




Introduction
Despite technological advances in diagnosis and treatment, stomach cancer remains the fourth most common cancer with the second highest mortality rate [1]. Although some prognostic factors, such as genes and the tumor microenvironment, have been evaluated, the exact mechanisms involved remain unclear [2–4]. The development of a prognostic marker that can accurately predict clinical results will better serve as a guide in the clinic. Long noncoding RNAs (lncRNAs) are more than 200 bp in length and lack coding ability. Several studies suggest that lncRNAs participate in the regulation of tumor progression and tumor biological behavior by interacting with microRNAs (miRNAs) or messenger RNAs (mRNAs). LncRNAs containing miRNA response elements can compete with miRNA target genes and regulate their expression by reducing free functional miRNAs. This kind of lncRNA is called a competitive endogenous RNA (ceRNA) [5–8]. This hypothesis has attracted increasing attention [9]. For example, Chen et al. explored whether the lncRNA PVT1 promotes tumor progression by regulating the miR-143/HK2 axis in gallbladder cancer [10]. Additionally, Wang et al. found that a novel lncRNA, MCM3AP-AS1, promotes the growth of hepatocellular carcinoma by targeting the miR-194-5p/FOXA1 axis [11]. Gastric cancer development in attributed to an imbalance between protein-coding and noncoding genes, and the regulatory mechanism of ceRNAs may be involved in this pathogenic process.
To identify ceRNAs associated with prognosis in GA and guide clinical applications, we integrated RNA-seq data from TCGA and GEO datasets and 112 GA specimens to establish a signature based on ceRNAs. Functional enrichment analysis and gene set enrichment analysis (GSEA) were performed to predict the potential functions of the genes in the ceRNA network.
Results
Data acquisition and preprocessing
We collected gene matrix and clinical information from a TCGA data set, including 376 tumor tissues and 32 normal tissues. We excluded patients with a survival time of zero or incomplete clinical and pathological data. After screening, 349 tumor and 32 normal samples remained. After annotation, we obtained 19,754 mRNAs and 14,848 LncRNAs. The GSE84437 dataset was used as a validation cohort and contains 431 GA patients with a nonzero survival time.
Moreover, from June 2017 to August 2019, a total of 112 frozen, surgically resected tumor tissues were obtained from patients with a pathological diagnosis of GA at the Department of Pathology, Chengdu Third People's Hospital. The clinicopathological data of the TCGA and real-time quantitative PCR (qRT-PCR) datasets are presented in Table 1.
Table 1. The clinicopathological data of TCGA and qRT-PCR datasets.
| AJCC | TCGA dataset (n=349) | qRT-PCR dataset (n=112) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Vital status | Alive | Dead | Alive | Dead | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| T Stage | 207 | 142 | 67 | 45 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| T1a | 6 | 4 | 9 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| T1b | 2 | 7 | 4 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| T2 | 44 | 30 | 10 | 4 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| T3 | 113 | 49 | 2 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| T4a | 30 | 36 | 29 | 19 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| T4b | 12 | 16 | 13 | 20 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N Stage | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N0 | 64 | 44 | 36 | 11 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N1 | 60 | 35 | 21 | 16 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N2 | 42 | 33 | 9 | 15 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N3a | 37 | 24 | 1 | 3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N3b | 4 | 6 | 0 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| M Stage | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| M0 | 188 | 124 | 67 | 39 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| M1 | 19 | 18 | 0 | 6 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Pathological stage | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IA | 6 | 8 | 13 | 2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IB | 20 | 16 | 8 | 3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IIA | 43 | 22 | 3 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IIB | 47 | 28 | 11 | 4 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IIIA | 36 | 22 | 22 | 15 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IIIB | 26 | 18 | 10 | 13 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IIIC | 10 | 10 | 0 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IV | 19 | 18 | 0 | 6 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviations: AJCC: American Joint Committee on Cancer. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Differential expression analysis
After obtaining the expression data, we identified differentially expressed genes among tumorous and normal GA samples in the TCGA dataset using the software package edgeR and selected genes that were at least 2-fold higher in GA samples than in normal samples (Poisson model FDR < 0.05). Ultimately, we obtained 4721 differentially expressed mRNAs/lncRNAs (Supplementary Figure 1A, 1B). For miRNAs, if their expression deviated by more than 1.2 among these samples, they were subjected to WGCNA. Ultimately, we obtained 486 miRNAs for subsequent analyses.
WGCNA
We used the expression profiles of 4721 mRNAs/lncRNAs and 486 mRNAs/lncRNAs to construct a coexpression network with the WGCNA software package in R software. In the coexpression network analysis, the β values of lncRNAs/mRNAs and miRNAs were 3 and 8, respectively (Figure 1A, and 1B). Ultimately, we obtained 17 and 10 modules in the coexpression network of lncRNAs/mRNAs and miRNAs, respectively (Figure 1C and 1D). Moreover, we calculated and plotted the relationship between each module and clinical features. Figure 1E shows a strong negative correlation between the turquoise module and tumor characteristics (module-trait weighted correlation =-0.61). However, as shown in Figure 1F, there is a significant positive correlation between the brown module and tumor characteristics (module-feature weighted correlation = 0.75).
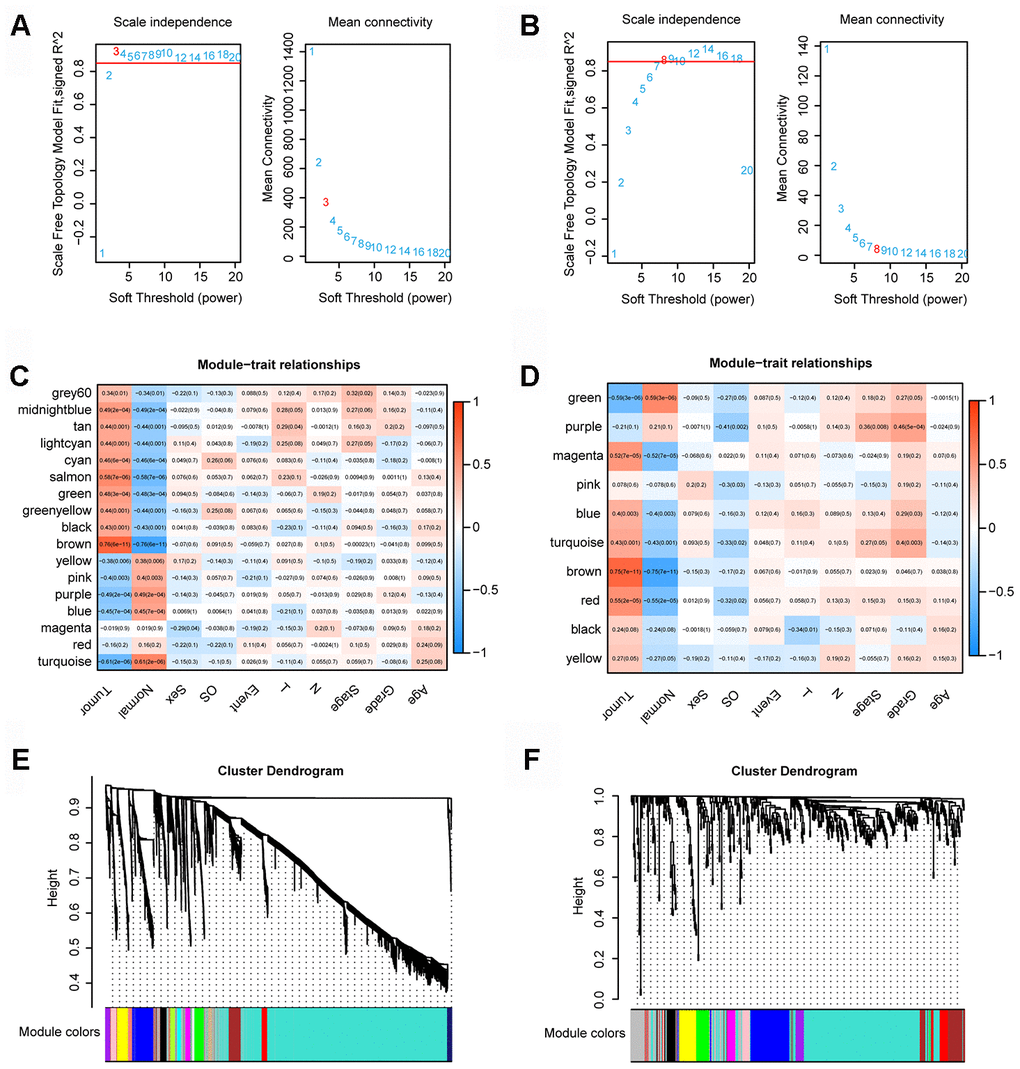
Figure 1. WGCNA. (A) Determination of the soft-thresholding power in the lncRNA/mRNA WGCNA. (B) Determination of the soft-thresholding power in the miRNAs WGCNA. (C) Module-trait associations of lncRNAs and mRNAs were evaluated by correlations between MEs and clinical traits. (D) Module-trait associations of miRNAs were evaluated by correlations between MEs and clinical traits. (E) Clustering dendrogram of lncRNAs and mRNAs. (F) Clustering dendrogram of miRNAs.
CeRNA network in GA
Through the prediction of online database and screening of matrix internal relationship pairs, 234 pairs of lncRNAs-miRNAs were obtained, including 55 lncRNAs (12 upregulated and 43 downregulated) and 17 miRNAs. Ninety pairs of miRNAs-mRNAs were also obtained, including 73 mRNAs (11 upregulated and 62 downregulated) (Supplementary Figure 2).
Functional enrichment analysis
Functional enrichment analysis was performed to explore the Gene Ontology (GO) database terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways associated with genes in the turquoise module. The results indicated that the enriched biological processes mainly involved nuclear division, DNA replication, chromosome segregation, organelle fission and so on (Figure 2A). The cell components that were correlated with the resulting terms included chromosome, centromeric region, condensed chromosome, spindle and so on (Figure 2B). The results also showed that the molecular functions were related to DNA helicase activity, catalytic activity, acting on DNA, DNA-dependent ATPase activity, actin binding and so on (Figure 2C). KEGG pathway functional enrichment analysis showed that Cell cycle, p53 signaling pathway, cAMP signaling pathway, cGMP-PKG signaling pathway and DNA replication were the main pathways related to the genes in this module (Figure 2D).
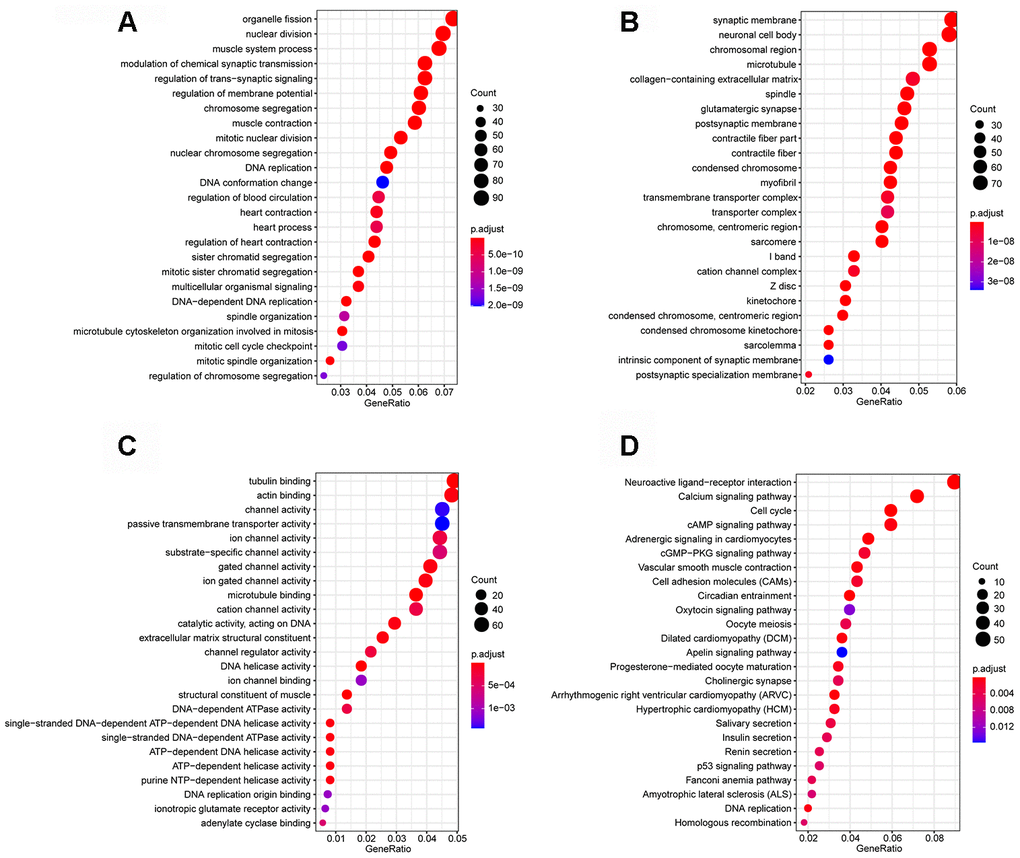
Figure 2. Enrichment analyses. (A) Biological process; (B) cellular component; (C) molecular function; (D) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways.
Kaplan-Meier analysis
Amo the 55 lncRNAs and 73 mRNAs, KM analysis revealed that 3 lncRNAs (LINC01106, FOXD2-AS1, and AC103702.2) and 3 mRNAs (CCDC34, ORC6, and SOX4) were identified as crucial prognostic factors. As shown in Figure 3A–3F, the survival time of GA patients with high LINC01106, FOXD2-AS1, AC103702.2, CCDC34, ORC6 and SOX4 expression was significantly shorter than that of patients with low expression. Therefore, the overexpression of LINC01106, FOXD2-AS1, AC103702.2, CCDC34, ORC6 and SOX4 may lead to a poor prognosis.
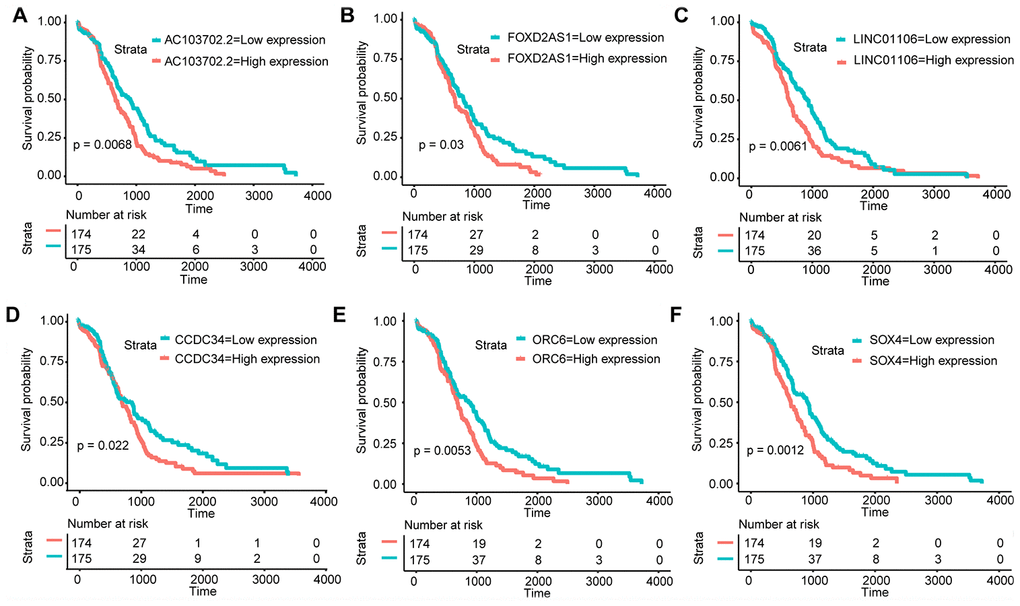
Figure 3. KM analysis. KM survival curves of the hub RNAs in the ceRNA network. (A) AC103702.2; (B) FOXD2-AS1; (C) LINC01106; (D) CCDC34; (E) ORC6; (F) SOX4.
Analysis of survival-related biomarkers in the GEPIA2 database
Compared with normal gastric tissues, SRY-box transcription factor 4 (SOX4) is significantly overexpressed in GA tissues (Figure 4A). SOX4 also appears to be upregulated in various tumor tissues compared with corresponding normal tissues (Figure 4B). Similar situations were also obtained for CCDC34 and ORC6 (Figure 4C–4F). The three lncRNAs (LINC01106, FOXD2-AS1, and AC103702.2) also showed highly similar results (Supplementary Figure 3).
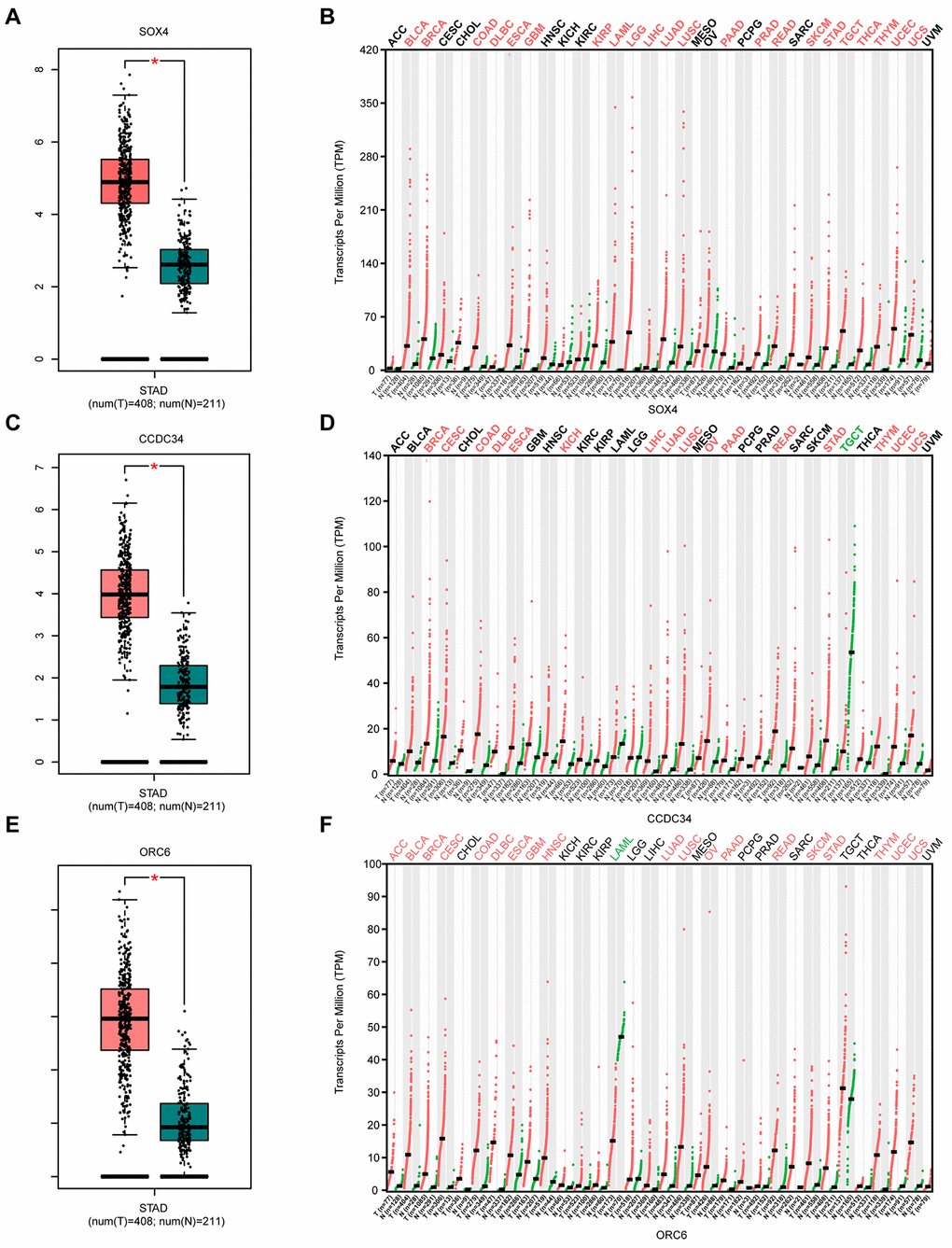
Figure 4. Analysis of 3 survival-related mRNAs in the GEPIA2 database. (A) Box plot of SOX4 expression in GA and normal gastric tissues. Red represents tumor tissue, while green represents normal tissue. (B) Dot diagram of SOX4 expression in various cancer tissues and corresponding normal tissues. Red indicates high expression, while green indicates low expression (C). Box plot of CCDC34 expression in GA and normal gastric tissues. (D) Dot diagram of CCDC34 expression in various cancer tissues and corresponding normal tissues. (E). Box plot of ORC6 expression in GA and normal gastric tissues. (F) Dot diagram of ORC6 expression in various cancer tissues and corresponding normal tissues. Abbreviations: num, Number; T, Tumor; N, Normal; ACC, Adrenocortical carcinoma; BLCA, Bladder urothelial carcinoma; BRCA, Breast invasive carcinoma; CESC, Cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, Cholangiocarcinoma; COAD, Colon adenocarcinoma; DLBC, Diffuse large B-cell lymphoma; ESCA, Esophageal carcinoma; GBM, Glioblastoma multiforme; HNSC, Head and neck squamous cell carcinoma; KICH, Kidney chromophobe; KIRC, Kidney renal clear cell carcinoma; KIRP, Kidney renal papillary cell carcinoma; AML, Acute myeloid leukemia; LGG, Low grade glioma; LIHC, Liver hepatocellular carcinoma; LUAD, Lung adenocarcinoma; LUSC, Lung squamous cell carcinoma; MESO, Mesothelioma; OV, Ovarian serous cystadenocarcinoma; PAAD, Pancreatic adenocarcinoma; PCPG, Pheochromocytoma and paraganglioma; PRAD, Prostate adenocarcinoma; READ, Rectum adenocarcinoma; SARC, Sarcoma; SKCM, Skin Cutaneous Melanoma; STAD, Stomach adenocarcinoma; TGCT, Testicular germ cell tumors; THCA, Thyroid carcinoma; THYM, Thymoma; UCEC, Uterine corpus endometrial carcinoma; UCS, Uterine carcinosarcoma; UVM, Uveal melanoma.
Construction of the survival - specific ceRNA network and prognostic signature
As shown in Figure 5A, the upregulated lncRNAs (LINC01106, FOXD2-AS1, and AC103702.2) associate with the same upregulated mRNAs (CCDC34, ORC6, and SOX4) via hsa-miR-17-5p and hsa-miR-7-5p. To further verify the functions of the hub RNAs, we conducted multivariate Cox regression analysis and calculated the risk score. The results are presented in Table 2. All samples were randomly separated into high- and low-risk groups with the median risk score as the cut-off value. Patients in the high-risk group had significantly worse OS than those in the low-risk group (Figure 5C). In addition, receiver operating characteristic (ROC) curves were used to explore whether the prognostic ability of the ceRNA-based signature was better than that of the traditional TNM pathological staging system. The AUC values of the signature assessed for 5-year (AUC = 0.801) and 7-year (AUC = 0.853) OS were more accurate than those of the pathological stage (5-year AUC = 0.609) (Figure 5B and 5D).
Table 2. The results of multivariate Cox analysis.
| β | HR | lower 95%CI | upper 95%CI | P-value | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ORC6 | 0.244 | 1.277 | 1.014 | 1.608 | 0.038* | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CCDC34 | 0.175 | 1.191 | 0.929 | 1.527 | 0.169 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SOX4 | 0.588 | 1.801 | 1.377 | 2.355 | 1.71e-05*** | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LINC01106 | 0.256 | 1.292 | 0.899 | 1.857 | 0.167 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FOXD2-AS1 | 0.384 | 1.469 | 1.106 | 1.951 | 0.008** | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AC103702.2 | 0.440 | 1.553 | 1.143 | 2.111 | 0.005** | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviations: β, coefficient; HR, Hazard ratio; CI, Confidence interval; *P<0.05; ** P<0.01; *** P<0.001. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
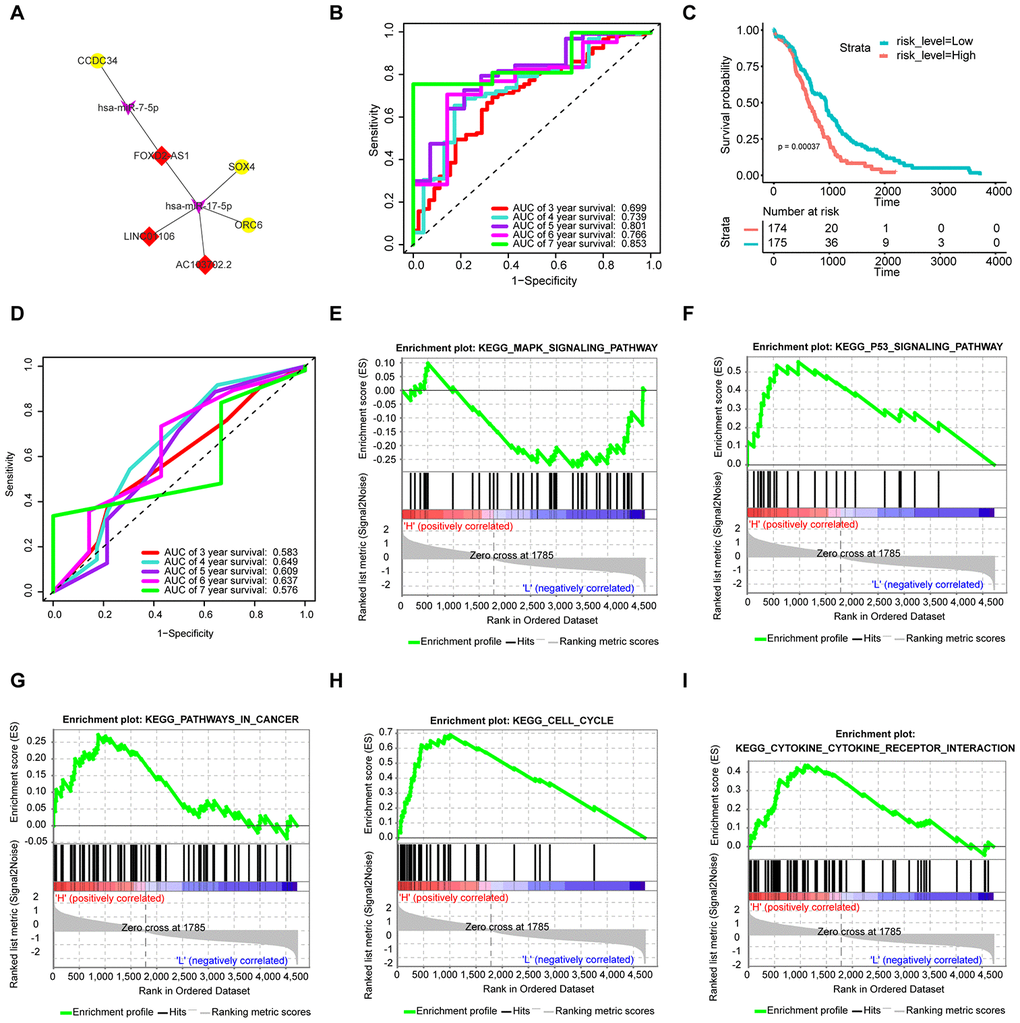
Figure 5. Construction of the prognostic signature based on the survival-specific ceRNA network and GSEA. (A) Hub ceRNA network. Notes: Red diamonds represent upregulated lncRNAs, purple arrows represent miRNAs, and gold circles represent upregulated mRNAs. (B) ROC curve analyses based on the signature. (C) KM curves of OS based on the signature. (D) ROC curve analyses based on the traditional TNM pathological staging system. (E–I) GSEA. Notes: H denotes a high signature score, while L denotes a low signature score.
GSEA
As shown in Figure 5E–5I, Cell cycle and P53 signaling pathway were enriched in the high-risk group. In addition, tumor-related pathways such as the MAPK signaling pathway, pathways in cancer and cytokine receptor interaction, were also enriched.
Validation of the prognostic value of the ceRNA-based signature
To determine the stability of the nomogram, we performed a similar process in the qRT-PCR validation cohort (n = 112). First, KM analysis was performed to identify the prognostic value of FOXD2-AS1, LINC01106 and ORC6 (Figure 6A–6C). Then, with the median risk score as the cut-off point, the patients were divided into the high-risk group (n = 56) and the low-risk group (n = 56) (Figure 6D). The AUC value for 3- and 4-year OS reached 0.809 and 0.820, respectively (Figure 6E), which were still higher than the AUC values of the traditional TNM pathological staging system (Figure 6F). The KM survival curves suggested that the OS of patients in the high-risk group was significantly worse than that of patients in the low-risk grop (Figure 6G).
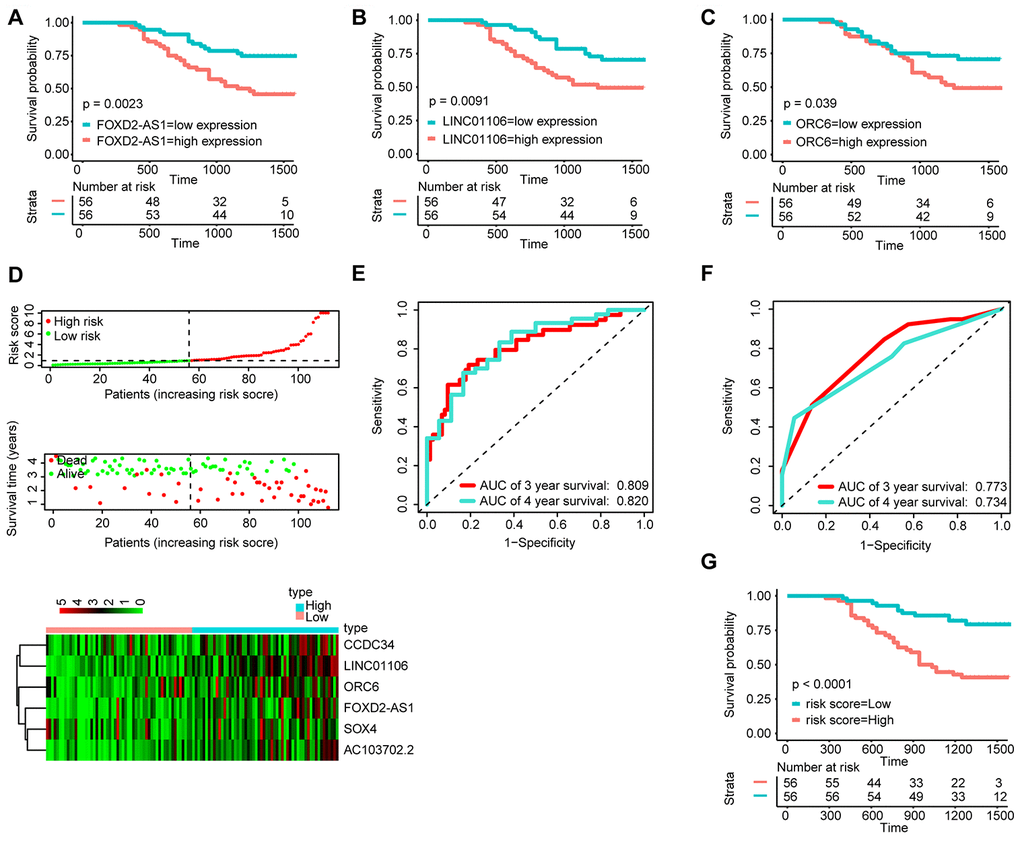
Figure 6. Validation of the signature by qRT-PCR (n=112). KM survival curves of FOXD2-AS1 (A), LINC01106 (B), and ORC6 (C); (D) Correlation between the prognostic signature and the OS of patients in the qRT-PCR cohort. Distribution of the signature scores (top), survival times (middle) and lincRNA expression levels (bottom). Black dotted lines represent the median signature score (cut-off) that was used to divide patients into the low- and high-risk groups. Red dots and lines represent patients in the high-risk group. Green dots and lines represent patients in the low-risk group. (E) ROC curve analyses based on the signature. (F) ROC curve analyses based on the traditional TNM pathological staging system. (G)KM curves of OS based on the signature.
We also validated the robustness of the signature in the GSE84437 dataset (n = 431). The KM OS curves showed that the high expression of FOXD2-AS1 and SOX4 could be related to a poor prognosis (Figure 7A and 7B). Moreover, the median risk score was used as the cut-off point to divide patients into the high-risk group (n = 215) and the low-risk group (n = 216) (Figure 7C). The AUC value for 5-year OS reached 0.755 (Figure 7D). The KM OS curves indicated that the OS of patients in the high-risk group was significantly worse than that of patients in the low-risk group (Figure 7E). These results suggest that the new signature can effectively evaluate the prognosis of GA patients.
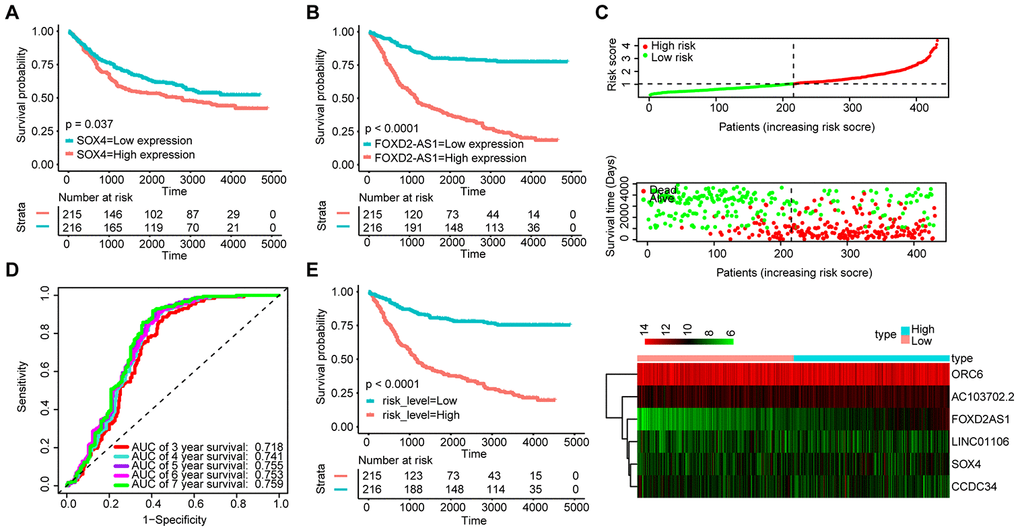
Figure 7. Validation of the signature with the GSE84437 dataset (n = 431). KM survival curves of FOXD2-AS1 (A) and SOX4 (B); (C) Distribution of the ceRNA-based signature scores, lncRNA expression levels and patient survival durations in the GSE84437 validation set. (D) ROC curve analyses based on the signature. (E) KM curves of OS according to the risk score.
Discussion
Despite advances in diagnosis, prognosis, and treatment, GA remains a worldwide public health concern. While some lncRNAs and mRNAs dysregulated in GA and their clinical value as potential biomarkers for prognosis have been previously reported[12, 13], this study provided additional data on two novel lncRNAs (LINC01106, and AC103702.2) and two novel mRNAs (CCDC34 and ORC6) contributing to GA and constructed a ceRNA-based signature that can be used to predict the prognosis of GA. Most importantly, the qRT-PCR validation cohort of 112 GA patients successfully verified its stability.
In the present study, we identified a turquoise module and a brown module (Brown) that were markedly associated with the GA tumor status by WGCNA. Next, we constructed a ceRNA network to identify potential prognostic lncRNA and mRNA biomarkers. The results revealed that LINC01106, FOXD2-AS1, AC103702.2, CCDC34, ORC6, and SOX4 whose high expression may indicate a poor OS. We also revealed the ceRNA relationship between these lncRNAs and mRNAs, that likely represents its mechanism of action in GA. Next, multivariate Cox regression analysis was carried out among these molecules, and the risk score was calculated. All samples were randomly separated into high- and low-risk groups with the median risk score as the cut-off value. KM analyses, ROC curves and AUC values showed that the signature based on the ceRNAs objectively and accurately predicted the prognosis of patients with GA. The results were successfully verified in the qRT-PCR validation cohort and the GSE84437 dataset.
LINC01106 is broadly distributed in the brain (RPKM 31.9), stomach (RPKM7.0) and 17 other tissues. Sun et al. [14] found that LINC01106 was overexpressed in colon cancer, and survival analysis showed that LINC01106 was strongly associated with the OS of colorectal cancer patients. Similarly, we found that LINC01106 was overexpressed in GA tissue and associated with a poor prognosis.
The protein encoded by ORC6 is a subunit of the ORC complex, which includes a core complex consisting of ORC2, ORC3, ORC4, and ORC5, loosely interacting with ORC6. Gene silencing studies with siRNAs demonstrated that this protein plays an essential role in coordinating chromosome replication and segregation with cytokinesis [15]. Research has also shown that the expression of ORC6 in colon cancer tissues is high and associated with invasion depth [16]. Moreover, a reduction in ORC6 expression sensitizes human colon cancer cells to 5-fluorouracil and cisplatin [17]. Our results suggest that ORC6 overexpression in GA likely causes poor OS.
Coiled-coil domain containing 34 (CCDC34), also known as renal carcinoma antigen NY-REN-41, is a protein-coding and disease related gene. Current research have shown that CCDC34 is upregulated in a variety of tumors and contributes to the malignant behaviors of cancer cells [18–22]. Recently, Gong et al. [19] found that CCDC34 was overexpressed in bladder cancer and promoted cell proliferation and migration. Lin et al. [21] found that it was also upregulated in hepatocellular carcinoma and contributes to cell proliferation and metastasis. However, its expression and role in GA have not been reported. In our study, we found that CDCC34 was highly expressed in GA tissues and not conducive to prognosis. However, little is known about its specific role in GA. Current studies have confirmed that SOX4 and FOXD2-AS1 are overexpressed in GA tissues and promote the proliferation and migration of GA cells [23–25]. These results further validate the authenticity of the current study. In addition, by constructing a ceRNA network, we also found that FOXD2-AS1 may increase the expression of SOX4, ORC6 and CCDC34 through hsa-miR-17-5p and hsa-miR-7-5p, respectively, and thus play a regulatory role in the growth, differentiation and migration of GA cells. In turn, SOX4 may be increased by LINC01106, FOXD2-AS1 and AC103702.2 through hsa-miR-17-5p, and activate the biological behavior of GA.
To validate our conclusion, we conducted qRT-PCR experiments to demonstrate that LINC01106, FOXD2-AS1, AC103702.2, CCDC34, ORC6, and SOX4 are differentially expressed in GA and corresponding normal tissues. Our results showed that the expression of LINC01106, FOXD2-AS1, AC103702.2, CCDC34, ORC6, and SOX4 was higher in GA tissues than in corresponding normal tissues (Supplementary Figure 4). We also found that SOX4, ORC6, and CCDC34 were more highly expressed in adenocarcinoma tissue than in normal gastric tissue using Western blot (WB) assays and immunohistochemistry (IHC). Moreover, SOX4, ORC6, and CCDC34 are located mainly in the nucleus of adenocarcinoma tissues (Supplementary Figure 5). It is worth noting that the lncRNA-miRNA relationship pair were constructed based on the correlation of the internal expression of the matrix and not the results of the analysis of the online databases miRcode and starBase, because very few relationship pairs remained.
In summary, we identified two novel lncRNAs (LINC01106 and AC103702.2) and two novel mRNAs (ORC6 and CCDC34) related to the prognosis of GA. We also constructed a survival-specific ceRNA network between these lncRNAs and mRNAs, and developed a prognostic signature for GA, that shows better accuracy than the traditional TNM pathological staging system. Our research will help further understand the molecular mechanism of GA and provide new insights for the treatment and prognosis of GA.
Materials and Methods
Data acquisition and preprocessing
Open data sets were downloaded from the TCGA (https://portal.gdc.cancer.gov/) and GEO (https://www.ncbi.nlm.nih.gov/gds/) databases, including the lncRNA, mRNA and miRNA expression profiles of GA specimens and the corresponding clinical follow-up data. The RNA expression profile was downloaded in fragments per kilobase million (FPKM) format. The data used in this study met the following criteria: (1) RNAs with nonzero expression levels (accounting for 75% of all samples); (2) the median and SD of the RNAs were larger than 1.2, and (3) exact follow-up times.
Differential expression analysis
Differentially expressed RNAs were identified by the edgeR package in R software [26]. Significantly expressed RNAs were identified by setting the adjusted P value to < 0.05 and the |log2FC (fold change) | > 1(|log2FC > 1| and adjusted FDR < 0.05).
Construction of the weighted gene coexpression network
The WGCNA package implemented in R software [27] was used to build a gene coexpression network based on the gene, lncRNA and miRNA expression characteristics. A scale-free plot was used to evaluate whether the network exhibited scale-free topology. The power value of the soft threshold of the adjacency matrix met the scale-free topology criterion. On this basis, we built a scale-free network and topological overlap matrix (TOM). The dynamic tree cutting method was used to generate modules with the following main parameters: deepSplit of 2 and min module size of 30 (for miRNAs, the min module size was set as 10). The height cut-off was set to 0.25, and if the module's similarity was > 0.8, the modules were merged. Based on Pearson's tests, we further determined the association between module eigengenes (MEs) and external clinical information, including sample status. If the P-value was < 0.05 and the correlation coefficient was>0.9, it was considered a significant correlation.
CeRNA network construction and analysis
According to the results of the WGCNA, we selected all the mRNAs/lncRNAs in the most negative correlation module and the miRNAs in the most positive correlation module to build the ceRNA network. In short, the related ceRNA network in GA was constructed through four stages: First, we identified the internal network relationship based on the lncRNA/mRNA and miRNA expression matrices via the WGCNA package in R software, in which the weight coefficient was controlled to be greater than 0.15. Second, miRTarBase (http://mirtarbase.cuhk.edu.cn/)[28], TargetScan (www.targetscan.org/), miRDB (mirdb.org/)[29] and miRWalk (http://mirwalk.umm.uni-heidelberg.de/)[30] were used to predict the target genes of the miRNAs. Then, to further ensure the reliability of the constructed ceRNA network, we compared the predicted relationship pairs with the internal network, and we retained only the data of overlapping interaction pairs for further analysis. Finally, Cytoscape 3.7.2 software was used to construct and visualize the ceRNA network based upon the remaining interaction pairs.
Module function annotation
GO and KEGG analyses were realized through the org.Hs.eg.db package and clusterProfiler in R software. GO consists of three terms: biological process (BP), molecular function (MF), and cellular composition (CC). All important GO terms and KEGG pathways were filtered according to a P < 0.05 and at least two associated mRNAs.
Survival analysis
KM survival analysis was performed to evaluate the association between disease prognosis and lncRNAs and mRNAs. We also analyzed the clinical data of 349 GA patients with a nonzero survival time from the TCGA database and drew the survival curves of all lncRNAs and mRNAs in the ceRNA network using the R package “survival”. When the P-value of lncRNA or mRNA was < 0.05, it was considered statistically significant and indicated that the lncRNA or mRNA has potential prognostic value.
Construction of the survival-specific ceRNA network and prognostic signature
We obtained the connections between 3 lncRNAs (LINC01106, FOXD2-AS1, and AC103702.2) and 3 mRNAs (CCDC34, ORC6, and SOX4), whose P-value was < 0.05 in the survival analysis, from the lncRNA-miRNA and mRNA-miRNA relationship pairs. The results were visualized using Cytoscape 3.7.2 software. Next, multivariate Cox regression was used to identify the corresponding coefficients of the GA prognostic signature and to calculate the risk score (risk score = expGene1 ×βGene1 + expGene2 × βGene2 + expGenen × βGenen (exp, prognostic gene expression level; β, multivariate Cox regression model regression coefficients)) by using the R packages “glmnet”, “survminer” and “survival”. All samples were randomly separated into high- and low-risk groups with the median risk score as the cut-off value. Survival for each group was evaluated by the KM analysis and the log-rank test. The ROC curve and AUC were drawn with the R package “timeROC”.
qRT-PCR
Total RNA was dreverse transcribed into cDNA with random primers using a Transcriptor First Strand cDNA Synthesis Kit (Roche, Penzberg, Germany) following the manufacturer’s instructions. The expression levels of the RNAs were measured by qRT-PCR using FastStart Essential DNA Green Master Mix (Roche, Penzberg, Germany) on a Roche LightCycler 480 (Roche, Penzberg, Germany). RNA expression was normalized to that of GAPDH. All quantitative PCRs were conducted in triplicate. Divergent primers, rather than the more commonly used convergent primers, were designed for the RNAs. We verified the specificity of the PCR primers using BLAST. A single peak in the melting curve indicated that the PCR products were specific. The primers used in the study are presented in Supplementary Table 1.
Abbreviations
GA: gastric adenocarcinoma; TCGA: The Cancer Genome Atlas; GEO: Gene Expression Omnibus; lncRNAs: long noncoding RNAs; ROC: receiver operating characteristic; OS: overall survival; AUC: area under the curve; WGCNA: weighted gene coexpression network analysis; KEGG: Kyoto Encyclopedia of Genes and Genomes; qRT-PCR: real-time quantitative reverse transcription polymerase chain reaction; ceRNA: competing endogenous RNA.
Author Contributions
M.-R., Z.-T.T. and L.-Y.J. conceived the project and designed the experiments. W.-Z., Z.-Y.C., and M.-R. carried out the experiments. M.-R, W.-Z., and Z.-Y.C. contributed equally to this work. L.-Y.J., Z.-T.T., and M.-R. wrote the manuscript. M.-R, C.-Y.Y., and L.-Q. carried out the statistical analysis and assisted in collecting tissue samples. L.-Y.J. contributed to manuscript revision. All authors provided suggestions during manuscript preparation and read the final version.
Acknowledgments
The authors would like to thank the staff of the National Center for Biotechnology Information, National Cancer Institute, and the Pathology Department of Chengdu Third People's Hospital for their efforts.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81502075) and the Foundation of Science and Technology of Sichuan Province (2019YJ0635). The funders had no role in the study design or implementation.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017; 67:7–30. https://doi.org/10.3322/caac.21387 [PubMed]
- 2. Fuse N, Kuboki Y, Kuwata T, Nishina T, Kadowaki S, Shinozaki E, Machida N, Yuki S, Ooki A, Kajiura S, Kimura T, Yamanaka T, Shitara K, et al. Prognostic impact of HER2, EGFR, and c-MET status on overall survival of advanced gastric cancer patients. Gastric Cancer. 2016; 19:183–91. https://doi.org/10.1007/s10120-015-0471-6 [PubMed]
- 3. Zhang J, Wu Y, Lin YH, Guo S, Ning PF, Zheng ZC, Wang Y, Zhao Y. Prognostic value of hypoxia-inducible factor-1 alpha and prolyl 4-hydroxylase beta polypeptide overexpression in gastric cancer. World J Gastroenterol. 2018; 24:2381–91. https://doi.org/10.3748/wjg.v24.i22.2381 [PubMed]
- 4. Thompson ED, Zahurak M, Murphy A, Cornish T, Cuka N, Abdelfatah E, Yang S, Duncan M, Ahuja N, Taube JM, Anders RA, Kelly RJ. Patterns of PD-L1 expression and CD8 T cell infiltration in gastric adenocarcinomas and associated immune stroma. Gut. 2017; 66:794–801. https://doi.org/10.1136/gutjnl-2015-310839 [PubMed]
- 5. Fachel AA, Tahira AC, Vilella-Arias SA, Maracaja-Coutinho V, Gimba ER, Vignal GM, Campos FS, Reis EM, Verjovski-Almeida S. Expression analysis and in silico characterization of intronic long noncoding RNAs in renal cell carcinoma: emerging functional associations. Mol Cancer. 2013; 12:140. https://doi.org/10.1186/1476-4598-12-140 [PubMed]
- 6. Hu Y, Chen HY, Yu CY, Xu J, Wang JL, Qian J, Zhang X, Fang JY. A long non-coding RNA signature to improve prognosis prediction of colorectal cancer. Oncotarget. 2014; 5:2230–42. https://doi.org/10.18632/oncotarget.1895 [PubMed]
- 7. Li J, Chen Z, Tian L, Zhou C, He MY, Gao Y, Wang S, Zhou F, Shi S, Feng X, Sun N, Liu Z, Skogerboe G, et al. LncRNA profile study reveals a three-lncRNA signature associated with the survival of patients with oesophageal squamous cell carcinoma. Gut. 2014; 63:1700–10. https://doi.org/10.1136/gutjnl-2013-305806 [PubMed]
- 8. Wang F, Yuan JH, Wang SB, Yang F, Yuan SX, Ye C, Yang N, Zhou WP, Li WL, Li W, Sun SH. Oncofetal long noncoding RNA PVT1 promotes proliferation and stem cell-like property of hepatocellular carcinoma cells by stabilizing NOP2. Hepatology. 2014; 60:1278–90. https://doi.org/10.1002/hep.27239 [PubMed]
- 9. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the rosetta stone of a hidden RNA language? Cell. 2011; 146:353–58. https://doi.org/10.1016/j.cell.2011.07.014 [PubMed]
- 10. Chen J, Yu Y, Li H, Hu Q, Chen X, He Y, Xue C, Ren F, Ren Z, Li J, Liu L, Duan Z, Cui G, Sun R. Long non-coding RNA PVT1 promotes tumor progression by regulating the miR-143/HK2 axis in gallbladder cancer. Mol Cancer. 2019; 18:33. https://doi.org/10.1186/s12943-019-0947-9 [PubMed]
- 11. Wang Y, Yang L, Chen T, Liu X, Guo Y, Zhu Q, Tong X, Yang W, Xu Q, Huang D, Tu K. A novel lncRNA MCM3AP-AS1 promotes the growth of hepatocellular carcinoma by targeting miR-194-5p/FOXA1 axis. Mol Cancer. 2019; 18:28. https://doi.org/10.1186/s12943-019-0957-7 [PubMed]
- 12. Huang LY, Zhao J, Chen H, Wan L, Inuzuka H, Guo J, Fu X, Zhai Y, Lu Z, Wang X, Han ZG, Sun Y, Wei W. SCFFBW7-mediated degradation of Brg1 suppresses gastric cancer metastasis. Nat Commun. 2018; 9:3569. https://doi.org/10.1038/s41467-018-06038-y [PubMed]
- 13. Yang Z, Guo X, Li G, Shi Y, Li L. Long noncoding RNAs as potential biomarkers in gastric cancer: opportunities and challenges. Cancer Lett. 2016; 371:62–70. https://doi.org/10.1016/j.canlet.2015.11.011 [PubMed]
- 14. Sun F, Liang W, Tang K, Hong M, Qian J. Profiling the lncRNA-miRNA-mRNA ceRNA network to reveal potential crosstalk between inflammatory bowel disease and colorectal cancer. PeerJ. 2019; 7:e7451. https://doi.org/10.7717/peerj.7451 [PubMed]
- 15. Prasanth SG, Prasanth KV, Stillman B. Orc6 involved in DNA replication, chromosome segregation, and cytokinesis. Science. 2002; 297:1026–31. https://doi.org/10.1126/science.1072802 [PubMed]
- 16. Hu Y, Wang L, Li Z, Wan Z, Shao M, Wu S, Wang G. Potential prognostic and diagnostic values of CDC6, CDC45, ORC6 and SNHG7 in colorectal cancer. Onco Targets Ther. 2019; 12:11609–21. https://doi.org/10.2147/OTT.S231941 [PubMed]
- 17. Gavin EJ, Song B, Wang Y, Xi Y, Ju J. Reduction of Orc6 expression sensitizes human colon cancer cells to 5-fluorouracil and cisplatin. PLoS One. 2008; 3:e4054. https://doi.org/10.1371/journal.pone.0004054 [PubMed]
- 18. Geng W, Liang W, Fan Y, Ye Z, Zhang L. Overexpression of CCDC34 in colorectal cancer and its involvement in tumor growth, apoptosis and invasion. Mol Med Rep. 2018; 17:465–73. https://doi.org/10.3892/mmr.2017.7860 [PubMed]
- 19. Gong Y, Qiu W, Ning X, Yang X, Liu L, Wang Z, Lin J, Li X, Guo Y. CCDC34 is up-regulated in bladder cancer and regulates bladder cancer cell proliferation, apoptosis and migration. Oncotarget. 2015; 6:25856–67. https://doi.org/10.18632/oncotarget.4624 [PubMed]
- 20. Hu DD, Li PC, He YF, Jia W, Hu B. Overexpression of coiled-coil domain-containing protein 34 (CCDC34) and its correlation with angiogenesis in esophageal squamous cell carcinoma. Med Sci Monit. 2018; 24:698–705. https://doi.org/10.12659/msm.908335 [PubMed]
- 21. Lin Z, Qu S, Peng W, Yang P, Zhang R, Zhang P, Guo D, Du J, Wu W, Tao K, Wang J. Up-regulated CCDC34 contributes to the proliferation and metastasis of hepatocellular carcinoma. Onco Targets Ther. 2020; 13:51–60. https://doi.org/10.2147/OTT.S237399 [PubMed]
- 22. Liu LB, Huang J, Zhong JP, Ye GL, Xue L, Zhou MH, Huang G, Li SJ. High expression of CCDC34 is associated with poor survival in cervical cancer patients. Med Sci Monit. 2018; 24:8383–90. https://doi.org/10.12659/MSM.913346 [PubMed]
- 23. Ding L, Zhao Y, Dang S, Wang Y, Li X, Yu X, Li Z, Wei J, Liu M, Li G. Circular RNA circ-DONSON facilitates gastric cancer growth and invasion via NURF complex dependent activation of transcription factor SOX4. Mol Cancer. 2019; 18:45. https://doi.org/10.1186/s12943-019-1006-2 [PubMed]
- 24. Pang L, Li B, Zheng B, Niu L, Ge L. miR-138 inhibits gastric cancer growth by suppressing SOX4. Oncol Rep. 2017; 38:1295–302. https://doi.org/10.3892/or.2017.5745 [PubMed]
- 25. Xu TP, Wang WY, Ma P, Shuai Y, Zhao K, Wang YF, Li W, Xia R, Chen WM, Zhang EB, Shu YQ. Upregulation of the long noncoding RNA FOXD2-AS1 promotes carcinogenesis by epigenetically silencing EphB3 through EZH2 and LSD1, and predicts poor prognosis in gastric cancer. Oncogene. 2018; 37:5020–36. https://doi.org/10.1038/s41388-018-0308-y [PubMed]
- 26. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–40. https://doi.org/10.1093/bioinformatics/btp616 [PubMed]
- 27. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008; 9:559. https://doi.org/10.1186/1471-2105-9-559 [PubMed]
- 28. Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG, Ma’ayan A. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database (Oxford). 2016; 2016:baw100. https://doi.org/10.1093/database/baw100 [PubMed]
- 29. Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020; 48:D127–31. https://doi.org/10.1093/nar/gkz757 [PubMed]
- 30. Sticht C, De La Torre C, Parveen A, Gretz N. miRWalk: an online resource for prediction of microRNA binding sites. PLoS One. 2018; 13:e0206239. https://doi.org/10.1371/journal.pone.0206239 [PubMed]
- 31. Tang Z, Kang B, Li C, Chen T, Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019; 47:W556–60. https://doi.org/10.1093/nar/gkz430 [PubMed]