



Introduction
Parkinson’s disease (PD) is a common neurodegenerative disorder that is pathologically characterized by the loss of midbrain dopaminergic (DA) neurons in the substantia nigra and the accumulation of Lewy body aggregate [1]. PD is a multifactorial disease with a complex relationship with genetic and environmental factors.
Leucine-rich repeat kinase 2 (LRRK2) is a large complex protein that consists of armadillo repeats, ankryn-like repeats and leucine-rich repeats at the N-terminal domain, a central catalytic core that contains a GTP-binding Ras of complex (Roc) domain, a carboxy-terminal of Roc (COR) domain and a kinase domain belonging to the serine/threonine kinases, and a WD40 domain at the C-terminus [2, 3]. It is associated with a diverse set of cellular functions including mitochondrial function [4–6], cytoskeletal function [7, 8], autophagy [9, 10] and various signaling pathways [11, 12]. Oxidative stress and neuroinflammation are associated with PD pathophysiology [13, 14] which ultimately leads to dopaminergic neuron (DA) degeneration. Mutations in LRRK2 are the frequent cause of autosomal dominant familial PD and have been shown to have increased susceptibility to oxidative stress. G2019S is one of the most common LRRK2 mutation, affecting 5-6% of familial PD [15, 16]. Studies have shown that G2019S has enhanced kinase activity [17] and this can lead to increased sensitivity to stress [18]. G2019S mutation carriers also showed mitochondrial and autophagy impairments [6, 19] as well as increased alpha synuclein inclusion [20].
Peroxiredoxins (PRDX) are a family of six anti-oxidative proteins that share a common reactive cysteine residue in the N-terminal region and their role to inactivate cellular hydroperoxides [21, 22]. They are classified into 2-Cys, atypical 2-Cys, and 1-Cys Prx subfamilies depending on the number of cys protein and its location [23]. PRDX2 belongs to the 2-Cys family and peroxidase activity of this family is regulated via tyrosine and threonine phosphorylation [24].
The PRDX family protects cells from oxidative stress-induced apoptosis by acting as free radical scavengers and has been associated with neurodegeneration [25, 26]. It helps to regulate the peroxide levels in the cells [27] and removes reactive oxygen species (ROS). On the other side of the scale, excessive peroxidase activity have also been implicated in cell and tissue damage [28]. It can also act as a redox sensor by binding to other proteins and regulating their signaling activities [29, 30]. That is why it is critical to regulate the peroxidase activity to have fine balance between beneficial and adverse effect.
We previously found that mutant LRRK2 enhanced phosphorylation of mitochondrial PRDX3, which led to a suppression of cellular peroxidase activity and aggravated toxicity [31] leading to suppression of LRRK2 mutant phenotype. It highlighted the potential of peroxidases as a neuroprotective vehicle for PD patients with LRRK2 mutations. Per, belonging to the same family, was shown to preserve cognitive function against age-linked hippocampal oxidative damage via signaling pathways involving CREB, CaMKII and ERK [32]. They are also believed to play a role in neuroprotection [33, 34]. Importantly, Peroxiredoxin 2 (PRDX2) is a cytoplasmic protein and is detected in the different neuronal population [35], similar to LRRK2, which localizes predominantly in the cytosol.
Here, we studied the interaction between LRRK2 and PRDX2 and the neuroprotective function of PRDX2 on LRRK2 with in vitro and in vivo experiments. We showed that LRRK2 interacts with PRDX2 through cell-based studies, and using Drosophila as an animal model, we demonstrated that G2019S mutation in LRRK2 increased the phosphorylation of PRDX2. Transgenic expression of PRDX2 and its mimic, Chetomin, were able to rescue the pathogenic phenotype of G2019S in Drosophila. Our data provides evidence that PRDX2 confers a neuroprotection on pathogenic LRRK2 and that PRDX2 mimic can be further studied as a potential clinical drug for PD.
Results
LRRK2 interacts with PRDX2 in vitro and in vivo
We explored the relationship of LRRK2 with PRDX2 in vitro, determining if there is a physical interaction between both proteins. The interaction of these two proteins was assessed by co-immunoprecipitation in HEK-293T cells co-transfected with GFP-tagged LRRK2 and FLAG-tagged PRDX2. Following immunoprecipitation of LRRK2 by GFP protein, and immunoblotting for both GFP and FLAG, we found that LRRK2 interacts with PRDX2 in vitro (Figure 1A).
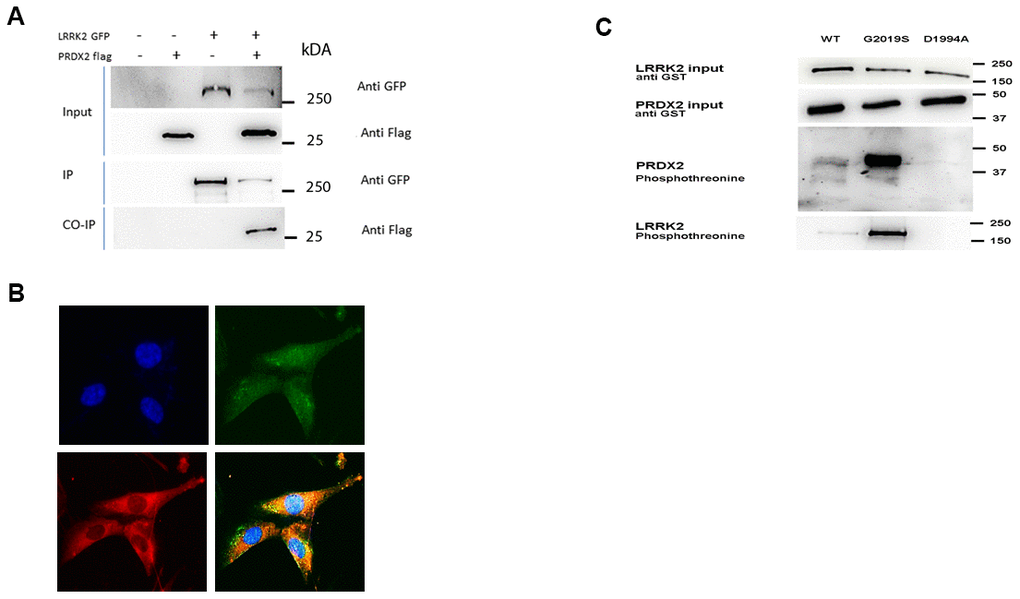
Figure 1. LRRK2 interacts and phosphorylates PRDX2. (A) LRRK2-GFP and PRDX2-flag are co transfected in HEK293T cells. The lysates were collected and subjected to immunoprecipitation with anti-GFP antibody. The protein was subjected to western blot with anti-flag. Un-transfected cells, LRRK2 GFP alone and PRDX flag alone was transfected into HEK293T cells to serve as negative control. (B) Endogenous colocalization of LRRK2 (green) and PRDX2 (red) protein in SKNSH neuronal cells (C) In vitro kinase assay on SDS-PAGE gel show phosphorylation of PRDX2 by LRRK2 wildtype and G2019S.
We also stained endogenous LRRK2 (Alexa Fluor 488-green) and endogenous PRDX2 (Alexa Fluor 546 dye-red) and showed their colocalization in SKNSH. (Figure 1B) In vitro kinase assay was carried out showing that G2019S LRRK2 phosphorylates PRDX2 more than LRRK2 wild-type, with negligible phosphorylation in the presence of kinase dead version of LRRK2 D1994A (Figure 1C).
To study their interaction in vivo, we generated GFP-tagged UAS-LRRK2 (wild-type and G2019S) and UAS-PRDX2 expressing transgenic flies and isogenized them. The ddc-GAL4 driver were used to drive expression of the various UAS-LRRK2 transgenes in tyrosine hydroxylase positive (TH+) neurons. Expression levels were verified via immunoblot to ensure that LRRK2 variants and PRDX2 are expressed (Figure 2A). To check for PRDX2 phosphorylation in vivo, we used an ELISA assay with a generic Phospho-Threonine/Serine antibody to detect the levels of phosphorylated PRDX2. This method was chosen, as it is more sensitive to detect phosphorylation from the small amount of protein extracted from flies. We found significant percentage increase of PRDX2 phosphorylation in PRDX2 + G2019S double transgenic flies compared to PRDX2 alone (p<0.05). LRRK2 wild-type + PRDX2 showed no substantial increase in phosphorylation (Figure 2B). These results indicated that G2019S mutation in LRRK2 increased the phosphorylation of PRDX2.
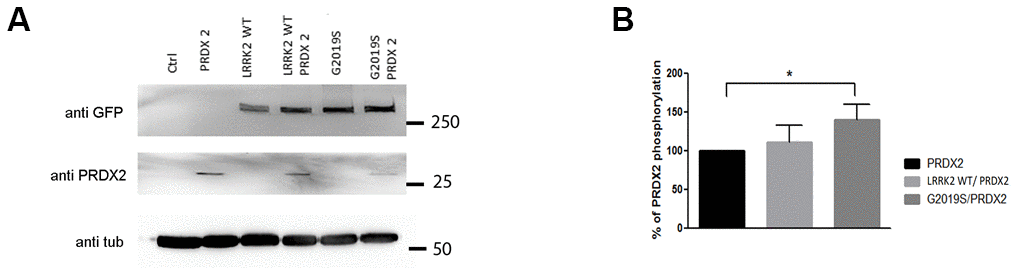
Figure 2. LRRK2 and PRDX2 transgenic flies. (A) LRRK2 GFP and PRDX2 expression driven by ddc-GAL-4 in transgenic fly head (B) Bar graphs show mean percentage and standard deviation of phosphorylation of PRDX2 in LRRK2 wildtype and G2019S flies (n = 3, cohort of 20).
PRDX2 suppress LRRK2 mutant phenotype in vivo
In flies, the expression of PRDX2 was able to partially suppress the loss of TH+ neurons caused by expression of the LRRK2 G2019S protein, as shown by an increase in TH+ neurons in the PPL2 cluster in G2019S + PRDX2 flies (p<0.05). Interestingly, there were also more TH+ neurons in PPM1/2 and PPM3 clusters in wild-type LRRK2 + PRDX2 double transgenic flies compared to wild-type LRRK2 flies (p <0.05) (Figure 3A, 3B). At 60 days, G2019S flies displayed significantly lowered climbing ability as compared to control flies. G2019S + PRDX2 flies also displayed a significantly better climbing score compared to G2019S flies (p <0.05) after 60 days (Figure 3C). G2019S flies showed reduced lifespan when compared to control flies. The rescue by PRDX2 was also observed in the lifespan assay where aged 60 days old G2019S-PRDX2 and wild-type LRRK2-PRDX2 flies had a significant increase in survival compared to age-matched G2019S and LRRK2 wild-type expressing flies respectively (Figure 3D). These results strongly suggest PRDX2 helps rescue and protect against the pathogenic phenotypes caused by G2019S mutation. LRRK2 wild type flies showed a significant reduction in peroxidase activity when compared to control flies. When co-expressed with PRDX2, there was a significant restoration of peroxidase activity similar to control (p <0.05) (Figure 4A). G2019S flies showed an increased peroxidase activity when compared to control flies. The peroxidase activity was restored to normal levels when co-expressed with PRDX2 (Figure 4B). These results suggest that the presence of PRDX2 can help regulate extreme peroxidase activity (either hypo or hyper level) to a basal level.
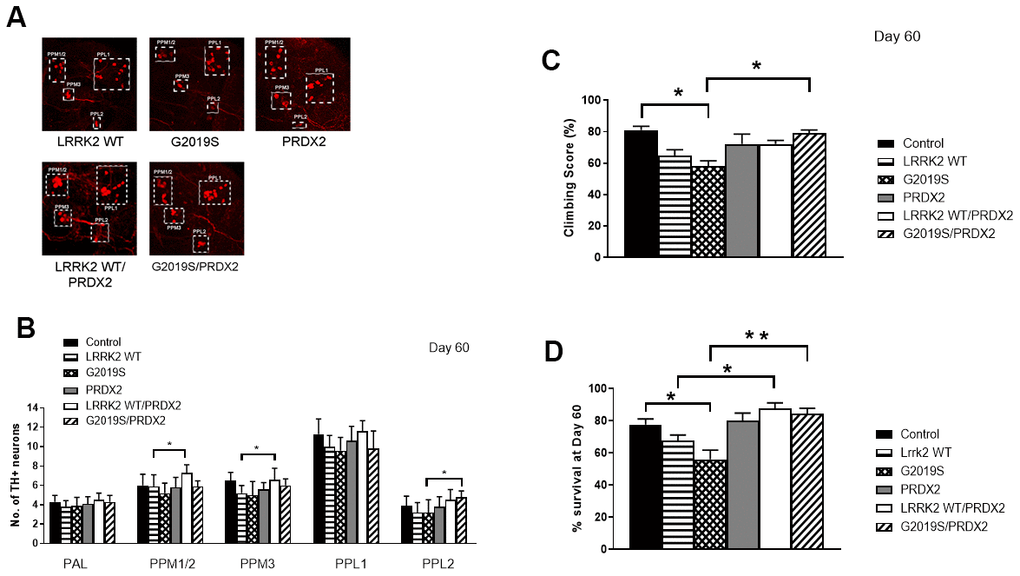
Figure 3. PRDX2 is able to rescue G2019S pathogenic phenotype. (A) Representative magnified confocal images of whole mount brains 60 days after eclosion. The different clusters of TH+ neurons are boxed up and labeled. (B) Bar graphs show number of TH-positive DA neuron clusters in flies at 60 days after eclosion (n = 3, cohort of 10). (C) Bar graph shows climbing scores of male flies at 60 days after eclosion. Percentage of flies that reached the top of the column after 1 min was counted (n = 3, cohort of 20). (D) Bar graph shows number of flies that survive after 60 days. Age-matched ddc-GAL4/+ flies were used as controls. Percentage of flies was tabulated. Significance indicated on the graph: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
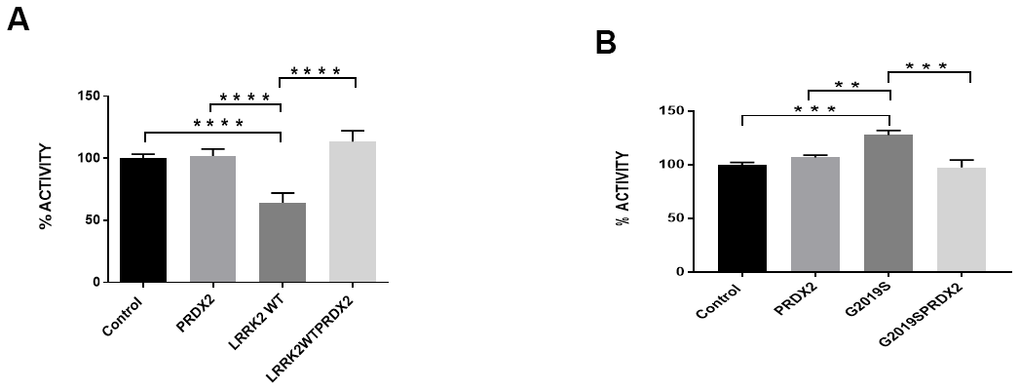
Figure 4. LRRK2 peroxidase activity. (A) Bar graph show LRRK2 WT peroxidase activity expressed as a percentage normalize to control. (B) Bar graph show G2019S peroxidase activity expressed as a percentage normalize to control. Significance indicated on the graph: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Chetomin drug treatment rescues G2019S pathology in vivo
Chetomin belongs to a class of epidithio-diketopiperazine (ETP) fungal secondary metabolites that contains a dithiol group that exhibits hydrogen peroxide-reducing activity relying on the thioredoxin system [36]. Thus, as PRDX2 is the only mammalian peroxidase coupled with a thioredoxin/thioredoxin reductase system, Chetomin is a natural compound that can mimic PRDX2 [37, 38]. Hence, we conducted Chetomin drug treatment on G2019S flies to detect if there is a rescue of G2019S pathogenic phenotype.
Chetomin partially rescued the integrity of DA neurons in PPL2 cluster as we observed a significant increase in the number of TH+ neurons in Chetomin treated G2019S flies compared to without treatment (p<0.05) (Figure 5A, 5B). Chetomin treated G2019S flies fared better in climbing compared to G2019S flies without drug treatment, indicating a rescue of pathogenic phenotype by Chetomin (p<0.05) (Figure 5C). There was also an improvement in the number of flies that survived after 60 days in Chetomin treated G2019S flies compared to G2019S flies without treatment (Figure 5D). The results collectively show that Chetomin is able to rescue the pathogenic phenotype of G2019S LRRK2 mutation in flies.
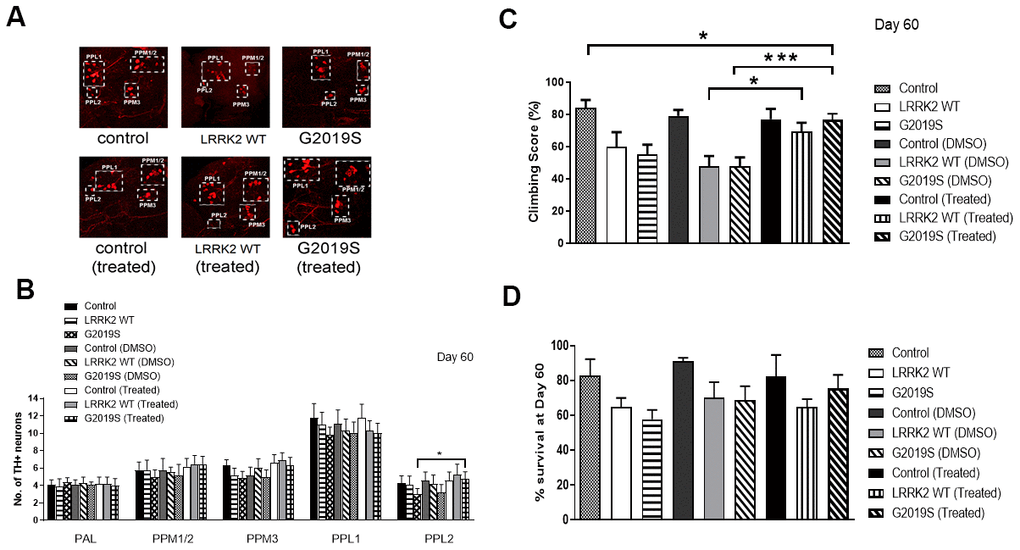
Figure 5. Challenge by Chetomin rescues G2019S phenotype. (A) Representative magnified confocal images of whole mount brains 60 days after eclosion. The different clusters of TH+ neurons are boxed up and labeled. (B) Bar graphs show number of TH-positive DA neuron clusters in flies at 60 days after eclosion (n = 3, cohort of 10). (C) Bar graph shows climbing scores of male flies at 60 days after eclosion. Percentage of flies that reached the top of the column after 1 min was counted (n = 3, cohort of 20). (D) Bar graph shows number of flies that survive after 60 days. Percentage of flies was tabulated. Significance indicated on the graph: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Discussion
Mutations in the kinase domain of LRRK2 were shown to increase its kinase activity and worsen the degeneration of DA neurons [18, 39]. These mutations lead to increase susceptibility to oxidative stress, although mechanisms linking both are still unclear. Oxidative stress is the imbalance between free radicals and antioxidants which disrupts key biological processes that leads to cell damage or death. Here, we showed that overexpression of mutant LRRK2 leads to DA neuronal death, decrease climbing ability, shortened lifespan and altered peroxidase activity. The overexpression of transgenic PRDX2 is able to suppress pathogenic LRRK2 phenotypes. We further demonstrated that Chetomin, PRDX2 mimic was able to rescue DA neurodegeneration associated with LRRK2 mutation.
We observed that LRRK2-G2019S kinase mutation causes an increase in the levels of phosphorylated PRDX2. For this mutation, a decreased number of DA neurons and lower climbing ability in the transgenic Drosophila were observed. We also observed a higher level of peroxidase activity in G2019S flies but this was restored to a basal level in the presence of PRDX2. Qu et al. showed that PRDX2 binds to Cdk5/p35 and is phosphorylated causing a reduction of PRDX2 peroxidase activity. These led to a downregulation of PRDX2 and increase in oxidative stress that affected DA neuronal loss [40]. Similarly another family of the 2-cys family PRDX1 peroxidase activity was showed to be inactivated by phosphorylation [41]. In G2019S flies there was excessive peroxidase activity and this could contribute to cell and tissue damage [28]. The presence of PRDX2 phosphorylation might serve to regulate peroxidase activity to a normalized level to protect the neurons. This further substantiates the importance of PRDX2 on oxidative stress and its involvement in the PD LRRK2 pathway.
The PRDX family protects cells from oxidative stress-induced apoptosis and has been associated with neurodegeneration [25, 26]. Studies previously have shown that the overexpression of PRDX2 in MN9D neuronal cells led to a decrease in reactive oxygen species [42]. PRDX2 has been shown to preserve cognitive function against hippocampal oxidative damage [32]. It is known that G2019S mutation in LRRK2 enhances oxidative stress-induced neurotoxicity and causes DA neurons to be more susceptible to oxidative stress [43, 44]. Endogenous PRDX2 might be part of this equation as with a lowered expression in LRRK2 G2019S mutation compared to wild-type LRRK2 because it is being phosphorylated, it fails to protect DA neurons from oxidative stress, and thus makes them more vulnerable. When PRDX2 is over expressed, there are more unphosphorylated PRDX2 to provide protection for the DA neurons. This supports our results that showed in vivo rescue of G2019S causing phenotypes when PRDX2 is over-expressed.
As proof of principle, challenges with Chetomin, a PRDX2 mimic drug, also recapitulated similar rescue of these phenotypic features in G2019S-mutant expressing flies. Chetomin has been shown to disrupt the interaction between hypoxia-inducible factor-1 (HIF-1) inhibitor by and p300 [37] and also exhibits anti-cancer activities [45, 46]. It was only recently found that the dithiol group in ETP family which Chetomin belongs to, displays hydrogen peroxide-reducing peroxidase activity, and played an effective role in regulating vascular endothelial growth factor receptor-β and vascular endothelial growth factor receptor mediated signaling in vascular cells, promoting healing of vascular injury [38]. Our study is the first to show that Chetomin can be used as a drug to mimic PRDX2 in rescuing the pathogenic phenotypes caused by G201S LRRK2 mutation in vivo. This provides the basis to further study the mechanism of Chetomin on PRDX2 and LRRK2.
In conclusion, we provided in vitro and in vivo evidence of PRDX2 interaction with LRRK2 and phosphorylation of PRDX2 by mutant G2019S LRRK2. We demonstrated that transgenic expression of PRDX2 with LRRK2 ameliorated G2019S induced loss of DA neurons, climbing ability and shortened lifespan. Challenging with Chetomin led to a similar rescue in Drosophila. Our findings suggest that chetomin can be a potential therapeutic compound in LRRK2 linked PD. Further studies to validate the effect of PRDX2 on PD patients with LRRK2 kinase mutations and to investigate the neuroprotective effects of Chetomin in other neurodegenerative diseases will be warranted.
Materials and Methods
Fly stocks
The following flies were used in this study: dopa decarboxylase (ddc)-GAL4, yellow white (yw) (Bloomington Drosophila Stock Center). Flies were raised on standard yeast-cornmeal-agar medium at 25ºC with 12-hour light and dark cycle.
Generation of transgenic strains
Human LRRK2-expressing flies were created by generating transgenic human LRRK2 wild type and variants and point mutations were introduced into LRRK2 using XL Quik change site directed mutagenesis kit and verified by sequencing to ensure the integrity of the cloned ORFs. PRDX2 plasmid (RC207413) was purchased from Origene. LRRK2 wild-type and G2019S cDNA containing a GFP tag at the C-terminus was inserted into the pUAST-attB plasmid, which will allow the UAS constructs to land into a chosen attP site in the fly genome during microinjection. Constructs were sent for microinjection into Drosophila embryos (Best Gene).
Co-immunoprecipitation
HEK293T cells were co-transfected with the plasmids using Turbofect (Thermo Scientific). Cells were collected 24h after transfection for western blot. Transfected HEK293T cells were washed with PBS and lysed in M-PER mammalian protein extraction reagent buffer (Thermo Scientific) supplemented with protease inhibitor and Phos Stop (Roche). The lysates are then incubated with anti GFP conjugated beads (Chemotek) O/N at 4ºC. The precipitates were then washed 5 times using NP40 buffer (50mM Tris pH7.4, 150mM NaCl and 1% NP40) and resuspended in 4X SDS loading buffer.
Western blot
40 to 50 heads were collected from flies and grinded in M-PER mammalian protein extraction reagent buffer supplemented with protease inhibitor and Phos Stop and placed on ice for 30 mins. They were centrifuged at maximum speed for 15mins and the supernatant was collected for western blot. Protein was extracted from fly head homogenates and equal amounts of protein from the various genotypes were resolved by SDS-PAGE. The following antibodies are used: For LRRK2: anti-GFP (Sigma Aldrich G1544), For PRDX2: anti DYKDDDK/Flag (Cell signaling 2368S) and anti PRDX2 (Sigma Aldrich SAB1406520 and EMD Millipore 07-610)
Phospho ELISA
PRDX antibody was coated on white 96 wells plate overnight and then blocked in BSA. The fly lysates were collected from 20 fly heads and protein concentration was quantified by thermo-scientific Bradford assay. Equal amount of proteins was added into the plate and incubated with the antibody. After which the plate was washed then it was incubated with generic phosphothreonine/serine HRP (Enzo Lifesciences ADI-KAP-ST2103-E and cell signaling 6949), exposed to chemiluminescence substrate and read by plate reader to detect PRDX2 phosphorylation levels. Background readings from ddc/+ flies were substracted from the phospho readings.
In vitro kinase assay
LRRK2 kinase assay was carried out using recombinant truncated LRRK2 protein (Invitrogen PV4873), ATP (Invitrogen PV3227), Kinase buffer (cell signaling) and purified PRDX2-GST protein. The kinase assay was carried out for 2 hours at 30 degrees Celsius. It was stopped by adding 2x SDS loading buffer and boiled for 5 min at 95°C. The protein was loaded on SDS page gel and PRDX2 phosphorylation was detected by phosphor-threonine (cell signaling 9386S).
Immuno-fluorescence and confocal microscopy
For endogenous cell staining, anti LRRK2 and anti PRDX2 (Sigma Aldrich L9918 and SAB1406520) were used. Flies were aged to day 60 after eclosion, before fly brains were dissected, fixed and stained according to published protocols [26]. Brains were probed with anti GFP and anti-flag for expression immune-florescence. Brains were probed with rabbit anti-tyrosine hydroxylase (1:500, Sigma-Aldrich T2928).
Climbing and lifespan assays
Climbing assay was performed as described previously [26]. To determine adult lifespan, 100 flies from each genotype under the direction of ddc-GAL4 were maintained on standard media. Newly eclosed adult flies were transferred into vials containing fresh media every 3 days and mortality was scored at day 60. Age-matched ddc-GAL4/+ flies used as controls.
Peroxidase assay
About 20 fly heads were homogenized lysis buffer and the peroxidase assay was performing according Amplex® Red Hydrogen Peroxide/Peroxidase Assay Kit Catalog no. A22188 from Invitrogen. The results were normalized against the control flies and expressed as a percentage.
Drug treatment
In drug treated flies, flies were fed with cornmeal-agar medium containing 10uM Chetomin (Sigma), which were first dissolved in DMSO, immediately after eclosion and throughout the entire experimental period.
Statistical analysis
Quantitative data are expressed as mean ± SEM, unless otherwise stated. Statistical significance for climbing assay, differences in the number of TH-positive DA neurons and peroxidase assay were analyzed using one-way Anova with Bonferroni’s post hoc test, unless otherwise stated. Significance indicated on the graph: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding
We like to thank the Singapore Ministry of Health’s National Medical Research Council for their support. This work was supported by the NMRC Open Fund Large Collaborative Grant (MOH-OFLCG18May-0002) and Singapore Translational Research (STaR) Investigator Award (NMRC/STaR/0030/2018).
References
- 1. Jankovic J, Tan EK. Parkinson’s disease: etiopathogenesis and treatment. Journal of Neurology, Neurosurgery and Psychiatry. 2020; http://jnnp.bmj.com/lookup/doi/10.1136/jnnp-2019-322338.
- 2. Bosgraaf L, Van Haastert PJ. Roc, a ras/GTPase domain in complex proteins. Biochim Biophys Acta. 2003; 1643:5–10. https://doi.org/10.1016/j.bbamcr.2003.08.008 [PubMed]
- 3. Mills RD, Mulhern TD, Liu F, Culvenor JG, Cheng HC. Prediction of the repeat domain structures and impact of Parkinsonism-associated variations on structure and function of all functional domains of leucine-rich repeat kinase 2 (LRRK2). Hum Mutat. 2014; 35:395–412. https://doi.org/10.1002/humu.22515 [PubMed]
- 4. Saha S, Guillily MD, Ferree A, Lanceta J, Chan D, Ghosh J, Hsu CH, Segal L, Raghavan K, Matsumoto K, Hisamoto N, Kuwahara T, Iwatsubo T, et al. LRRK2 modulates vulnerability to mitochondrial dysfunction in caenorhabditis elegans. J Neurosci. 2009; 29:9210–18. https://doi.org/10.1523/JNEUROSCI.2281-09.2009 [PubMed]
- 5. Wang X, Yan MH, Fujioka H, Liu J, Wilson-Delfosse A, Chen SG, Perry G, Casadesus G, Zhu X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet. 2012; 21:1931–44. https://doi.org/10.1093/hmg/dds003 [PubMed]
- 6. Juárez-Flores DL, González-Casacuberta I, Ezquerra M, Bañó M, Carmona-Pontaque F, Catalán-García M, Guitart-Mampel M, Rivero JJ, Tobias E, Milisenda JC, Tolosa E, Marti MJ, Fernández-Santiago R, et al. Exhaustion of mitochondrial and autophagic reserve may contribute to the development of LRRK2G2019S–Parkinson’s disease. J Transl Med. 2018; 16:160. https://doi.org/10.1186/s12967-018-1526-3 [PubMed]
- 7. Gillardon F. Interaction of elongation factor 1-alpha with leucine-rich repeat kinase 2 impairs kinase activity and microtubule bundling in vitro. Neuroscience. 2009; 163:533–39. https://doi.org/10.1016/j.neuroscience.2009.06.051 [PubMed]
- 8. Parisiadou L, Xie C, Cho HJ, Lin X, Gu XL, Long CX, Lobbestael E, Baekelandt V, Taymans JM, Sun L, Cai H. Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J Neurosci. 2009; 29:13971–80. https://doi.org/10.1523/JNEUROSCI.3799-09.2009 [PubMed]
- 9. Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, Raya A, Sulzer D, Cuervo AM. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. 2013; 16:394–406. https://doi.org/10.1038/nn.3350 [PubMed]
- 10. Bravo-San Pedro JM, Niso-Santano M, Gómez-Sánchez R, Pizarro-Estrella E, Aiastui-Pujana A, Gorostidi A, Climent V, López de Maturana R, Sanchez-Pernaute R, López de Munain A, Fuentes JM, González-Polo RA. The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell Mol Life Sci. 2013; 70:121–36. https://doi.org/10.1007/s00018-012-1061-y [PubMed]
- 11. Berwick DC, Harvey K. LRRK2 functions as a Wnt signaling scaffold, bridging cytosolic proteins and membrane-localized LRP6. Hum Mol Genet. 2012; 21:4966–79. https://doi.org/10.1093/hmg/dds342 [PubMed]
- 12. Sancho RM, Law BM, Harvey K. Mutations in the LRRK2 roc-COR tandem domain link Parkinson’s disease to Wnt signalling pathways. Hum Mol Genet. 2009; 18:3955–68. https://doi.org/10.1093/hmg/ddp337 [PubMed]
- 13. Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis. 2013; 3:461–91. https://doi.org/10.3233/JPD-130230 [PubMed]
- 14. Tan EK, Chao YX, West A, Chan LL, Poewe W, Jankovic J. Parkinson disease and the immune system – associations, mechanisms and therapeutics. Nat Rev Neurol. 2020; 16:303–18. https://doi.org/10.1038/s41582-020-0344-4 [PubMed]
- 15. Di Fonzo A, Rohé CF, Ferreira J, Chien HF, Vacca L, Stocchi F, Guedes L, Fabrizio E, Manfredi M, Vanacore N, Goldwurm S, Breedveld G, Sampaio C, et al, and Italian Parkinson Genetics Network. A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson’s disease. Lancet. 2005; 365:412–15 https://doi.org/10.1016/S0140-6736(05)17829-5 [PubMed]
- 16. Chan SL, Tan EK. Targeting LRRK2 in Parkinson’s disease: an update on recent developments. Expert Opin Ther Targets. 2017; 21:601–10. https://doi.org/10.1080/14728222.2017.1323881 [PubMed]
- 17. Refai FS, Ng SH, Tan EK. Evaluating LRRK2 genetic variants with unclear pathogenicity. Biomed Res Int. 2015; 2015:678701. https://doi.org/10.1155/2015/678701 [PubMed]
- 18. Long S, Guo W, Hu S, Su F, Zeng Y, Zeng J, Tan EK, Ross CA, Pei Z. G2019S LRRK2 increases stress susceptibility through inhibition of DAF-16 nuclear translocation in a 14-3-3 associated-manner in Caenorhabditis elegans. Front Neurosci. 2018; 12:782. https://doi.org/10.3389/fnins.2018.00782 [PubMed]
- 19. Howlett EH, Jensen N, Belmonte F, Zafar F, Hu X, Kluss J, Schüle B, Kaufman BA, Greenamyre JT, Sanders LH. LRRK2 G2019S-induced mitochondrial DNA damage is LRRK2 kinase dependent and inhibition restores mtDNA integrity in Parkinson’s disease. Hum Mol Genet. 2017; 26:4340–51. https://doi.org/10.1093/hmg/ddx320 [PubMed]
- 20. Volpicelli-Daley LA, Abdelmotilib H, Liu Z, Stoyka L, Daher JP, Milnerwood AJ, Unni VK, Hirst WD, Yue Z, Zhao HT, Fraser K, Kennedy RE, West AB. G2019S-LRRK2 expression augments α-synuclein sequestration into inclusions in neurons. J Neurosci. 2016; 36:7415–27. https://doi.org/10.1523/JNEUROSCI.3642-15.2016 [PubMed]
- 21. Fujii J, Ikeda Y. Advances in our understanding of peroxiredoxin, a multifunctional, mammalian redox protein. Redox Rep. 2002; 7:123–30. https://doi.org/10.1179/135100002125000352 [PubMed]
- 22. Seaver LC, Imlay JA. Alkyl hydroperoxide reductase is the primary scavenger of endogenous hydrogen peroxide in escherichia coli. J Bacteriol. 2001; 183:7173–81. https://doi.org/10.1128/JB.183.24.7173-7181.2001 [PubMed]
- 23. Rhee SG, Kang SW, Chang TS, Jeong W, Kim K. Peroxiredoxin, a novel family of peroxidases. IUBMB Life. 2001; 52:35–41. https://doi.org/10.1080/15216540252774748 [PubMed]
- 24. Rhee SG, Woo HA. Multiple functions of peroxiredoxins: peroxidases, sensors and regulators of the intracellular messenger H2O2, and protein chaperones. Antioxid Redox Signal. 2011; 15:781–94. https://doi.org/10.1089/ars.2010.3393 [PubMed]
- 25. Lee YM, Park SH, Shin DI, Hwang JY, Park B, Park YJ, Lee TH, Chae HZ, Jin BK, Oh TH, Oh YJ. Oxidative modification of peroxiredoxin is associated with drug-induced apoptotic signaling in experimental models of Parkinson disease. J Biol Chem. 2008; 283:9986–98. https://doi.org/10.1074/jbc.M800426200 [PubMed]
- 26. Angeles DC, Ho P, Chua LL, Wang C, Yap YW, Ng C, Zhou Zd, Lim KL, Wszolek ZK, Wang HY, Tan EK. Thiol peroxidases ameliorate LRRK2 mutant-induced mitochondrial and dopaminergic neuronal degeneration in Drosophila. Hum Mol Genet. 2014; 23:3157–65. https://doi.org/10.1093/hmg/ddu026 [PubMed]
- 27. Perkins A, Nelson KJ, Parsonage D, Poole LB, Karplus PA. Peroxiredoxins: guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem Sci. 2015; 40:435–45. https://doi.org/10.1016/j.tibs.2015.05.001 [PubMed]
- 28. Vlasova II. Peroxidase activity of human hemoproteins: keeping the fire under control. Molecules. 2018; 23:2561. https://doi.org/10.3390/molecules23102561 [PubMed]
- 29. Hopkins BL, Neumann CA. Redoxins as gatekeepers of the transcriptional oxidative stress response. Redox Biol. 2019; 21:101104. https://doi.org/10.1016/j.redox.2019.101104 [PubMed]
- 30. Liu J, Su G, Gao J, Tian Y, Liu X, Zhang Z. Effects of peroxiredoxin 2 in neurological disorders: a review of its molecular mechanisms. Neurochem Res. 2020; 45:720–30. https://doi.org/10.1007/s11064-020-02971-x [PubMed]
- 31. Angeles DC, Gan BH, Onstead L, Zhao Y, Lim KL, Dachsel J, Melrose H, Farrer M, Wszolek ZK, Dickson DW, Tan EK. Mutations in LRRK2 increase phosphorylation of peroxiredoxin 3 exacerbating oxidative stress-induced neuronal death. Hum Mutat. 2011; 32:1390–97. https://doi.org/10.1002/humu.21582 [PubMed]
- 32. Kim SU, Jin MH, Kim YS, Lee SH, Cho YS, Cho KJ, Lee KS, Kim YI, Kim GW, Kim JM, Lee TH, Lee YH, Shong M, et al. Peroxiredoxin II preserves cognitive function against age-linked hippocampal oxidative damage. Neurobiol Aging. 2011; 32:1054–68. https://doi.org/10.1016/j.neurobiolaging.2009.05.017 [PubMed]
- 33. Botia B, Seyer D, Ravni A, Bénard M, Falluel-Morel A, Cosette P, Jouenne T, Fournier A, Vaudry H, Gonzalez BJ, Vaudry D. Peroxiredoxin 2 is involved in the neuroprotective effects of PACAP in cultured cerebellar granule neurons. J Mol Neurosci. 2008; 36:61–72. https://doi.org/10.1007/s12031-008-9075-5 [PubMed]
- 34. Fang J, Nakamura T, Cho DH, Gu Z, Lipton SA. S-nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson’s disease. Proc Natl Acad Sci USA. 2007; 104:18742–47. https://doi.org/10.1073/pnas.0705904104 [PubMed]
- 35. Goemaere J, Knoops B. Peroxiredoxin distribution in the mouse brain with emphasis on neuronal populations affected in neurodegenerative disorders. J Comp Neurol. 2012; 520:258–80. https://doi.org/10.1002/cne.22689 [PubMed]
- 36. Gardiner DM, Waring P, Howlett BJ. The epipolythiodioxopiperazine (ETP) class of fungal toxins: distribution, mode of action, functions and biosynthesis. Microbiology. 2005; 151:1021–32. https://doi.org/10.1099/mic.0.27847-0 [PubMed]
- 37. Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, Cornell-Kennon S, Lee J, Wang B, Wang J, Memmert K, Naegeli HU, Petersen F, et al. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell. 2004; 6:33–43. https://doi.org/10.1016/j.ccr.2004.06.009 [PubMed]
- 38. Kang DH, Lee DJ, Kim J, Lee JY, Kim HW, Kwon K, Taylor WR, Jo H, Kang SW. Vascular injury involves the overoxidation of peroxiredoxin type II and is recovered by the peroxiredoxin activity mimetic that induces reendothelialization. Circulation. 2013; 128:834–44. https://doi.org/10.1161/CIRCULATIONAHA.113.001725 [PubMed]
- 39. West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA. 2005; 102:16842–47. https://doi.org/10.1073/pnas.0507360102 [PubMed]
- 40. Qu D, Rashidian J, Mount MP, Aleyasin H, Parsanejad M, Lira A, Haque E, Zhang Y, Callaghan S, Daigle M, Rousseaux MW, Slack RS, Albert PR, et al. Role of Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity and Parkinson’s disease. Neuron. 2007; 55:37–52. https://doi.org/10.1016/j.neuron.2007.05.033 [PubMed]
- 41. Chu KL, Lew QJ, Rajasegaran V, Kung JT, Zheng L, Yang Q, Shaw R, Cheong N, Liou YC, Chao SH. Regulation of PRDX1 peroxidase activity by Pin1. Cell Cycle. 2013; 12:944–52. https://doi.org/10.4161/cc.23916 [PubMed]
- 42. Hu X, Weng Z, Chu CT, Zhang L, Cao G, Gao Y, Signore A, Zhu J, Hastings T, Greenamyre JT, Chen J. Peroxiredoxin-2 protects against 6-hydroxydopamine-induced dopaminergic neurodegeneration via attenuation of the apoptosis signal-regulating kinase (ASK1) signaling cascade. J Neurosci. 2011; 31:247–61. https://doi.org/10.1523/JNEUROSCI.4589-10.2011 [PubMed]
- 43. Heo HY, Park JM, Kim CH, Han BS, Kim KS, Seol W. LRRK2 enhances oxidative stress-induced neurotoxicity via its kinase activity. Exp Cell Res. 2010; 316:649–56. https://doi.org/10.1016/j.yexcr.2009.09.014 [PubMed]
- 44. Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, Kee K, Schüle B, Dolmetsch RE, Langston W, Palmer TD, Pera RR. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell. 2011; 8:267–80. https://doi.org/10.1016/j.stem.2011.01.013 [PubMed]
- 45. Staab A, Loeffler J, Said HM, Diehlmann D, Katzer A, Beyer M, Fleischer M, Schwab F, Baier K, Einsele H, Flentje M, Vordermark D. Effects of HIF-1 inhibition by chetomin on hypoxia-related transcription and radiosensitivity in HT 1080 human fibrosarcoma cells. BMC Cancer. 2007; 7:213. https://doi.org/10.1186/1471-2407-7-213 [PubMed]
- 46. Viziteu E, Grandmougin C, Goldschmidt H, Seckinger A, Hose D, Klein B, Moreaux J. Chetomin, targeting HIF-1α/p300 complex, exhibits antitumour activity in multiple myeloma. Br J Cancer. 2016; 114:519–23. https://doi.org/10.1038/bjc.2016.20 [PubMed]