



Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative illness and the leading cause of dementia in elderly people. Inflammation, insoluble protein deposition and neuronal cell loss are important features in the brains of AD patients [1]. At present, it is widely believed that maladaptive astrocytic activation constitutes a pathogenic mechanism of AD [2]. S100B, expressed primarily by astrocytes, is associated with the neuropathological hallmarks of AD; S100B also causes neuroinflammation and neurotoxicity [3, 4].
S100B belongs to the large family of S100 proteins, which are EF-hand calcium-binding proteins that exert both intracellular and extracellular effects on a variety of cellular processes [5–7]. Intracellularly, S100B modulates microtubule assembly and regulates the cell cycle. Extracellularly, the action of S100B is strongly dependent on its concentration [8]. Extracellular S100B shows a neuroprotective effect at the nanomolar level. However, at micromolar levels, extracellular S100B can stimulate the receptor for advanced glycation end products (RAGE) in neurons, leading to an overproduction of reactive oxygen species and, ultimately, resulting in apoptosis [9, 10] and upregulation of several proinflammatory cytokines [11]. At high concentrations, S100B also upregulates nitric oxide (NO) synthase, and stimulates NO release by microglia through synergy with bacterial endotoxin and IFN-γ, thereby participating in microglia activation [12].
In addition to its functional relevance, the human S100B gene is located on chromosome 21q22.3 [13] within or near risk regions for familial late-onset AD. To date, several single nucleotide polymorphisms (SNPs) of the S100B gene—particularly rs9722 and rs1051169—have been shown to affect the S100B protein level. Recently, Hohoff suggested the important role of S100B polymorphisms in S100B serum concentrations and S100B mRNA expression [14]. Moreover, S100B SNPs are significantly associated with several neuropathological and psychiatric disorders, such as Parkinson's disease [15, 16], depressive disorder [17], schizophrenia [18, 19], stroke [20] and dementia [21]. However, very few investigations of connecting S100B polymorphisms with AD risk have been performed, especially in the Chinese Han population. Thus, herein, we conducted a case-control study in that population to examine the potential role of S100B polymorphisms in AD risk.
An increasing number of experimental studies and clinical examinations have reported elevated levels of S100B in the brains or cerebrospinal fluid of AD patients [22–25], which implies that S100B is closely tied to the pathogenesis of AD. Additional supporting evidence comes from a recent study that used double transgenic mice that overexpressed S100B and carried the APP mutation (Tg2576/S100B), which promotes brain inflammation characterized as astrogliosis and microgliosis and enhances Aβ generation [3]. Further, many previous animal studies have demonstrated that S100B dysregulation can alter Aβ deposits, plaques, and gliosis [3, 26, 27]. Owing to such evidence, S100B may act as an unconventional cytokine, playing a role in the pathophysiology of AD; S100B is therefore a plausible biological candidate as a susceptibility gene for AD.
MicroRNAs (miRNAs) are a class of single-stranded RNA molecules responsible for post-transcriptional gene silencing, usually by binding to the 3′ untranslated region (3′-UTR) of target genes that play critical roles in the regulation of target gene expression [28]. Recent studies have found that approximately 70% of experimentally detectable miRNAs are expressed in the brain, and some studies suggest that miRNAs are intimately involved in synaptic function and specific signals during memory formation. Increasing evidence implies the possible involvement of miRNAs in AD [29–32]. Using genome-wide profiling, Chang (2017) found five differently expressed genes (DEGs) regulated by four differently expressed miRNAs (DEmiRNAs) in AD. Swarbrick (2019) reported on 10 miRNAs that could be deregulated early in the peripheral blood of Alzheimer’s patients, nearly 20 years before the onset of clinical symptoms [33]. These miRNAs could serve as specific AD biomarkers, which may provide the basis for a novel, effective diagnostic approach and new targets for pharmaceutical development.
Limited studies have focused on the miRNAs that regulate the S100B level. Chen (2018) showed that S100B genes are the targets of miR-330-3p, and lncRNA X-inactive specific transcript (XIST) promotes S100B expression by harboring complementary binding sites with miR-330-3p, eventually preventing cardiac hypertrophy [34]. Using a cerebral palsy rat model, Wen (2020) discovered that the overexpression of miR-135b can downregulate S100B, which helps to inactivate the signal transducer and activator of transcription-3 (STAT3) pathway, and promotes neural stem cell (NSC) differentiation and proliferation but inhibits NSC apoptosis [35]. Recently, Chen (2020) reported that plasma miR-340-3p and S100B levels differ significantly among various rs9722 genotypes and that the S100B rs9722 locus SNP is associated with the risk of chronic heart failure. This finding signals that miR-340-3p may play different roles in regulating the S100B level among different S100B rs9722 genotypes [36].
In this study, we analyzed the plasma S100B levels in AD patients with different S100B genotypes. To explore in detail how SNPs are related to AD, we determined whether the SNPs in the 3′-UTR of the S100B gene could affect the S100B level by altering the combination of miRNAs and S100B mRNAs.
Results
Distributions of the genotype and allele frequencies of S100B polymorphisms
In total, we recruited 280 AD patients and 400 healthy control individuals. Using the SNaPshot method, we chose and genotyped the four most reported phenotypically relevant SNPs of the S100B gene (rs2839364 in the promoter region, rs1051169 in the second exon, rs2300403 in the second intron and rs9722 in the 3′UTR of the S100B gene) (Figure 1).
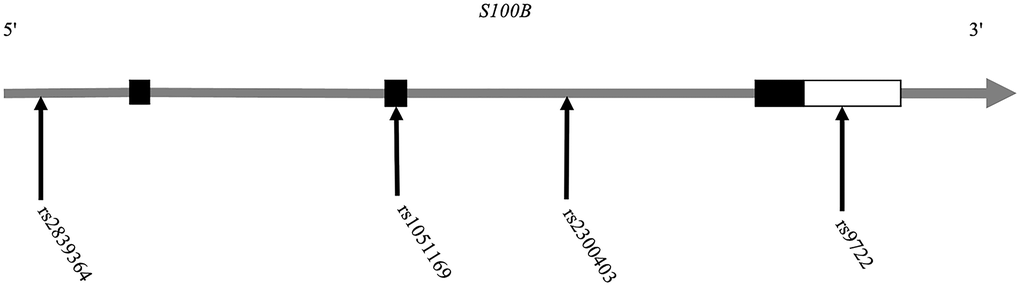
Figure 1. Position distribution of the SNPs in the S100B gene.
Table 1 summarizes the genotypes and allele frequencies of the four polymorphisms. We did not observe deviations from Hardy–Weinberg equilibrium (HWE) for the candidate polymorphisms in the control group. We noted significant differences in the genotype frequencies of rs9722 between the AD patients and the controls (OR = 2.029, 95% CI = 1.243–3.311, P = 0.005). Likewise, we observed significant differences in the allele distributions of rs9722 between the AD patients and the controls (OR = 1.342, 95% CI = 1.085–1.661, P = 0.008). The mutant alleles (rs9722 T) were more frequent in the AD population than in the control population. However, no significant differences in the allele distributions of rs1051169 between the AD patients and the controls was observed. In addition, neither the genotype nor allele frequency displayed significant differences between the AD patients and the controls for the rs2839364 and rs2300403 polymorphisms.
Table 1. Genotype and allele frequency distributions of the S100B SNPs in the AD patients and healthy controls.
| SNPs | genotype | AD | control | p | OR (95% CI) | allele | AD | control | p | OR (95% CI) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs2839364 | CC | 283 | 291 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CT | 91 | 90 | 0.865 | 1.040 (0.745–1.451) | C | 657 | 672 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TT | 25 | 19 | 0.352 | 1.353 (0.729–2.513) | T | 141 | 128 | 0.385 | 1.126 (0.867–1.464) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ApoE ɛ4(−) | CC | 175 | 235 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CT | 59 | 77 | 0.920 | 1.029 (0.695–1.522) | C | 409 | 547 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TT | 20 | 13 | 0.067 | 2.066 (1.055–4.273) | T | 99 | 103 | 0.119 | 1.285 (0.949–1.742) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ApoE ɛ4(+) | CC | 108 | 56 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CT | 32 | 13 | 0.593 | 1.277 (0.621–2.625) | C | 248 | 125 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TT | 5 | 6 | 0.200 | 0.432 (0.126–1.477) | T | 42 | 25 | 0.577 | 0.847 (0.494–1.453) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs1051169 | GG | 166 | 179 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GC | 158 | 168 | 0.938 | 1.021 (0.754–1.381) | G | 490 | 526 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CC | 75 | 53 | 0.049 | 1.526 (1.012–2.300) | C | 313 | 274 | 0.063 | 1.215 (0.992–1.489) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ApoE ɛ4(−) | GG | 97 | 144 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GC | 104 | 139 | 0.581 | 1.111 (0.774–1.595) | G | 298 | 427 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CC | 42 | 42 | 0.126 | 1.485 (0.901–2.446) | C | 188 | 223 | 0.135 | 1.208 (0.946–1.542) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ApoE ɛ4(+) | GG | 69 | 35 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GC | 54 | 29 | 0.878 | 0.945 (0.515–1.734) | G | 192 | 99 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CC | 33 | 11 | 0.336 | 1.522 (0.688–3.368) | C | 126 | 51 | 0.262 | 1.274 (0.849–1.911) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs2300403 | AA | 189 | 194 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AG | 156 | 168 | 0.764 | 0.953 (0.709–1.282) | A | 534 | 556 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GG | 52 | 37 | 0.128 | 1.443 (0.905–2.299) | G | 260 | 242 | 0.306 | 1.119 (0.905–1.383) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ApoE ɛ4(−) | AA | 115 | 157 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AG | 98 | 144 | 0.720 | 0.929 (0.654–0.321) | A | 328 | 458 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GG | 30 | 24 | 0.099 | 1.706 (0.948–3.077) | G | 158 | 192 | 0.299 | 1.149 (0.891–1.481) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ApoE ɛ4(+) | AA | 74 | 37 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AG | 58 | 24 | 0.639 | 1.208 (0.651–2.242) | A | 206 | 98 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GG | 22 | 13 | 0.687 | 0.846 (0.384–1.866) | G | 102 | 50 | 0.916 | 0.971 (0.641–1.471) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs9722 | GG | 182 | 209 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GA | 165 | 161 | 0.294 | 1.177 (0.877–1.580) | G | 529 | 579 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AA | 53 | 30 | 0.005 | 2.029 (1.243–3.311) | A | 271 | 221 | 0.008 | 1.342 (1.085–1.661) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ApoE ɛ4(−) | GG | 111 | 169 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GA | 94 | 138 | 0.857 | 1.037 (0.727–1.479) | G | 316 | 468 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AA | 31 | 18 | 0.003 | 2.622 (1.399–4.915) | A | 156 | 166 | 0.013 | 1.393 (1.072–1.808) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ApoE ɛ4(+) | GG | 71 | 40 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GA | 71 | 41 | 0.929 | 0.976 (0.565–1.684) | G | 213 | 121 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AA | 22 | 12 | 0.937 | 1.033 (0.463–2.306) | A | 93 | 65 | 0.320 | 0.813 (0.552–1.198) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| P values under 0.0167 were indicated in bold font. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Stratified analyses
We also carried out stratified analyses according to the ApoE ε4 allele. An increased risk of AD was more evident among ApoE ε4(-) subjects carrying the rs9722 AA-genotype (OR = 2.622, 95% CI = 1.399–4.915, P = 0.003). Likewise, the allele distributions of rs9722 between the AD patients and the controls was significantly different in the ApoE ε4(-) subjects (OR = 1.393, 95% CI = 1.072–1.808, P = 0.013). Nevertheless, we did not find any association between the different genotypes of rs1051169 and AD risk after performing stratified analyses based on the ApoE ε4 allele. In addition, we did not observe more apparent associations between rs2839364 or rs2300403 and AD risk among the subgroups via the ApoE ε4 allele (Table 1).
Haplotype association analyses
We performed haplotype association analyses of the four polymorphic loci. We identified and evaluated the haplotypes of the four SNPs. We found four main haplotypes; the C-G-A-G haplotype (in the order of rs2839364, rs1051169, rs2300403 and rs9722) was the most prevalent in both the AD population and the controls. Assigning the most common haplotype C-G-A-G as the reference, the haplotype C-C-G-A showed significant differences between the AD patients and the controls (OR = 1.605, 95% CI = 1.189–2.165, P = 0.002). We did not find any more significant differences between the AD patients and the controls for the other two main haplotypes (Table 2).
Table 2. Haplotype frequencies of the S100B SNPs in the AD patients and controls.
| Haplotypes | Control (freq) | AD (freq) | P | OR 95% CI | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C-G-A-G | 485.31 (0.607) | 421.79 (0.527) | 1 | − | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C-C-A-G | 121.94 (0.152) | 125.88 (0.157) | 0.251 | 1.188 (0.896–1.1.572) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C-C-G-A | 91.02 (0.114) | 127.36 (0.159) | 0.002 | 1.605 (1.189–2.165) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C-G-G-A | 89.14 (0.111) | 75.31 (0.094) | 0.865 | 0.968 (0.693–1.351) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Haplotypes consisted of S100B SNPs rs2839364, rs1051169, rs2300403 and rs9722. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Serum S100B levels analyses
Since AD is closely linked to the in vivo S100B level, we compared the serum S100B levels in the AD patients and the healthy controls. The data revealed the average serum S100B level in AD patients to be significantly higher than that in the controls (P = 0.036) (Figure 2A). We also investigated the association between the S100B polymorphisms and the serum S100B levels. As shown in Figure 2B, the serum S100B levels were significantly upregulated in the rs9722 AA genotype compared to the rs9722 GG genotype in the AD patients (P = 0.003). Similarly, the serum S100B levels were significantly upregulated in the rs1051169 CC genotype compared to the rs1051169 GG genotype in the AD patients (P < 0.001). We did not observe any significant differences between patients carrying the mutated genotypes and wild-type genotypes of the rs2839364 and rs2300403 polymorphisms.
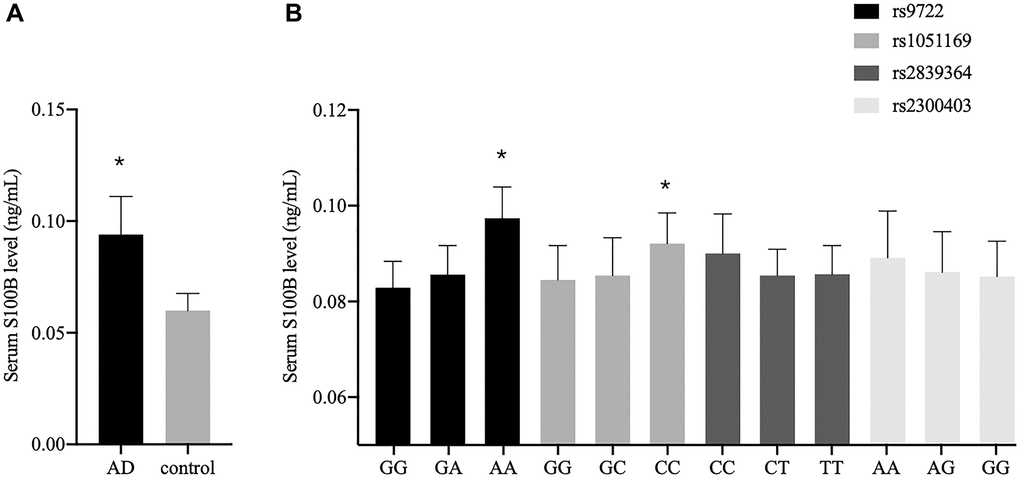
Figure 2. Serum S100B levels among different groups. (A) Serum S100B levels between the AD patients and the controls. (B) Serum S100B levels among different genotypes in the AD patients (*P < 0.05).
Luciferase assay detection
After using online software prediction, we identified three miRNAs (miR-340-3p, miR-593-3p, and miR-6827-3p) in which the seed match region covered locus rs9722 (Figure 3A). A subsequent luciferase assay indicated that miR-340-3p and miR-6827-3p stimulation could significantly reduce the fluorescence intensity of 293T cells that contained the rs9722 G allele (rather than the rs9722 A/MUT allele) (Figure 3B). We did not observe any significant decrease in fluorescence intensity following stimulation by miR-593 (Figure 3C, 3D).
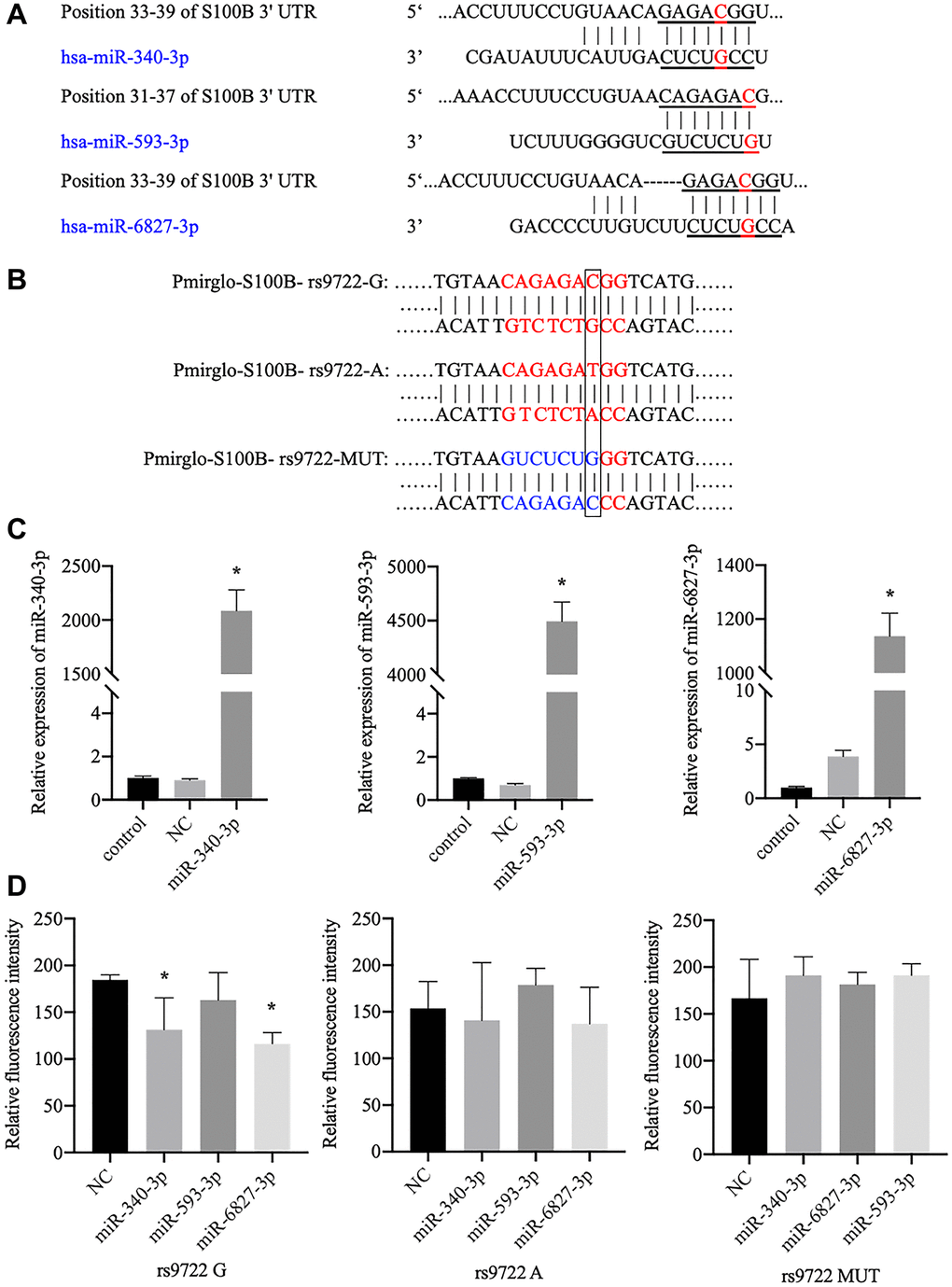
Figure 3. Verification of the interaction between candidate miRNAs and the 3′-UTR of the S100B gene, which contains the rs9722 loci, using the luciferase assay. (A) The candidate miRNAs that can bind to the rs9722 locus in the 3′-UTR of the S100B gene. The red letters show the rs9722 locus. (B) Plasmids containing different genotypes and artificial mutations. The black frame indicates the rs9722 locus. (C) Detection of miRNA levels in the 293T cells after transfection. (D) Fluorescence intensity after transfection of the miRNAs and the plasmids containing the different rs9722 alleles or mutations (*P < 0.05).
Western blot detection
The subsequent western blot data showed that miR-6827-3p could significantly reduce the S100B level of the SH-SY5Y cells containing the rs9722 G allele (P = 0.009). We did not observe a significant difference, although the S100B level decreased following stimulation by miR-340-3p. Likewise, we did not detect a significant difference in the S100B protein level following stimulation by miR-593-3p (Figure 4).
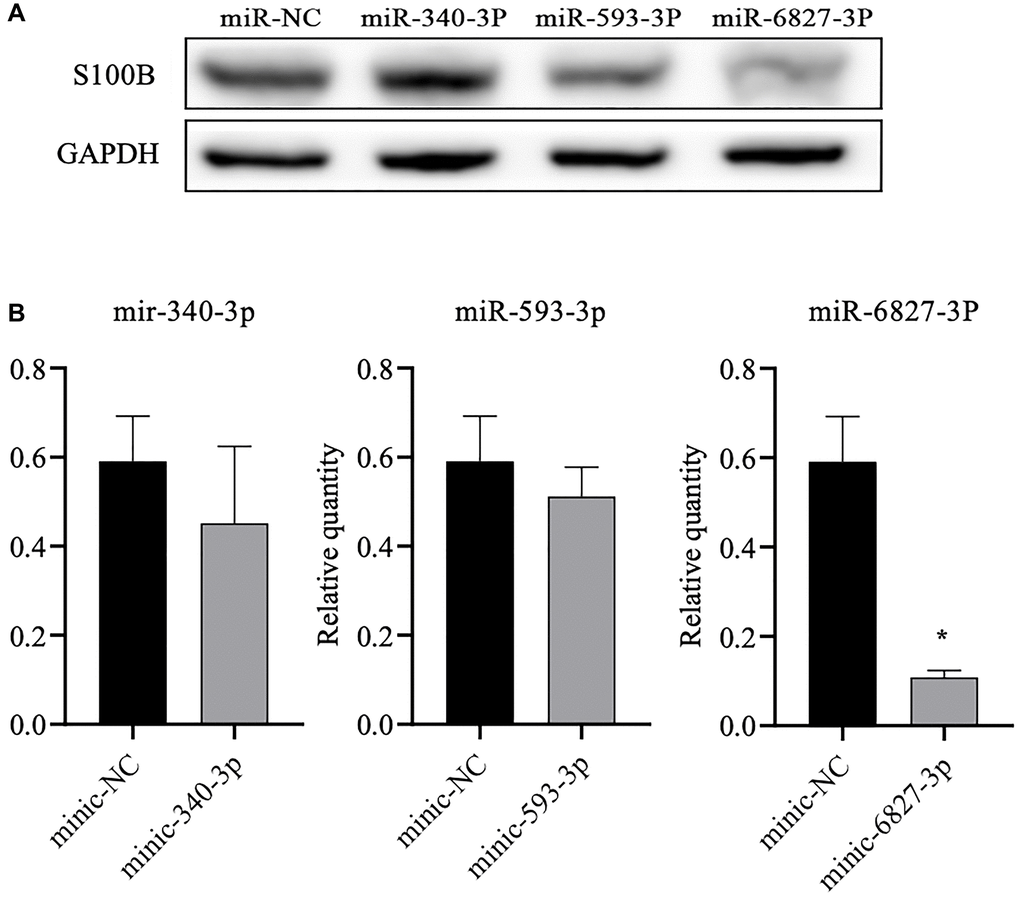
Figure 4. Western blot analysis of the S100B levels in SH-SY5Y cells after transfection of the three different miRNAs. (A) We detected the S100B protein level using western blot analysis. (B) Densitometry analysis to determine the ratio of S100B to GAPDH. All data are the average of three measurements (*P < 0.05).
Discussion
Some studies have verified that S100B may contribute to the pathogenesis of AD, revealing that S100B persists in the extracellular space and upregulates chronically activate RAGE in astrocytes or microglia, thereby amplifying the inflammatory response [37–39]. Extracellular S100B promotes RAGE-dependent hyperphosphorylation of tau protein and the development of neurofibrillary tangles (NFTs) [40]. Moreover, a large number of clinical studies have demonstrated that the S100B levels are elevated in the CSF and/or serum of patients with various neuropsychiatric diseases, including schizophrenia [41, 42], bipolar disorder [43], ischemic stroke [20], multiple sclerosis [44] and AD [22, 24]. We found the serum S100B level of AD patients to be significantly higher than that of healthy controls (Figure 2A). Increasing S100B levels in the brain can accelerate cerebral amyloidosis, possibly by promoting the cleavage of APP to Aβ [4, 45]. Additionally, S100B may promote the conversion of diffuse, non-fibrillar Aβ deposits into neuritic Aβ plaques, thus exacerbating the progression of AD pathology [4].
In this case-control study, we found the S100B SNP loci rs9722 to be associated with AD risk. We also examined the association of the serum S100B level with respect to the corresponding S100B polymorphisms in AD patients. The data revealed that the AA genotype of the rs9722, as well as the CC genotype of the rs1051169 locus, are significantly more associated with higher serum S100B levels. This outcome confirmed the finding reported by Hohoff (2010) regarding the potential role of S100B SNPs in S100B serum concentrations in a healthy population [14]. In addition, the expression quantitative trait loci (eQTL) data of S100B in whole blood from the GTEx Portal showed that the serum S100B levels were significantly upregulated in the AA genotype compared to the GG genotype of the rs9722 locus. Likewise, the serum S100B levels were significantly upregulated in the CC genotype compared to the GG genotype of the rs1051169 locus (Supplementary Figure 1). This finding is in line with our results. The alleles of the two SNPs mentioned above, which were shown to influence S100B gene expression, became risk factors that were also indirectly confirmed by the haplotype analysis. Thus, the haplotype C-C-G-A contained both the rs1051169 C allele and the rs9722 A allele, indicating a significant association with AD risk (Table 2).
Lambert (2007) found that the S100B polymorphism rs2300403 was correlated with low cognitive performance and dementia in elderly people, thereby underlining the importance of S100B in genetic susceptibility to AD [21]. However, we did not detect any significant association of rs2300403 with AD in our study, probably because Lambert analyzed the S100B mRNA level, while we focused on the serum S100B protein level. Hence, we speculate that post-transcriptional regulation may play a role in the S100B translation process.
The SNP rs9722 is located in the 3′-UTR of S100B (Figure 1); 3′-UTRs typically contain important regulatory elements, such as U-rich motifs, AU-rich elements, or microRNA target sites, which modulate gene expression and may also apply to rs9722 [46]. We examined the microRNA binding sites in the 3′-UTR of the S100B gene and identified three miRNAs (miR-593-3p, miR-340-3p and miR-6827-3p) that bind precisely to the rs9722G allele (Figure 3A).
The subsequent luciferase assay data verified that miR-6827-3p can bind to the 3′-UTR of S100B mRNA (Figure 3D). In addition, the western blot assay signaled a significant decreasing S100B protein level in SH-SY5Y cells containing rs9722 G after stimulation by miR-6827-3p (Figure 4). Therefore, we believe that miR-6827-3p can specifically bind to the 3′-UTR of S100B mRNA in individuals carrying the rs9722 G allele, which results in the degradation of S100B mRNA and a subsequently lower S100B protein level. The rs9722 A allele reduced the stability of miR-6827-3p in binding to the S100B mRNA, thereby preventing the S100B protein from decreasing, which may explain why the rs9722 A allele seems to be a risk factor for AD.
We found that the GG genotype of the rs1051169 locus is also significantly related to the increased serum S100B level in AD patients. However, after stratified analyses based on the ApoE ε4 allele, we did not observe any significant genotypes/alleles frequencies differences between the AD patients and the controls of rs1051169 (Table 1). We speculate that the varying S100B levels between different rs1051169 genotypes may be due to a genetic linkage or other nongenetic influence factors.
Taking these pieces of evidence together, we can hypothesize that a possible intrinsic factor within the S100B gene may play a role in regulating S100B expression. Although S100B levels might be mediated by a possible feedback loop induced by the binding receptors of S100B (like RAGE), we observed indications that an intrinsic genetic factor could be partially involved in regulating S100B expression. The mechanism of AD is still unclear, but it may at least be partially explained by the increased S100B expression due to different S100B genotype carriers.
In addition, we observed an interesting phenomenon in which the distribution of the rs9722 genotype was quite different between our population and the German population. The genotype frequency of AA+AG in our healthy population was 47.8% (191/400), which is much higher than in the German population (18.4%, 36/160) reported by Hohoff [14]. Given that the distribution of the S100B gene frequencies might vary among different ethnic groups, this discrepancy suggests that studies need to be performed with a larger ethnically diverse population to clarify whether the association is limited to the Chinese Han population or whether it could also apply to other ethnic groups.
In sum, our study implies a significant association between the genotypes of S100B polymorphisms and increased incidence of AD in the Chinese Han population. Our findings highlight the rs9722 variant of S100B as an important risk factor for AD, which may act by regulating S100B expression. Further investigation revealed that different rs9722 genotypes may alter the combination of miR-6827-3p and S100B mRNA, which could subsequently decrease the S100B protein level. This is the first study to indicate an association between polymorphisms of the S100B gene and AD in an Asian cohort. In addition, our study signals the epigenetic regulation of S100B expression during the pathogenesis of AD. More research is necessary to substantiate these results and to shed light on the importance of interethnic differences affecting the outcomes of the complex nature of AD.
Materials and Methods
Study subjects
We recruited all participants from the First Affiliated Hospital of Harbin Medical University in North China. The patient group consisted of 280 individuals (mean age at onset: 69.68 ± 8.27 years; mean age: 71.86 ± 9.07 years; 49.6% male). All patients met the criteria of the National Institute of Neurological and Communicative Disorders and Stroke as well as of the Alzheimer’s Disease and Related Disorders Association for probable AD. We excluded patients with a family history of dementia. The control group was composed of 400 healthy individuals (mean age: 74.21 ± 8.33 years; 51.5% male) recruited from individuals who underwent a regular health examination at the same hospital. The participants were confirmed to be healthy and neurologically normal using the Mini-Mental State Examination, the Revised Hasegawa Dementia Scale and general examinations. All participants were representative of the northern Chinese Han population living in North China, as defined by the geography of the Yellow River. Both groups were matched for geographic location, ethnicity, sex and age. We obtained informed consent either directly from the participants or their guardians. The Medical Ethical Committee of the Affiliated Hospital of Guangdong Medical University reviewed and approved of the study’s protocol.
Sample collection and SNPs genotyping
We took a 5-mL venous whole blood sample from each participant for DNA extraction and SNP genotyping (including rs2839364, rs1051169, rs2300403 and rs9722). We isolated genomic DNA from venous blood samples using the Blood Genomic DNA Extraction Kit (Tiangen, China). We genotyped polymorphisms of the samples using the ABI PRISM SNaPshot method (Applied Biosystems, Foster, CA). The assay conditions followed the manufacturer’s protocols. We genotyped 10% of the samples in duplicate to verify the accuracy of the genotyping data.
Cell lines acquisition and culture
We obtained the 293T and SH-SY5Y cell lines from ATCC (Manassas, VA, USA). We cultured cells in the DMEM, to which we added 10% FBS (Gibco, USA) at 37°C, and supplemented it with 5% CO2 using the Thermo HERAcell 150i incubator (Thermo, USA).
Luciferase assay
All the miRNAs that could bind to the SNP loci in the 3′UTR of the S100B gene were predicted using the online software Targetscan (http://www.targetscan.org/vert_71/). Moreover, we employed the luciferase assay to evaluate the targeted binding relationship between the candidate miRNAs and the different SNP genotypes. We amplified the 3′UTR region sequences covering the SNP locus through specific PCR. We obtained the amplicons containing the different alleles from different homozygote individuals carrying different SNP genotypes. Then, we cloned the amplicons into the psi-CHECK2 vector (Promega, USA) and transfected them into 293T cells using lipofectamine3000 (Life Technology, USA). Additionally, we cotransferred the miRNA negative control and mimics (RiboBio, China) into 293T cells. After 48 hours of culture, we harvested the cells and subjected them to the luciferase activity tests of both firefly and renilla using the Dual-Luciferase Reporter Assay System (Promega, USA).
Transfection and western blot
We constructed overexpressed plasmids containing different rs9722 genotypes and transfected them into SH-SY5Y cells using Lipofectamine™ 3000 Transfection Reagent (Invitrogen™, USA) according to the recommended protocol. We detected the S100B protein levels of SH-SY5Y cells with different genotypes using the western blot method following stimulation by the candidate miRNA mimics 48 hours later.
RNA extraction and real-time quantitative PCR
We extracted total RNA using Trizol Regent (Invitrogen, USA) according to the recommended protocol. We detected the relative transcription levels of the gene/miRNAs through real-time quantitative PCR (qPCR) analysis. We adopted the 2-ΔΔCT method and selected GAPDH and U6 as internal reference for genes and miRNAs, respectively. All the primers were summarized in the Supplementary Table 1.
ELISA assay and S100B level detection
We used an enzyme immunoassay quantitative measurement, an ELISA Kit (KA0037, Abnova), for S100B to determine the serum S100B levels in 90 randomly selected venous blood samples. We also examined the eQTL data with the different genotypes of the four candidate SNPs in whole blood, and we obtained the eQTL data from the GTEx Portal (https://gtexportal.org/home/).
Statistical analysis
We performed all analyses using SPSS version 19.0 (IBM, NY, USA). We counted and estimated the genotype and allele frequency distributions in the groups using the chi-square or Fisher’s exact test, and results were adjusted for multiple comparisons by Bonferroni correction. We assessed deviations of the genotype or allele frequency using HWE. We calculated the odds ratio (OR) and the 95% confidence interval (CI) to establish the correlation between the S100B genotype and AD.
Author Contributions
JF Wang and YL Zhou performed most of the experimental work and drafted the manuscript. YX Yang and X Gao participated in part of the experimental work. YX Yang, X Gao, ZB Liu, GH Hong, LF Yao and JW Yin collected the specimens. XF Gu helped to design the study and revise the manuscript. KS Li helped to design the study, revise the manuscript and contributed reagents and materials. All the authors read and approved of the final manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding
This study was supported by the National Natural Science Foundation of China (81471294, 81600445, 81971079), the Natural Science Foundation of Guangdong Province (2018A030307036), the Medical Scientific Research Foundation of Guangdong Province (No. A2016522, A2020376), the Science and Technology Program of Zhanjiang (2019A01011, 2019A01032), the Guangdong Medical University Research Fund (M2017009), and the Doctoral Research Fund of the Affiliated Hospital of Guangdong Medical College (BJ201512). Science and Technology Innovation Strategy of Special Fund of Guangdong Province (No. Pdjh2020b0258).
References
- 1. Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. 2006; 368:387–403. https://doi.org/10.1016/S0140-6736(06)69113-7 [PubMed]
- 2. Nilsen LH, Witter MP, Sonnewald U. Neuronal and astrocytic metabolism in a transgenic rat model of Alzheimer's disease. J Cereb Blood Flow Metab. 2014; 34:906–14. https://doi.org/10.1038/jcbfm.2014.37 [PubMed]
- 3. Mori T, Koyama N, Arendash GW, Horikoshi-Sakuraba Y, Tan J, Town T. Overexpression of human S100B exacerbates cerebral amyloidosis and gliosis in the Tg2576 mouse model of Alzheimer's disease. Glia. 2010; 58:300–14. https://doi.org/10.1002/glia.20924 [PubMed]
- 4. Mrak RE, Griffinbc WS. The role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer's disease. Neurobiol Aging. 2001; 22:915–22. https://doi.org/10.1016/s0197-4580(01)00293-7 [PubMed]
- 5. Rothermundt M, Peters M, Prehn JH, Arolt V. S100B in brain damage and neurodegeneration. Microsc Res Tech. 2003; 60:614–32. https://doi.org/10.1002/jemt.10303 [PubMed]
- 6. Sorci G, Riuzzi F, Agneletti AL, Marchetti C, Donato R. S100B inhibits myogenic differentiation and myotube formation in a RAGE-independent manner. Mol Cell Biol. 2003; 23:4870–81. https://doi.org/10.1128/mcb.23.14.4870-4881.2003 [PubMed]
- 7. Marenholz I, Heizmann CW, Fritz G. S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature). Biochem Biophys Res Commun. 2004; 322:1111–22. https://doi.org/10.1016/j.bbrc.2004.07.096 [PubMed]
- 8. Nishiyama H, Takemura M, Takeda T, Itohara S. Normal development of serotonergic neurons in mice lacking S100B. Neurosci Lett. 2002; 321:49–52. https://doi.org/10.1016/s0304-3940(01)02549-6 [PubMed]
- 9. Huttunen HJ, Kuja-Panula J, Sorci G, Agneletti AL, Donato R, Rauvala H. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J Biol Chem. 2000; 275:40096–105. https://doi.org/10.1074/jbc.M006993200 [PubMed]
- 10. Businaro R, Leone S, Fabrizi C, Sorci G, Donato R, Lauro GM, Fumagalli L. S100B protects LAN-5 neuroblastoma cells against Abeta amyloid-induced neurotoxicity via RAGE engagement at low doses but increases Abeta amyloid neurotoxicity at high doses. J Neurosci Res. 2006; 83:897–906. https://doi.org/10.1002/jnr.20785 [PubMed]
- 11. Donato R, Sorci G, Riuzzi F, Arcuri C, Bianchi R, Brozzi F, Tubaro C, Giambanco I. S100B's double life: intracellular regulator and extracellular signal. Biochim Biophys Acta. 2009; 1793:1008–22. https://doi.org/10.1016/j.bbamcr.2008.11.009 [PubMed]
- 12. Adami C, Bianchi R, Pula G, Donato R. S100B-stimulated NO production by BV-2 microglia is independent of RAGE transducing activity but dependent on RAGE extracellular domain. Biochim Biophys Acta. 2004; 1742:169–77. https://doi.org/10.1016/j.bbamcr.2004.09.008 [PubMed]
- 13. Allore R, O'Hanlon D, Price R, Neilson K, Willard HF, Cox DR, Marks A, Dunn RJ. Gene encoding the beta subunit of S100 protein is on chromosome 21: implications for Down syndrome. Science. 1988; 239:1311–13. https://doi.org/10.1126/science.2964086 [PubMed]
- 14. Hohoff C, Ponath G, Freitag CM, Kästner F, Krakowitzky P, Domschke K, Koelkebeck K, Kipp F, von Eiff C, Deckert J, Rothermundt M. Risk variants in the S100B gene predict elevated S100B serum concentrations in healthy individuals. Am J Med Genet B Neuropsychiatr Genet. 2010; 153B:291–97. https://doi.org/10.1002/ajmg.b.30950 [PubMed]
- 15. Fardell C, Zettergren A, Ran C, Carmine Belin A, Ekman A, Sydow O, Bäckman L, Holmberg B, Dizdar N, Söderkvist P, Nissbrandt H. S100B polymorphisms are associated with age of onset of Parkinson's disease. BMC Med Genet. 2018; 19:42. https://doi.org/10.1186/s12881-018-0547-3 [PubMed]
- 16. Guo Y, Yang H, Deng X, Song Z, Yang Z, Xiong W, Yuan L, Xu H, Deng S, Deng H. Genetic analysis of the S100B gene in Chinese patients with Parkinson disease. Neurosci Lett. 2013; 555:134–36. https://doi.org/10.1016/j.neulet.2013.09.037 [PubMed]
- 17. Yang K, Xie GR, Hu YQ, Mao FQ, Su LY. Association study of astrocyte-derived protein S100B gene polymorphisms with major depressive disorder in Chinese people. Can J Psychiatry. 2009; 54:312–19. https://doi.org/10.1177/070674370905400505 [PubMed]
- 18. Zhai J, Cheng L, Dong J, Shen Q, Zhang Q, Chen M, Gao L, Chen X, Wang K, Deng X, Xu Z, Ji F, Liu C, et al. S100B gene polymorphisms predict prefrontal spatial function in both schizophrenia patients and healthy individuals. Schizophr Res. 2012; 134:89–94. https://doi.org/10.1016/j.schres.2011.09.029 [PubMed]
- 19. Zhai J, Zhang Q, Cheng L, Chen M, Wang K, Liu Y, Deng X, Chen X, Shen Q, Xu Z, Ji F, Liu C, Dong Q, et al. Risk variants in the S100B gene, associated with elevated S100B levels, are also associated with visuospatial disability of schizophrenia. Behav Brain Res. 2011; 217:363–68. https://doi.org/10.1016/j.bbr.2010.11.004 [PubMed]
- 20. Lu YL, Wang R, Huang HT, Qin HM, Liu CH, Xiang Y, Wang CF, Luo HC, Wang JL, Lan Y, Wei YS. Association of S100B polymorphisms and serum S100B with risk of ischemic stroke in a Chinese population. Sci Rep. 2018; 8:971. https://doi.org/10.1038/s41598-018-19156-w [PubMed]
- 21. Lambert JC, Ferreira S, Gussekloo J, Christiansen L, Brysbaert G, Slagboom E, Cottel D, Petit T, Hauw JJ, DeKosky ST, Richard F, Berr C, Lendon C, et al. Evidence for the association of the S100beta gene with low cognitive performance and dementia in the elderly. Mol Psychiatry. 2007; 12:870–80. https://doi.org/10.1038/sj.mp.4001974 [PubMed]
- 22. Peskind ER, Griffin WS, Akama KT, Raskind MA, Van Eldik LJ. Cerebrospinal fluid S100B is elevated in the earlier stages of Alzheimer's disease. Neurochem Int. 2001; 39:409–13. https://doi.org/10.1016/s0197-0186(01)00048-1 [PubMed]
- 23. Petzold A, Jenkins R, Watt HC, Green AJ, Thompson EJ, Keir G, Fox NC, Rossor MN. Cerebrospinal fluid S100B correlates with brain atrophy in Alzheimer's disease. Neurosci Lett. 2003; 336:167–70. https://doi.org/10.1016/s0304-3940(02)01257-0 [PubMed]
- 24. Chaves ML, Camozzato AL, Ferreira ED, Piazenski I, Kochhann R, Dall'Igna O, Mazzini GS, Souza DO, Portela LV. Serum levels of S100B and NSE proteins in Alzheimer's disease patients. J Neuroinflammation. 2010; 7:6. https://doi.org/10.1186/1742-2094-7-6 [PubMed]
- 25. Hov KR, Bolstad N, Idland AV, Zetterberg H, Blennow K, Chaudhry FA, Frihagen F, Ræder J, Wyller TB, Watne LO. Cerebrospinal Fluid S100B and Alzheimer's Disease Biomarkers in Hip Fracture Patients with Delirium. Dement Geriatr Cogn Dis Extra. 2017; 7:374–85. https://doi.org/10.1159/000481853 [PubMed]
- 26. Shapiro LA, Bialowas-McGoey LA, Whitaker-Azmitia PM. Effects of S100B on Serotonergic Plasticity and Neuroinflammation in the Hippocampus in Down Syndrome and Alzheimer's Disease: Studies in an S100B Overexpressing Mouse Model. Cardiovasc Psychiatry Neurol. 2010; 2010:153657. https://doi.org/10.1155/2010/153657 [PubMed]
- 27. Cirillo C, Capoccia E, Iuvone T, Cuomo R, Sarnelli G, Steardo L, Esposito G. S100B Inhibitor Pentamidine Attenuates Reactive Gliosis and Reduces Neuronal Loss in a Mouse Model of Alzheimer's Disease. Biomed Res Int. 2015; 2015:508342. https://doi.org/10.1155/2015/508342 [PubMed]
- 28. Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009; 460:479–86. https://doi.org/10.1038/nature08170 [PubMed]
- 29. Silvestro S, Bramanti P, Mazzon E. Role of miRNAs in Alzheimer's Disease and Possible Fields of Application. Int J Mol Sci. 2019; 20:3979. https://doi.org/10.3390/ijms20163979 [PubMed]
- 30. Gupta P, Bhattacharjee S, Sharma AR, Sharma G, Lee SS, Chakraborty C. miRNAs in Alzheimer Disease - A Therapeutic Perspective. Curr Alzheimer Res. 2017; 14:1198–206. https://doi.org/10.2174/1567205014666170829101016 [PubMed]
- 31. Chang WS, Wang YH, Zhu XT, Wu CJ. Genome-Wide Profiling of miRNA and mRNA Expression in Alzheimer's Disease. Med Sci Monit. 2017; 23:2721–31. https://doi.org/10.12659/msm.905064 [PubMed]
- 32. Putteeraj M, Fairuz YM, Teoh SL. MicroRNA Dysregulation in Alzheimer's Disease. CNS Neurol Disord Drug Targets. 2017; 16:1000–09. https://doi.org/10.2174/1871527316666170807142311 [PubMed]
- 33. Swarbrick S, Wragg N, Ghosh S, Stolzing A. Systematic Review of miRNA as Biomarkers in Alzheimer's Disease. Mol Neurobiol. 2019; 56:6156–67. https://doi.org/10.1007/s12035-019-1500-y [PubMed]
- 34. Chen Y, Liu X, Chen L, Chen W, Zhang Y, Chen J, Wu X, Zhao Y, Wu X, Sun G. The long noncoding RNA XIST protects cardiomyocyte hypertrophy by targeting miR-330-3p. Biochem Biophys Res Commun. 2018; 505:807–15. https://doi.org/10.1016/j.bbrc.2018.09.135 [PubMed]
- 35. Wen L, Sun J, Chen X, Du R. miR-135b-dependent downregulation of S100B promotes neural stem cell differentiation in a hypoxia/ischemia-induced cerebral palsy rat model. Am J Physiol Cell Physiol. 2020; 319:C955–66. https://doi.org/10.1152/ajpcell.00481.2019 [PubMed]
- 36. Chen Y, Chen X, Yao M, Chen L, Chen W, Liu X. Association of S100B 3′UTR polymorphism with risk of chronic heart failure in a Chinese Han population. Medicine (Baltimore). 2020; 99:e21018. https://doi.org/10.1097/MD.0000000000021018 [PubMed]
- 37. Li RL, Zhang ZZ, Peng M, Wu Y, Zhang JJ, Wang CY, Wang YL. Postoperative impairment of cognitive function in old mice: a possible role for neuroinflammation mediated by HMGB1, S100B, and RAGE. J Surg Res. 2013; 185:815–24. https://doi.org/10.1016/j.jss.2013.06.043 [PubMed]
- 38. Michetti F, Donno C, D’Ambrosi N. The S100B-RAGE pathway is dysregulated in the ALS-linked neuroinflammatory process. SpringerPlus. 2015 (Suppl 1); 4:P27. https://doi.org/10.1186/2193-1801-4-S1-P27
- 39. Bianchi R, Giambanco I, Donato R. S100B/RAGE-dependent activation of microglia via NF-kappaB and AP-1 Co-regulation of COX-2 expression by S100B, IL-1beta and TNF-alpha. Neurobiol Aging. 2010; 31:665–77. https://doi.org/10.1016/j.neurobiolaging.2008.05.017 [PubMed]
- 40. Esposito G, Scuderi C, Lu J, Savani C, De Filippis D, Iuvone T, Steardo L
Jr , Sheen V, Steardo L. S100B induces tau protein hyperphosphorylation via Dickopff-1 up-regulation and disrupts the Wnt pathway in human neural stem cells. J Cell Mol Med. 2008; 12:914–27. https://doi.org/10.1111/j.1582-4934.2008.00159.x [PubMed] - 41. Liu J, Shi Y, Tang J, Guo T, Li X, Yang Y, Chen Q, Zhao X, He G, Feng G, Gu N, Zhu S, Liu H, He L. SNPs and haplotypes in the S100B gene reveal association with schizophrenia. Biochem Biophys Res Commun. 2005; 328:335–41. https://doi.org/10.1016/j.bbrc.2004.12.175 [PubMed]
- 42. Steiner J, Myint AM, Schiltz K, Westphal S, Bernstein HG, Walter M, Schroeter ML, Schwarz MJ, Bogerts B. S100B Serum Levels in Schizophrenia Are Presumably Related to Visceral Obesity and Insulin Resistance. Cardiovasc Psychiatry Neurol. 2010; 2010:480707. https://doi.org/10.1155/2010/480707 [PubMed]
- 43. Dagdan E, Morris DW, Campbell M, Hill M, Rothermundt M, Kästner F, Hohoff C, von Eiff C, Krakowitzky P, Gill M, McKeon P, Roche S. Functional assessment of a promoter polymorphism in S100B, a putative risk variant for bipolar disorder. Am J Med Genet B Neuropsychiatr Genet. 2011; 156B:691–99. https://doi.org/10.1002/ajmg.b.31211 [PubMed]
- 44. Barateiro A, Afonso V, Santos G, Cerqueira JJ, Brites D, van Horssen J, Fernandes A. S100B as a Potential Biomarker and Therapeutic Target in Multiple Sclerosis. Mol Neurobiol. 2016; 53:3976–91. https://doi.org/10.1007/s12035-015-9336-6 [PubMed]
- 45. Sheng JG, Mrak RE, Bales KR, Cordell B, Paul SM, Jones RA, Woodward S, Zhou XQ, McGinness JM, Griffin WS. Overexpression of the neuritotrophic cytokine S100beta precedes the appearance of neuritic beta-amyloid plaques in APPV717F mice. J Neurochem. 2000; 74:295–301. https://doi.org/10.1046/j.1471-4159.2000.0740295.x [PubMed]
- 46. Wilk G, Braun R. regQTLs: Single nucleotide polymorphisms that modulate microRNA regulation of gene expression in tumors. PLoS Genet. 2018; 14:e1007837. https://doi.org/10.1371/journal.pgen.1007837 [PubMed]