




Introduction
Amyotrophic lateral sclerosis (ALS) is the most common adult-onset progressive neurodegenerative disease and is characterized by selective degeneration of upper and lower motor neurons in the brain and spinal cord [1]. The ALS median incidence rate is approximately two per 100,000 persons [2]. Median age of symptom onset is 55 years, and the average survival time is 3-5 years [3]. Approximately 10% of ALS cases are patients with familial ALS (fALS), whereas the remaining 90% are sporadic ALS (sALS) [4].
To date, more than 30 genes have been linked to ALS. However, only a few, including SOD1, C9ORF72, FUS, and TARDBP, have been identified as causative ALS genes [5, 6]. The pathogenesis of ALS remains unclear; however, one of the possible mechanisms underlying ALS is defective RNA metabolism and homeostasis [7]. Recent studies have shown that FUS and TARDBP RNA-binding proteins may contribute to ALS pathogenesis [8], and that most of these causative FUS and TARDBP mutations cluster in the C-terminus of proteins, called mutation hotspot regions [9, 10].
Sanger sequencing and next-generation sequencing for mutation screening in large samples are time consuming and expensive. In contrast, here we screen the FUS and TARDBP mutation hotspot regions in 146 ALS cases using the rapid, highly sensitive screening method, high-resolution melting (HRM) analysis. HRM analysis is a recently developed genetic analysis method for fast, high-throughput post-PCR analysis. During high-resolution melting analysis, melting curves are produced using dyes that fluoresce in the presence of double-stranded DNA (dsDNA) and specialized instruments designed to monitor fluorescence during heating; as the temperature increases, the fluorescence decreases, producing a characteristic melting profile. The shape differences in melting curves, obtained as fluorescence difference plots, are used to distinguish between mutations and controls [11, 12].
In this study, we demonstrated that highly sensitive HRM analysis can be used for screening FUS and TARDBP mutations and identified two novel mutations in Chinese sALS and fALS cases.
Results
Sensitivity and HRM analysis
Excluding synonymous mutations and normal single-nucleotide polymorphisms, HRM analysis identified eight pathogenic mutations in 146 patients (Table 1). Similarly, targeted sequencing also detected the eight pathogenic mutations, confirming the mutation detection sensitivity of HRM analysis was 100% in this cohort. These mutations included two different FUS pathogenic mutations in two different ALS patients and five TARDBP pathogenic mutations in six ALS patients, which included two novel mutations. Therefore, FUS mutations were present in 1.4% of Chinese ALS patients, whereas TARDBP mutations were responsible for 4.1% of Chinese ALS cases.
Table 1. Clinical data of ALS patients carrying pathogenic mutations in FUS and TARDBP genes.
| Gene | Nucleotide change | Amino acid change | Type of mutation | Onset; age of onset;gender | Disease duration | ALS family history | References | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FUS | c.1541+1G>A | - | splicing | Bulbar; 51; M | 1 y | Yes | This study | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FUS | c.1562G>A | p. R521H | missense | Spinal; 41; M | 3 y | Yes | [14] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TARDBP | c.881G>T | p. G294V | missense | Bulbar, Spinal; 61; M | 0.5 y | No | [15, 16] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Bulbar; 53; M | 1.5+y | No | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TARDBP | c.1009A>G | p. M337V | missense | Spinal; 58; M | 1+y | Yes | [17] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TARDBP | c.1043G>T | p. G348V | missense | Spinal; 24; M | 10+y | No | [18] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TARDBP | c.1147A>G | p. I383V | missense | Spinal; 45; F | 3 y | No | [19] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TARDBP | c. *731A>G | - | 3’UTR | -;N/A;M | - | No | This study | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| The symbol + indicates disease in progress. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Key: ALS, amyotrophic lateral sclerosis; F, female; M, male; N/A, information not available | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
FUS mutations and clinical characteristics
Figure 1A shows the detected novel splicing mutation, c.1541+1G>A (Figure 1A), located at the splice donator site of the FUS intron 14. The guanine of the wild-type donator site is highly conserved across species (Figure 1B). A functional splicing reporter minigene assay confirmed the novel splicing mutation result in a frameshift, leading to truncated FUS protein p.G466VfsX14 (Figure 1C).
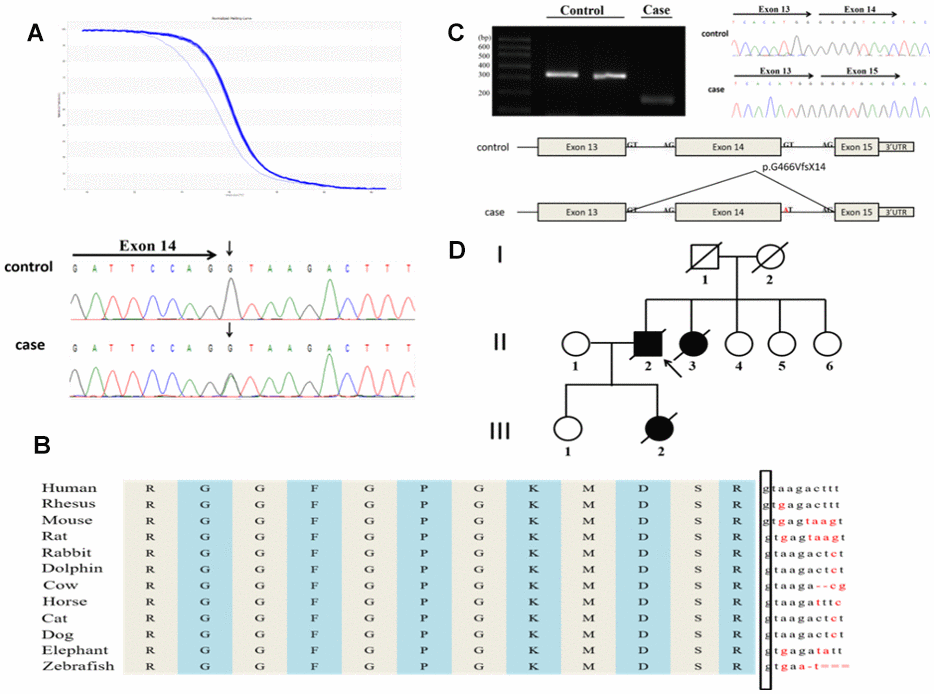
Figure 1. A novel splicing mutation, c.1541+1G>A in the FUS gene identified in a Chinese ALS patient. (A) The novel splicing mutation was identified by HRM analysis and direct sequencing. (B) The evolutionary conservation of the splicing mutation c.1541+1G>A are shown. (C) Minigene splicing analysis of the novel splice mutation. (D) Pedigree of the family. An arrowhead indicates the proband.
The novel splicing mutation was identified in a 51-year-old male patient whose daughter previously died from ALS. Initially, he presented with neck weakness and dysphagia. After 5 months, lower right limb weakness emerged and gradually increased without cognitive impairment. The patient died 13 months after onset of symptoms. A 20-year-old female patient, the daughter of the 51-year-old patient, experienced lower right limb weakness, which quickly developed in all four extremities within 1 year. After 12 months, she died of respiratory failure. Figure 1D shows the same mutation was detected in the sister of the 51-year-old patient’s sister and shared a similar clinical phenotype; the disease duration of this patient was only 8 months.
A heterozygous missense substitution, c.1562G>A (p. R521H, Figure 2A), was identified in an index fALS patient who began suffering from progressive weakness in the upper-right limb at 41 years of age. Twelve months later, the weakness developed in other limbs. Then, he presented with dysarthria and dysphagia after 13 months and without cognitive impairment. The patient died 3 years after onset of symptoms. The patient’s brother who shared the same mutation (patient III1) also had a similar clinical phenotype and disease duration. Patient III2, as well as several family members of patients III1 and III3, also share the mutation; however, they did not show any clinical characteristics of ALS until now (Figure 2B).
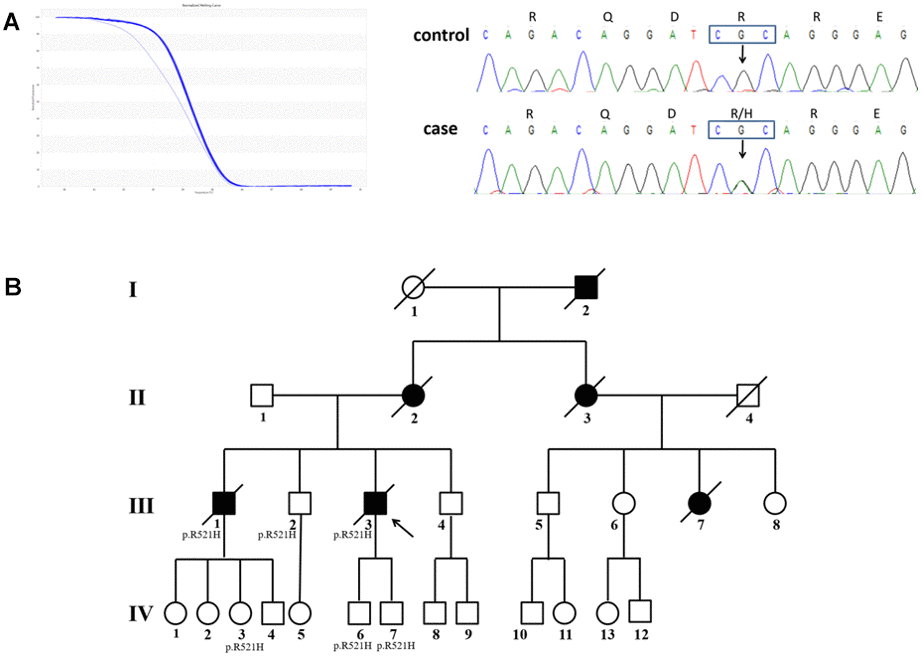
Figure 2. The pedigree with FUS mutation p. R521H. (A) Results of HRM analysis and direct sequencing of patients. (B) Pedigree of the family. An arrowhead indicates the proband. Patients III2, IV3, IV6, and IV7 shared the same mutation, but they did not appear as ALS phenotypes until now.
TARDBP mutations and clinical characteristics
Five TARDBP mutations were observed in six unrelated ALS patients (Figure 3A), which included a novel 3′UTR mutation (c. *731A>G) and four known causative mutations (p.G294V, p.G348V, p.M337V, and p.I383V). According to the UCSC Genome Browser, the TARDBP 3′-UTR site c. *731 is highly conserved through the evolutionary spectrum from human to chicken (Figure 3B), supporting its functional importance. MicroRNA (miRNA) binding searches of TARDBP through the TargetScanHuman database showed binding of miR-376a directly to the 3′UTR variant region. The novel c. *731A>G TARDBP mutation was expected to interfere with the normal binding of miR-376, and then disorder the expression of TARDBP mRNA. However, we sequenced this region of the patient’s parents and confirmed this was a de novo mutation. In addition, the mutation was not observed in available variation databases, including mutation database and SNP databases such as the National Center for Biotechnology Information (NCBI) Single Nucleotide Polymorphism Database (http://www.ncbi.nlm.nih.gov/projects/SNP/), Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org), and ALS Online Genetics Database (http://alsod.iop.kcl.ac.uk/) [13]. According to American College of Medical Genetics and Genomics guidelines, it meets de novo criteria PS2 (in a patient with the disease and no family history), PM2 (absent from controls or at extremely low frequency if recessive in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium), and PP4 (patient’s phenotype or family history is highly specific for a disease with a single genetic etiology) for classifying pathogenic variants. We suggest this mutation is a “likely pathogenic” for the previously stated reason.
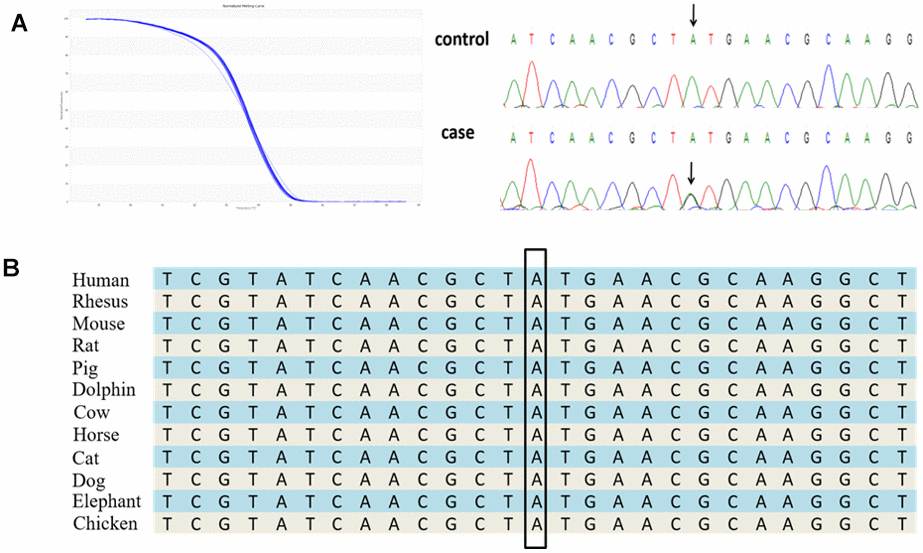
Figure 3. A novel 3′UTR mutation, c. *731A>G in the TARDBP gene identified in a Chinese ALS patient. (A) The novel 3′UTR mutation was identified by HRM analysis and direct sequencing. (B) The evolutionary conservation of the TARDBP 3′UTR mutation c. *731A>G.
The TARDBP p.G294V mutation is located in the sixth exon of the gene and was present in two unrelated male sALS patients. The patient began undergoing dysarthria with weakness in the upper right limb. Five months later, the weakness developed in the upper left limb, and dysphagia occasionally occurred. He died 6 months after onset of symptoms. At age 53, the second patient experienced symptoms primarily characterized by dysarthria. Neither ALS patient carrying the p.G294V mutation showed signs of cognitive dysfunction.
The p.G348V mutation was identified in a 24-year-old juvenile-onset male patient who reported no family history of ALS. During the initial onset of symptoms, the patient showed slight fasciculation in the upper right limb. During the next 3 years, the fasciculation was progressive and developed in the upper left limb. The MRI images show that the anisotropy of cortical spinal tract scores in the left midbrain was indicative of white matter degeneration.
To date, fasciculation and muscle atrophy have been observed in all extremities.
A 45-year-old female patient carrying the p.I383V mutation presented with progressive weakness in the upper limb, and electromyography results suggested spinal anterior of the lower cervical was injured. After 3 years, the patient died of respiratory failure.
Discussion
Following rapid development of molecular genetics, many genes have been identified with ALS pathogenesis. Although the exact mechanism for ALS is still unknown, several hypotheses include defective RNA metabolism, glutamate excitotoxicity, disruption of membrane trafficking, endoplasmic reticulum stress, mitochondrial dysfunction, and protein misfolding and aggregation [20]. Some mutations in FUS and TARDBP genes, which both play an important role in mRNA transport, axonal maintenance, and motor neuron development, have been reported as causative ALS mutations for disturbing RNA homeostasis [4, 9, 21]. Interestingly, because the vast majority of these causative mutations in FUS and TARDBP cluster in the C-terminus of the proteins, also known as mutation hotspot regions, [9, 10] this suggested we could focus on mutation detection at the mutational hotspot regions, instead of the whole gene.
Although Sanger sequencing and next-generation sequencing are two of most common mutation detection methods for large-scale genomics sequencing samples, they can be cost prohibitive and slow. In this study, we first identified the FUS and TARDBP mutations in Chinese ALS patients by the fast and highly sensitive screening method, HRM analysis.
Using HRM analysis, we successfully identified five known possible causative mutations, and two novel mutations. The mutation frequency was 1.4% for FUS, and 4.1% for TARDBP in Chinese ALS patients. However, the frequency of our study is inconsistent with previously reported results, [22] which may be due to small sample size and population difference.
We identified two FUS pathogenic mutations in this study. Previous reports identified mutation p.R521H as the highest FUS mutation frequency, accounting for 30%, and experimental results show that the FUS R521H missense mutation could lead to aberrant trafficking and retention of the mutant FUS protein in the cytoplasm [14, 23, 24]. Interestingly, several relatives and a sibling of the proband also have the p.R521H mutation; however, they did not present with clinically relevant ALS symptoms, which may be related to age or the presence of a recessive inheritance pattern [14, 25].
A novel splicing mutation, c.1541+1G>A, the third splicing mutation linked to ALS in FUS gene [26, 27], produced a truncated FUS protein, p.G466VfsX14, eliminating the nuclear localization sequence (NLS), where most previously published mutations are located [9], and then impair its nuclear import. Previous results show that overexpression of cytoplasmic FUS with deleted NLS results in recapitulation of ALS-like motor neuron abnormalities in mice, [28] indicating this mutation is significant. Furthermore, mutation of a truncated protein lacking the NLS was reported to cause juvenile ALS with rapid disease progression [29, 30], which was consistent with the clinical symptoms in our study. Subsequent research is still needed to study the function of the mutation. The novel splicing mutation we found confirms the C-terminal NLS of the FUS gene plays a key role in ALS pathology.
We identified five TARDBP mutations in our ALS cohort, four of which have previously been reported as causative mutations. The p.G294V mutation was previously identified in a sALS patient from Italy and in a fALS patient from Australia, and was predicted to disrupt the glycine-rich domain in the C-terminus, which plays a role in RNA binding and is required for the exon-skipping activity of TARDBP [15, 31]. Previous reports showed the percentages of cells with positive nuclear immunostaining were significantly lower in patients with TARDBP p.G294V mutations than in controls, indicating its importance [16]. In this study, we identified p.G294V mutations in two unrelated sALS patients, for whom the onset of symptoms is characterized by dysarthria. To our knowledge, this is the first Asian population study for this mutation. The p.G348V mutation, which increases the amount of TARDBP protein at the cellular level, was detected in ALS patients from various populations [18, 32–34]. However, there are some differences for the ALS clinical symptoms between the western and Chinese populations. The age of onset for Chinese ALS cases were much younger than previously published western cases. Additionally, the mutation carriers in the Chinese population experienced slower disease progression and longer survival time [34]. Our study confirmed these differences. The third mutation we identified, p.I383V, has been detected in fALS patients from the United States, Turkey, and Taiwan. However, the onset and progression of the disease for the carrier p.I383V are different [19, 35–37]. In our study, the mutation p.I383V was identified from a sALS woman who suffered progressive weakness in the upper limb at the age of 45, and died 3 years later.
The final mutation we identified, the novel 3'UTR mutation c. *731A>G, was a de novo and was not observed in the parents of patients. Additionally, it has not be reported in any mutation databases, and the site c. *731 is highly conserved. According to the software, the mutation may affect the combination of mir-376a for TARDBP and then lead to increasing mRNA levels, which may accelerate disease progression by inducing the formation of cytoplasmic inclusions and/or the dysregulation of RNA metabolism [38]. In addition, a previous study reported a similar 3'UTR mutation linked to ALS [39]. According to the guideline of ACMG, we suggested c. *731A>G as a likely pathogenic mutation for ALS. Unfortunately, we did not have access to detailed medical records and fresh blood samples.
In conclusion, to our knowledge, this is the first time that HRM analysis was used to identify FUS and TARDBP mutations in Chinese patients with ALS, demonstrating its high sensitivity and accuracy. Our data suggests that HRM analysis is a fast and easy method for mutation screening in large samples. Most important, however, we identified two novel pathogenic ALS mutations, which further expands the ALS-related gene mutation spectrum, and provides data for subsequent studies. We also investigated the frequency of FUS and TARDBP mutations in the Chinese population.
Materials and Methods
Subjects
The study included 146 patients with sALS and nine fALS index cases (male: 62.3%; age at onset: 47.9 ± 11.7 years). All patients were recruited from the Department of Neurology, Southwest Hospital, Third Military Medical University, Chongqing between 2012 and 2017. All patients were Han Chinese, and the patients met the El Escorial criteria for ALS diagnosis [40]. Family history was considered positive if the patient had at least one affected relative within three generations [41]. All participants provided informed consent before blood donation. Protocols were in accordance with the ethical standards of the responsible committee on human experimentation and the Helsinki Declaration of 1964 and approved by the institutional ethics committee of the Hospital (equivalent to an Institutional Review Board). This study was carried out in accordance with the approved guidelines. Written informed consent was obtained from all participants. All patients were tested for the C9orf72 GGGGCC expanding mutation with repeat-primed PCR, together with proper positive control and negative control. No C9orf72 GGGGCC expanding mutation was detected in this cohort.
Screening TARDBP and FUS gene mutations by PCR-HRM
We designed PCR primers for HRM analysis to screen all of the mutation hotspot regions containing OMIM Allelic Variants mutations related to ALS (from the UCSC Genome Browser) of FUS (NM_004960.3) and TARDBP (NM_007375.3). Table 2 shows the amplified primer sequence and the length of each expected fragment.
Table 2. PCR primer sequences of FUS and TARDBP gene and size of amplified fragment.
| Gene | Primer | Primer sequence | Fragment size |
| FUS | 1 | 5′-GAGGGTAACACTGGGTACAGGAC-3′ | 160 |
| 5′-GGCTTGGAGAGGCTGGTAAC-3′ | |||
| 2 | 5′-GGTTATGGCAATCAAGACCAGA-3′ | 149 | |
| 5′-CTGGGTTCATAGCCACCACT-3′ | |||
| 3 | 5′- GCTATGATCGAGGCGGCTA -3′ | 158 | |
| 5′-TGGCCTCTGTTCAACTGCTC -3′ | |||
| TARDBP | 1 | 5′-CGGAATATGAAACACAAGTGAAAG-3′ | 82 |
| 5′-GAATTAGGAAGTTTGCAGTCACAC-3′ | |||
| 2 | 5′-CCGAACCTAAGCACAATAGCA-3′ | 156 | |
| 5′-TCATCCCACCACCCATATTACT-3′ | |||
| 3 | 5′-AATATGGGTGGTGGGATGAAC -3′ | 160 | |
| 5′-TGTTGCCTTGGTTTTGGTTATT-3′ | |||
| 4 | 5′-AATAACCAAAACCAAGGCAACAT-3′ | 157 | |
| 5′-TTGAGCCAAAGCCTCCATTA- 3′ | |||
| 5 | 5′-GGGTTGTGGTTGGTTGGTATAG-3′ | 130 | |
| 5′-CTGCTGAATATACTCCACACTGAAC-3′ | |||
| 6 | 5′-GCCATAGGAATACTGTCTACATGCT-3′ | 152 | |
| 5′-CCATATCACAGCCTTGCGTT-3′ |
Genomic DNA was extracted from peripheral blood leukocytes using standard methods.
PCR-HRM was performed with Eco Real-Time PCR System (Illumina) in 10-μL reaction mixtures comprising 20 ng of DNA, 1×PCR buffer, 0.2 mM dNTP, Eva Green (Biotium),1U of Taq polymerase, and 0.25 μM each forward and reverse primers. Initial denaturation was performed at 95°C for 5 min, followed by 50 cycles of 95°C for 20 sec, 60°C for 20 sec, and 72°C for 20 sec. The melting curves were obtained under the following conditions: 95°C for 15 sec, 55°C for 15 sec, and with a ramping rate of +0.2°C/sec to 95°C.
Direct DNA sequencing and bioinformatics
To confirmed the genotype of mutations and the specificity of HRM analysis, All PCR products showing an HRM aberrant pattern were analyzed with direct DNA sequencing on a PRISM 377 full-automatic sequencing analyzer (ABI).
When the DNA sequencing result was determined, we first confirmed whether there was a mutation aimed to detect the specificity of HRM analysis. Then, we ensured it was a novel mutation using the HGMD database (http://www.hgmd.cf.ac.uk/ac) and the dbSNP database (http://www.ncbi.nlm.nih.gov/snp). Finally, to evaluate the potential functional effect of the novel splicing mutation and 3'UTR site mutation, we used the UCSC Genome Browser (http://genome.ucsc.edu/), Target (http://www.targetscan.org/vert_71), Human Splicing Finder (http://www.umd.be/HSF3/). To identify known variations, we used ALS Online Genetics Database (http://alsod.iop.kcl.ac.uk/) (13), the NCBI Database (http://www.ncbi.nlm.nih.gov), and Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org).
Targeted sequencing
To verify the sensitivity of HRM analysis, we sequenced all exons of the FUS and TARDBP gene in 146 patients by Illumina Hiseq 3000 system. The target coverage is 98% with an average depth of 100X. All nonsynonymous variants detected were filtered.
Functional splicing reporter minigene assay
Because of the lack of simple RNA, we used splicing reporter minigene assay to confirm the functional effect of the novel splicing mutation. The minigene was constructed by PCR amplification of mutant genomic DNA sequences as previously described [42]. The amplified sequences included exons 13 through 3’UTR of the FUS gene. FUS minigene products were digested with BamH I and Xho I (Thermo) and cloned into the pCDNA3.1(+) reporter vector. The resulting constructs were transfected into U87 cells. Total cellular RNA was extracted after 24 hours, followed by RT-PCR analysis.
Author Contributions
F.W., S.F. and J.L. performed experiments, analyzed the data, composed the figures, and wrote the manuscript. H.W. and S.F. assisted with data analysis, presentation and the manuscript. S.S., K.C. and J.H. as neurologic clinicians, diagnosed diseases, enrolled the participants and collected the blood samples. X.X., F.W. and J.H. conceived and supervised the project, analyzed the data, and wrote the manuscript. All the authors have contributed to and approved the final manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding
The study reported in the article was supported by the National Natural Science Foundation of China (grant no.30300116,30771950) and Shenzhen Municipal Government of China (JCYJ20180703093402288).
References
- 1. Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC. Amyotrophic lateral sclerosis. Lancet. 2011; 377:942–55. https://doi.org/10.1016/S0140-6736(10)61156-7 [PubMed]
- 2. Chiò A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, White LA. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology. 2013; 41:118–30. https://doi.org/10.1159/000351153 [PubMed]
- 3. Qureshi M, Schoenfeld DA, Paliwal Y, Shui A, Cudkowicz ME. The natural history of ALS is changing: improved survival. Amyotroph Lateral Scler. 2009; 10:324–31. https://doi.org/10.3109/17482960903009054 [PubMed]
- 4. Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014; 17:17–23. https://doi.org/10.1038/nn.3584 [PubMed]
- 5. Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K, Marroquin N, Nordin F, Hübers A, Weydt P, Pinto S, Press R, Millecamps S, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015; 18:631–36. https://doi.org/10.1038/nn.4000 [PubMed]
- 6. Vajda A, McLaughlin RL, Heverin M, Thorpe O, Abrahams S, Al-Chalabi A, Hardiman O. Genetic testing in ALS: a survey of current practices. Neurology. 2017; 88:991–99. https://doi.org/10.1212/WNL.0000000000003686 [PubMed]
- 7. Taylor JP, Brown RH
Jr , Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016; 539:197–206. https://doi.org/10.1038/nature20413 [PubMed] - 8. Shang Y, Huang EJ. Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res. 2016; 1647:65–78. https://doi.org/10.1016/j.brainres.2016.03.036 [PubMed]
- 9. Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat. 2013; 34:812–26. https://doi.org/10.1002/humu.22319 [PubMed]
- 10. Drepper C, Herrmann T, Wessig C, Beck M, Sendtner M. C-terminal FUS/TLS mutations in familial and sporadic ALS in Germany. Neurobiol Aging. 2011; 32:548.e1–4. https://doi.org/10.1016/j.neurobiolaging.2009.11.017 [PubMed]
- 11. Wang J, Ren X, Bai X, Zhang T, Wang Y, Li K, Li G. Identification of gene mutation in patients with osteogenesis imperfect using high resolution melting analysis. Sci Rep. 2015; 5:13468. https://doi.org/10.1038/srep13468 [PubMed]
- 12. Montgomery J, Wittwer CT, Palais R, Zhou L. Simultaneous mutation scanning and genotyping by high-resolution DNA melting analysis. Nat Protoc. 2007; 2:59–66. https://doi.org/10.1038/nprot.2007.10 [PubMed]
- 13. Abel O, Powell JF, Andersen PM, Al-Chalabi A. ALSoD: a user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum Mutat. 2012; 33:1345–51. https://doi.org/10.1002/humu.22157 [PubMed]
- 14. Kwiatkowski TJ
Jr , Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009; 323:1205–08. https://doi.org/10.1126/science.1166066 [PubMed] - 15. Del Bo R, Ghezzi S, Corti S, Pandolfo M, Ranieri M, Santoro D, Ghione I, Prelle A, Orsetti V, Mancuso M, Sorarù G, Briani C, Angelini C, et al. TARDBP (TDP-43) sequence analysis in patients with familial and sporadic ALS: identification of two novel mutations. Eur J Neurol. 2009; 16:727–32. https://doi.org/10.1111/j.1468-1331.2009.02574.x [PubMed]
- 16. Sabatelli M, Zollino M, Conte A, Del Grande A, Marangi G, Lucchini M, Mirabella M, Romano A, Piacentini R, Bisogni G, Lattante S, Luigetti M, Rossini PM, Moncada A. Primary fibroblasts cultures reveal TDP-43 abnormalities in amyotrophic lateral sclerosis patients with and without SOD1 mutations. Neurobiol Aging. 2015; 36:2005.e5–13. https://doi.org/10.1016/j.neurobiolaging.2015.02.009 [PubMed]
- 17. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008; 319:1668–72. https://doi.org/10.1126/science.1154584 [PubMed]
- 18. Kirby J, Goodall EF, Smith W, Highley JR, Masanzu R, Hartley JA, Hibberd R, Hollinger HC, Wharton SB, Morrison KE, Ince PG, McDermott CJ, Shaw PJ. Broad clinical phenotypes associated with TAR-DNA binding protein (TARDBP) mutations in amyotrophic lateral sclerosis. Neurogenetics. 2010; 11:217–25. https://doi.org/10.1007/s10048-009-0218-9 [PubMed]
- 19. Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF, Stewart H, Kelley BJ, Kuntz K, Crook RJ, Sreedharan J, Vance C, Sorenson E, et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008; 4:e1000193. https://doi.org/10.1371/journal.pgen.1000193 [PubMed]
- 20. Peters OM, Ghasemi M, Brown RH
Jr . Emerging mechanisms of molecular pathology in ALS. J Clin Invest. 2015; 125:2548. https://doi.org/10.1172/JCI82693 [PubMed] - 21. Nolan M, Talbot K, Ansorge O. Pathogenesis of FUS-associated ALS and FTD: insights from rodent models. Acta Neuropathol Commun. 2016; 4:99. https://doi.org/10.1186/s40478-016-0358-8 [PubMed]
- 22. Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2017; 88:540–49. https://doi.org/10.1136/jnnp-2016-315018 [PubMed]
- 23. Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, Neumann M, Haass C. ALS-associated fused in sarcoma (FUS) mutations disrupt transportin-mediated nuclear import. EMBO J. 2010; 29:2841–57. https://doi.org/10.1038/emboj.2010.143 [PubMed]
- 24. Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ
Jr , Sapp P, McKenna-Yasek D, Brown RHJr , Hayward LJ. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet. 2010; 19:4160–75. https://doi.org/10.1093/hmg/ddq335 [PubMed] - 25. Bertolin C, D’Ascenzo C, Querin G, Gaiani A, Boaretto F, Salvoro C, Vazza G, Angelini C, Cagnin A, Pegoraro E, Sorarù G, Mostacciuolo ML. Improving the knowledge of amyotrophic lateral sclerosis genetics: novel SOD1 and FUS variants. Neurobiol Aging. 2014; 35:1212.e7–10. https://doi.org/10.1016/j.neurobiolaging.2013.10.093 [PubMed]
- 26. DeJesus-Hernandez M, Kocerha J, Finch N, Crook R, Baker M, Desaro P, Johnston A, Rutherford N, Wojtas A, Kennelly K, Wszolek ZK, Graff-Radford N, Boylan K, Rademakers R. De novo truncating FUS gene mutation as a cause of sporadic amyotrophic lateral sclerosis. Hum Mutat. 2010; 31:E1377–89. https://doi.org/10.1002/humu.21241 [PubMed]
- 27. Belzil VV, St-Onge J, Daoud H, Desjarlais A, Bouchard JP, Dupré N, Camu W, Dion PA, Rouleau GA. Identification of a FUS splicing mutation in a large family with amyotrophic lateral sclerosis. J Hum Genet. 2011; 56:247–49. https://doi.org/10.1038/jhg.2010.162 [PubMed]
- 28. Shiihashi G, Ito D, Yagi T, Nihei Y, Ebine T, Suzuki N. Mislocated FUS is sufficient for gain-of-toxic-function amyotrophic lateral sclerosis phenotypes in mice. Brain. 2016; 139:2380–94. https://doi.org/10.1093/brain/aww161 [PubMed]
- 29. Ito D, Yagi T, Ikawa M, Suzuki N. Characterization of inclusion bodies with cytoprotective properties formed by seipinopathy-linked mutant seipin. Hum Mol Genet. 2012; 21:635–46. https://doi.org/10.1093/hmg/ddr497 [PubMed]
- 30. Hou L, Jiao B, Xiao T, Zhou L, Zhou Z, Du J, Yan X, Wang J, Tang B, Shen L. Screening of SOD1, FUS and TARDBP genes in patients with amyotrophic lateral sclerosis in central-southern China. Sci Rep. 2016; 6:32478. https://doi.org/10.1038/srep32478 [PubMed]
- 31. Williams KL, Durnall JC, Thoeng AD, Warraich ST, Nicholson GA, Blair IP. A novel TARDBP mutation in an Australian amyotrophic lateral sclerosis kindred. J Neurol Neurosurg Psychiatry. 2009; 80:1286–88. https://doi.org/10.1136/jnnp.2008.163261 [PubMed]
- 32. Brown JA, Min J, Staropoli JF, Collin E, Bi S, Feng X, Barone R, Cao Y, O’Malley L, Xin W, Mullen TE, Sims KB. SOD1, ANG, TARDBP and FUS mutations in amyotrophic lateral sclerosis: a United States clinical testing lab experience. Amyotroph Lateral Scler. 2012; 13:217–22. https://doi.org/10.3109/17482968.2011.643899 [PubMed]
- 33. Budini M, Romano V, Avendaño-Vázquez SE, Bembich S, Buratti E, Baralle FE. Role of selected mutations in the Q/N rich region of TDP-43 in EGFP-12xQ/n-induced aggregate formation. Brain Res. 2012; 1462:139–50. https://doi.org/10.1016/j.brainres.2012.02.031 [PubMed]
- 34. Zou ZY, Peng Y, Wang XN, Liu MS, Li XG, Cui LY. Screening of the TARDBP gene in familial and sporadic amyotrophic lateral sclerosis patients of Chinese origin. Neurobiol Aging. 2012; 33:2229.e11–18. https://doi.org/10.1016/j.neurobiolaging.2012.03.014 [PubMed]
- 35. Gendron TF, Rademakers R, Petrucelli L. TARDBP mutation analysis in TDP-43 proteinopathies and deciphering the toxicity of mutant TDP-43. J Alzheimers Dis. 2013 (Suppl 1); 33:S35–45. https://doi.org/10.3233/JAD-2012-129036 [PubMed]
- 36. Cheng YW, Lee MJ, Chen TF, Cheng TW, Lai YM, Hua MS, Chiu MJ. A single nucleotide TDP-43 mutation within a Taiwanese family: a multifaceted demon. Amyotroph Lateral Scler Frontotemporal Degener. 2016; 17:292–94. https://doi.org/10.3109/21678421.2015.1111905 [PubMed]
- 37. Özoğuz A, Uyan Ö, Birdal G, Iskender C, Kartal E, Lahut S, Ömür Ö, Agim ZS, Eken AG, Sen NE, Kavak P, Saygı C, Sapp PC, et al. The distinct genetic pattern of ALS in Turkey and novel mutations. Neurobiol Aging. 2015; 36:1764.e9–18. https://doi.org/10.1016/j.neurobiolaging.2014.12.032 [PubMed]
- 38. Koyama A, Sugai A, Kato T, Ishihara T, Shiga A, Toyoshima Y, Koyama M, Konno T, Hirokawa S, Yokoseki A, Nishizawa M, Kakita A, Takahashi H, Onodera O. Increased cytoplasmic TARDBP mRNA in affected spinal motor neurons in ALS caused by abnormal autoregulation of TDP-43. Nucleic Acids Res. 2016; 44:5820–36. https://doi.org/10.1093/nar/gkw499 [PubMed]
- 39. Gitcho MA, Bigio EH, Mishra M, Johnson N, Weintraub S, Mesulam M, Rademakers R, Chakraverty S, Cruchaga C, Morris JC, Goate AM, Cairns NJ. TARDBP 3'-UTR variant in autopsy-confirmed frontotemporal lobar degeneration with TDP-43 proteinopathy. Acta Neuropathol. 2009; 118:633–45. https://doi.org/10.1007/s00401-009-0571-7 [PubMed]
- 40. Brooks BR, Miller RG, Swash M, Munsat TL, and World Federation of Neurology Research Group on Motor Neuron Diseases. El escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000; 1:293–99. https://doi.org/10.1080/146608200300079536 [PubMed]
- 41. Tsai PC, Liu YC, Lin KP, Liu YT, Liao YC, Hsiao CT, Soong BW, Yip PK, Lee YC. Mutational analysis of TBK1 in Taiwanese patients with amyotrophic lateral sclerosis. Neurobiol Aging. 2016; 40:191.e11–16. https://doi.org/10.1016/j.neurobiolaging.2015.12.022 [PubMed]
- 42. Cooper TA. Use of minigene systems to dissect alternative splicing elements. Methods. 2005; 37:331–40. https://doi.org/10.1016/j.ymeth.2005.07.015 [PubMed]