



Introduction
Cardiovascular diseases, including acute myocardial infarction, are global leading cause of death [1, 2]. Currently, rapid and safe restoration of blood supply to ischemic myocardium is considered the best and the most effective treatment for acute myocardial infarction [3]. However, revascularization may also aggravate myocardial damage, produce a second blow to the myocardium, and cause myocardial reperfusion injury after ischemia [4]. Exploring prevention measures and action mechanisms of ischemia/reperfusion (I/R) injury is of crucial research significance for improving the prognosis and survival rate of patients with cardiovascular disease. Simple ischemic pre-conditioning and post-conditioning usually affect and block the vascular system, thus causing different degrees of physical damage. So far, several drugs have been shown to have a cardioprotective effect, preventing adverse effects of ischemic treatment on blood vessels [5]. Because of the unpredictability of ischemia, pharmacological postconditioning after ischemia-reperfusion has shown to be convenient and feasible. Our previous research revealed that metformin postconditioning could protect against myocardial I/R injury [6, 7].
Metformin is widely used for the treatment of type 2 diabetes and metabolic syndrome due to its strong ability to enhance insulin sensitivity and its safety [8]. Previous studies have demonstrated that metformin can inhibit I/R injury through multiple signaling pathways in organs such as intestines, kidneys, heart, and brain [9–12]. Moreover, the anti-inflammatory properties of metformin have been described in several models of autoimmune inflammation, such as arthritis, uveitis, and hepatitis [13–15]. For example, a recent study showed that metformin effectively improves intestinal I/R injury by inhibiting pyroptosis through an adenosine monophosphate-activated protein kinase (AMPK) [16]. Similarly, multiple drugs exert the effect of resisting pyroptosis by activating AMPK.
Pyroptosis is a newly discovered programmed cell death process, which frequently occurs in stressful environments of various organs and tissues [17]. Pyroptosis can also induce immoderate cell inflammatory damage [18]. In addition to apoptosis, necrosis, and autophagy, pyroptosis has a crucial role in myocardial I/R injury [19]. NOD-like receptor protein 3 (NOD-like receptor protein 3, NLRP3) inflammasome, as a participant of the inflammatory immune response, is closely related to cardiovascular diseases [20]. Numerous studies have confirmed that NLRP3 inflammasome is involved in the occurrence and development of myocardial I/R injury, cardiomyopathy, arrhythmia, and other diseases [21–23]. However, there are no reports on the role of NLRP3 inflammasome activation in myocardial I/R injury and metformin-related intervention studies.
In this study, we investigated the cardioprotective effect of metformin against myocardial I/R injury through inhibition of NLRP3 inflammasome and activation of AMPK. Metformin post-conditioning in animal tissue and cell models was used to investigate the effect and mechanism of metformin-mediated AMPK activation on intracellular NLRP3 inflammasome when resisting myocardial I/R injury. Compared with drug pre-conditioning, drug post-conditioning is easier to implement in clinic. Moreover, drug post-conditioning is an important measure to prevent myocardial I/R injury.
Results
Metformin reduces myocardial infarction size, suppresses myocardial tissue enzyme content and improves hemodynamic performance
To confirm the cardioprotective effect of metformin postconditioning on myocardial I/R injury, myocardial infarction size was measured by using 1%TTC. Consistent with our previous studies [6, 24, 25], I/R injury increased myocardial infarction size in I/R group (P < 0.05). Compared with the I/R group, myocardial infarction size was decreased in the MET group (P < 0.05).
Next, we examined the effect of metformin postconditioning on the activation of AMPK. Isolated hearts were treated with Compound C (CC), a small molecule compound functioning as a direct inhibitor of AMPK, in addition to metformin. As shown in Figure 1A, 1B, myocardial infarction size was increased in the MET+CC group compared to hearts treated with metformin alone (P < 0.05). Furthermore, the contents of myocardial tissue enzyme LDH, CK-MB, and cTnl (P < 0.05, Figure 1C–1E) were up-regulated after the I/R injury, but down-regulated after using metformin (P < 0.05, Figure 1C–1E). Compared with the MET group, myocardial tissue enzyme LDH, CK-MB, and cTnl were increased in the MET+CC group (P < 0.05, Figure 1C–1E).
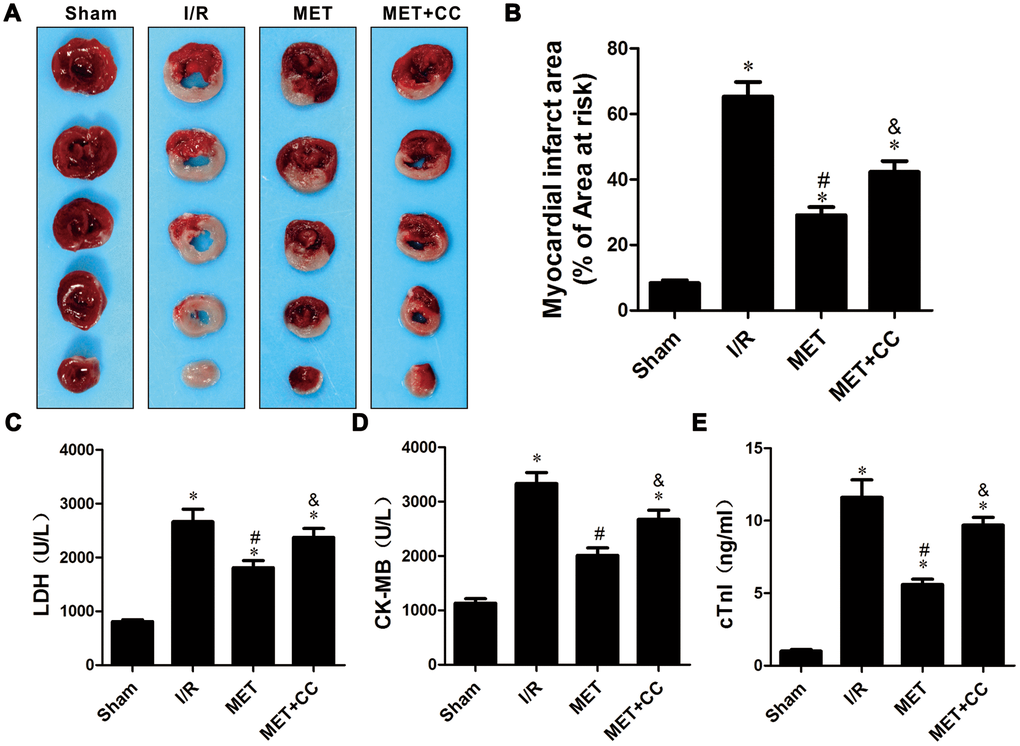
Figure 1. Metformin ameliorated IR-induced cardiac tissue damage in a rat I/R injury model. (A) Representative images of myocardial infarct size stained by TTC staining. (B) Myocardial infarct volumes presented as percentage of infarct area/area at risk (n = 6–8 per group). (C) Mean levels of lactate dehydrogenase (LDH) in all groups (n = 6 per group). (D) Mean levels of creatine kinase-MB (CK-MB) in all groups (n = 6 per group). (E) Mean levels of cardiac troponin I (cTnI) in all groups (n = 6 per group). Values are expressed as the mean ± SEM. * P < 0.05 vs. Sham. # P < 0.05 vs. I/R. & P < 0.05 vs. MET.
To further understand the role that metformin-induced AMPK activation has in the protection of cardiac function, the hemodynamics indexes in all groups were monitored and recorded at T0, T1, T2, T3, and T4. The hemodynamics indexes at T0 in all groups were significantly different (P > 0.05, Table 1). Compared with T0, HR and LVSP were decreased, while LVEDP was increased at T1, T2, T3, and T4 in the I/R, MET, and MET+CC groups (P < 0.05, Table 1). Compared with the Sham group, HR and LVSP were also decreased, while LVEDP was increased at T1, T2, T3, T4 in the other groups (P < 0.05, Table 1). As expected, metformin postconditioning significantly inhibited the changes caused by myocardial I/R injury in the MET group (P < 0.05, Table 1). However, compared with the MET group, decreased HR, LVSP, and increased LVEDP were observed in the MET+CC group (P < 0.05, Table 1). To sum up, these results indicated that AMPK is associated with reduced I/R injury-induced myocardial infarction and cardiac dysfunction caused by metformin post-conditioning.
Table 1. Hemodynamics in vitro experiments.
| Group | T0 | T1 | T2 | T3 | T4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HR (min-1) | Sham | 267 ± 21 | 273 ± 19 | 302 ± 23 | 298 ± 20 | 331 ± 31 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| I/R | 298 ± 19 | 220 ± 22*# | 201 ± 17*# | 183 ± 30*# | 156± 32*# | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MET | 303 ± 20 | 245 ± 23*#& | 260± 17*#& | 231 ± 25*#& | 209 ± 20*#& | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MET+CC | 309 ± 25 | 224 ± 20*# | 209 ± 25*#† | 179 ± 15*#† | 147± 28*#† | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LVSP (mmHg) | Sham | 103 ± 7 | 113 ± 6 | 110 ± 8 | 109 ± 8 | 115 ± 11 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| I/R | 108 ± 7 | 93 ± 7*# | 63 ± 9*# | 51 ± 7*# | 44 ± 6*# | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MET | 111 ± 11 | 101 ± 9#& | 84 ± 10*#& | 71 ± 13*#& | 61± 9*#& | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MET+CC | 112 ± 13 | 89 ± 7*#† | 61 ± 7*#† | 53 ± 8*#† | 46± 7*#† | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LVEDP (mmHg) | Sham | 7.8 ± 0.9 | 7.4 ± 0.7 | 8.2 ± 1.3 | 7.9 ± 0.9 | 8.3 ± 1.1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| I/R | 8.4 ± 1.2 | 27.6 ± 3.9*# | 39.7 ± 9.4*# | 51.8 ± 5.9*# | 73.3 ± 8.0*# | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MET | 7.3 ± 0.8 | 13.5 ± 2.9*#& | 25.5 ± 7.5*#& | 40.6 ± 5.5*#& | 53.9 ± 8.6*#& | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MET+CC | 7.1 ± 0.7 | 24.9 ± 6.3*#† | 41.9 ± 7.1*#† | 53.3 ± 7.9*# | 71.5 ± 6.1*#† | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| * P < 0.05 vs T0; # P < 0.05 vs SHAM group; & P < 0.05 vs IR group; †P < 0.05 vs IR group. Values are means ± standard deviation. n = 6 /group. HR, heart rate; LVSP, left ventricular peak pressure; LVEDP, left ventricular end diastolic pressure. T0, equilibration; T1, 30 min after reperfusion; T2, 60 min after reperfusion; T3, 90 min after reperfusion; T4, 2h after reperfusion. SHAM, sham control; IR, ischemia reperfusion; MET, metformin postconditioning; CC, compound C, the metformin inhibitor. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Metformin alleviates the degree of cardiac cell damage and fibrosis by reducing the expression of Col-I and Col-III
To further examine the cardioprotective effect of metformin postconditioning, the degree of damaged myocardium and fibrotic scar tissue was determined. As shown in Figure 2A, HE staining indicated that the structure of myocardial tissue in the Sham group was clear; the cardiomyocytes were normally arranged, and there was no cardiomyocytes necrosis, inflammatory cell infiltration, and fibrous hyperplasia. In the I/R group, the cardiomyocytes arrangement was messy, and the cardiomyocytes were enlarged. Among the cardiomyocytes, small blood vessels were dilated and congested, and a large number of inflammatory cells and fibrous hyperplasia were found (Figure 2A). Compared with the I/R group, the MET group had a clear myocardial tissue structure and lighter myocardial damage. Also, a small number of inflammatory cells in the myocardial interstitium, and a buildup of collagen fibers between the myocardium, during which the small blood vessels dilated and the congestion was lighter, were detected (Figure 2A). In the MET+CC group, the myocardial tissue was aggravated compared to the MET group but reduced compared to the I/R group (Figure 2A).
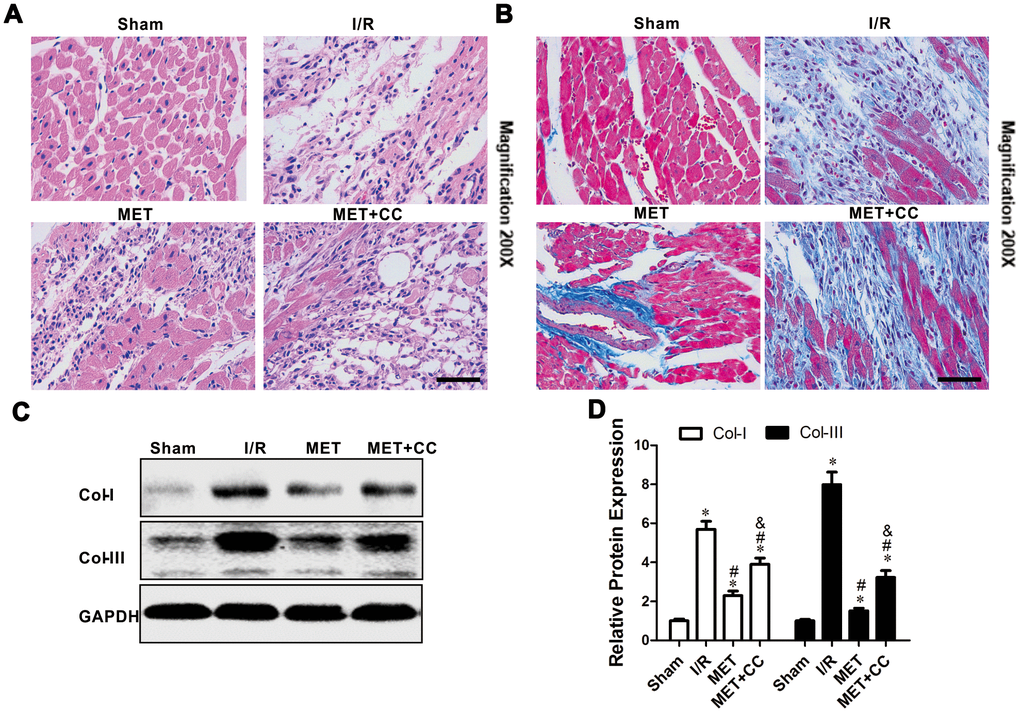
Figure 2. Metformin protected I/R injury-induced myocardial injury by suppressing collagen synthesis. (A) Representative pictures of H&E-stained cardiac sections and (B) representative images of myocardial fibrosis stained with the Masson trichrome method (n = 4 per group). Magnification 200x, Scale bar = 100μm; (C) Collagen-related proteins in the ischemic area, including COL-I, COL-III and GAPDH were examined by Western blot analysis. (D) Quantitative analysis of COL-I and COL-III expression (n = 4 per group). Values are expressed as the mean ± SEM. * P < 0.05 vs. Sham. # P < 0.05 vs. I/R. & P < 0.05 vs. MET.
Mason staining was further used to detect myocardial collagen deposition in all groups. Only a small amount of collagen was found in the myocardial interstitium in the Sham group (Figure 2B). Collagen deposition in I/R group significantly increased compared with the Sham group (Figure 2B). Compared with I/R group, the collagen fibers accumulation between cardiomyocytes was significantly reduced in the MET group (Figure 2B). In comparison, the positive staining of myocardial collagen in the MET+CC group was significantly higher than that in the MET group (Figure 2B). Collagen type I (Col-I) and collagen type III (Col-III) have an important role in the entire collagen network of myocardial tissue and can reflect the degree of myocardial fibrosis.
Next, we examined whether metformin down-regulates collagen synthesis through AMPK activation. Compared with the Sham group, the expression levels of Col-I and Col-III were up-regulated in other groups (P < 0.05, Figure 2C, 2D). Moreover, metformin administration down-regulated the expression levels of Col-I and Col-III (P < 0.05, Figure 2C, 2D). Compared with the MET group, the expression levels of Col-I and Col-III were significantly up-regulated in the MET+CC group (P < 0.05, Figure 2C, 2D). Taken together, these data indicate that metformin substantially reduces histopathological necrotic areas and improves cardiac fibrosis.
Metformin restrains apoptotic cardiomyocytes and apoptosis-related protein expression level involved by activating the AMPK pathway
Apoptosis of myocardial cells has a crucial part in I/R injury [26]. In this study, we measured apoptosis cells using TUNEL staining. Our results showed that the green fluorescence of myocardial tissue in the IR group was significantly higher than in the Sham group (P < 0.05, Figure 3A, 3B). After metformin administration, the green fluorescence was significantly reduced (P < 0.05, Figure 3A, 3B). These suggested that metformin effectively reduces cardiomyocytes apoptosis. In the MET+CC group, the fluorescence was increased compared with the MET group (P < 0.05, Figure 3A, 3B).
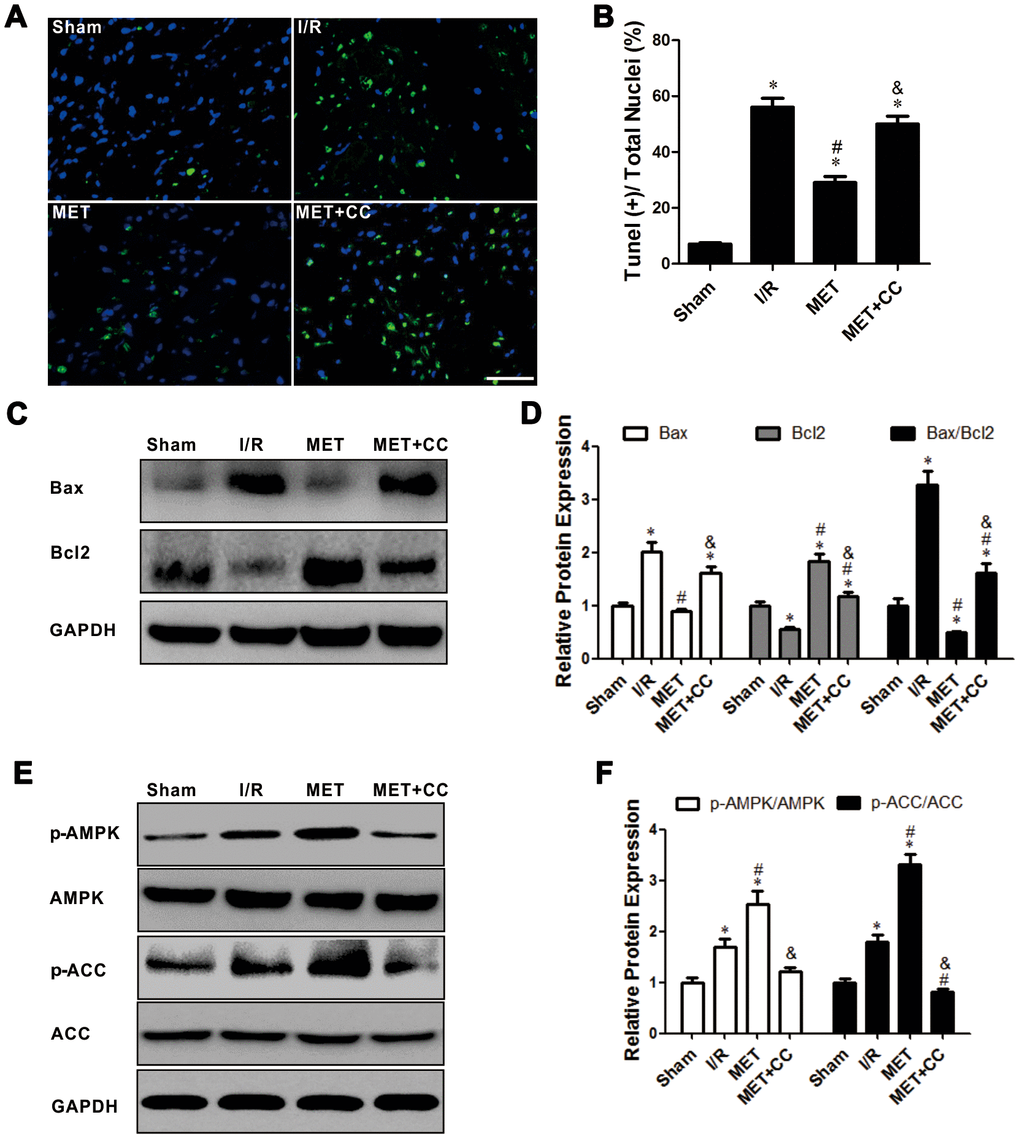
Figure 3. Activation of AMPK with Metformin protected against myocardial I/R injury induced apoptosis. (A) Top representative TUNEL-stained (green fluorescence) and DAPI-stained (blue fluorescence) photomicrographs are shown (Magnification 200x, Scale bar = 50 μm). (B) Bar graph represents the quantification of apoptotic cells (green fluorescence)/the total number of nucleated cells (blue fluorescence, n = 6 per group). (C) Apoptosis-related proteins in the ischemic area, including Bax, Bcl2 and GAPDH were examined by Western blot analysis. (D) Quantitative analysis of Bax, Bcl2 and calculate the ratio of Bax/Bcl2 (n = 4 per group). (E) AMPK pathway-related proteins in the ischemic area, including p-AMPK, AMPK, p-ACC, ACC and GAPDH were examined by Western blot analysis. (F) Quantitative analysis of p-AMPK, AMPK, p-ACC, ACC expression and calculate the ratio of p-AMPK/AMPK and p-ACC/ACC (n = 4 per group). Values are expressed as the mean ± SEM. * P < 0.05 vs. Sham. # P < 0.05 vs. I/R. & P < 0.05 vs. MET.
Next, the apoptosis-related proteins were analyzed by Western Blot. As shown in Figure 3C, 3D, the expression levels of Bax and Bax/Bcl2 were increased, while the expression of Bcl2 was more decreased in I/R group than in the Sham group (P < 0.05); this reaction was reversed in the MET group (P < 0.05, Figure 3C, 3D). After the administration of CC, the variation induced by metformin was receded; the expression levels of Bax and Bax/Bcl2 were higher, and the expression level of Bcl2 was lower in the MET+CC group than in the MET group (P < 0.05, Figure 3C, 3D).
As shown in Figure 3E, 3F, the results from Western Blot demonstrated that the expression level ratios of p-AMPK/AMPK and p-ACC/ACC were higher in the MET group than in the I/R group (P < 0.05). Moreover, co-administration of CC with metformin largely abolished the activation of the AMPK/ACC axis (P < 0.05, Figure 3E, 3F). Thus, our results indicated that the activation of AMPK by metformin lessened IR-injury-induced apoptosis, which is consistent with previous findings [6, 7].
Metformin suppresses pyroptosis after I/R injury, which is partially depended on AMPK activation
AMPK has been reported to mediate the activation of NLRP3 inflammasome [21]. Subsequently, we examined whether metformin-mediated AMPK phosphorylation reduces pyroptosis in vivo. As shown in Figure 4A–4F, the I/R injury-induced pyroptosis, indicated by elevated expression levels of NLRP3, ASC, cleaved-caspase 1, IL-1β and IL-18, were significantly decreased by metformin (P < 0.05). Metformin further inhibited I/R injury-induced activation of NLRP3 inflammasome and its downstream proteins. Moreover, the administration of CC lessened the effect caused by metformin. The expression levels of NLRP3, ASC, cleaved-caspase 1, IL-1β, and IL-18 were higher in the MET+CC group than in the MET group (P < 0.05, Figure 4A–4F).
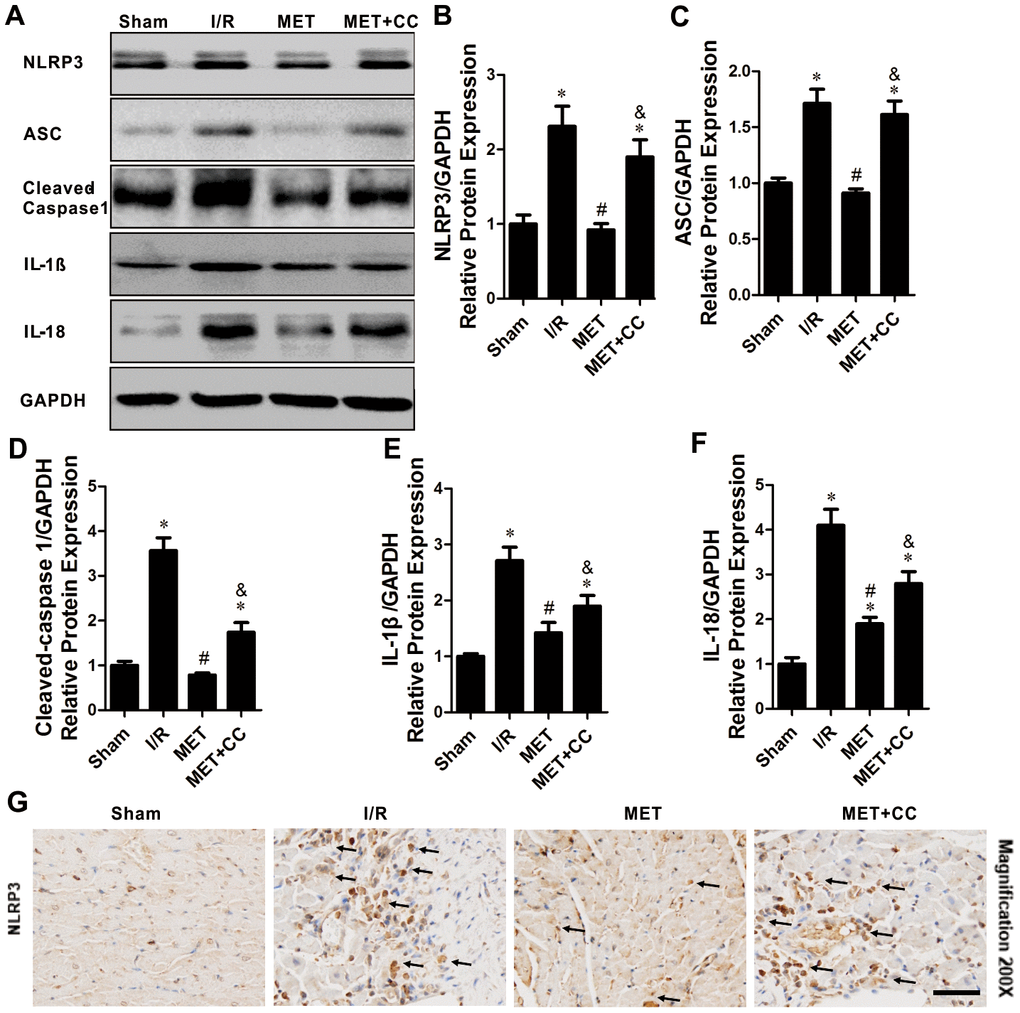
Figure 4. The inhibiting effects of Metformin on NLRP3 inflammasome activation within the infarct area following myocardial I/R injury. (A) Apoptosis-related proteins in the ischemic area, including NLRP3, ASC, cleaved-caspase 1, IL-1β, IL-18 and GAPDH were examined by Western blot analysis. (B–F) Quantitative analysis of NLRP3, ASC, cleaved-caspase 1, IL-1β and IL-18 expression (n = 4 per group). (G) Bottom representative immunohistochemical-stained NLRP3 in cardiac tissue of each group are shown (n = 4 per group). Magnification 200x, Scale bar = 100 μm; Values are expressed as the mean ± SEM. * P < 0.05 vs. Sham. # P < 0.05 vs. I/R. & P < 0.05 vs. MET.
The immunohistochemical staining of myocardium sections of all group hearts revealed the anticipated results. There was no positive staining for NLRP3 in the Sham, while in the I/R injury group, the NLRP3 staining increased (Figure 4G). In myocardium sections of the MET group, staining was decreased compared to the I/R group (Figure 4G). However, compared with the MET group, the NLRP3 staining in MET+CC myocardium sections was markedly increased (Figure 4G). Therefore, these data indicate that metformin leads to AMPK activation and NLRP3 mediated pyroptosis inhibition; this reaction could be reversed by CC treatment.
Metformin inhibits the release of pro-inflammatory factors by regulating AMPK activation
In this experiment, we investigated the effects of metformin on I/R injury-induced release of cytokines associated with inflammation. ELISA estimated the levels of IL-1β, IL-18, and TNF-α in cardiac tissue homogenates. As shown in Figure 5A–5C, the levels of IL-1β, IL-18, and TNF-α were elevated in all groups expect the Sham group (P < 0.05). Compared with the I/R group, the levels of IL-1β, IL-18, and TNF-α were reduced in the MET group (P < 0.05, Figure 5A–5C), whereas accompanied by metformin and CC, the levels of IL-1β, IL-18, and TNF-α in the MET+CC group were elevated compared with the MET group (P < 0.05, Figure 5A–5C).
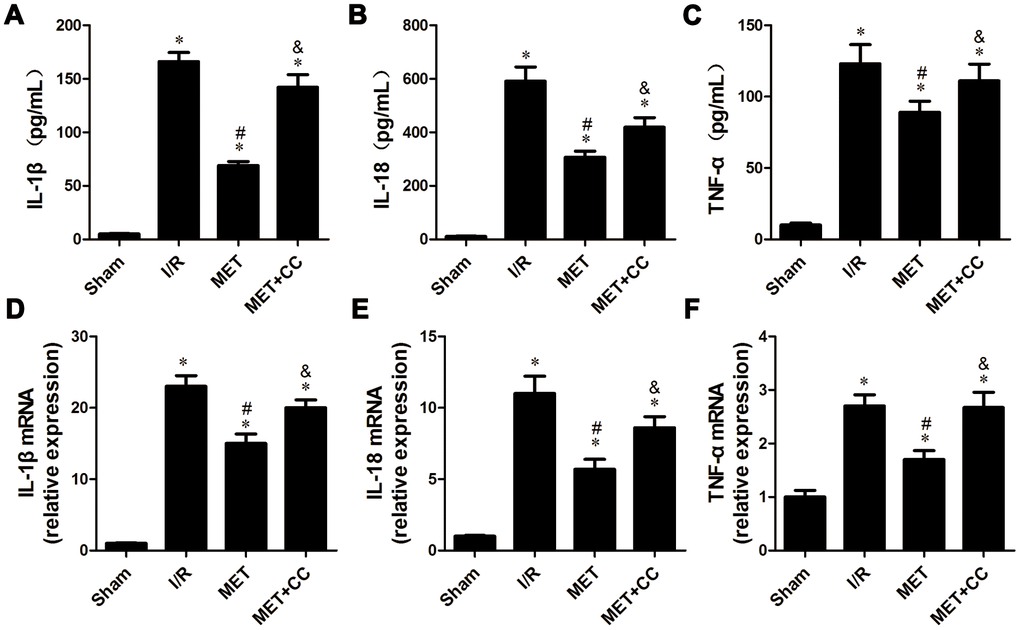
Figure 5. Metformin inhibited inflammatory cytokines release following myocardial I/R injury. (A) The IL-1β content, (B) IL-18 content and (C) TNF-α content were detected by ELISA (n = 6–7 per group). (D) The mRNA levels of IL-1β, (E) IL-18 and (F) TNF-α were measured using quantitative RT-PCR (n = 6–7 per group). The housekeeping gene β-actin was used for normalization. Values are expressed as the mean ± SEM. * P < 0.05 vs. Sham. # P < 0.05 vs. I/R. & P < 0.05 vs. MET.
Next, changes in mRNA expression of IL-1β, IL-18, and TNF-α were examined by RT-PCR. Similar to the above results, we observed that metformin significantly prevented I/R injury-induced the up-regulation of mRNA levels of IL-1β, IL-18, and TNF-α (P < 0.05, Figure 5E, 5F). Additionally, the mRNA levels of IL-1β, IL-18, and TNF-α were mildly increased in the MET+CC group compared to that in the single metformin treatment group (P < 0.05, Figure 5E, 5F).
Activation of NLRP3 with nigericin abrogates metformin-induced cardioprotection in NCVMs
To further determine the impact of metformin on H/R injury in an NLRP3-dependent way in vitro, NCVMs were treated with a special stimulator of NLRP3 (nigericin). We found that the treatment of nigericin reversed the protective effects of metformin. Consistent with our animal data, we observed that metformin substantially increased cell viability (P < 0.05, Figure 6A), reduced LDH release (P < 0.05, Figure 6B), and decreased cardiomyocyte apoptosis (P < 0.05, Figure 6C, 6D). However, metformin did not show cardioprotective effect against H/R injury when the activation of NLRP3 was suppressed (P < 0.05, Figure 6A–6D).
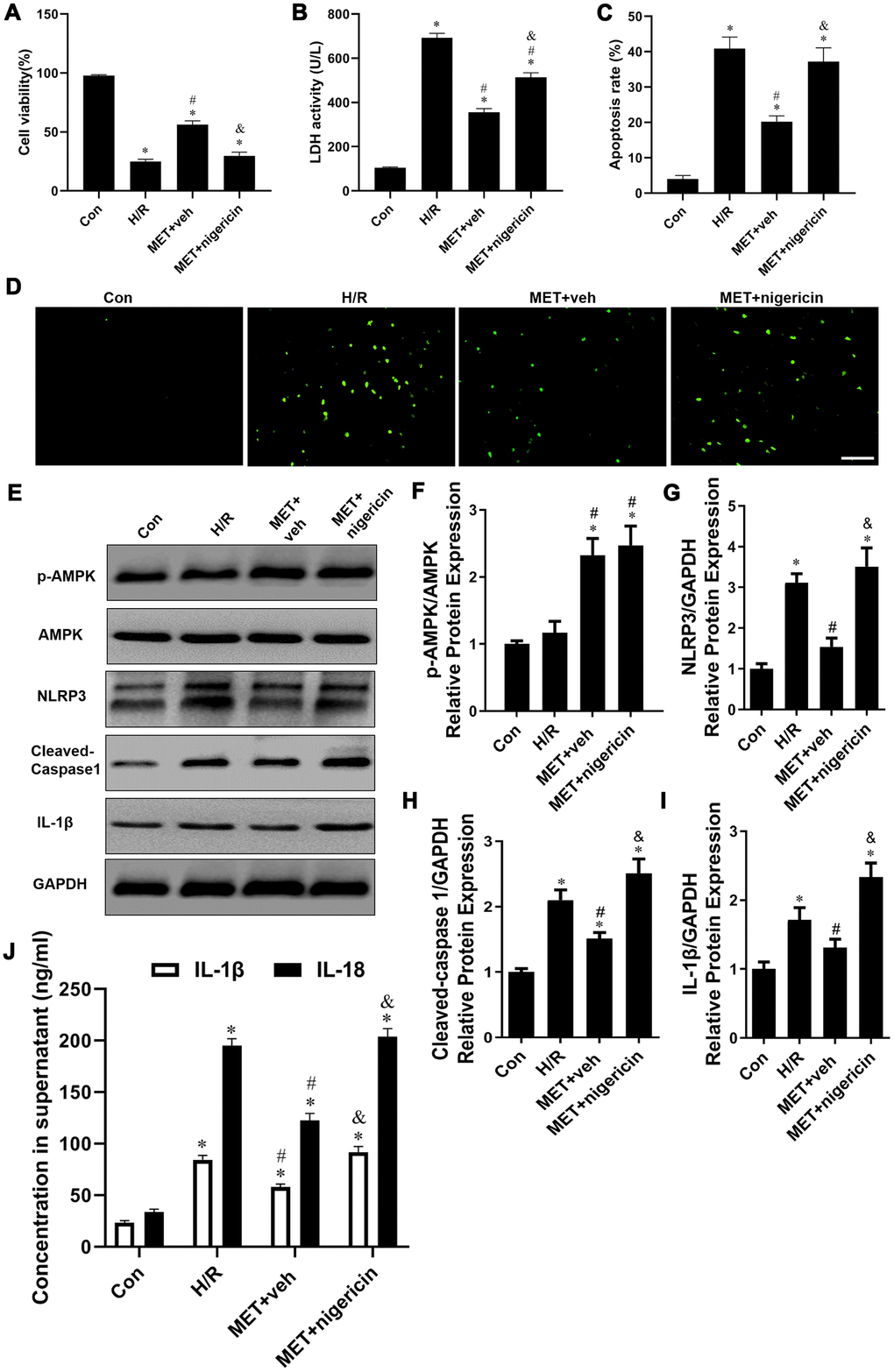
Figure 6. Activation of NLRP3 with nigericin abolished Metformin-inhibited the release of pro-inflammatory factor in vitro. (A) Cell viability was measured by MTT assay (n = 6 per group). (B) The supernatant was collected and used to determine the LDH activity (n = 6 per group). (C) Statistical results of TUNEL-positive cells per field. (D) Tunel assay (apoptotic cells stained in green fluorescence) was performed to assess examine the apoptosis rate of NRVM cells in all groups (n = 6 per group). Magnification 200x, Scale bar = 100 μm; (E) Western blots were performed to determine p-AMPK, AMPK, NLRP3, cleaved-caspase 1, IL-1β and GAPDH in the total cell lysates. (F–I) Quantitative analysis of p-AMPK, AMPK, NLRP3, cleaved-caspase 1, IL-1β expression and calculate the ratio of p-AMPK/AMPK (n = 4 per group). (J) The levels of IL-1β, IL-18 and TNF-α in the supernatant from NCVMs were detected by ELISA (n = 6 per group). Values are expressed as the mean ± SEM. * P < 0.05 vs. Con. # P < 0.05 vs. H/R. & P < 0.05 vs. MET+veh.
Furthermore, we determined the expression of AMPK and the interaction of AMPK-NLRP3 in vitro. As shown in Figure 6E–6I, the Western blot results showed that compared with the Con group, the ratios of p-AMPK/AMPK, NLRP3/GAPDH, cleaved-caspase 1/GAPDH and IL-1β/GAPDH in the other groups were increased (P < 0.05). After metformin treatment, the ratio of p-AMPK/AMPK in the MET+veh group was up-regulated, and the ratios of NLRP3/GAPDH, cleaved-caspase 1/GAPDH and IL-1β/GAPDH in MET+veh group were markedly down-regulated compared with the H/R group (P < 0.05, Figure 6E–6I), whereas, the ratios of NLRP3/GAPDH, cleaved-caspase 1/GAPDH, and IL-1β/GAPDH in MET+nigericin group were obviously up-regulated compared to the MET+veh group (P < 0.05, Figure 6E–6I).
Consistent with the western blot results, the levels of IL-1β and IL-18 in all groups in the supernatant from NCVMs showed a similar trend. Compared to the Con group, the levels of IL-1β and IL-18 in the other groups were significantly enhanced (P < 0.05, Figure 6J). Metformin significantly inhibited the levels of IL-1β and IL-18 in the MET+veh group compared with H/R group (P < 0.05, postconditioning). Nonetheless, compared to the MET+veh group, the levels of IL-1β and IL-18 in the MET+nigericin group were significantly increased when the activation of NLRP3 was suppressed (P < 0.05, Figure 6J). These results suggest that metformin protects cardiomyocytes against H/R-induced injury by suppressing pyroptosis via the AMPK/NLRP3 inflammasome pathway.
Discussion
To the best of our knowledge, this is the first study that examined the regulatory effect of metformin post-conditioning on the AMPK/NLRP3 inflammasome pathway following myocardial ischemia-reperfusion injury. We found that Metformin exerts cardioprotective effect by alleviating myocardial structural lesions, suppressing myocardial apoptosis, and inhibiting inflammatory response in vivo and in vitro. In clinical practice, Metformin is a widely used oral hypoglycemic drug. Previous studies have demonstrated that metformin exerts its cardioprotective effect by regulating multiple molecular pathways. Notably, several studies have shown that metformin pre-conditioning and postconditioning could effectively reduce myocardial I/R injury by activating AMPK and its downstream signaling pathways, such as PI3K/Akt, GRP94, PGC-1α and eNOS signaling pathway [27–30].
The Langendorff heart is an ex vivo technique that allows the examination of cardiac contractile strength and heart rate without the complications of an intact animal or human. In this study, the degree of myocardial I/R injury was measured by analyzing myocardial infarction size, myocardial injury-related enzymes, and hemodynamics indexes. We found that metformin post-conditioning inhibits myocardial infarction size, reduces the contents of myocardial injury-related enzyme LDH, CK-MB, and cTnl, and improves hemodynamics indexes. The results from HE staining, mason staining, and Western Blot further indicated that metformin post-conditioning reduces myocardial inflammatory cell infiltration, myocardial collagen deposition, and improves myocardial histopathological changes. In addition, it suppresses the up-regulation of myocardial fibrosis-related proteins Col-I and Col-III expression levels. Our data further indicated that metformin post-conditioning could significantly inhibit the augments of myocardial apoptosis and the ratio of Bax/Bcl2 induced by myocardial I/R injury, which also provided further confirmation of the cardioprotection effect of metformin on myocardial I/R injury, suggesting that metformin reduces tissue necrosis and myocardial cell apoptosis.
With the administration of the AMPK inhibitor CC, the area of myocardial infarction and the levels of the myocardial injury-related enzyme were increased, while the hemodynamic indicators significantly deteriorated. CC is an effective, reversible, and selective AMPK inhibitor that exerts its effects by allosteric regulation and inhibiting AMPK phosphorylation activation at Thr172. After using CC to inhibit AMPK activation, the protective effect of metformin postconditioning on myocardium was abolished, and the degree of myocardial cell necrosis was increased. More importantly, this protective effect was eliminated by the AMPK inhibitor CC. Consistent with the latest research results [27–30], our data suggested that metformin postconditioning effectively reduced myocardial I/R injury by activating AMPK.
Previous studies have shown that AMPK is present in almost all tissues and cells of mammals [31]. It has an essential role in regulating energy and material metabolism and participates in various biological functions, such as cell proliferation, apoptosis, and inflammation [32]. Drugs pre-conditioning or post-conditioning exerts its cardioprotective effects against myocardial I/R injury through a variety of mechanisms, such as activating AMPK and downstream signaling pathways, regulating energy metabolism, reducing myocardial cell inflammation, inhibiting myocardial apoptosis, and regulating autophagy [33–35]. In this study, we demonstrated that metformin postconditioning could lead to the activation of AMPK and its downstream protein ACC. Meanwhile, the results from ELISA and RT-PCR indicated that metformin postconditioning effectively inhibited the up-regulation of myocardial inflammatory cytokines IL-1β, IL-18, and TNF-α induced by myocardial I/R injury. Our data confirmed that metformin has an anti-inflammatory effect on myocardial I/R injury.
Many pharmacological studies have focused on suppressing inflammation response to reduce cardiac I/R injury [36, 37]. A large number of studies have confirmed that the inflammatory response could aggravate the tissue necrosis and apoptosis in myocardial I/R injury, and the NLRP3 inflammasome has an important role in it [20, 21, 38]. NLRP3, as one of the representatives of the inflammasome nodding-like receptor (NLR) family, is currently the most extensively studied inflammatory complex protein in the body. NLRP3 can be activated by different molecules, bacteria, and viruses. After NLRP3 is activated, its N-terminal heat protein domain (PYD) binds to the ASC containing the caspase recruitment domain and aggregates caspase1 to form the NLRP3 inflammasome [39]. Under the action of autocatalysis, the precursor of caspase-1 is activated. The activated caspase-1 can then cleave the precursors of IL-1β and IL-18. Subsequently, activated IL-1β and IL-18 as main effectors have an important role in inflammation and participate in the development of myocardial I/R injury [40].
According to a recent study, adiponectin upregulation of the phosphorylation of AMPK can reduce inflammation, inhibit the activation of the NLRP3 inflammasome, while also having a beneficial role in cerebral I/R injury [41]. It is worthy of paying attention that Metformin protects against intestinal I/R injury by decreasing cell pyroptosis via the NLRP3-GSDMD axis [9]. In this study, we examined whether metformin exerts its cardioprotective effect against myocardial I/R injury by activating AMPK and inhibiting NLRP3 inflammasome activation. The expression levels of NLRP3, ASC, cleaved-caspase 1, IL-1β, and IL-18 were measured by Western Blot. We found that metformin activated AMPK, inhibited the expressions of NLRP3 and ASC, and significantly reduced the levels of cleaved caspase1, IL-1β, and IL-18, and ultimately restrained NLRP3-mediated cardiomyocyte pyroptosis. Moreover, after treatment with AMPK inhibitor CC, the expressions of NLPR3 and ASC were up-regulated, and the expressions of cleaved-caspase1, IL-1β, and IL-18 proteins increased. CC inhibited AMPK and myocardial I/R injury-induced inflammatory response and led to a significant increase in myocardial cell necrosis. Hence, our results suggest that the AMPK/NLPR3 inflammasome signaling pathway has a crucial role in resisting myocardial I/R injury-induced inflammatory response induced by metformin.
The molecular mechanism of metformin against myocardial I/R injury is complex and involves multiple molecular pathways. As shown in Figure 7, our study suggests that metformin can prevent myocardial I/R injury and suppress inflammatory response through the activation of AMPK and its regulated NLRP3 inflammasome. Thus, AMPK and its regulated NLRP3 inflammasome can be used as potential new therapeutic targets for clinical prevention and treatment of ischemic heart disease.
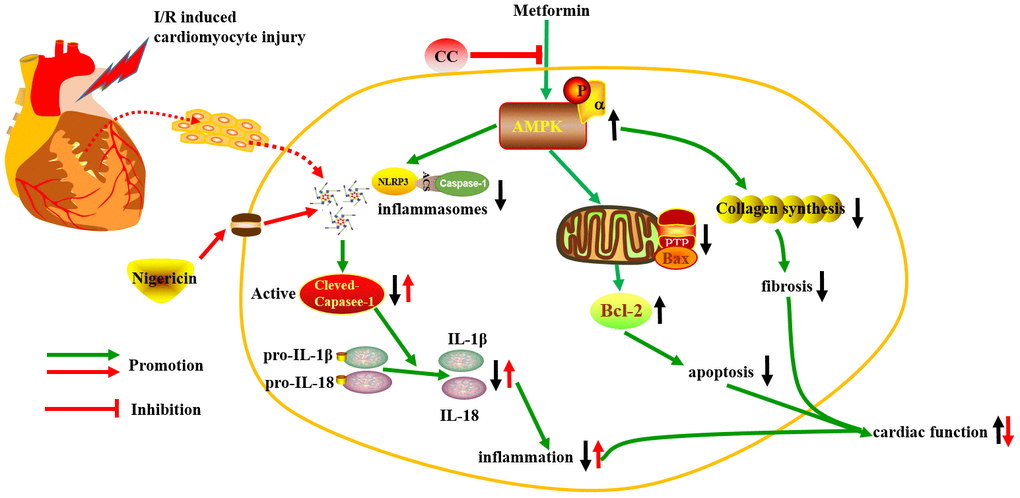
Figure 7. The schematic diagram of the protective properties of Metformin against myocardial I/R injury via the AMPK/NLRP3 inflammasome pathway.
Materials and Methods
Animals and neonatal rat ventricle myocytes culture
The Sprague-Dawley rats (male, 180-230g) were maintained and obtained from medical laboratory animal center of Nanchang University. All the animal experiments were approved by the Institutional Animal Care and Use Committee of Nanchang University and performed in compliance with the guidelines for the Principles of Laboratory Animal Care and Use of Laboratory Animals published by NIH (NIH Publication, 8th Edition, 2011).
Neonatal rat ventricle myocytes (NRVMs) were isolated from the 2-day-old Sprague–Dawley rat pups via enzymatic digestion with pancreatin and type-2 collagenase, as described before [42]. The cell suspension was preplated for 90 mins in serum-free PC-1 medium (BioWhittaker, Walkersville, MD). After removing cardiac fibroblasts, the unattached cells were NRVMs. The separated and purified NRVMs were cultured with in a mixture of 10% fetal bovine serum and antibiotics (100 μg/ml penicillin and 100 μg/ml streptomycin) in dulbecco's modified eagle medium (DMEM). The combined treatment of the experimental group was illustrated in Supplementary Figure 1 and Supplementary Figure 2. All hearts and NRVMs were randomly divided into four groups. The specific information was shown in Supplemental Materials.
Langendorff isolated heart perfusion model
According to the previous research, the K-H solution was configured [6]. NaCl 118.0, KCl 4.8, KH2PO4 1.2, NaHCO3 25.0, MgSO4 1.2, CaCl2 2.5 and glucose 11.0. Adjusted the PH value to 7.35-7.45, and advance to inject into mixed gas (95% O2-5% CO2) for 30 min to fully mix the K-H liquid and the gas, keeping the circulation temperature at 37° C. Rats were intraperitoneally injected with pentobarbital sodium (50 mg/kg) for anesthesia and femoral vein heparin (500 U/kg) for heparinization, and quickly opened the chest to take the heart, placed it in 4° C K-H solution, drained the residual blood and hung on the Langendoff perfusion device. K-H buffer is continuously perfused at constant pressure (80 mmHg). A latex balloon connected to the sensor was inserted into the left ventricle through the left atrium incision, and the balloon pressure was maintained at 0-10 mmHg via injecting water. In the meantime, the hemodynamics indexes (heart rate, HR; left ventricular peak pressure, LVSP; left ventricular end diastolic pressure, LVEDP) at 30 min of equilibration (T0), 30 min (T1), 60 min (T2), 90 min (T3), and 2 h (T4) after reperfusion were monitored and recorded by using biological signal acquisition and processing system (U/4C501H Med Lab, Shanghai).
In vitro H/R model
When NRVMs reached 80% confluence, the cells were treated with hypoxi/reoxygenation (H/R) to mimic the I/R model in vitro. The culture medium (90% DMEM+10% FBS) was replaced with serum-free Earle’s medium (CaCl2 0.18 mmol/L, MgSO4·7H2O 0.08 mmol/L, KCl 0.05 mmol/L, NaCl 11.43 mmol/L, NaHCO3 2.62 mmol/L, and NaH2PO4 0.10 mmol/L), and then incubated for 3h in an incubator with 94% N2, 5% CO2, and 1% O2 at 37° C. Thereafter, earle’s medium was then replaced with normal medium, and the cells were incubated for 6 h reoxygenation in 95% air and 5% CO2 at 37° C.
Myocardial infarction size measurement
At the end of reperfusion, the hearts were immediately removed from the Langendoff perfusion device. After freezing at -80° C for 5 min, 5-6 pieces of 2mm thick tissue were made with a heart cutter, and then placed these pieces in 1% triphenyltetrazolium chloride (TTC, lot number: T8877, Sigma, USA) to stain evenly. Avoid light, incubate at constant temperature for 20 min. After washing in phosphate buffered saline (PBS) buffer, 10% formalin was fixed for 24 h. The infarcted area and the non-infarcted area were stained off-white and brick red respectively. Alpha View gel image software was used to evaluate the volume of the left ventricular infarction and the total volume of left myocardium (myocardial infarction size).
Cell viability and LDH activity assay
The cell viability of NRVMs was determined by MTT assay as describe previously [6]. Cell viability was measured with a cell counting kit-8 assay (CCK8, Shanghai Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s instructions. Briefly, NRVMs were plated in 96-well plates at a density of 5,000 cells per well. After proper treatment, the wells were incubated with a mixture of 10 μL CCK-8 solution and 90 μL serum-free DMEM at room temperature for 2 h. Then, the optical density (OD) value was detected at a wavelength of 450 nm with a microplate reader.
Cardiac lactate dehydrogenase (LDH) activity was measured using a LDH assay kit (Nanjing Jiancheng Biotechnology, China), according to the manufacturer’s instructions. LDH release was expressed as units per liter (U/L).
CK-MB and cTnI measurement
After the rat heart reperfusion for 2h, the left ventricular tissue was taken. 10% tissue homogenate was made of normal saline, the supernatant was obtained after centrifugation at 4° C, 3 000 r /min for 15 min. Creatine kinase-MB (CK-MB) and cardiac troponin I (cTnl) levels were detected using commercial kits from Nanjing Jiancheng Bioengineering Institute according to the instructions, expressed as units per liter (U/L).
Myocardial histopathological staining
At the end of reperfusion, 1 mm3 of left ventricle was immediately cut and placed in 4% paraformaldehyde for fixation to prepare frozen sections. The rat heart tissue was soaked in 4% paraformaldehyde solution for more than 24h, dehydrated with ethanol and embedded in paraffin, and then sliced into 4-6μm thick slices. These slices were performed by routine hematoxylin-eosin (H and E) and mason staining as described in our previous studies [24, 25]. And the immunohistochemical (IHE) staining was performed on the next adjacent slice with a heat-induced antigen-retrieval step. All tissue sections were deparaffinized and hydrated, and endogenous peroxidase was blocked by hydrogen dioxide. The tissue sections were incubated with primary antibody against NLRP3 (1:200; Abcam, Cambridge, UK) overnight at 4° C. The negative control was the primary antibody replaced by phosphate-buffered saline (PBS). After washing with PBS, the slides were incubated using 3,3′-diaminobenzidine tetrahydrochloride (DAB, ZSGB-BIO, Beijing, China) to visualize the antigen-antibody compound. Finally, images were obtained by using a light microscope (LM, BX51, Olympus Co., Japan).
Cell apoptosis detection
Apoptosis were detected in the myocardium and NRVMs by TUNEL staining. At 4° C, 4% paraformaldehyde was used to fix rat myocardial tissue, dehydrated with ethanol and embedded in paraffin for sectioning. And then washed with PBS (pH:7.35) for two times, the components of PBS solution are NaCl, Na2HPO4, KCl, KH2PO4. According to the operation steps of the kit, added TUNEL mixed solution and incubated at 37° C for 1h in the dark. Continue to stain the nuclei with 4,6-biamidine-2-phenylindole (DAPI), incubate for 10 min at room temperature, and rinse twice with PBS. The myocardial tissues were observed and photographed under a fluorescent microscope. The apoptotic cells showed green fluorescence in TUNEL staining, and the nucleus showed blue fluorescence. Apoptosis rate of cardiomyocytes appertained to the percentage value of the number of apoptotic cardiomyocytes and the total number of cardiomyocytes.
Inflammatory cytokines measurement
The concentrations of inflammatory cytokines interleukin-1β (IL-1β), interleukin-18 (IL-18), and tumor necrosis factor-α (TNF-α) in cell supernatant or serum were detected using an enzyme-linked immuno sorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN, USA). The specific operation steps of ELISA are strictly in accordance with the instructions of the kit, and the data was acquired by the microplate reader.
RNA extraction and real-time PCR
The rat heart tissues were immediately taken from in vitro myocardial I/R injury model. According to the manufacturer’s instructions, total RNAs were extracted from rat heart tissues or NRVMs using the RNAsimple total RNA extraction kit (Tiangen, Beijing, China). The RNA concentration and purity were measured. The cDNA was synthesize using reverse transcription kit (Thermo Fisher Scientific, MA, USA) and amplified using TransStarts Green qPCR SuperMix (Transgen, Beijing, China). β-actin was used as an internal control. After cDNA synthesis, the expression levels of IL-1β, IL-18, TNF-α and the housekeeping gene β-actin were determined by real time-PCR using the FastStart Universal SYBR Green Master (Roche, Indianapolis, USA). The experiment was repeated 3 times. The primers used in the experiments were listed in Table 2 and β-actin served as an internal control.
Table 2. Real-time PCR primer sequences.
| Target gene | Sequences (5’-3’) |
| IL-1β (RAT) | forword:5-TGAAAGCTCTCCACCTCAATGGAC-3 |
| reverse: 5-TGCAGCCATCTTTAGGAAGACACG -3 | |
| IL-18 (RAT) | forword:5-GACTCTTGCGTCAACTTCAAGG-3 |
| reverse: 5- CAGGCTGTCTTTTGTCAACGA-3 | |
| TNF-α (RAT) | forword: 5-CATCTTCTCAAAATTCGAGTGACAA-3 |
| reverse:5-TGGGAGTAGACAAGGTACAACCC-3 | |
| β-actin (RAT) | forword:5-TAAAGACCTCTATGCCAACACAGT-3 |
| reverse:5-CACGATGGAGGGGCCGGACTCATC-3 |
Western blot analysis
Cardiomyocytes were collected and lysed by using cell lysate, including RIPA lysis buffer and protease inhibitors. After taking the supernatant and measuring the protein content by the BCA method, a protein sample was prepared. 30 μg protein sample was taken and mixed well with the buffer, and heat in the bath for 5 min. The proteins were transferred to polyvinylidene fluoride membranes (PVDF) after electrophoresis. The PVDF membranes were blocked in Tris-buffered saline with Tween-20 (TBST, 20 mM Tris, 137 mM NaCl, 0.1% Tween-20) containing 5% non-fat milk for 1 h, and then incubated with primary antibodies against Col-I (1:1000, Cell Signaling Technology, Danvers, MA, USA), Col-III (1:1000, Cell Signaling Technology, Danvers, MA, USA), pho-AMPK (1:1000, Cell Signaling Technology, Danvers, MA, USA), AMPK (1:1000, Cell Signaling Technology, Danvers, MA, USA), pho- acetyl CoA carboxylase (ACC, 1:1000, Cell Signaling Technology, Danvers, MA, USA), ACC (1:1000, Cell Signaling Technology, Danvers, MA, USA), Bax (1:1000, Abcam, Cambridge, UK), Bcl2 (1:1000, Abcam, Cambridge, UK), NLRP3 (1:1000, Abcam, Cambridge, UK), apoptosis associated speck like protein containing a CARD (ASC, 1:1000, Abcam, Cambridge, UK), Cleaved caspase-1 (1:1000, Cell Signaling Technology, Danvers, MA, USA), IL-1β(1:1000, Cell Signaling Technology, Danvers, MA, USA), IL-18 (1:500, Abcam, Cambridge, UK) and GAPDH (1:2000, BOSTER, Wuhan, China) at 4° C overnight. After being washed with TBST, the PVDF membranes were incubated with secondary antibodies for 2 h at room temperature. Subsequently, after washing, developing, grayscale scanning, the gray values of the band were analyzed by using Image J software. The protein expression was reflected by the ratio of the gray value of the target band to the gray value of GAPDH.
Statistical analysis
Data were expressed as means ± SEM. All experimental data were analyzed by using one-way analysis of variance (ANOVA) followed by Tukey multiple comparison test. A level of P<0.05 was considered to be statistically significant difference.
Author Contributions
JZ, LLH, XS, LY, FZH, JYM, and XL performed nearly all the experiments. RX and WGZ performed Hemodynamic study and analyzed the data. YFS, JPL, XYL and PY designed the whole study. PY and ZJ were the major contributors in writing the manuscript, and all the experiments were performed under their guidance. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This study was supported by grants from the Natural Science Foundation in Jiangxi Province grant (20192ACBL21037 to P.Y.), the National Natural Science Foundation of China (81760048 to J.Z. and 81760050 to P.Y.).
References
- 1. Tamis-Holland JE, Jneid H, Reynolds HR, Agewall S, Brilakis ES, Brown TM, Lerman A, Cushman M, Kumbhani DJ, Arslanian-Engoren C, Bolger AF, Beltrame JF, and American Heart Association Interventional Cardiovascular Care Committee of the Council on Clinical Cardiology, and Council on Cardiovascular and Stroke Nursing, and Council on Epidemiology and Prevention, and Council on Quality of Care and Outcomes Research. Contemporary diagnosis and management of patients with myocardial infarction in the absence of obstructive coronary artery disease: a scientific statement from the American heart association. Circulation. 2019; 139:e891–908. https://doi.org/10.1161/CIR.0000000000000670 [PubMed]
- 2. Okwuosa IS, Lewsey SC, Adesiyun T, Blumenthal RS, Yancy CW. Worldwide disparities in cardiovascular disease: challenges and solutions. Int J Cardiol. 2016; 202:433–40. https://doi.org/10.1016/j.ijcard.2015.08.172 [PubMed]
- 3. Pepine CJ. New concepts in the pathophysiology of acute myocardial infarction. Am J Cardiol. 1989; 64:2B–8B. https://doi.org/10.1016/s0002-9149(89)80002-5 [PubMed]
- 4. Neri M, Riezzo I, Pascale N, Pomara C, Turillazzi E. Ischemia/reperfusion injury following acute myocardial infarction: a critical issue for clinicians and forensic pathologists. Mediators Inflamm. 2017; 2017:7018393. https://doi.org/10.1155/2017/7018393 [PubMed]
- 5. Sposito AC, Berwanger O, de Carvalho LS, Saraiva JF. Correction to: GLP-1RAs in type 2 diabetes: mechanisms that underlie cardiovascular effects and overview of cardiovascular outcome data. Cardiovasc Diabetol. 2019; 18:23. https://doi.org/10.1186/s12933-019-0825-1 [PubMed]
- 6. Wang YS, Yu P, Wang Y, Zhang J, Hang W, Yin ZX, Liu G, Chen J, Werle KD, Quan CS, Gao H, Zeng Q, Cui R, et al. AMP-activated protein kinase protects against necroptosis via regulation of Keap1-PGAM5 complex. Int J Cardiol. 2018; 259:153–62. https://doi.org/10.1016/j.ijcard.2018.01.036 [PubMed]
- 7. Kanugula AK, Thodeti CK. AMP-activated kinase "Keaps" ischemia/reperfusion-induced necroptosis under control. Int J Cardiol. 2018; 259:168–169. https://doi.org/10.1016/j.ijcard.2018.02.053 [PubMed]
- 8. Sanchez-Rangel E, Inzucchi SE. Metformin: clinical use in type 2 diabetes. Diabetologia. 2017; 60:1586–93. https://doi.org/10.1007/s00125-017-4336-x [PubMed]
- 9. Jia Y, Cui R, Wang C, Feng Y, Li Z, Tong Y, Qu K, Liu C, Zhang J. Metformin protects against intestinal ischemia-reperfusion injury and cell pyroptosis via TXNIP-NLRP3-GSDMD pathway. Redox Biol. 2020; 32:101534. https://doi.org/10.1016/j.redox.2020.101534 [PubMed]
- 10. Wang ZS, Liu XH, Wang M, Jiang GJ, Qiu T, Chen ZY, Wang L. Metformin attenuated the inflammation after renal ischemia/reperfusion and suppressed apoptosis of renal tubular epithelial cell in rats. Acta Cir Bras. 2015; 30:617–23. https://doi.org/10.1590/S0102-865020150090000006 [PubMed]
- 11. Leech T, Chattipakorn N, Chattipakorn SC. The beneficial roles of metformin on the brain with cerebral ischaemia/reperfusion injury. Pharmacol Res. 2019; 146:104261. https://doi.org/10.1016/j.phrs.2019.104261 [PubMed]
- 12. Jo W, Kang KK, Chae S, Son WC. Metformin alleviates left ventricular diastolic dysfunction in a rat myocardial ischemia reperfusion injury model. Int J Mol Sci. 2020; 21:1489. https://doi.org/10.3390/ijms21041489 [PubMed]
- 13. Kim EK, Lee SH, Lee SY, Kim JK, Jhun JY, Na HS, Kim SY, Choi JY, Yang CW, Park SH, Cho ML. Metformin ameliorates experimental-obesity-associated autoimmune arthritis by inducing FGF21 expression and brown adipocyte differentiation. Exp Mol Med. 2018; 50:e432. https://doi.org/10.1038/emm.2017.245 [PubMed]
- 14. Kalariya NM, Shoeb M, Ansari NH, Srivastava SK, Ramana KV. Antidiabetic drug metformin suppresses endotoxin-induced uveitis in rats. Invest Ophthalmol Vis Sci. 2012; 53:3431–40. https://doi.org/10.1167/iovs.12-9432 [PubMed]
- 15. Gong X, Duan R, Ao JE, Ai Q, Ge P, Lin L, Zhang L. Metformin suppresses intrahepatic coagulation activation in mice with lipopolysaccharide/D-galactosamine-induced fulminant hepatitis. Mol Med Rep. 2015; 12:6384–90. https://doi.org/10.3892/mmr.2015.4206 [PubMed]
- 16. Saber S, El-Kader EM. Novel complementary coloprotective effects of metformin and MCC950 by modulating HSP90/NLRP3 interaction and inducing autophagy in rats. Inflammopharmacology. 2020. [Epub ahead of print]. https://doi.org/10.1007/s10787-020-00730-6 [PubMed]
- 17. Bortolotti P, Faure E, Kipnis E. Inflammasomes in tissue damages and immune disorders after trauma. Front Immunol. 2018; 9:1900. https://doi.org/10.3389/fimmu.2018.01900 [PubMed]
- 18. Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ. 2019; 26:99–114. https://doi.org/10.1038/s41418-018-0212-6 [PubMed]
- 19. Rauf A, Shah M, Yellon DM, Davidson SM. Role of caspase 1 in ischemia/reperfusion injury of the myocardium. J Cardiovasc Pharmacol. 2019; 74:194–200. https://doi.org/10.1097/FJC.0000000000000694 [PubMed]
- 20. Liu D, Zeng X, Li X, Mehta JL, Wang X. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res Cardiol. 2017; 113:5. https://doi.org/10.1007/s00395-017-0663-9 [PubMed]
- 21. Yue RC, Lu SZ, Luo Y, Wang T, Liang H, Zeng J, Liu J, Hu HX. Calpain silencing alleviates myocardial ischemia-reperfusion injury through the NLRP3/ASC/Caspase-1 axis in mice. Life Sci. 2019; 233:116631. https://doi.org/10.1016/j.lfs.2019.116631 [PubMed]
- 22. Luo B, Huang F, Liu Y, Liang Y, Wei Z, Ke H, Zeng Z, Huang W, He Y. NLRP3 inflammasome as a molecular marker in diabetic cardiomyopathy. Front Physiol. 2017; 8:519. https://doi.org/10.3389/fphys.2017.00519 [PubMed]
- 23. Yao C, Veleva T, Scott L
Jr , Cao S, Li L, Chen G, Jeyabal P, Pan X, Alsina KM, Abu-Taha IDr , Ghezelbash S, Reynolds CL, Shen YH, et al. Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation. 2018; 138:2227–42. https://doi.org/10.1161/CIRCULATIONAHA.118.035202 [PubMed] - 24. Zhang J, Wang C, Yu S, Luo Z, Chen Y, Liu Q, Hua F, Xu G, Yu P. Sevoflurane postconditioning protects rat hearts against ischemia-reperfusion injury via the activation of PI3K/AKT/mTOR signaling. Sci Rep. 2014; 4:7317. https://doi.org/10.1038/srep07317 [PubMed]
- 25. Zhang J, Zhang J, Yu P, Chen M, Peng Q, Wang Z, Dong N. Remote ischaemic preconditioning and sevoflurane postconditioning synergistically protect rats from myocardial injury induced by ischemia and reperfusion partly via inhibition TLR4/MyD88/NF-κB signaling pathway. Cell Physiol Biochem. 2017; 41:22–32. https://doi.org/10.1159/000455815 [PubMed]
- 26. Charununtakorn ST, Shinlapawittayatorn K, Chattipakorn SC, Chattipakorn N. Potential roles of humanin on apoptosis in the heart. Cardiovasc Ther. 2016; 34:107–14. https://doi.org/10.1111/1755-5922.12168 [PubMed]
- 27. Calvert JW, Gundewar S, Jha S, Greer JJ, Bestermann WH, Tian R, Lefer DJ. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes. 2008; 57:696–705. https://doi.org/10.2337/db07-1098 [PubMed]
- 28. Lee SY, Ku HC, Kuo YH, Chiu HL, Su MJ. Pyrrolidinyl caffeamide against ischemia/reperfusion injury in cardiomyocytes through AMPK/AKT pathways. J Biomed Sci. 2015; 22:18. https://doi.org/10.1186/s12929-015-0125-3 [PubMed]
- 29. Ashabi G, Khodagholi F, Khalaj L, Goudarzvand M, Nasiri M. Activation of AMP-activated protein kinase by metformin protects against global cerebral ischemia in male rats: interference of AMPK/PGC-1α pathway. Metab Brain Dis. 2014; 29:47–58. https://doi.org/10.1007/s11011-013-9475-2 [PubMed]
- 30. Zhang Y, Liu X, Zhang L, Li X, Zhou Z, Jiao L, Shao Y, Li M, Leng B, Zhou Y, Liu T, Liu Q, Shan H, Du Z. Metformin protects against H2O2-induced cardiomyocyte injury by inhibiting the miR-1a-3p/GRP94 pathway. Mol Ther Nucleic Acids. 2018; 13:189–97. https://doi.org/10.1016/j.omtn.2018.09.001 [PubMed]
- 31. Bairwa SC, Parajuli N, Dyck JR. The role of AMPK in cardiomyocyte health and survival. Biochim Biophys Acta. 2016; 1862:2199–210. https://doi.org/10.1016/j.bbadis.2016.07.001 [PubMed]
- 32. Griffiths HR, Gao D, Pararasa C. Redox regulation in metabolic programming and inflammation. Redox Biol. 2017; 12:50–57. https://doi.org/10.1016/j.redox.2017.01.023 [PubMed]
- 33. Liu Z, Chen JM, Huang H, Kuznicki M, Zheng S, Sun W, Quan N, Wang L, Yang H, Guo HM, Li J, Zhuang J, Zhu P. The protective effect of trimetazidine on myocardial ischemia/reperfusion injury through activating AMPK and ERK signaling pathway. Metabolism. 2016; 65:122–30. https://doi.org/10.1016/j.metabol.2015.10.022 [PubMed]
- 34. Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q, Jin Q, Cao F, Tian F, Chen Y. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J Pineal Res. 2017; 63:e12413. https://doi.org/10.1111/jpi.12413 [PubMed]
- 35. Li RL, Wu SS, Wu Y, Wang XX, Chen HY, Xin JJ, Li H, Lan J, Xue KY, Li X, Zhuo CL, Cai YY, He JH, et al. Irisin alleviates pressure overload-induced cardiac hypertrophy by inducing protective autophagy via mTOR-independent activation of the AMPK-ULK1 pathway. J Mol Cell Cardiol. 2018; 121:242–55. https://doi.org/10.1016/j.yjmcc.2018.07.250 [PubMed]
- 36. Kosuru R, Kandula V, Rai U, Prakash S, Xia Z, Singh S. Pterostilbene decreases cardiac oxidative stress and inflammation via activation of AMPK/Nrf2/HO-1 pathway in fructose-fed diabetic rats. Cardiovasc Drugs Ther. 2018; 32:147–63. https://doi.org/10.1007/s10557-018-6780-3 [PubMed]
- 37. Rameshrad M, Soraya H, Maleki-Dizaji N, Vaez H, Garjani A. A-769662, a direct AMPK activator, attenuates lipopolysaccharide-induced acute heart and lung inflammation in rats. Mol Med Rep. 2016; 13:2843–49. https://doi.org/10.3892/mmr.2016.4821 [PubMed]
- 38. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, D’Agostino D, Planavsky N, Lupfer C, Kanneganti TD, Kang S, Horvath TL, Fahmy TM, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015; 21:263–69. https://doi.org/10.1038/nm.3804 [PubMed]
- 39. Green JP, Yu S, Martín-Sánchez F, Pelegrin P, Lopez-Castejon G, Lawrence CB, Brough D. Chloride regulates dynamic NLRP3-dependent ASC oligomerization and inflammasome priming. Proc Natl Acad Sci USA. 2018; 115:E9371–80. https://doi.org/10.1073/pnas.1812744115 [PubMed]
- 40. Grebe A, Hoss F, Latz E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ Res. 2018; 122:1722–40. https://doi.org/10.1161/CIRCRESAHA.118.311362 [PubMed]
- 41. Liu H, Wu X, Luo J, Zhao L, Li X, Guo H, Bai H, Cui W, Guo W, Feng D, Qu Y. Adiponectin peptide alleviates oxidative stress and NLRP3 inflammasome activation after cerebral ischemia-reperfusion injury by regulating AMPK/GSK-3β. Exp Neurol. 2020; 329:113302. https://doi.org/10.1016/j.expneurol.2020.113302 [PubMed]
- 42. Liu WJ, Deng JX, Wang G, Gao KP, Lin ZX, Liu SY, Wang YH, Liu J. Manipulation of KCNE2 expression modulates action potential duration and ito and IK in rat and mouse ventricular myocytes. Am J Physiol Heart Circ Physiol. 2015; 309:H1288–302. https://doi.org/10.1152/ajpheart.00757.2014 [PubMed]