



Introduction
Sepsis is a systemic inflammatory response syndrome and the main cause of death in critically ill patients [1, 2]. Cardiac dysfunction with left ventricular (LV) dilation and reduced LV ejection fraction (LVEF) accompanied by sepsis is the main cause of death in intensive care units. The infectious organisms and dead or damaged cells released pathogen-related molecular patterns and damage-related molecular patterns (DAMPs) activate inflammatory responses. Overproduction of inflammatory cytokines including tumor necrosis factor (TNF)-α, interleukin (IL)-1α, IL-1β, and IL-6 also leads to cardiac dysfunction in sepsis [3–5]. Treatments for septic cardiomyopathy include optimizing volume loading and blood pressure, improving cardiac contractility, and alleviating inflammation. Inhibiting inflammation is considered a potential therapeutic strategy for sepsis-induced cardiomyopathy [6–8].
Accumulating evidence demonstrates that autophagy is involved in the regulation of inflammatory responses, in different cell types in the heart and thus plays crucial roles in sepsis-induced cardiomyopathy [9–11]. So far, the effects of autophagy in the pathogenesis of sepsis remain unclear. The effects of autophagy on inflammatory cardiac diseases could be protective or adverse depending on the different conditions [12–15]. On the one hand, autophagy controls cardiac inflammatory responses by limiting the inflammasome activators and/or components [16–18] and by decreasing DAMPs from mitochondria [19]. On the other hand, overactive autophagy in the context of myocardial infarction or ischemia/reperfusion stress may exacerbate adverse results due to the degradation of cellular components, leading to a positive inflammatory response [20].
Natural products from plants and their derivatives are valuable sources of therapeutic agents [21, 22]. Plants in the Amaryllidaceae family are known for their pharmacologically active alkaloids. Traditionally, corm extracts from chicory bulbs have been used to treat inflammation-related diseases [23]. Narciclasine is an isocyanide alkaloid that is present in the bulbs of Coneflower extract and has been found to have estrogen-killing properties [24]. Narcissus has been shown to improve the prognosis of neonatal rats with sepsis by inhibiting calprotectin and reducing inflammation [25, 26]. Narciclasine has also been shown to improve outcomes in septic neonatal rats by alleviating inflammatory responses [27].
The activation of multiple stress signaling cascade like mitogen-activated protein kinase (MAPK) is believed to play a key role in sepsis-induced cardiomyopathy [28, 29]. C-jun N-terminal kinase (JNK) is a member of the MAPK family that is important in numerous human diseases, including sepsis-induced cardiomyopathy [30, 31]. Autophagy plays an important role in inflammation-induced AMI. However, the role of autophagy and the mechanism of narciclasine-afforded cardioprotection remain unclear. In this study, we aimed to study whether narciclasine could improve LPS-induced AMI by regulating autophagy and whether the regulation of autophagy in AMI is mediated by the JNK signaling pathway.
Results
Narciclasine attenuates LPS-induced cardiomyocyte inflammation
We stimulated isolated neonatal rat cardiomyocytes (NRCMs) with LPS and treated these NRCMs with narciclasine (0, 30, 100, and 300 nM). Cell viability of NRCMs was not affected by narciclasine (Figure 1A), but the LPS-induced decrease in cell viability was attenuated by narciclasine treatment in a dose-dependent manner (Figure 1B). In addition, narciclasine suppressed LPS-induced inflammatory cytokine (TNF-α, IL-1β, and IL-6) release from NRCMs in a dose-dependent manner (Figure 1C–1E). Narciclasine also inhibited LPS-induced apoptosis in NRCMs in a dose-dependent manner (Figure 1F). These data indicated that narciclasine inhibited the LPS-induced inflammation in cardiomyocytes.
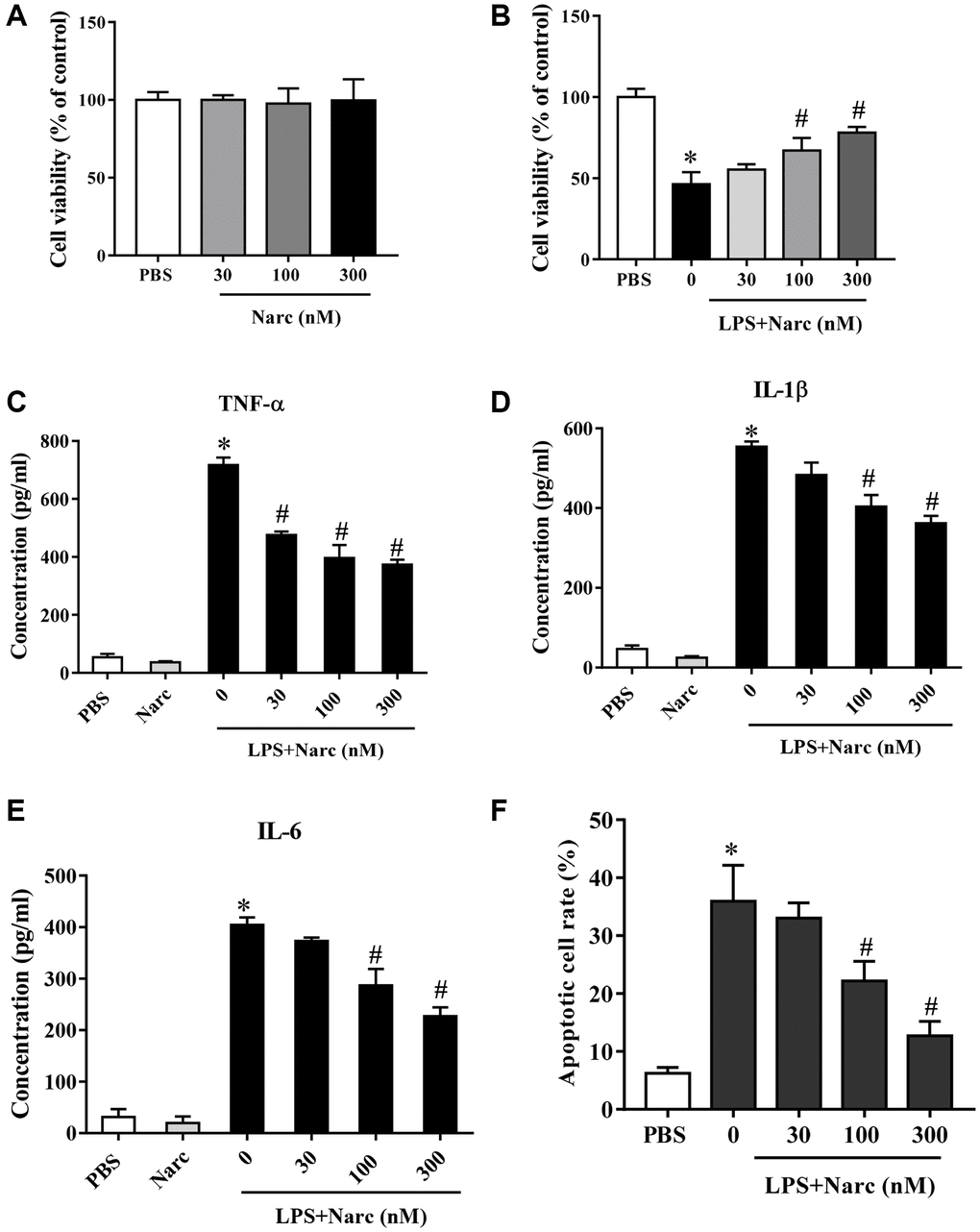
Figure 1. Narciclasine attenuates LPS-induced cardiomyocyte inflammation. (A) The viability of cardiomyocytes treated with different concentrations of narciclasine (0, 30, 100, and 300 nM) was measured by CCK8 assays. (B) The viability of cardiomyocytes stimulated with LPS and treated with different concentrations of narciclasine (0, 30, 100, and 300 nM) was measured by CCK8 assays. (C–E) The concentration of inflammatory cytokines (TNF-α, IL-1β, and IL-6) in cardiomyocytes stimulated with LPS and treated with different concentrations of narciclasine (0, 30, 100, and 300 nM) was measured by ELISA kits. (F) Quantification of LPS-induced neonatal rat cardiomyocyte apoptosis was analyzed by FACS. n = 3 per group. Data represent the mean ± SEM. *P < 0.05 vs. the PBS group. #P < 0.05 vs. the LPS group.
Narciclasine attenuates LPS-induced myocardial dysfunction
To investigate the role of narciclasine in LPS-induced AMI, mice were administered narciclasine before LPS injection. Echocardiography was performed to evaluate cardiac functions. There was no significant difference between the control group and narciclasine groups in the absence of LPS stimulation. After 12 h of LPS stimulation, the mice showed significant cardiac dysfunction, as evidenced by increased LVIDs and LVIDd, decreased ejection fraction, and fractional shortening compared with those of the control group. Interestingly, narciclasine treatment (LPS + Narc) partly improved these parameters (Figure 2A). In addition, treatment with narciclasine elevated the survival rates of LPS-induced mice (Figure 2B).
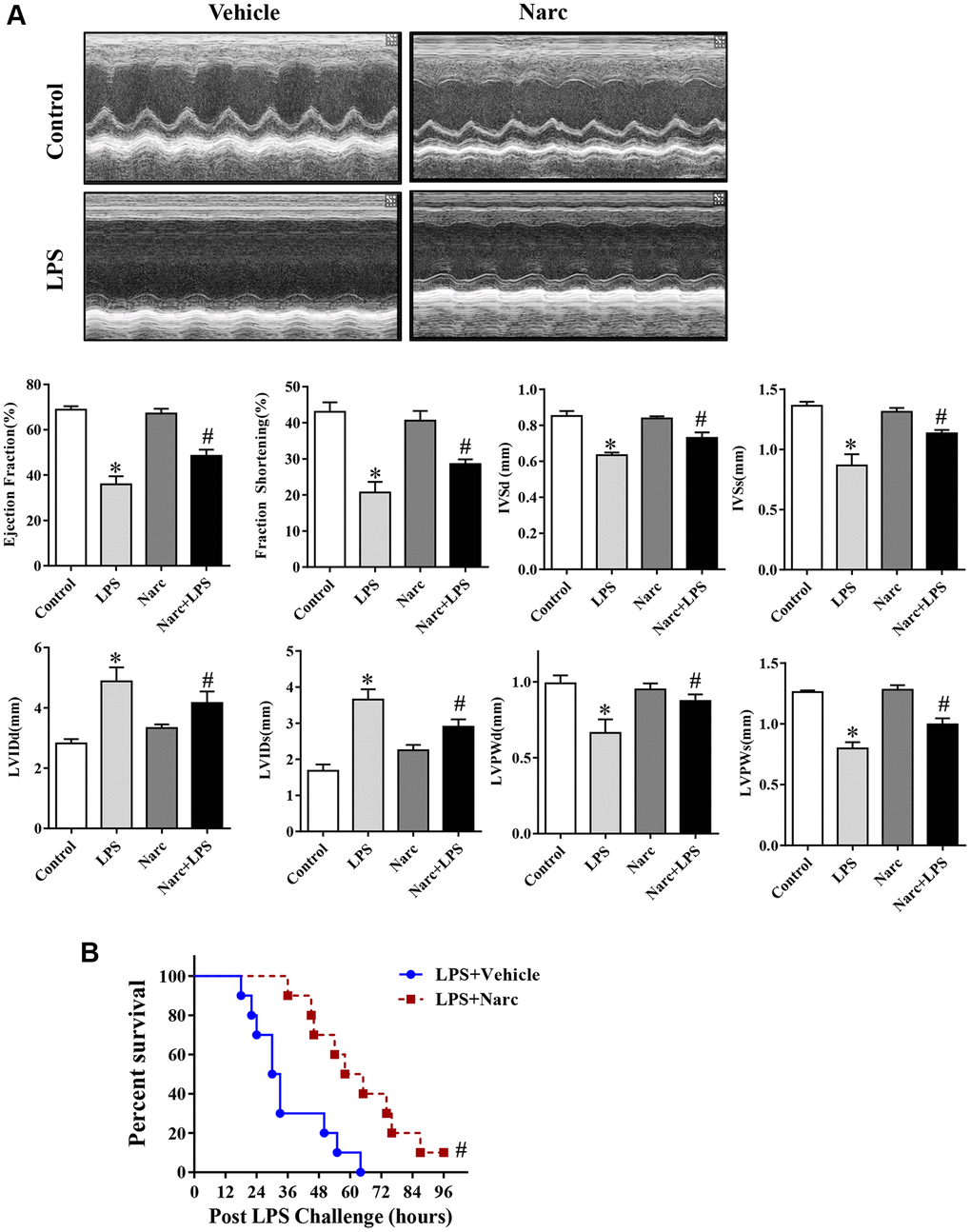
Figure 2. Narciclasine attenuates LPS-induced myocardial dysfunction. (A) Echocardiographic analysis of LVEF, LVFS, IVSd, IVSs, LVIDs, LVIDd, LVPWd, and LVPWs after LPS challenge for 12 h. (B) The survival rates of LPS-injected mice throughout the 96-h study period. LVEF, left ventricular ejection fraction; LVFS, left ventricular fractional shortening; IVSd and IVSs, interventricular septal end diastole and end systole, respectively; LVIDd and LVIDs, left ventricular internal diameter end diastole and end systole, respectively; LVPWd and LVPWs, left ventricular posterior wall end diastole and end systole, respectively. n = 8 per group. The data are shown as the means ± SEM. *P < 0.05 vs. the PBS group. #P < 0.05 vs. the LPS group.
Narciclasine attenuates the LPS-induced inflammatory response in vivo
To evaluate the cardioprotective effects of narciclasine on inflammation in LPS-induced AMI, the levels of inflammatory indicators were measured. An indicator of neutrophil infiltration, MPO, was increased in the LPS group, and narciclasine effectively abrogated this increase (Figure 3A). Additionally, the mRNA expression levels of IL-6, IL-1β, TNF-α, and VEGF in heart tissues were significantly increased in LPS-induced AMI but were notably attenuated by narciclasine treatment (Figure 3B). These results demonstrated that narciclasine could attenuate heart inflammation in LPS-induced AMI.
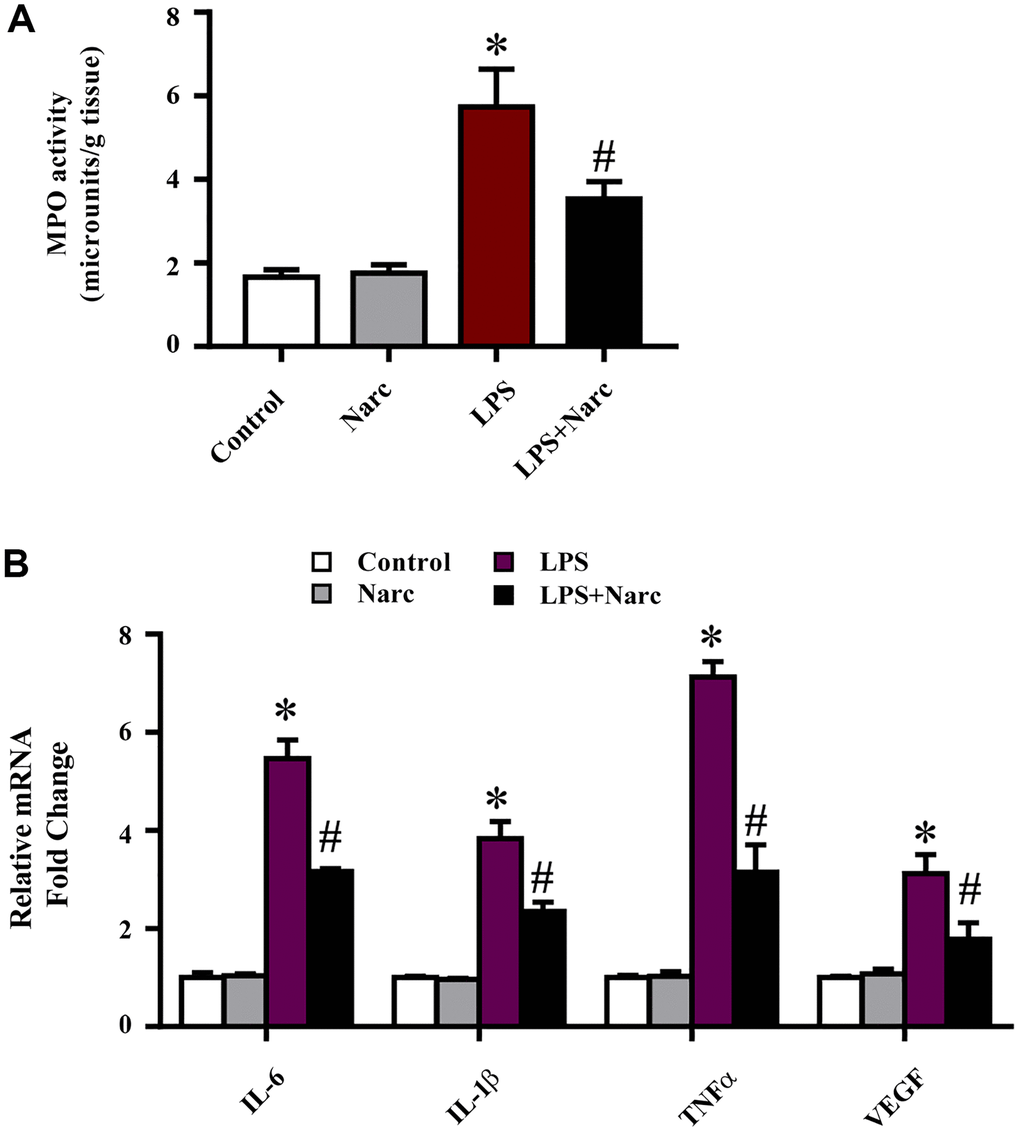
Figure 3. Narciclasine attenuates the LPS-induced inflammatory response in vivo. (A) Myeloperoxidase (MPO) activity was measured by an MPO assay kit. (B) The mRNA expression levels of IL-6, IL-1β, TNF-α, and VEGF in heart tissues were measured by q-PCR. n = 3 per group. The data are shown as the means ± SEM. *P < 0.05 vs. the PBS group. #P < 0.05 vs. the LPS group.
Autophagy is upregulated during narciclasine-mediated attenuation of LPS-induced AMI
To determine whether narciclasine could activate autophagy in LPS-induced AMI, we examined several autophagy indicators, such as microtubule-associated light chain (LC)-3, p62, and Beclin-1. Beclin-1 and LC3-II were increased while the p62 expression level was decreased in the LPS group compared with the sham group (Figure 4A). However, narciclasine administration further upregulated Beclin-1 and LC3-II expression and further downregulated p62 expression in response to LPS-induced AMI (Figure 4A). Furthermore, autophagy was measured by live-cell imaging using a GFP-LC3 adenovirus (Figure 4B). We found that narciclasine treatment upregulated the autophagy level, as indicated by increased GFP+ puncta. These data indicated that narciclasine could improve autophagy in the heart during LPS-induced AMI.
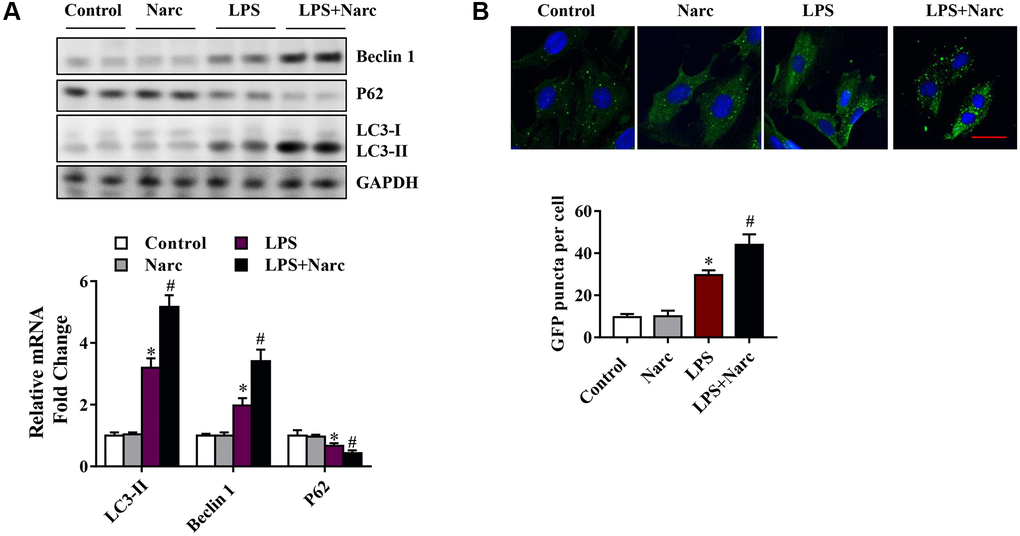
Figure 4. Narciclasine upregulates autophagy in a mouse model of LPS-induced myocardial injury. (A) Western blot analysis of autophagy markers, including Beclin-1, p62 and microtubule-associated light chain (LC)-3, in heart tissue. (B) The autophagy level was measured by live-cell imaging using Ad-GFP-LC3. Scale bar, 20 μm. n = 3 per group. The data are shown as the means ± SEM. *P < 0.05 vs. the PBS group. #P < 0.05 vs. the LPS group.
Autophagy inhibition abrogates narciclasine-mediated protection against LPS-induced myocardial injury
To study the effects of narciclasine-induced autophagy in AMI model, C57BL/6 male mice were pretreated with autophagy inhibitor 3-methyladenine (3-MA) before LPS administration. Echocardiography was performed to evaluate heart functions. Based on the ejection fraction and fractional shortening parameters, 3-MA did not influence heart function in normal hearts but abrogated the protective effect of narciclasine on LPS-induced myocardial injury (Figure 5A). In addition, 3-MA increased the mortality rate in narciclasine-pretreated LPS-induced mice (Figure 5C). Similar results were found in mice in which endogenous ATG5 was knocked down, which is a genetic strategy that interferes with autophagy (Figure 5B and 5D). Taken together, these results indicated that narciclasine has cardioprotective effects in LPS-induced AMI via autophagy.
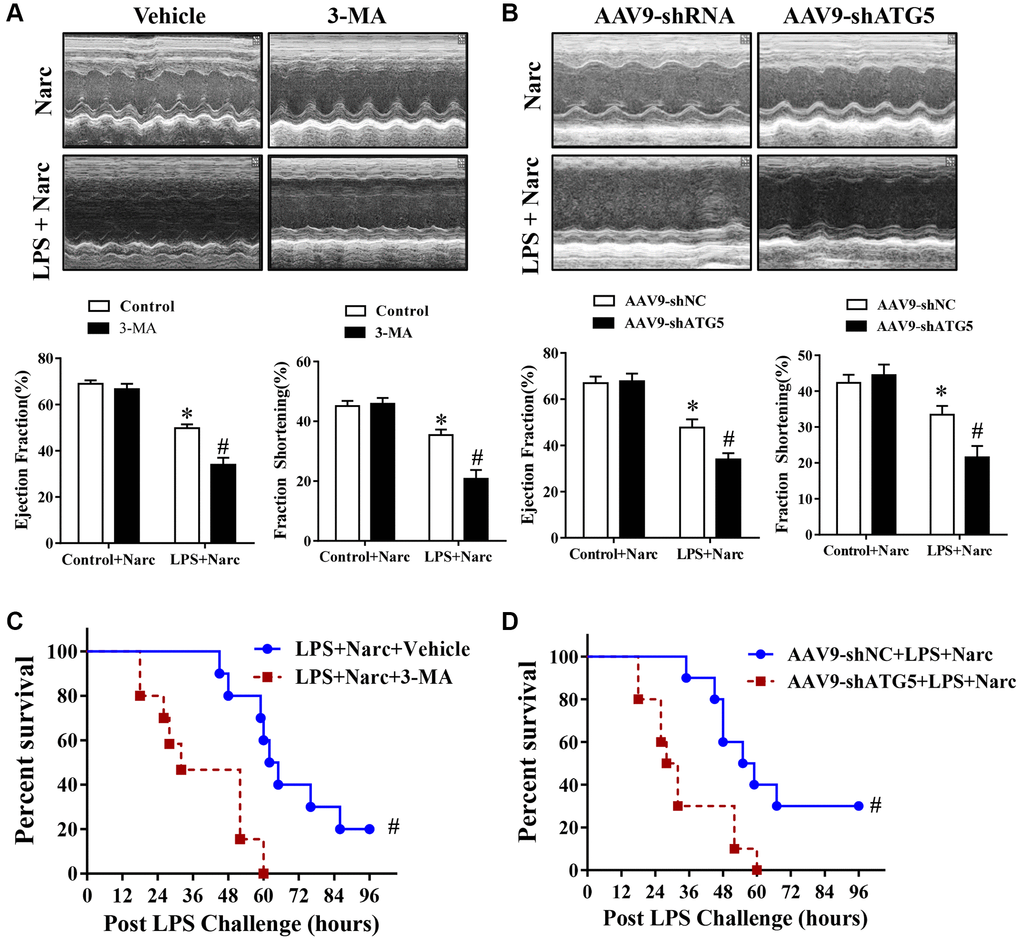
Figure 5. The autophagy inhibitor 3-MA and knockdown of ATG5 abrogates narciclasine-mediated protection against LPS-induced myocardial injury. (A–B) Echocardiographic analysis of the LVEF and LVFS in mouse hearts after LPS challenge for 12 h. (C–D) The survival rates of LPS-injected mice throughout the 96-h study period. n = 8 per group. The data are shown as the means ± SEM. *P < 0.05 vs. the Narc group. #P < 0.05 vs. the Narc+LPS group.
The JNK signaling pathway regulates narciclasine-mediated protection against sepsis-induced myocardial injury
To determine the role of JNK signaling pathway in narciclasine-induced autophagy, we measured the phosphorylation of JNK. Interestingly, LPS treatment significantly increased the phosphorylation of JNK while narciclasine treatment reduced the phosphorylation by LPS (Figure 6A). SP600125, an inhibitor of JNK activity, was treated on mice prior to LPS stimulation to further study the contribution of JNK signaling pathway in autophagy. We found that SP600125 treatment suppressed the phosphorylation of JNK and increased the autophagy-associated proteins expression (Figure 6B). Additionally, SP600125 pretreatment ameliorated heart function and improved the survival rates of LPS-induced AMI mice (Figure 6C–6D). The above findings indicated that narciclasine-induced autophagy was mediated by inhibiting the phosphorylation of JNK.
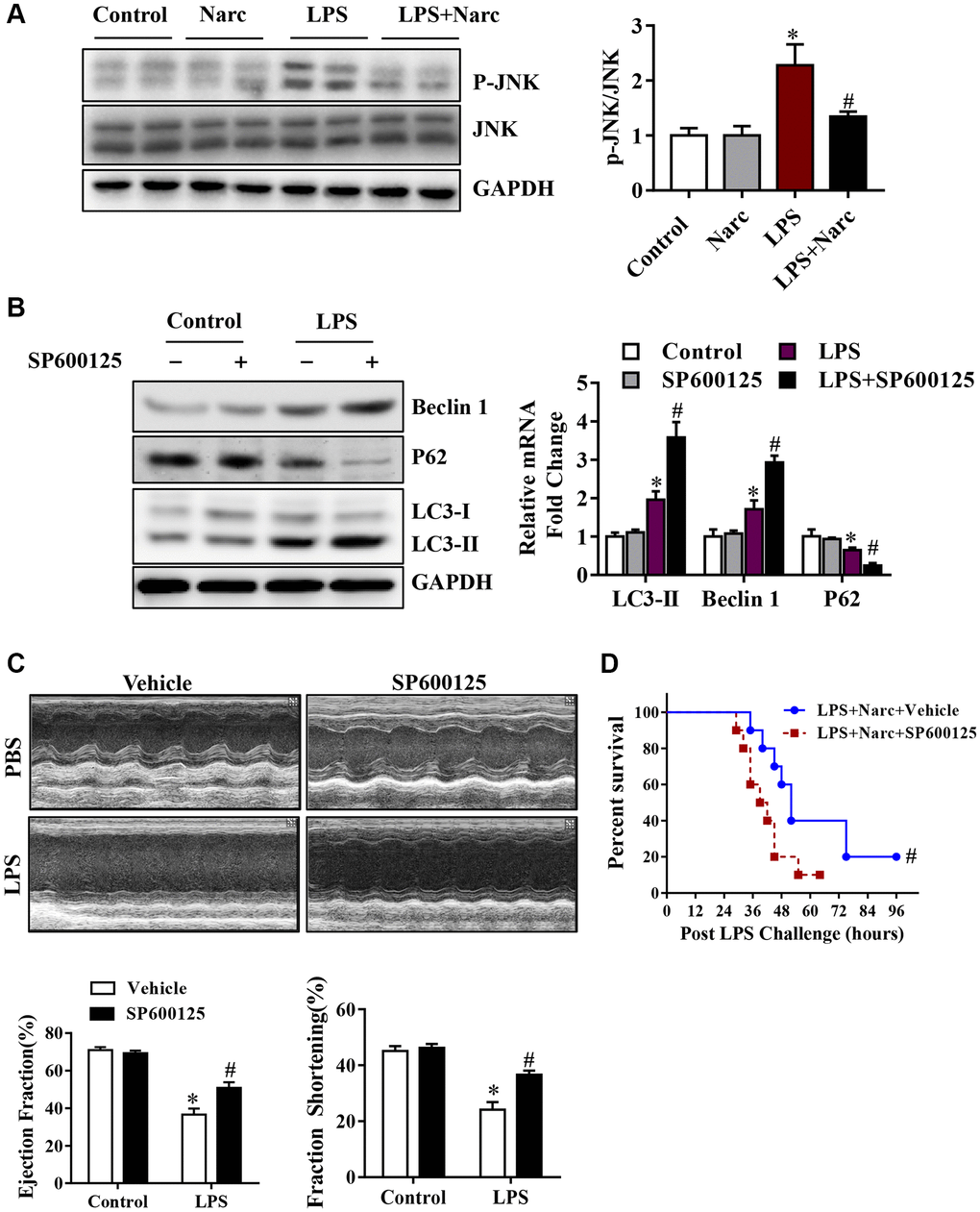
Figure 6. The JNK signaling pathway is responsible for narciclasine-mediated protection against sepsis-induced myocardial injury. (A) The expression levels of total JNK and phosphorylated JNK (p-JNK) were measured by western blotting. Mice were treated with SP600125, an inhibitor of JNK activity, prior to LPS stimulation to further investigate the role of JNK in autophagy. (B) The levels of autophagy-associated proteins, p-JNK and JNK were assessed by western blotting. (C) The LVEF and LVFS of mouse hearts after LPS challenge for 96 h were measured by echocardiography. (D) The survival rates of LPS-injected mice throughout the 96-h study period. n = 3–8 per group. The data are shown as the means ± SEM. *P < 0.05 vs. the PBS group. #P < 0.05 vs. the LPS group.
Narciclasine-induced autophagy is mediated by JNK signaling pathway in cardiomyocytes
To investigate the mechanism of narciclasine in AMI, we stimulated NRCMs with 1000 ng/ml LPS to simulate the AMI model in vitro. SP600125 is also used on NRCMs to inhibit JNK activity, thereby explain the regulatory role of JNK in autophagy. Narciclasine decreased JNK phosphorylation levels in response to LPS stress in NRCMs, which was similar to the effect in the in vivo LPS-induced AMI model (Figure 7A–7B). Besides, the phosphorylation of JNK was inhibited by SP600125, while LC3B-II/Iand Beclin-1 levels were increased and p62 levels were decreased in NRCMs (Figure 7A–7B). We also confirmed this result by live-cell imaging using a GFP-LC3 adenovirus and found that SP600125 increased the autophagy level compared to that in the LPS group. These results demonstrated that inhibition of JNK could enhance autophagic activity in cardiomyocytes.
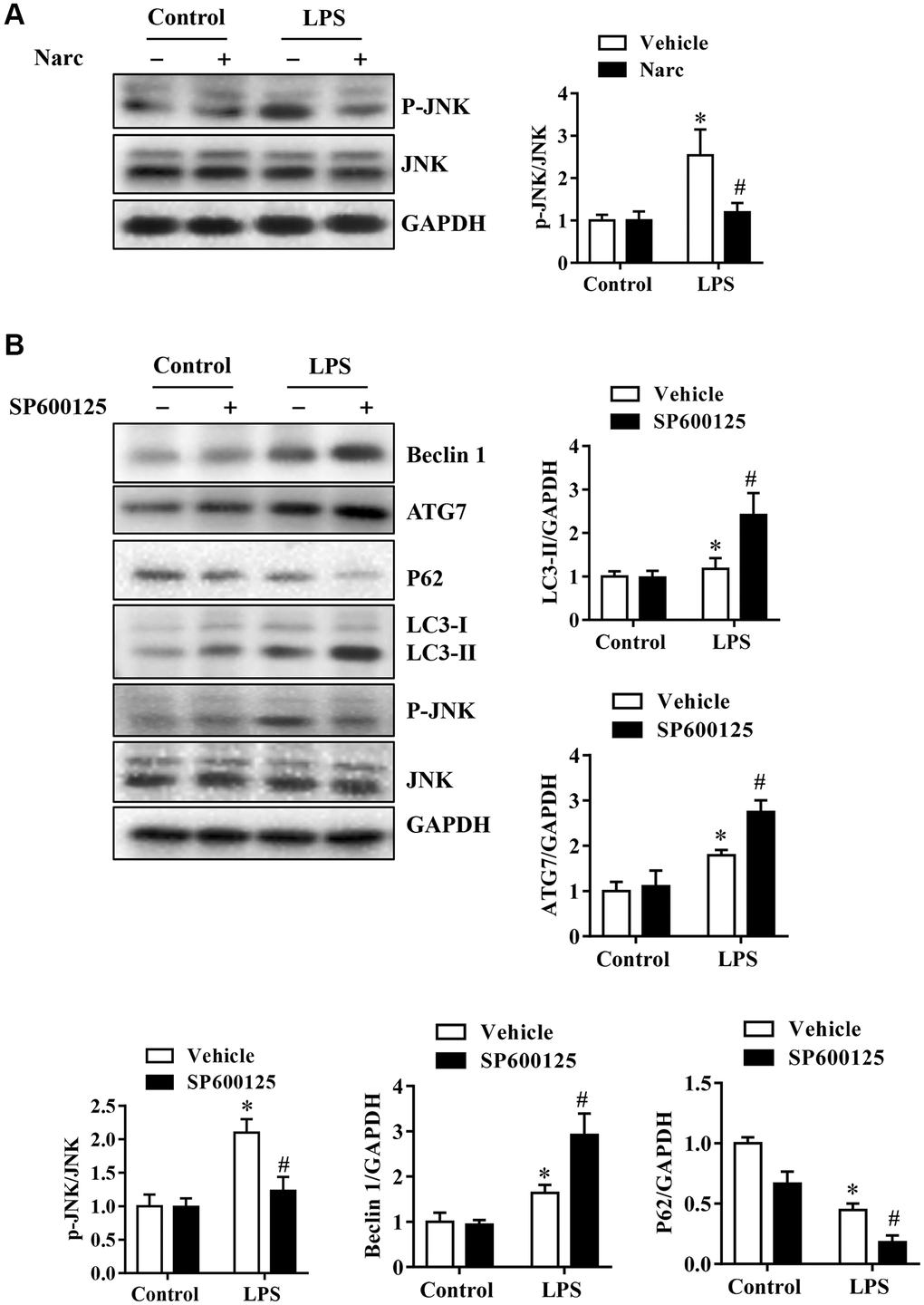
Figure 7. Narciclasine-induced autophagy is associated with JNK activity in cardiomyocytes. Neonatal rat cardiomyocytes were pretreated with narciclasine (300 nmol/L, 30 min) or SP600125 (SP, 20 μmol/L, 30 min) before LPS challenge. (A) JNK activity in the presence or absence of narciclasine was measured by western blotting. (B) The levels of autophagy-associated proteins, p-JNK, and JNK were examined by western blotting. n = 3 per group. The data are shown as the means ± SEM. *P < 0.05 vs. the PBS group. #P < 0.05 vs. the LPS group.
Discussion
Narciclasine is an isocyanide alkaloid that is present in the bulbs of coneflower extract and has been shown to have estrogen-killing properties. Narciclasine has also been shown to improve the prognosis of neonatal rats with sepsis by reducing inflammation [25–27]. In the present study, we evaluated the role and the underlying mechanism of narciclasine on the pathogenesis of AMI. We demonstrated that narciclasine attenuated LPS-induced cardiomyocyte inflammation, protected heart function and suppressed inflammation in LPS-induced AMI. Mechanistically, narciclasine protected against LPS-induced AMI by inducing JNK-dependent autophagy.
The heart is an important organ that is frequently affected by sepsis [32–34]. Sepsis-induced systemic inflammatory syndrome is a result of the combined actions of PAMPs (pathogen-associated molecular patterns) and DAMPs, which initiate the overwhelming production of inflammatory cytokines [35, 36]. LPS triggers inflammatory cytokine secretion, resulting in the release of DAMPs from activated, injured, or necrotic cells [37, 38]. The LPS-induced AMI model was treated with or without narciclasine, and we measured the expression of inflammatory cytokines and found that narciclasine inhibited neutrophil infiltration and decreased the expression of IL-6, IL-1β, and TNF-α. Our results indicated that narciclasine attenuates cardiac inflammation in LPS-induced AMI.
Multiple researches indicate that sepsis initiates autophagy in multiple organs, including the heart. Autophagy is critical for cell survival and death, homeostasis, and development [39, 40]. Evidence from CLP and LPS-induced sepsis models suggests that inducing autophagy pharmacologically protects the heart, providing evidence that autophagy is a cellular adaptive response [9, 41]. In line with these observations, we showed that narciclasine can increase Beclin-1 and LC3B-II level, decrease p62 level, and promote autophagic activity in LPS-induced AMI mice. In order to determine the protective role of narciclasine-induced autophagy, 3-MA was used to inhibit narciclasine-induced autophagy. We found that 3-MA abolished the beneficial effect of narciclasine on LPS-induced AMI in mice by blocking autophagy. These data suggested that narciclasine confers its cardioprotective effects via induction of autophagy.
JNK signaling pathway is important for the regulation of autophagy [42]. It was reported that JNK phosphorylation was increased in sepsis-induced acute lung injury [43, 44]. Our study also showed that in the LPS-induced AMI mouse model, p-JNK levels in the heart increased after LPS stimulation, while narcissus reduced pJNK levels and upregulated autophagy. More importantly, the inhibitory effect of SP600125 on JNK also triggered autophagy and reduced heart damage. Similar findings were observed in vitro. The inhibitory effect of SP600125 on JNK further enhanced the effect of autophagy in NRCMs on LPS attack. Although these results indicate that narciclasine-induced autophagy on AMI is mediated by the inhibition of JNK signaling pathway, the mechanism by which narciclasine inhibits JNK still needs further study.
In summary, we demonstrated that narciclasine could alleviate LPS-induced AMI by increasing autophagy, and uncovered the mechanism by which autophagy protects against AMI by the inhibition of JNK signaling pathway. These data suggest that narciclasine acts as a promising agent to protect the heart against sepsis injury, and autophagy may also be a therapeutic target for AMI treatment.
Materials and Methods
Animals
Adult male C57BL/6 mice were used in the experiments. All procedures involving animals were performed in accordance with the Guidelines of the National Institutes of Health for Animal Care and Use and were approved by the Institutional Animal Care and Use Committee and Ethics Committee of Harbin Medical University. The saline injected mice or the LPS-treated mice (a single 6 mg/kg injection of LPS) were pretreated with or without narciclasine (0.1 mg/kg body weight) administered by gavage for 7 days. Control mice were given equal volumes of saline by gavage. The mice were killed, and the hearts were collected after 6 hours.
Echocardiography
Transthoracic echocardiography (VEVO 2100, Visual Sonics) with a 25-MHz imaging transducer was performed on anesthetized animals as previously described [18]. Left ventricular (LV) ejection fraction (LVEF), LV fractional shortening (LVFS), interventricular septal end diastole and end systole (IVSd, IVSs), LV systolic dimension (LVDs), LV posterior wall diastolic dimensions (LVPWd) and LV posterior wall systolic dimensions (LVPWs) were measured.
Isolation and culture of NRCMs
The isolation and culture of NRCMs were performed as described previously [7]. Briefly, cardiomyocytes were collected from the freshly dissected ventricles of 1–3-day-old Sprague Dawley rats were plated on gelatin-coated dishes in Dulbecco’s modified Eagle’s medium, 20% M199, 15% FBS and 1% penicillin/streptomycin (P/S). The cells were transferred to low-serum maintenance media containing DMEM, 18.5% M199, 5% horse serum, 1% P/S after 24 hours and maintained at 37°C and 5% CO2 in a humidified environment.
Real-time polymerase chain reaction
The real-time PCR was performed as described previously [20]. The following primer sequences were used: TNF-α, forward: CATCTTCTCAAAATTCGAGTGACAA, reverse: TGGGAGTAGACAAGGTACAACCC; IL-1β, forward: GTGGCTGTGGAGAAGCTGTG, reverse: GAAGGTCCACGGGAAAGACAC; IL-6, forward: GAGGATACCACTCCCAACAGACC, reverse: AAGTGCATCATCGTTGTTCATACA; and GAPDH, forward: TGACCTCAACTACATGGTCTACA, reverse: CTTCCCATTCTCGGCCTTG.
Western blotting
The heart tissues and neonatal rat cardiomyocytes proteins were extracted with RIPA lysis buffer (containing protease and phosphatase inhibitors, Thermo Scientific), after which the lysate was centrifuged at 12,000 rpm, 30 min at 4°C. The protein supernatant was then collected and analyzed by standard immunoblotting. Protein samples were prepared for PAGE after the concentrations were measured by a BCA assay (Thermo Scientific). The proteins were then transferred to a PVDF membrane (0.45 μm, BioRad) and probed with the indicated antibodies. The primary antibodies used were Beclin 1 (3738S, Cell Signaling Technology), P62 (23214, Cell Signaling Technology), LC3 (ab192890, Abcam), JNK (ab199380, Abcam), p-JNK (ab215208, Abcam) and GAPDH (2118, Cell Signaling Technology).
Enzyme-linked immunosorbent assay (ELISA) and myeloperoxidase (MPO) activity measurement
TNF-α, IL-1β, and IL-6 levels in cardiomyocyte lysates were quantitatively measured by commercial ELISA kits according to the manufacturer's instructions (ABclonal, Wuhan, China; Dakewe, Shenzhen, China). MPO activity in heart lysates was measured according to the manufacturer's procedure (Nanjing Jiancheng Corp., Nanjing, China).
Quantification of apoptosis by fluorescence-activated cell sorting (FACS)
The high affinity of Annexin-V (AV) for phosphatidylserine (PI) (Beyotime, China), which is exposed on the surface of apoptotic cells, was used to examine apoptosis as described previously [27]. The data are reported as the percentages of early apoptotic cells (FITC+/PI-) and late apoptotic cells (FITC+/PI+).
AAV9 vector generation and transfection
We prepared AAV9-shNC and AAV9-shATG5 vectors as described [45]. We injected 4- to 5-week-old male C57 mice with 1 × 1012 vg of AAV9-shNC and AAV9-shATG5 intravenously via the tail vein as described previously [46].
Statistical analysis
Results are expressed as the mean ± standard error of the mean (SEM). GraphPad Prism 8.0 (GraphPad Software Inc., CA, USA) is used to determine the statistical significance among multiple groups by one-way ANOVA with the Bonferroni post hoc test. A 2-tailed p-value of less than 0.05 was considered statistically significant.
Author Contributions
Pingping Qi and Junbo Zheng designed the study, designed and performed experiments, analyzed and interpreted data, and wrote the manuscript. Rong Tang performed experiments, analyzed data, and edited the manuscript; Liu Jia and Yunlong Li assisted with study design and data analysis. All authors read and approved the manuscript.
Editorial Note
This corresponding author has a verified history of publications using a personal email address for correspondence
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Funding
This work was supported by Nn10 program and Distinguished Young Scholars Fund of Harbin Medical University Cancer Hospital.
Editorial Note
This corresponding author has a verified history of publications using a personal email address for correspondence
References
- 1. Azevedo LC. The many facets of sepsis pathophysiology and treatment. Shock. 2013 (Suppl 1); 39:1–2. https://doi.org/10.1097/SHK.0b013e31828fad4a [PubMed]
- 2. Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003; 348:138–50. https://doi.org/10.1056/NEJMra021333 [PubMed]
- 3. Nedeva C, Menassa J, Puthalakath H. Sepsis: Inflammation Is a Necessary Evil. Front Cell Dev Biol. 2019; 7:108. https://doi.org/10.3389/fcell.2019.00108 [PubMed]
- 4. Trinder M, Boyd JH, Brunham LR. Molecular regulation of plasma lipid levels during systemic inflammation and sepsis. Curr Opin Lipidol. 2019; 30:108–16. https://doi.org/10.1097/MOL.0000000000000577 [PubMed]
- 5. Zhao H, Chen H, Xiaoyin M, Yang G, Hu Y, Xie K, Yu Y. Autophagy Activation Improves Lung Injury and Inflammation in Sepsis. Inflammation. 2019; 42:426–39. https://doi.org/10.1007/s10753-018-00952-5 [PubMed]
- 6. Liu Y, Yang W, Sun X, Xie L, Yang Y, Sang M, Jiao R. SS31 Ameliorates Sepsis-Induced Heart Injury by Inhibiting Oxidative Stress and Inflammation. Inflammation. 2019; 42:2170–80. https://doi.org/10.1007/s10753-019-01081-3 [PubMed]
- 7. Lohner R, Schwederski M, Narath C, Klein J, Duerr GD, Torno A, Knuefermann P, Hoeft A, Baumgarten G, Meyer R, Boehm O. Toll-like receptor 9 promotes cardiac inflammation and heart failure during polymicrobial sepsis. Mediators Inflamm. 2013; 2013:261049. https://doi.org/10.1155/2013/261049 [PubMed]
- 8. Wang F, Jin Z, Shen K, Weng T, Chen Z, Feng J, Zhang Z, Liu J, Zhang X, Chu M. Butyrate pretreatment attenuates heart depression in a mice model of endotoxin-induced sepsis via anti-inflammation and anti-oxidation. Am J Emerg Med. 2017; 35:402–09. https://doi.org/10.1016/j.ajem.2016.11.022 [PubMed]
- 9. Sun Y, Yao X, Zhang QJ, Zhu M, Liu ZP, Ci B, Xie Y, Carlson D, Rothermel BA, Sun Y, Levine B, Hill JA, Wolf SE, et al. Beclin-1-Dependent Autophagy Protects the Heart During Sepsis. Circulation. 2018; 138:2247–62. https://doi.org/10.1161/CIRCULATIONAHA.117.032821 [PubMed]
- 10. Ceylan-Isik AF, Zhao P, Zhang B, Xiao X, Su G, Ren J. Erratum to "Cardiac overexpression of metallothionein rescues cardiac contractile dysfunction and endoplasmic reticulum stress but not autophagy in sepsis"[J. Mol. Cell Cardiol. 48 (2010) 367-378]. J Mol Cell Cardiol. 2016; 97:306–07. https://doi.org/10.1016/j.yjmcc.2016.06.063 [PubMed]
- 11. Zhao P, Gao J, Jiang J, Peng X, Wu W, Zheng L, Yao L. [Myocardial cells and mitochondrial autophagy in sepsis mice induced by lipopolysaccharide]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2016; 32:177–81. [PubMed]
- 12. Hu Z, Su H, Zeng Y, Lin C, Guo Z, Zhong F, Jiang K, Yuan G, He S. Tetramethylpyrazine ameliorates hepatic fibrosis through autophagy-mediated inflammation. Biochem Cell Biol. 2020; 98:327–37. https://doi.org/10.1139/bcb-2019-0059 [PubMed]
- 13. Liu J, Wang YG, Yu SY, Li CE, Kang SM. Protective effect of acacetin in human periodontal ligament cells via regulation of autophagy and inflammation. Pharmazie. 2020; 75:436–39. [PubMed]
- 14. Painter JD, Galle-Treger L, Akbari O. Role of Autophagy in Lung Inflammation. Front Immunol. 2020; 11:1337. https://doi.org/10.3389/fimmu.2020.01337 [PubMed]
- 15. Zhu Q, Wang R, Nemecio D, Liang C. How autophagy is tied to inflammation and cancer. Mol Cell Oncol. 2020; 7:1717908. https://doi.org/10.1080/23723556.2020.1717908 [PubMed]
- 16. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013; 13:722–37. https://doi.org/10.1038/nri3532 [PubMed]
- 17. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011; 469:323–35. https://doi.org/10.1038/nature09782 [PubMed]
- 18. Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007; 117:1782–93. https://doi.org/10.1172/JCI27523 [PubMed]
- 19. Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012; 485:251–55. https://doi.org/10.1038/nature10992 [PubMed]
- 20. Mukhopadhyay P, Eid N, Abdelmegeed MA, Sen A. Interplay of Oxidative Stress, Inflammation, and Autophagy: Their Role in Tissue Injury of the Heart, Liver, and Kidney. Oxid Med Cell Longev. 2018; 2018:2090813. https://doi.org/10.1155/2018/2090813 [PubMed]
- 21. Koehn FE, Carter GT. The evolving role of natural products in drug discovery. Nat Rev Drug Discov. 2005; 4:206–20. https://doi.org/10.1038/nrd1657 [PubMed]
- 22. Mishra BB, Tiwari VK. Natural products: an evolving role in future drug discovery. Eur J Med Chem. 2011; 46:4769–807. https://doi.org/10.1016/j.ejmech.2011.07.057 [PubMed]
- 23. Ingrassia L, Lefranc F, Mathieu V, Darro F, Kiss R. Amaryllidaceae isocarbostyril alkaloids and their derivatives as promising antitumor agents. Transl Oncol. 2008; 1:1–13. https://doi.org/10.1593/tlo.08100 [PubMed]
- 24. van Wyk BE. A review of Khoi-San and Cape Dutch medical ethnobotany. J Ethnopharmacol. 2008; 119:331–41. https://doi.org/10.1016/j.jep.2008.07.021 [PubMed]
- 25. Fuchs S, Hsieh LT, Saarberg W, Erdelmeier CA, Wichelhaus TA, Schaefer L, Koch E, Fürst R. Haemanthus coccineus extract and its main bioactive component narciclasine display profound anti-inflammatory activities in vitro and in vivo. J Cell Mol Med. 2015; 19:1021–32. https://doi.org/10.1111/jcmm.12493 [PubMed]
- 26. Mikami M, Kitahara M, Kitano M, Ariki Y, Mimaki Y, Sashida Y, Yamazaki M, Yui S. Suppressive activity of lycoricidinol (narciclasine) against cytotoxicity of neutrophil-derived calprotectin, and its suppressive effect on rat adjuvant arthritis model. Biol Pharm Bull. 1999; 22:674–78. https://doi.org/10.1248/bpb.22.674 [PubMed]
- 27. Kingsley MK, Bhat BV, Badhe BA, Dhas BB, Parija SC. Narciclasine improves outcome in sepsis among neonatal rats via inhibition of calprotectin and alleviating inflammatory responses. Sci Rep. 2020; 10:2947. https://doi.org/10.1038/s41598-020-59716-7 [PubMed]
- 28. Shi N, Chen F, Zhang X, Clinton SK, Tang X, Sun Z, Chen T. Suppression of Oxidative Stress and NFκB/MAPK Signaling by Lyophilized Black Raspberries for Esophageal Cancer Prevention in Rats. Nutrients. 2017; 9:413. https://doi.org/10.3390/nu9040413 [PubMed]
- 29. Senoner T, Dichtl W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients. 2019; 11:2090. https://doi.org/10.3390/nu11092090 [PubMed]
- 30. Tan Y, Ouyang H, Xiao X, Zhong J, Dong M. Irisin ameliorates septic cardiomyopathy via inhibiting DRP1-related mitochondrial fission and normalizing the JNK-LATS2 signaling pathway. Cell Stress Chaperones. 2019; 24:595–608. https://doi.org/10.1007/s12192-019-00992-2 [PubMed]
- 31. Joseph LC, Kokkinaki D, Valenti MC, Kim GJ, Barca E, Tomar D, Hoffman NE, Subramanyam P, Colecraft HM, Hirano M, Ratner AJ, Madesh M, Drosatos K, Morrow JP. Inhibition of NADPH oxidase 2 (NOX2) prevents sepsis-induced cardiomyopathy by improving calcium handling and mitochondrial function. JCI Insight. 2017; 2:e94248. https://doi.org/10.1172/jci.insight.94248 [PubMed]
- 32. Kumar A, Haery C, Parrillo JE. Myocardial dysfunction in septic shock. Crit Care Clin. 2000; 16:251–87. https://doi.org/10.1016/s0749-0704(05)70110-x [PubMed]
- 33. Bouhemad B, Nicolas-Robin A, Arbelot C, Arthaud M, Féger F, Rouby JJ. Acute left ventricular dilatation and shock-induced myocardial dysfunction. Crit Care Med. 2009; 37:441–47. https://doi.org/10.1097/CCM.0b013e318194ac44 [PubMed]
- 34. Gibot S, Lévy B, Nevière R, Cariou A, Lesur O. Dysfonctionnement myocardique et choc septique [Myocardial dysfunction and septic shock]. Med Sci (Paris). 2004; 20:1115–18. https://doi.org/10.1051/medsci/200420121115 [PubMed]
- 35. Kang JW, Kim SJ, Cho HI, Lee SM. DAMPs activating innate immune responses in sepsis. Ageing Res Rev. 2015; 24:54–65. https://doi.org/10.1016/j.arr.2015.03.003 [PubMed]
- 36. Gentile LF, Moldawer LL. DAMPs, PAMPs, and the origins of SIRS in bacterial sepsis. Shock. 2013; 39:113–14. https://doi.org/10.1097/SHK.0b013e318277109c [PubMed]
- 37. Hung YL, Fang SH, Wang SC, Cheng WC, Liu PL, Su CC, Chen CS, Huang MY, Hua KF, Shen KH, Wang YT, Suzuki K, Li CY. Corylin protects LPS-induced sepsis and attenuates LPS-induced inflammatory response. Sci Rep. 2017; 7:46299. https://doi.org/10.1038/srep46299 [PubMed]
- 38. Jamal Uddin M, Joe Y, Kim SK, Oh Jeong S, Ryter SW, Pae HO, Chung HT. IRG1 induced by heme oxygenase-1/carbon monoxide inhibits LPS-mediated sepsis and pro-inflammatory cytokine production. Cell Mol Immunol. 2016; 13:170–79. https://doi.org/10.1038/cmi.2015.02 [PubMed]
- 39. Das G, Shravage BV, Baehrecke EH. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb Perspect Biol. 2012; 4:a008813. https://doi.org/10.1101/cshperspect.a008813 [PubMed]
- 40. Kourtis N, Tavernarakis N. Autophagy and cell death in model organisms. Cell Death Differ. 2009; 16:21–30. https://doi.org/10.1038/cdd.2008.120 [PubMed]
- 41. De Meyer GR, Martinet W. Autophagy in the cardiovascular system. Biochim Biophys Acta. 2009; 1793:1485–95. https://doi.org/10.1016/j.bbamcr.2008.12.011 [PubMed]
- 42. Chaanine AH, Jeong D, Liang L, Chemaly ER, Fish K, Gordon RE, Hajjar RJ. JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis. 2012; 3:265. https://doi.org/10.1038/cddis.2012.5 [PubMed]
- 43. Zheng Q, Wang YC, Liu QX, Dong XJ, Xie ZX, Liu XH, Gao W, Bai XJ, Li ZF. FK866 attenuates sepsis-induced acute lung injury through c-jun-N-terminal kinase (JNK)-dependent autophagy. Life Sci. 2020; 250:117551. https://doi.org/10.1016/j.lfs.2020.117551 [PubMed]
- 44. Lou L, Hu D, Chen S, Wang S, Xu Y, Huang Y, Shi Y, Zhang H. Protective role of JNK inhibitor SP600125 in sepsis-induced acute lung injury. Int J Clin Exp Pathol. 2019; 12:528–38. [PubMed]
- 45. Xie C, Zhang YP, Song L, Luo J, Qi W, Hu J, Lu D, Yang Z, Zhang J, Xiao J, Zhou B, Du JL, Jing N, et al. Genome editing with CRISPR/Cas9 in postnatal mice corrects PRKAG2 cardiac syndrome. Cell Res. 2016; 26:1099–111. https://doi.org/10.1038/cr.2016.101 [PubMed]
- 46. Somanathan S, Jacobs F, Wang Q, Hanlon AL, Wilson JM, Rader DJ. AAV vectors expressing LDLR gain-of-function variants demonstrate increased efficacy in mouse models of familial hypercholesterolemia. Circ Res. 2014; 115:591–99. https://doi.org/10.1161/CIRCRESAHA.115.304008 [PubMed]