of lymph node metastases. The patients had not received any chemotherapy or radiotherapy prior to surgery. The BRE-0192 (ER+/PR+/HER2-) PDX model was generated from a clinical sample obtained from a 45-year-old female with luminal-B invasive lobular breast carcinoma with the presence of lymph node metastases [50]. The patient was a poor responder to prior therapies, including epirubicin, 5-fluorouracil, cyclophosphamide, taxotere, paclitaxel, bevacizumab, and gemcitabine.
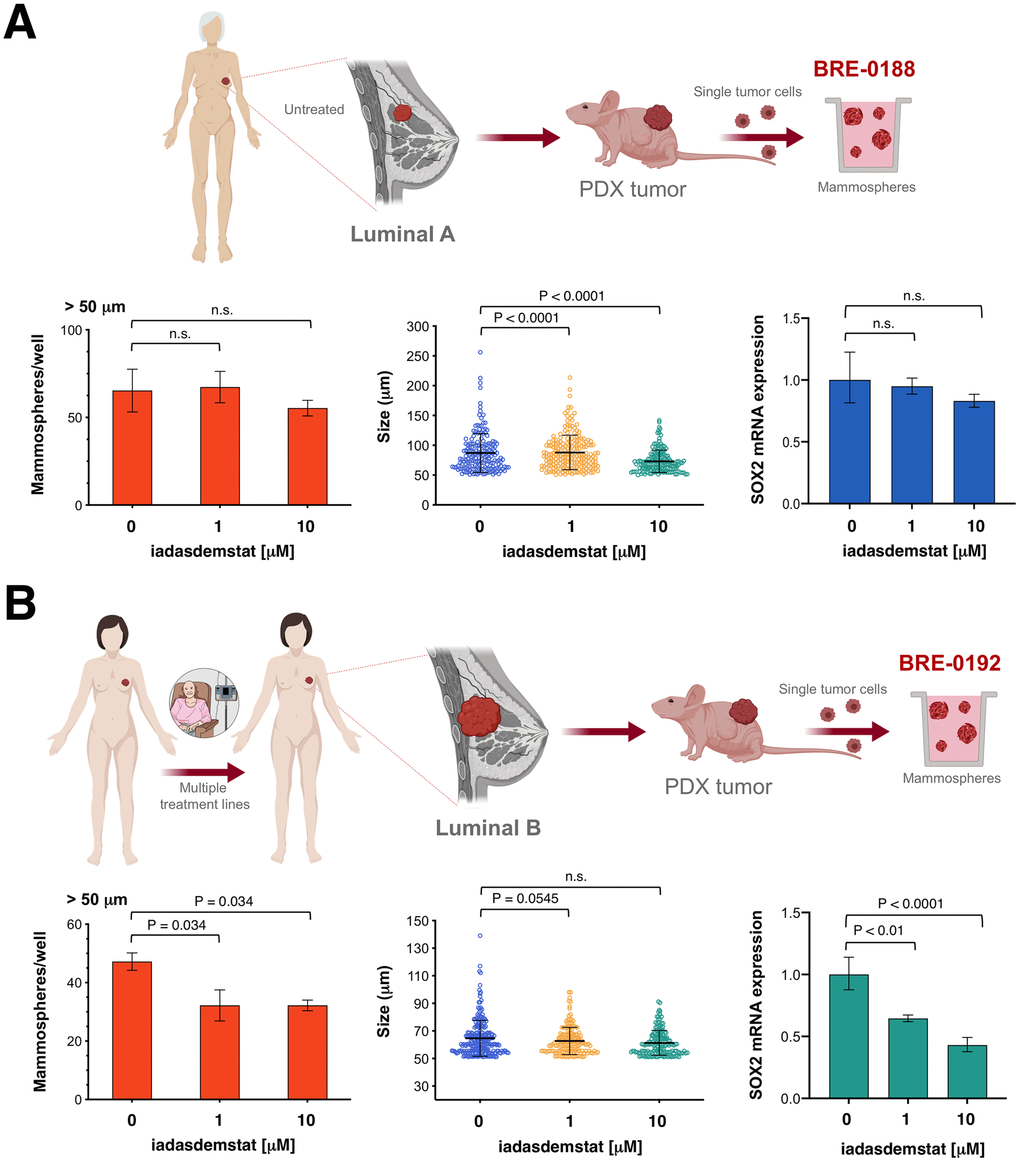
Figure 5. Iadademstat targets SOX2-driven CSC in breast cancer patient-derived xenografts. Cell2Sphere™ assays using BRE-0188 (A) and BRE-0192 (B) PDXs were performed as per the manufacturer’s instructions (http://stemtektherapeutics.com/en/cell2sphere#cell2 sphere_kit). Increasing concentrations of iadademstat were added to sextuplicate sets of wells on day 1. ImageJ was used to quantify the number (left panels) and size (middle panels; central lines indicate mean values) of 9-day-old mammospheres. Right panels. Total RNA from untreated and iadademstat-treated mammosphere cells was evaluated in technical triplicates for the abundance of SOX2 (Hs01053049_s1) relative to housekeeping gene 18S (Hs99999901_s1). The transcript abundance was calculated using the delta Ct method (i.e., the difference of Ct value between the target SOX2 gene and the endogenous 18S control) and presented as relative quantification.
Iadademstat treatment failed to decrease the number of mammospheres originated from CSC-like cells derived from the luminal-A BRE-0188 PDX (Figure 5A); the size of BRE-0188 mammospheres, however, was significantly decreased after treatment with iadademstat. Treatment of BRE-0188 mammospheres cultures with iadademstat failed to alter SOX2 expression. Conversely, iadademstat treatment significantly decreased the number but not the size of mammospheres originated from the multidrug-resistant CSC-like cells derived from the luminal-B BRE-0192 PDX (Figure 5B). Moreover, treatment with iadademstat notably reduced the expression of SOX2 in mammospheres collected from the BRE-0192 PDX.
Discussion
We provide the first evidence that the clinically proven inhibitor of LSD1 iadademstat can be used to circumvent the challenge of pharmacologically manipulating the epigenetic re-activation of SOX2 that causes a subpopulation of tumor cells to shift towards a CSC state, which has constrained the clinical relevance of SOX2-centered therapeutic strategies in major cancer types such as breast cancer. The therapeutic potential of targeting SOX2-driven CSC supports the clinical use of iadademstat as a novel anti-SOX2 epigenetic breast cancer therapy, particularly in SOX2-enriched luminal-B and HER2-positive subtypes.
Some malignant tumors such as breast carcinoma depend on SOX2 for their tumor-initiating ability [34, 51–53]. Elevated LSD1 levels are associated with enhanced SOX2 expression, and SOX2-overexpressing cells are particularly sensitive to LSD1 inhibitors. Accordingly, LSD1 has been proposed as a selective epigenetic target for therapy in SOX2-expressing cancers, particularly in those carcinomas in which high SOX2 expression occurs via 3q26 chromosomal amplification (e.g., lung SCLC, serous ovarian carcinoma, cervical, head and neck, oral, and esophageal carcinomas [6]). However, such LSD1 inhibitor-based therapeutic strategies might become even more relevant as cancer is entering a new era where the concept of cellular/phenotypic plasticity involving the dynamic interconversion of cells with and without CSC states is challenging how we treat and understand tumors relapse [54–61]. LSD1 controls SOX2 expression by binding to its distal enhancer, which drives SOX2 activation in pluripotent stem-like cells including CSC [15, 33, 62]. Pharmacological blockade of LSD1 selectively promotes methylation of H3K4 and H3K9 within the regulatory (enhancer) region of the SOX2 promoter, suppressing SOX2 activity and stimulating cell differentiation by augmenting H3K4 methylation on the promoters of differentiation genes [15]. In a process that is not mutually exclusive, the differentiation genes that are directly controlled by LSD1 can indirectly inhibit the expression of SOX2 that confers stem cell-like traits to breast cancer cells [63]. Indeed, the therapeutic implications of our current findings with the LSD1 iadademstat might involve molecular scenarios in which the epigenetic re-activation of SOX2 within a group of transformed cells in response to microenvironmental cues, stochastic genetic and epigenetic alterations, and/or treatment-imposed selective pressures can cause them to shift toward a CSC state, LSD1-driven aberrant activation of SOX2 via stemness-specific enhancers irrespective of their mutational landscape (e.g., BRCA1-mutated basal-like, HER2 gene-amplified), but also in certain breast cancer intrinsic subtypes that might be intrinsically addicted to the major role of SOX2 in self-renewal growth and expansion of CSC-like cells (i.e., luminal-B and HER2-positive). Such clinically relevant specificity of the LSD1 inhibitor iadademstat against SOX2-driven CSC was confirmed not only in established breast cancer cell lines that are dependent on SOX2 expression to maintain their CSC phenotype [31, 34] but also in CSC-enriched mammospheres generated by growing single-cell suspensions from excised PDX tumors.
The fact that the SOX2 reporter assay detects variations in SOX2 transcription regardless of the proportion of cells expressing a CSC marker along with the ability of iadademstat-induced knockdown of SOX2 to sharply decrease the quantity of CSC-like cells capable of self-renewal in vitro but not to completely suppress subpopulations with high enzymatic activity of ALDH1 (ALDH1high), which has been shown to mark a breast cancer population enriched for proliferating CSC, strongly suggest that the anti-CSC activity of iadademstat might be restricted to SOX2-driven CSC bio-behaviors (e.g., 1-3% of all luminal-B/HER2+ BT-474 cells) that do not necessarily overlap with that of ALDHhigh proliferating cells. Using CSC-enriched mammospheres obtained by growing single-cell suspensions from excised PDX tumor under adherent-free conditions, treatment with iadademstat resulted in significantly reduced mammosphere formation (but not reduced mammosphere size) in a multi-drug resistant luminal-B breast carcinoma and in significantly reduced mammosphere size (but not reduced mammosphere number) in a treatment-naïve luminal-A breast cancer. The number of mammospheres reflects the quantity of CSC-like cells capable of self-renewal in vitro, while the size of mammospheres is an indirect measure of the self-renewal capacity of each mammosphere-generating cell; thus, cell proliferation during mammosphere growth determines the size of the mammospheres [64–66]. Taking together, these results indicate that iadademstant inhibited CSC-driven mammosphere formation efficiency through suppressing LSD1/SOX2 axis in a luminal-B breast carcinoma, whereas the size of mammospheres in a luminal-A tumor was regulated through other underlying proliferative mechanism via LSD1. Accordingly, treatment of mammospheres from the luminal-B (but not the luminal-A) breast cancer patient with iadademstat significantly reduced the expression of SOX2, likely indicating a selective targeting of SOX2-driven CSC. The therapeutic potential of targeting SOX2-driven CSC supports the clinical use of iadademstat as a novel anti-SOX2 epigenetic breast cancer therapy, particularly in endocrine therapy-resistant luminal-B cases -which are known employ SOX2 to increase the proportion of CSC-like cells, rendering them insensitive to tamoxifen [34]- and in HER2-positive disease –in which SOX2 overexpression correlates with poor differentiation [67] and HER2-targeted therapies such as trastuzumab fail to eliminate SOX2-overexpressing CSC [68]. We have to acknowledge that the LSD1/SOX2 axis may represent a provocative potential target for CSC elimination not only in luminal-B and HER2-positive tumor but also in the basal-like subtype, which can also be found in the SOX2-overexpressing IntClust5 [46]. In this regard, because SOX2 reactivation has been shown to depend on the recruitment of the tumor suppressor protein BRCA1 to the pluripotency-related distal enhancer in the SOX2 promoter and concomitant modification of H3K4 and H3K9 at the same enhancer [62], our findings in basal-like MDA-MB-436 cells support the idea that BRCA1-mediated predisposition to breast and ovarian cancer [69] might involve a controlled loss of SOX2 expression, establishing a direct link between LSD1-regulated expression of SOX2, acquisition of stem-like cell phenotypes, and BRCA1-related breast/ovarian cancer initiation. Indeed, SOX2 is frequently gained in BRCA1 germline mutated tumors and is preferentially expressed in sporadic basal-like phenotypes having similar phenotypic and clinical characteristics to breast cancer arising in BRCA1 mutation carriers. Accordingly, SOX2 might play a driver role in the development of their less differentiated/stem cell-like phenotypic traits characteristics of the basal-like breast cancer phenotype [70]. Moreover, SOX2 overexpression occurs not only in high-grade serous ovarian tumors, but also in pre-malignant, fallopian tube epithelial cells from BRCA1/BRCA2 mutation carriers who underwent prophylactic salpingo-oophorectomy [71] – opening the possibility for considering LSD1-targeted epigenetic approaches such as iadademstat for breast/ovarian cancer prevention.
SOX2 activation has proved instrumental for the plastic acquisition of aberrant stemness properties in cancer cells. Accordingly, certain CSC bio-behaviors can be defined as rare therapy-resistant, self-renewing cancer cells that aberrantly express SOX2, which might provide specificity for CSC-targeted drug screening. The use of fluorescence protein expression-based transcriptional reporters for activation of the LSD1-regulated enhancer element of the SOX2 gene promoter can specifically identify cells with tumor-initiating activity; compounds that would be capable of impeding SOX2 activation might be viewed as valuable candidates for drugs aimed to target CSC. In this regard, our findings unravel for the first time the ability of iadademstat to inhibit an LSD1-targeted distal enhancer that specifically controls the expression of the stem cell transcription factor SOX2 in pluripotent stem cells, thereby suppressing the re-activation of SOX2 exclusively occurring in mammosphere-initiating breast CSC. The ability of iadademstat to efficiently and specifically target the on/off LSD1-driven SOX2 regulatory process that provides higher tumorigenic potential to cells with an epigenetically acquired CSC phenotype [61, 72] might open new therapeutic horizons that incorporate iadademstat in the anti-breast cancer armamentarium. We further propose that iadademstat might functionally deplete tumor-initiating CSC-like cellular states that sustain tumorigenicity by impacting on fundamental controllers of cell fate choice, an epigenetic mechanism involving both the downregulation of SOX2 and the re-activation of epigenetically suppressed differentiation programs in SOX2-enriched breast cancer subtypes such as Luminal-B and HER2-positive.
Furthermore, we are rapidly appreciating that nuclear reprogramming-like phenomena inducing the acquisition of epigenetic plasticity and phenotype malleability should be viewed as a fundamental element of a tissue’s capacity to undergo successful repair, aging degeneration or malignant transformation [73–77]. Thus, chronic or unrestrained cell plasticity would drive aging phenotypes by impairing the repair or the replacement of damaged cells and such uncontrolled phenomena of in vivo reprogramming might also generate CSC-like cellular states [73–77]. Pharmacological tools selectively targeting the LSD1-SOX2 axis might be appropriate to experimentally uncouple the apparently counterintuitive capacity of LSD1 blockade to promote reprogramming phenomena by regulating the balance between pluripotency and differentiation [78–83] while preventing SOX2-driven cancer stemness. This would raise the possibility of pharmacologically managing, in the appropriate direction and intensity, the physiological versus pathological processes of SOX2-related reparative cellular reprogramming in aging and cancer.
Materials and Methods
Molecular docking
The human histone demethylase LSD1 (UniProt code O60341)/REST corepressor 1 (UniProt code Q9UKL0)/histone H3 peptide (UniProt code P68431) ternary complex structure (3 Å resolution, PDB code 2X0L) was obtained from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (PDB). The molecular structure of iadademstat was obtained from PubChem (PubChem_ID: 71543365). The specific edition of the LSD1 protein structure involving the removal of water, FAD, and histone H3 peptide, was made using PyMol 2.0 software (PyMOL Molecular Graphics System, v2.0 Schrödinger, LLC, at http://www.pymol.org/) without further optimization.
Molecular docking analysis of iadademstat against LSD1 was performed as previously described [84–88]. The selected protein structure was subjected to geometry optimization using the repair function of the FoldX algorithm [89]. To search for potential binding sites of iadademstat, a global molecular docking procedure was performed with AutoDock/Vina using YASARA v19.4.27 software [90, 91], where a total of 999 flexible docking runs were set and clustered (7 Å) around the putative binding sites. The YASARA pH command was set to 7.4. The docking software has a scoring function to give an approximate calculation of the Gibbs free energy variation (ΔG, kcal/mol) between LSD1 and iadademstat in each binding pose, with more positive energy values indicating stronger binding [92]. All the values were included in the corresponding table with a negative sign; only the ΔG value for the best compound docked in each cluster is shown in Table 1. To calculate this parameter, which is used to rank compounds, Autodock Vina uses a force field scoring function that considers the strength of electrostatic interactions, hydrogen bonding between all atoms of the two binding partners in the complex, intermolecular van der Waals forces, and also solvation and entropy contributions [93]. Docking results usually cluster around certain hot spot conformations. Two complexed compounds were considered to belong to different clusters if the ligand Root-Mean-Square Deviation of their atomic positions was greater than a minimum of 6 Å. Dissociation constants were recalculated from the average binding energy of all compounds of each cluster. The number of iadademstat-docked molecules included in each compound cluster is indicated as "members", as a percentage in Table 1. The key residues of LSD1 interacting with iadademstat in each cluster were detected using also YASARA v19.4.27 software [90, 91]. All of the figures were prepared using PyMol 2.0 software and all interactions were detected using the PLIP algorithm [94].
Molecular dynamics simulations
YASARA dynamics v19.4.27 was also used for all the MD simulations with AMBER14 as a force field. The simulation cell was allowed to include 20 Å surrounding the protein and filled with water at a density of 0.997 g/mL. Initial energy minimization was carried out under relaxed constraints using steepest descent minimization. Simulations were performed in water at constant pressure-constant temperature (25°C) conditions. To mimic physiological conditions, counter ions were added to neutralize the system; Na+ or Cl- were added in replacement of water to give a total NaCl concentration of 0.9% and pH was maintained at 7.4. Hydrogen atoms were added to the protein structure at the appropriate ionizable groups according to the calculated pKa in relation to the simulation pH (i.e., a hydrogen atom will be added if the computed pKa is higher than the pH). The pKa was computed for each residue according to the Ewald method [95]. All simulation steps were run by a preinstalled macro (md_run.mcr) within the YASARA suite. Data were collected every 100 ps.
TMM/PBSA was implemented with the YASARA macro md_analyzebindenergy.mcr to calculate the binding free energy with solvation of iadademstat, complex, and free protein for the LSD1 form complexes. The binding free energy (kcal/mol) was expressed according to the following equation:
ΔEbinding = [poterec(i) + solverec(i) + potelig + solvelig] - [potecmp(i) + solvecmp(i)]
where i is the position number, “pote” is the potential energy for the complex (potecmp), free protein (poterec), or free ligand (potelig), and “solve” is the solvation energy for the complex (solvecmp), free protein (solverec), or free ligand (solvelig). More positive binding free values indicate better binding.
LSD1 enzymatic activity
Enzymatic reactions were performed in an AlphaScreen format in duplicate at room temperature for 60 minutes in a 10 μL mixture containing assay buffer, histone H3 peptide substrate, LSD1 (BPS#50103, lot#130806-D) enzyme, and iadademstat (RG-6016, Cat. No. S7795, Selleckchem.com). The 10-μL reactions were carried out in 384-well Optiplates (Perkin Elmer Life Sciences, Waltham, MA). A serial dilution of the compounds was first performed in 3.3% DMSO/assay buffer. From this step, 3 μL of iadademstat was added to 4 μL of enzyme and incubated for 30 minutes at room temperature. After this incubation, 3 μL of substrate was added to initiate the reaction. The final DMSO concentration was 1%. After the reaction, 5 μL of anti-mouse acceptor beads (Perkin Elmer, diluted 1:500 with 1× detection buffer) or 5 μL of anti-rabbit acceptor beads (Perkin Elmer, diluted 1:500 with 1× detection buffer) and 5 μL of primary antibody (BPS#52140E,F, diluted 1:200 with 1x detection buffer) were added to the reaction mix. After brief shaking, the plate was incubated for 30 minutes. Finally, 10 μL of AlphaScreen streptavidin-conjugated donor beads (Perkin Elmer, diluted 1:125 with 1× detection buffer) were added. After 30 minutes, the samples were measured in the AlphaScreen microplate reader (EnSpire Alpha 2390 Multilabel reader, Perkin Elmer).
The AlphaScreen intensity data were analyzed and compared using Graphpad Prism software (GraphPad Software Inc., San Diego, CA). In the absence of iadademstat, the AlphaScreen or fluorescence intensity (Ft) was defined as 100% activity. In the absence of enzyme, the intensity (Fb) was defined as 0% activity. The percent activity in the presence of iadademstat was calculated according to the following equation: %activity = (F-Fb)/(Ft-Fb), where F=the A-screen intensity in the presence of iadademstat. Once A-screen data were converted to LSD1 activity (%), those values were then plotted against a series of iadademstat concentrations using non-linear regression analysis of sigmoidal dose-response curves generated with the equation Y=B+(T-B)/1+10((LogEC50-X)×Hill Slope), where Y=percent activity, B=minimum percent activity, T=maximum percent activity, X=logarithm of compound and Hill Slope=slope factor or Hill coefficient. The IC50 value was determined as the concentration of iadademstat causing a half-maximal inhibition of control activity.
SOX2 profiling in breast cancer datasets
We interrogated the publicly available METABRIC breast cancer dataset in the United Kingdom and Canada [40], in which mRNA expression was measured using the Illumina HT-12v13 platform and CNA with the Affymetrix SNP 6.0 array. Gene-level expression files from METABRIC were downloaded from the cBioportal for Cancer Genomics (https://www.cbioportal.org/). We used the 3-gene, PAM50, and integrative clusters subtypes provided in the METABRIC dataset.
Breast cancer xenograft models
For generation of BRE-0188 and BRE-0192 PDX models, all clinical samples were collected under written informed consent (according to the Declaration of Helsinki) and a declaration for commercial use of the samples from the Consultative Committee for the Protection of Persons in Biomedical Research (CCPPRB) of Dijon University Hospital under authorization by a French Ministry of Higher Education, Research and Innovation for human tissue collection, and redistribution (CSP articles L 1243-3, L 1243-4, and L 1245-5). PDX tumors were passaged by serial transplantation in immunocompromised mice [50].
Mammosphere formation
Mammosphere formation was monitored using Cell2Sphere™ assays (StemTek Therapeutics, Bilbao, Spain) as per the manufacturer’s instructions [96, 97]. Graded concentrations of iadademstat were added to triplicate sets of wells on day 1 and the number of either 6- (MDA-MB-436, BT-474) or 9-day-old (BRE-0188, BRE-0192 PDX) mammospheres was recorded as a measurement of CSC content. Images were recorded using a BioTek Cytation 5 image cytometer at 2.5× magnification. Prior to image acquisition, spheroid cultures were stained with a fluorescent vital dye to increase the accuracy of spheroid detection and analysis. The system was then set to count number, size, and aspect ratio of the objects. Thresholds were set to >100 μm in size and 0.4 as aspect ratio (with 1 being the aspect ratio of a perfect circle). Aspect ratio did not vary upon iadademstat dosage.
Cell viability
Cell viability was determined using a standard colorimetric MTT-based reduction assay 72 h after exposure to graded concentrations of iadademstat.
Aldefluor® activity assay
The Aldefluor® assay was performed as per the manufacturer’s instructions (StemCell Technologies, Vancouver, BC, Canada), with or without the addition of graded concentrations of iadademstat for 72 h. Analysis was performed using a MACSQuant® Analyzer 10 flow cytometer (Miltenyi Biotec, Bergisch Gladbach, Germany) for data acquisition.
SOX2 enhancer reporter assay
BT-474 cells were transfected with 5 μg of pGL3 Luc control (Promega, Madison, WI, USA) or pGL2-Sox2-enhancer-Luc reporter plasmids [32, 33] using Lipofectamine Plus (Invitrogen, Carslbad, CA). Twenty-four hours after transfection, the culture was split into two parts: one part was seeded in two-dimensional adherent culture plates and the other part was cultured in non-adherent culture conditions to allow mammosphere formation, in the absence or presence of graded concentrations of iadademstat. After 48 h, cells were harvested and luciferase activity was measured in duplicate with the Glomax 20/20 luminometer (Promega) and normalized by protein concentration in the extracts. Results were expressed as fold induction of sphere culture reporter activity above adherent culture control.
Quantitative real-time PCR
Total RNA was extracted from mammosphere cells using the Qiagen RNeasy Kit according to the manufacturer’s instructions. One microgram of total RNA was reverse-transcribed to cDNA using the Reaction Ready™ First Strand cDNA Synthesis Kit (SABiosciences, Frederick, MD). PCR arrays were processed according to the SABiosciences RT-PCR manual and analyzed using an Applied Biosystems 7500 Fast Real-Time PCR System with an automated baseline and threshold cycle detection. The data were interpreted using the web-based PCR array analysis tool from SABiosciences.
Statistical analysis
All statistical analyses were performed using GraphPad Prism software. Cell-based experimental data are presented as mean ± S.D. Comparisons of means of ≥ 3 groups were performed by analysis of variance (ANOVA) and the existence of individual differences, in case of significant F values at ANOVA, were assessed by multiple contrasts. The expression of SOX2 in the breast cancer subtypes (METABRIC dataset) was examined using one-way ANOVA (Kruskal-Wallis) with Dunnett’s test. P values < 0.05 were considered to be statistically significant (denoted as *). All statistical tests were two-sided.
Supplementary Materials
Acknowledgments
We are grateful to the Cluster of Scientific Computing (http://ccc.umh.es/) of the Miguel Hernández University (UNH) and the Centro de Supercomputación of the University of Granada (ALHAMBRA-CSIRC) for providing computing facilities. The authors would like to thank Dr. Kenneth McCreath for editorial support.
Conflicts of Interest
Stock ownership: Á.G.M., StemTek Therapeutics (CEO). All other authors have no competing interests to declare. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. Ethics approval was not required for this study as per the local legislation.
Funding
Work in the Menendez laboratory is supported by the Spanish Ministry of Science and Innovation (Grant SAF2016-80639-P, Plan Nacional de l+D+I, founded by the European Regional Development Fund, Spain) and by an unrestricted research grant from the Fundació Oncolliga Girona (Lliga catalana d’ajuda al malalt de càncer, Girona). The Spanish Ministry of Economy and Competitiveness (MINECO, Project AGL2015-67995-C3-1-R) and the Generalitat Valenciana (PROMETEO/ 2016/006) supports work in the Encinar laboratory.
References
- 1. Feng R, Wen J. Overview of the roles of Sox2 in stem cell and development. Biol Chem. 2015; 396:883–91. https://doi.org/10.1515/hsz-2014-0317 [PubMed]
- 2. Rizzino A, Wuebben EL. Sox2/Oct4: A delicately balanced partnership in pluripotent stem cells and embryogenesis. Biochim Biophys Acta. 2016; 1859:780–91. https://doi.org/10.1016/j.bbagrm.2016.03.006 [PubMed]
- 3. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006; 126:663–76. https://doi.org/10.1016/j.cell.2006.07.024 [PubMed]
- 4. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007; 131:861–72. https://doi.org/10.1016/j.cell.2007.11.019 [PubMed]
- 5. Novak D, Hüser L, Elton JJ, Umansky V, Altevogt P, Utikal J. SOX2 in development and cancer biology. Semin Cancer Biol. 2019. [Epub ahead of print]. https://doi.org/10.1016/j.semcancer.2019.08.007 [PubMed]
- 6. Mamun MA, Mannoor K, Cao J, Qadri F, Song X. SOX2 in Cancer Stemness: Tumor Malignancy and Therapeutic Potentials. J Mol Cell Biol. 2018. [Epub ahead of print]. https://doi.org/10.1093/jmcb/mjy080 [PubMed]
- 7. Hüser L, Novak D, Umansky V, Altevogt P, Utikal J. Targeting SOX2 in anticancer therapy. Expert Opin Ther Targets. 2018; 22:983–91. https://doi.org/10.1080/14728222.2018.1538359 [PubMed]
- 8. Zhang S, Sun Y. Targeting oncogenic SOX2 in human cancer cells: therapeutic application. Protein Cell. 2020; 11:82–84.. https://doi.org/10.1007/s13238-019-00673-x [PubMed]
- 9. Beltran AS, Blancafort P. Remodeling genomes with artificial transcription factors (ATFs). Methods Mol Biol. 2010; 649:163–82. https://doi.org/10.1007/978-1-60761-753-2_10 [PubMed]
- 10. Garcia-Bloj B, Moses C, Sgro A, Plani-Lam J, Arooj M, Duffy C, Thiruvengadam S, Sorolla A, Rashwan R, Mancera RL, Leisewitz A, Swift-Scanlan T, Corvalan AH, Blancafort P. Waking up dormant tumor suppressor genes with zinc fingers, TALEs and the CRISPR/dCas9 system. Oncotarget. 2016; 7:60535–54. https://doi.org/10.18632/oncotarget.11142 [PubMed]
- 11. Stolzenburg S, Rots MG, Beltran AS, Rivenbark AG, Yuan X, Qian H, Strahl BD, Blancafort P. Targeted silencing of the oncogenic transcription factor SOX2 in breast cancer. Nucleic Acids Res. 2012; 40:6725–40. https://doi.org/10.1093/nar/gks360 [PubMed]
- 12. Yin Y, Xie CM, Li H, Tan M, Chen G, Schiff R, Xiong X, Sun Y. The FBXW2-MSX2-SOX2 axis regulates stem cell property and drug resistance of cancer cells. Proc Natl Acad Sci USA. 2019; 116:20528–38. https://doi.org/10.1073/pnas.1905973116 [PubMed]
- 13. Pietrobono S, Morandi A, Gagliardi S, Gerlini G, Borgognoni L, Chiarugi P, Arbiser JL, Stecca B. Down-Regulation of SOX2 Underlies the Inhibitory Effects of the Triphenylmethane Gentian Violet on Melanoma Cell Self-Renewal and Survival. J Invest Dermatol. 2016; 136:2059–69. https://doi.org/10.1016/j.jid.2016.06.610 [PubMed]
- 14. Singh S, Trevino J, Bora-Singhal N, Coppola D, Haura E, Altiok S, Chellappan SP. EGFR/Src/Akt signaling modulates Sox2 expression and self-renewal of stem-like side-population cells in non-small cell lung cancer. Mol Cancer. 2012; 11:73. https://doi.org/10.1186/1476-4598-11-73 [PubMed]
- 15. Zhang X, Lu F, Wang J, Yin F, Xu Z, Qi D, Wu X, Cao Y, Liang W, Liu Y, Sun H, Ye T, Zhang H. Pluripotent stem cell protein Sox2 confers sensitivity to LSD1 inhibition in cancer cells. Cell Rep. 2013; 5:445–57. https://doi.org/10.1016/j.celrep.2013.09.018 [PubMed]
- 16. Mimasu S, Umezawa N, Sato S, Higuchi T, Umehara T, Yokoyama S. Structurally designed trans-2-phenylcyclopropylamine derivatives potently inhibit histone demethylase LSD1/KDM1. Biochemistry. 2010; 49:6494–503. https://doi.org/10.1021/bi100299r [PubMed]
- 17. Wang J, Lu F, Ren Q, Sun H, Xu Z, Lan R, Liu Y, Ward D, Quan J, Ye T, Zhang H. Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Res. 2011; 71:7238–49. https://doi.org/10.1158/0008-5472.CAN-11-0896 [PubMed]
- 18. Maes T, Mascaró C, Tirapu I, Estiarte A, Ciceri F, Lunardi S, Guibourt N, Perdones A, Lufino MM, Somervaille TC, Wiseman DH, Duy C, Melnick A, et al. ORY-1001, a Potent and Selective Covalent KDM1A Inhibitor, for the Treatment of Acute Leukemia. Cancer Cell. 2018; 33:495–511.e12. https://doi.org/10.1016/j.ccell.2018.02.002 [PubMed]
- 19. Maiques-Diaz A, Spencer GJ, Lynch JT, Ciceri F, Williams EL, Amaral FM, Wiseman DH, Harris WJ, Li Y, Sahoo S, Hitchin JR, Mould DP, Fairweather EE, et al. Enhancer Activation by Pharmacologic Displacement of LSD1 from GFI1 Induces Differentiation in Acute Myeloid Leukemia. Cell Rep. 2018; 22:3641–59. https://doi.org/10.1016/j.celrep.2018.03.012 [PubMed]
- 20. Somervaille T, Salamero O, Montesinos P, Willekens C, Perez Simon JA, Pigneux A, Recher C, Popat R, Molinero C, Mascaro C, Maes T, Bosch F. Safety, phamacokinetics (PK), pharmacodynamics (PD) and preliminary activity in acute leukemia of ORY-1001, a first-in-class inhibitor of lysine-specific histone demethylase 1A (LSD1/KDM1A): initial results from a first-in-human phase 1 study. Blood. 2016; 128:4060. https://doi.org/10.1182/blood.V128.22.4060.4060
- 21. Augert A, Eastwood E, Ibrahim AH, Wu N, Grunblatt E, Basom R, Liggitt D, Eaton KD, Martins R, Poirier JT, Rudin CM, Milletti F, Cheng WY, et al. Targeting NOTCH activation in small cell lung cancer through LSD1 inhibition. Sci Signal. 2019; 12:eaau2922. https://doi.org/10.1126/scisignal.aau2922 [PubMed]
- 22. Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, Li Y, Chen H, Yang H, Hsu PH, Van Allen EM, Freeman GJ, De Carvalho DD, et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell. 2018; 174:549–563.e19. https://doi.org/10.1016/j.cell.2018.05.052 [PubMed]
- 23. Yin F, Lan R, Zhang X, Zhu L, Chen F, Xu Z, Liu Y, Ye T, Sun H, Lu F, Zhang H. LSD1 regulates pluripotency of embryonic stem/carcinoma cells through histone deacetylase 1-mediated deacetylation of histone H4 at lysine 16. Mol Cell Biol. 2014; 34:158–79. https://doi.org/10.1128/MCB.00631-13 [PubMed]
- 24. Hino S, Kohrogi K, Nakao M. Histone demethylase LSD1 controls the phenotypic plasticity of cancer cells. Cancer Sci. 2016; 107:1187–92. https://doi.org/10.1111/cas.13004 [PubMed]
- 25. Manuel Iglesias J, Beloqui I, Garcia-Garcia F, Leis O, Vazquez-Martin A, Eguiara A, Cufi S, Pavon A, Menendez JA, Dopazo J, Martin AG. Mammosphere formation in breast carcinoma cell lines depends upon expression of E-cadherin. PLoS One. 2013; 8:e77281. https://doi.org/10.1371/journal.pone.0077281 [PubMed]
- 26. Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012; 21:283–96. https://doi.org/10.1016/j.ccr.2012.03.003 [PubMed]
- 27. Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012; 10:717–28. https://doi.org/10.1016/j.stem.2012.05.007 [PubMed]
- 28. Weiswald LB, Bellet D, Dangles-Marie V. Spherical cancer models in tumor biology. Neoplasia. 2015; 17:1–15. https://doi.org/10.1016/j.neo.2014.12.004 [PubMed]
- 29. Brooks MD, Burness ML, Wicha MS. Therapeutic Implications of Cellular Heterogeneity and Plasticity in Breast Cancer. Cell Stem Cell. 2015; 17:260–71. https://doi.org/10.1016/j.stem.2015.08.014 [PubMed]
- 30. Martin-Castillo B, Lopez-Bonet E, Cuyàs E, Viñas G, Pernas S, Dorca J, Menendez JA. Cancer stem cell-driven efficacy of trastuzumab (Herceptin): towards a reclassification of clinically HER2-positive breast carcinomas. Oncotarget. 2015; 6:32317–38. https://doi.org/10.18632/oncotarget.6094 [PubMed]
- 31. Bhat-Nakshatri P, Goswami CP, Badve S, Sledge GW
Jr , Nakshatri H. Identification of FDA-approved drugs targeting breast cancer stem cells along with biomarkers of sensitivity. Sci Rep. 2013; 3:2530. https://doi.org/10.1038/srep02530 [PubMed] - 32. Leis O, Eguiara A, Lopez-Arribillaga E, Alberdi MJ, Hernandez-Garcia S, Elorriaga K, Pandiella A, Rezola R, Martin AG. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene. 2012; 31:1354–65. https://doi.org/10.1038/onc.2011.338 [PubMed]
- 33. Takanaga H, Tsuchida-Straeten N, Nishide K, Watanabe A, Aburatani H, Kondo T. Gli2 is a novel regulator of sox2 expression in telencephalic neuroepithelial cells. Stem Cells. 2009; 27:165–74. https://doi.org/10.1634/stemcells.2008-0580 [PubMed]
- 34. Piva M, Domenici G, Iriondo O, Rábano M, Simões BM, Comaills V, Barredo I, López-Ruiz JA, Zabalza I, Kypta R, Vivanco M. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol Med. 2014; 6:66–79. https://doi.org/10.1002/emmm.201303411 [PubMed]
- 35. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007; 1:555–67. https://doi.org/10.1016/j.stem.2007.08.014 [PubMed]
- 36. Korkaya H, Paulson A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008; 27:6120–30. https://doi.org/10.1038/onc.2008.207 [PubMed]
- 37. Jiang G, Zhang S, Yazdanparast A, Li M, Pawar AV, Liu Y, Inavolu SM, Cheng L. Comprehensive comparison of molecular portraits between cell lines and tumors in breast cancer. BMC Genomics. 2016 (Suppl 7); 17:525. https://doi.org/10.1186/s12864-016-2911-z [PubMed]
- 38. Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, Speed T, Spellman PT, DeVries S, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006; 10:515–27. https://doi.org/10.1016/j.ccr.2006.10.008 [PubMed]
- 39. Kao J, Salari K, Bocanegra M, Choi YL, Girard L, Gandhi J, Kwei KA, Hernandez-Boussard T, Wang P, Gazdar AF, Minna JD, Pollack JR. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS One. 2009; 4:e6146. https://doi.org/10.1371/journal.pone.0006146 [PubMed]
- 40. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Gräf S, Ha G, Haffari G, et al, and METABRIC Group. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012; 486:346–52. https://doi.org/10.1038/nature10983 [PubMed]
- 41. Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ, Tsui DW, Liu B, Dawson SJ, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016; 7:11479. https://doi.org/10.1038/ncomms11479 [PubMed]
- 42. Desmedt C, Haibe-Kains B, Wirapati P, Buyse M, Larsimont D, Bontempi G, Delorenzi M, Piccart M, Sotiriou C. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin Cancer Res. 2008; 14:5158–65. https://doi.org/10.1158/1078-0432.CCR-07-4756 [PubMed]
- 43. Haibe-Kains B, Desmedt C, Rothé F, Piccart M, Sotiriou C, Bontempi G. A fuzzy gene expression-based computational approach improves breast cancer prognostication. Genome Biol. 2010; 11:R18. https://doi.org/10.1186/gb-2010-11-2-r18 [PubMed]
- 44. Tibshirani R, Hastie T, Narasimhan B, Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci USA. 2002; 99:6567–72. https://doi.org/10.1073/pnas.082099299 [PubMed]
- 45. Dawson SJ, Rueda OM, Aparicio S, Caldas C. A new genome-driven integrated classification of breast cancer and its implications. EMBO J. 2013; 32:617–28. https://doi.org/10.1038/emboj.2013.19 [PubMed]
- 46. Russnes HG, Lingjærde OC, Børresen-Dale AL, Caldas C. Breast Cancer Molecular Stratification: From Intrinsic Subtypes to Integrative Clusters. Am J Pathol. 2017; 187:2152–62. https://doi.org/10.1016/j.ajpath.2017.04.022 [PubMed]
- 47. Siolas D, Hannon GJ. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. 2013; 73:5315–19. https://doi.org/10.1158/0008-5472.CAN-13-1069 [PubMed]
- 48. Byrne AT, Alférez DG, Amant F, Annibali D, Arribas J, Biankin AV, Bruna A, Budinská E, Caldas C, Chang DK, Clarke RB, Clevers H, Coukos G, et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat Rev Cancer. 2017; 17:254–68. https://doi.org/10.1038/nrc.2016.140 [PubMed]
- 49. Ehmsen S, Pedersen MH, Wang G, Terp MG, Arslanagic A, Hood BL, Conrads TP, Leth-Larsen R, Ditzel HJ. Increased Cholesterol Biosynthesis Is a Key Characteristic of Breast Cancer Stem Cells Influencing Patient Outcome. Cell Rep. 2019; 27:3927–3938.e6. https://doi.org/10.1016/j.celrep.2019.05.104 [PubMed]
- 50. Asano M, Matsui J, Towle MJ, Wu J, McGonigle S, DE Boisferon MH, Uenaka T, Nomoto K, Littlefield BA. Broad-spectrum Preclinical Antitumor Activity of Eribulin (Halaven®): Combination with Anticancer Agents of Differing Mechanisms. Anticancer Res. 2018; 38:3375–85. https://doi.org/10.21873/anticanres.12604 [PubMed]
- 51. Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008; 40:499–507. https://doi.org/10.1038/ng.127 [PubMed]
- 52. Gangemi RM, Griffero F, Marubbi D, Perera M, Capra MC, Malatesta P, Ravetti GL, Zona GL, Daga A, Corte G. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells. 2009; 27:40–48. https://doi.org/10.1634/stemcells.2008-0493 [PubMed]
- 53. Wen Y, Hou Y, Huang Z, Cai J, Wang Z. SOX2 is required to maintain cancer stem cells in ovarian cancer. Cancer Sci. 2017; 108:719–31. https://doi.org/10.1111/cas.13186 [PubMed]
- 54. Yuan S, Norgard RJ, Stanger BZ. Cellular Plasticity in Cancer. Cancer Discov. 2019; 9:837–51. https://doi.org/10.1158/2159-8290.CD-19-0015 [PubMed]
- 55. Gupta PB, Pastushenko I, Skibinski A, Blanpain C, Kuperwasser C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell. 2019; 24:65–78. https://doi.org/10.1016/j.stem.2018.11.011 [PubMed]
- 56. Jolly MK, Celià-Terrassa T. Dynamics of Phenotypic Heterogeneity Associated with EMT and Stemness during Cancer Progression. J Clin Med. 2019; 8:E1542. https://doi.org/10.3390/jcm8101542 [PubMed]
- 57. Wainwright EN, Scaffidi P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer. 2017; 3:372–86. https://doi.org/10.1016/j.trecan.2017.04.004 [PubMed]
- 58. Scaffidi P, Misteli T. Cancer epigenetics: from disruption of differentiation programs to the emergence of cancer stem cells. Cold Spring Harb Symp Quant Biol. 2010; 75:251–58. https://doi.org/10.1101/sqb.2010.75.007 [PubMed]
- 59. Corominas-Faja B, Cufí S, Oliveras-Ferraros C, Cuyàs E, López-Bonet E, Lupu R, Alarcón T, Vellon L, Iglesias JM, Leis O, Martín ÁG, Vazquez-Martin A, Menendez JA. Nuclear reprogramming of luminal-like breast cancer cells generates Sox2-overexpressing cancer stem-like cellular states harboring transcriptional activation of the mTOR pathway. Cell Cycle. 2013; 12:3109–24. https://doi.org/10.4161/cc.26173 [PubMed]
- 60. Vazquez-Martin A, Cufí S, López-Bonet E, Corominas-Faja B, Cuyàs E, Vellon L, Iglesias JM, Leis O, Martín AG, Menendez JA. Reprogramming of non-genomic estrogen signaling by the stemness factor SOX2 enhances the tumor-initiating capacity of breast cancer cells. Cell Cycle. 2013; 12:3471–77. https://doi.org/10.4161/cc.26692 [PubMed]
- 61. Iglesias JM, Leis O, Pérez Ruiz E, Gumuzio Barrie J, Garcia-Garcia F, Aduriz A, Beloqui I, Hernandez-Garcia S, Lopez-Mato MP, Dopazo J, Pandiella A, Menendez JA, Martin AG. The Activation of the Sox2 RR2 Pluripotency Transcriptional Reporter in Human Breast Cancer Cell Lines is Dynamic and Labels Cells with Higher Tumorigenic Potential. Front Oncol. 2014; 4:308. https://doi.org/10.3389/fonc.2014.00308 [PubMed]
- 62. Kondo T, Raff M. Chromatin remodeling and histone modification in the conversion of oligodendrocyte precursors to neural stem cells. Genes Dev. 2004; 18:2963–72. https://doi.org/10.1101/gad.309404 [PubMed]
- 63. Wu Y, Wang Y, Yang XH, Kang T, Zhao Y, Wang C, Evers BM, Zhou BP. The deubiquitinase USP28 stabilizes LSD1 and confers stem-cell-like traits to breast cancer cells. Cell Rep. 2013; 5:224–36. https://doi.org/10.1016/j.celrep.2013.08.030 [PubMed]
- 64. Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003; 17:1253–70. https://doi.org/10.1101/gad.1061803 [PubMed]
- 65. Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J, Song E. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007; 131:1109–23. https://doi.org/10.1016/j.cell.2007.10.054 [PubMed]
- 66. Xu C, Sun X, Qin S, Wang H, Zheng Z, Xu S, Luo G, Liu P, Liu J, Du N, Zhang Y, Liu D, Ren H. Let-7a regulates mammosphere formation capacity through Ras/NF-κB and Ras/MAPK/ERK pathway in breast cancer stem cells. Cell Cycle. 2015; 14:1686–97. https://doi.org/10.1080/15384101.2015.1030547 [PubMed]
- 67. Yang F, Zhang J, Yang H. OCT4, SOX2, and NANOG positive expression correlates with poor differentiation, advanced disease stages, and worse overall survival in HER2+ breast cancer patients. Oncotargets Ther. 2018; 11:7873–81. https://doi.org/10.2147/OTT.S173522 [PubMed]
- 68. Oak PS, Kopp F, Thakur C, Ellwart JW, Rapp UR, Ullrich A, Wagner E, Knyazev P, Roidl A. Combinatorial treatment of mammospheres with trastuzumab and salinomycin efficiently targets HER2-positive cancer cells and cancer stem cells. Int J Cancer. 2012; 131:2808–19. https://doi.org/10.1002/ijc.27595 [PubMed]
- 69. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994; 266:66–71. https://doi.org/10.1126/science.7545954 [PubMed]
- 70. Rodriguez-Pinilla SM, Sarrio D, Moreno-Bueno G, Rodriguez-Gil Y, Martinez MA, Hernandez L, Hardisson D, Reis-Filho JS, Palacios J. Sox2: a possible driver of the basal-like phenotype in sporadic breast cancer. Mod Pathol. 2007; 20:474–81. https://doi.org/10.1038/modpathol.3800760 [PubMed]
- 71. Hellner K, Miranda F, Fotso Chedom D, Herrero-Gonzalez S, Hayden DM, Tearle R, Artibani M, KaramiNejadRanjbar M, Williams R, Gaitskell K, Elorbany S, Xu R, Laios A, et al. Premalignant SOX2 overexpression in the fallopian tubes of ovarian cancer patients: discovery and validation studies. EBioMedicine. 2016; 10:137–49. https://doi.org/10.1016/j.ebiom.2016.06.048 [PubMed]
- 72. Iglesias JM, Gumuzio J, Martin AG. Linking Pluripotency Reprogramming and Cancer. Stem Cells Transl Med. 2017; 6:335–39. https://doi.org/10.5966/sctm.2015-0225 [PubMed]
- 73. Mosteiro L, Pantoja C, Alcazar N, Marión RM, Chondronasiou D, Rovira M, Fernandez-Marcos PJ, Muñoz-Martin M, Blanco-Aparicio C, Pastor J, Gómez-López G, De Martino A, Blasco MA, et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science. 2016; 354:aaf4445. https://doi.org/10.1126/science.aaf4445 [PubMed]
- 74. Ocampo A, Reddy P, Martinez-Redondo P, Platero-Luengo A, Hatanaka F, Hishida T, Li M, Lam D, Kurita M, Beyret E, Araoka T, Vazquez-Ferrer E, Donoso D, et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell. 2016; 167:1719–1733.e12. https://doi.org/10.1016/j.cell.2016.11.052 [PubMed]
- 75. Menendez JA, Alarcón T. Senescence-Inflammatory Regulation of Reparative Cellular Reprogramming in Aging and Cancer. Front Cell Dev Biol. 2017; 5:49. https://doi.org/10.3389/fcell.2017.00049 [PubMed]
- 76. Folguera-Blasco N, Cuyàs E, Menéndez JA, Alarcón T. Epigenetic regulation of cell fate reprogramming in aging and disease: A predictive computational model. PLOS Comput Biol. 2018; 14:e1006052. https://doi.org/10.1371/journal.pcbi.1006052 [PubMed]
- 77. Folguera-Blasco N, Pérez-Carrasco R, Cuyàs E, Menendez JA, Alarcón T. A multiscale model of epigenetic heterogeneity-driven cell fate decision-making. PLOS Comput Biol. 2019; 15:e1006592. https://doi.org/10.1371/journal.pcbi.1006592 [PubMed]
- 78. Adamo A, Sesé B, Boue S, Castaño J, Paramonov I, Barrero MJ, Izpisua Belmonte JC. LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells. Nat Cell Biol. 2011; 13:652–59. https://doi.org/10.1038/ncb2246 [PubMed]
- 79. Whyte WA, Bilodeau S, Orlando DA, Hoke HA, Frampton GM, Foster CT, Cowley SM, Young RA. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature. 2012; 482:221–25. https://doi.org/10.1038/nature10805 [PubMed]
- 80. Yang P, Wang Y, Chen J, Li H, Kang L, Zhang Y, Chen S, Zhu B, Gao S. RCOR2 is a subunit of the LSD1 complex that regulates ESC property and substitutes for SOX2 in reprogramming somatic cells to pluripotency. Stem Cells. 2011; 29:791–801. https://doi.org/10.1002/stem.634 [PubMed]
- 81. Wang Q, Xu X, Li J, Liu J, Gu H, Zhang R, Chen J, Kuang Y, Fei J, Jiang C, Wang P, Pei D, Ding S, Xie X. Lithium, an anti-psychotic drug, greatly enhances the generation of induced pluripotent stem cells. Cell Res. 2011; 21:1424–35. https://doi.org/10.1038/cr.2011.108 [PubMed]
- 82. Hou P, Li Y, Zhang X, Liu C, Guan J, Li H, Zhao T, Ye J, Yang W, Liu K, Ge J, Xu J, Zhang Q, et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science. 2013; 341:651–54. https://doi.org/10.1126/science.1239278 [PubMed]
- 83. Sun H, Liang L, Li Y, Feng C, Li L, Zhang Y, He S, Pei D, Guo Y, Zheng H. Lysine-specific histone demethylase 1 inhibition promotes reprogramming by facilitating the expression of exogenous transcriptional factors and metabolic switch. Sci Rep. 2016; 6:30903. https://doi.org/10.1038/srep30903 [PubMed]
- 84. Encinar JA, Fernández-Ballester G, Galiano-Ibarra V, Micol V. In silico approach for the discovery of new PPARγ modulators among plant-derived polyphenols. Drug Des Devel Ther. 2015; 9:5877–95. https://doi.org/10.2147/DDDT.S93449 [PubMed]
- 85. Galiano V, Garcia-Valtanen P, Micol V, Encinar JA. Looking for inhibitors of the dengue virus NS5 RNA-dependent RNA-polymerase using a molecular docking approach. Drug Des Devel Ther. 2016; 10:3163–81. https://doi.org/10.2147/DDDT.S117369 [PubMed]
- 86. Cuyàs E, Verdura S, Lozano-Sánchez J, Viciano I, Llorach-Parés L, Nonell-Canals A, Bosch-Barrera J, Brunet J, Segura-Carretero A, Sanchez-Martinez M, Encinar JA, Menendez JA. The extra virgin olive oil phenolic oleacein is a dual substrate-inhibitor of catechol-O-methyltransferase. Food Chem Toxicol. 2019; 128:35–45. https://doi.org/10.1016/j.fct.2019.03.049 [PubMed]
- 87. Cuyàs E, Verdura S, Micol V, Joven J, Bosch-Barrera J, Encinar JA, Menendez JA. Revisiting silibinin as a novobiocin-like Hsp90 C-terminal inhibitor: computational modeling and experimental validation. Food Chem Toxicol. 2019; 132:110645. https://doi.org/10.1016/j.fct.2019.110645 [PubMed]
- 88. Cuyàs E, Gumuzio J, Lozano-Sánchez J, Carreras D, Verdura S, Llorach-Parés L, Sanchez-Martinez M, Selga E, Pérez GJ, Scornik FS, Brugada R, Bosch-Barrera J, Segura-Carretero A, et al. Extra Virgin Olive Oil Contains a Phenolic Inhibitor of the Histone Demethylase LSD1/KDM1A. Nutrients. 2019; 11:E1656. https://doi.org/10.3390/nu11071656 [PubMed]
- 89. Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005; 33:W382–8. https://doi.org/10.1093/nar/gki387 [PubMed]
- 90. Krieger E, Vriend G. YASARA View - molecular graphics for all devices - from smartphones to workstations. Bioinformatics. 2014; 30:2981–82. https://doi.org/10.1093/bioinformatics/btu426 [PubMed]
- 91. Krieger E, Darden T, Nabuurs SB, Finkelstein A, Vriend G. Making optimal use of empirical energy functions: force-field parameterization in crystal space. Proteins. 2004; 57:678–83. https://doi.org/10.1002/prot.20251 [PubMed]
- 92. Lionta E, Spyrou G, Vassilatis DK, Cournia Z. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr Top Med Chem. 2014; 14:1923–38. https://doi.org/10.2174/1568026614666140929124445 [PubMed]
- 93. Morris GM, Huey R, Olson AJ. Using AutoDock for ligand-receptor docking. Curr Protoc Bioinformatics. 2008; Chapter 8:Unit 8.14. https://doi.org/10.1002/0471250953.bi0814s24 [PubMed]
- 94. Salentin S, Schreiber S, Haupt VJ, Adasme MF, Schroeder M. PLIP: fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015; 43:W443–7. https://doi.org/10.1093/nar/gkv315 [PubMed]
- 95. Krieger E, Nielsen JE, Spronk CA, Vriend G. Fast empirical pKa prediction by Ewald summation. J Mol Graph Model. 2006; 25:481–86. https://doi.org/10.1016/j.jmgm.2006.02.009 [PubMed]
- 96. Corominas-Faja B, Cuyàs E, Lozano-Sánchez J, Cufí S, Verdura S, Fernández-Arroyo S, Borrás-Linares I, Martin-Castillo B, Martin ÁG, Lupu R, Nonell-Canals A, Sanchez-Martinez M, Micol V, et al. Extra-virgin olive oil contains a metabolo-epigenetic inhibitor of cancer stem cells. Carcinogenesis. 2018; 39:601–13. https://doi.org/10.1093/carcin/bgy023 [PubMed]
- 97. Cuyàs E, Verdura S, Folguera-Blasco N, Bastidas-Velez C, Martin ÁG, Alarcón T, Menendez JA. Mitostemness. Cell Cycle. 2018; 17:918–26. https://doi.org/10.1080/15384101.2018.1467679 [PubMed]
- 98. Moreno-Manzano V, Rodríguez-Jiménez FJ, Aceña-Bonilla JL, Fustero-Lardíes S, Erceg S, Dopazo J, Montaner D, Stojkovic M, Sánchez-Puelles JM. FM19G11, a new hypoxia-inducible factor (HIF) modulator, affects stem cell differentiation status. J Biol Chem. 2010; 285:1333–42. https://doi.org/10.1074/jbc.M109.008326 [PubMed]