



Introduction
There are approximately 15,000 sensory hair cells in the mouse inner ear cochlea [1]. The hair cells are anatomically and functionally divided into two distinct types, namely outer and inner hair cells [2]. All hair cells are located on a thin basilar membrane along the cochlea, and the perception of different sound frequencies is dependent on the localization of hair cells on this membrane [3]. Notably, hair cells in the basal region of the cochlea are remarkably sensitive to external stress inducers, including reactive oxygen species, ototoxic drugs, and excessive noise [4]. In particular, aging is highly associated with impairment of hair cells in the basal region of the cochlea. Age-related hearing loss (ARHL) is usually due to loss and/or loss of function of hair cells in this region, and these cells are responsible for the perception of high-frequency sound; therefore, changes in perception in the high-frequency range are the earliest indicator of ARHL [5, 6].
Aging is also a challenge for neuronal cells in the central nervous system; age accelerates the symptoms of age-related neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease, and Huntington's disease [7, 8]. AD is an epidemic brain disorder characterized by memory deficits, spatial disorientation, and other psychiatric problems in older people [9]. Amyloid precursor protein (APP) is thought to be one of the key agents in AD development [9]. The truncated peptide amyloid-β (Aβ) is produced from APP by sequential proteolysis by β-secretases (cysteine proteases and β-site APP-cleaving enzyme) and γ-secretase (a multimeric protein complex composed of presenilin, nicastrin, Aph-1 and Pen-2) [9, 10]. Aβ appears to produce neuronal toxicity through binding neuronal receptors, such as the prion-receptor, metabotropic glutamate receptors or N-methyl-d-aspartate receptors [11-13].
Many studies have shown that defects in sensory systems are highly correlated with age-related neurodegenerative diseases, including AD [14-18]. This raises the possibility that sensory neurons have some commonalities with neurons in the brain. In support of this hypothesis, it has been shown that hair cells in the inner ear cochleae have properties similar to those of neurons: they electronically signal incoming auditory stimuli, make afferent and efferent synaptic connections to the auditory nerves, and the connectivity between hair cells and auditory nerves is important for sound perception [19]. Hair cells contain ion channels, including calcium-activated SK2 channels coupled to α9α10 nicotinic acetylcholine receptors, whereas the pre-synaptic region of auditory neurons contains many types of glutamate receptors, including N-methyl-d-aspartate receptors [19-21]. Furthermore, it was recently reported that the liver kinase B1, which is involved in cellular events in several organs including the central nervous system and mediates sporadic AD progression, is required for the maintenance of stereo cilia in the inner ear hair cells [22, 23]. These reports raised an intriguing idea that the auditory system may provide a useful tool to examine the development and progression of AD.
Auditory system function can be monitored using electrophysiological methods, which provides an easy way to observe functional impairment of sensory cells over time [19]. To develop a new AD model that takes advantage of this aspect of the auditory system, we established transgenic (Tg) mouse lines in which Aβ derivatives are expressed in the cochlear hair cells. Electrophysiological assessment of the auditory brainstem response (ABR) revealed that these mice had impaired high-frequency sound perception at 4 months after birth. We also observed that there was a synergistic interaction between Aβ and tau in the auditory system. In addition, we found that there was loss of hair cells in the basal region of the cochlea. Our data indicate that Aβ expression in the auditory hair cells induces early-onset auditory defects, demonstrating that this system provides an easy and quick assessment of Aβ-dependent toxicity, making it an ideal platform with which to monitor the effect(s) of Aβ-induced neuronal dysfunction over time.
Results
Establishment of Tg mouse lines expressing Aβ42 or its derivative in hair cells of the cochlea
To express Aβ or its truncated or mutated forms in mouse cochlear hair cells, we established Tg mouse lines carrying an expression vector for Aβ, a truncated form of Aβ (Aβ42), or Aβ with a familial AD mutation (Aβ42Arc, E22G substitution) downstream of the minimal enhancer region of mouse Math1 (Math1E), which drives gene expression in many types of sensory neurons including cochlear hair cells [24] (Fig. 1A). These constructs were used for expression in mouse cochlear hair cells. The rat prepro-enkephalin signal sequence was fused upstream of the Aβ sequences to ensure their secretion [25]. Quantitative PCR (qPCR) showed that Aβ42 and Aβ42Arc mRNAs were well expressed in the cochleae of Tg mice compared with non-Tg control mice (Fig. 1B). Enzyme-linked immunosorbent assay (ELISA) revealed the presence of Tris-buffered saline–soluble and –insoluble (formic acid–soluble) forms of an Aβ protein in the cochleae of 4-month-old Tg(Math1E-Aβ42Arc)1Lt mice (Fig. 1C).
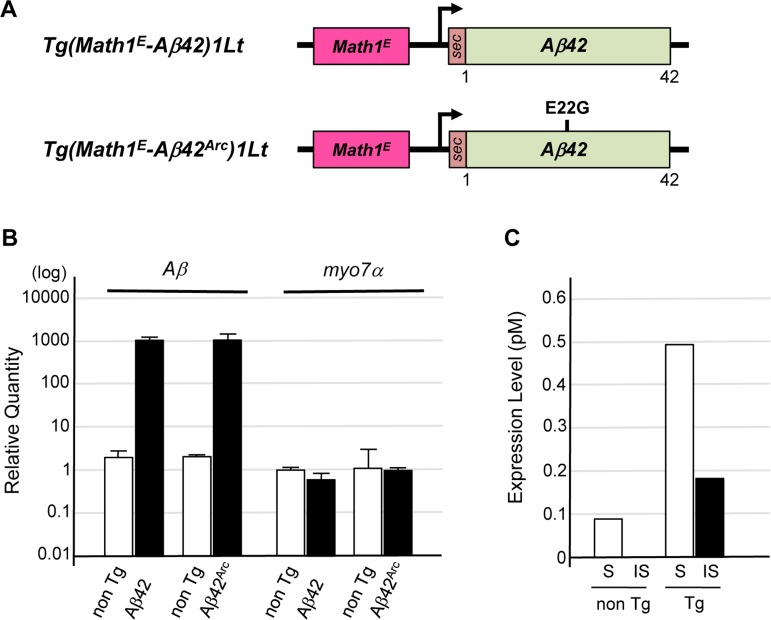
Figure 1. Establishment of Tg mice expressing Aβ in cochlear hair cells (A) Representative vector construction for the Tg mice used in this study. Tg(Math1E-Aβ42)1Lt contains Aβ42 fused to the rat prepro-enkephalin secretion signal (sec) under the control of the Math1 minimal enhancer (Math1E) and cytomegalovirus promoter. Tg(Math1E-Aβ42Arc)1Lt contains almost the same sequence as Tg(Math1E-Aβ42)1Lt except that the sec-Aβ42Arc sequence replaces sec-Aβ42. (B) Levels of Tg(Math1E-Aβ42)1Lt and Tg(Math1E-Aβ42Arc)1Lt mRNAs from cochleae were measured by qPCR and normalized to the control (b-actin) in non-Tg mice. (C) ELISA of cochleae extracts from Tg(Math1E-Aβ42Arc)1Lt and non-Tg mice at age 4 months. S; Tris-buffered saline–soluble form, IS; insoluble (formic acid–soluble) form.
Aβ42Arc is found around the hair cells of Tg mouse
We surveyed the cochlear hair cells of Tg(Math1E-Aβ42Arc)1Lt mice histochemically and found that the structures of hair cells of Tg-mice were not different from those of non-Tg siblings at age 4 months (Fig. 2A, B). Next, staining with anti-Aβ (6E10) revealed that Aβ proteins were distributed mainly around the cochlear hair cells of 4-month-old Tg(Math1E-Aβ42Arc)1Lt mice (Fig. 2C, D). Aggregated Aβ proteins were also found near these hair cells, although they were not as abundant (Fig. 2E, arrows). To identify the precise cellular location of Aβ in the Tg(Math1E-Aβ42Arc)1Lt mice, we performed immunoelectron microscopy. Aβ immuno-staining was observed in the endoplasmic reticulum, the stereo cilia, and the plasma membrane of the hair cells (Fig. 2F, G and H). Notably, Aβ was found as clusters comprised of 2–20 particles in the stereo cilia and plasma membrane (Fig. 2G, H, arrowheads). This is similar to previous results that Aβ tends to exist as oligomeric clusters in the central nervous system [26]. Interestingly, Aβ immunostaining was also observed within and on the surface of the surrounding cells, suggesting that Aβ-like protein might be secreted to the cells neighboring the hair cells (Fig. 2H; arrows). These finding are in agreement with previously observed properties of Aβ-related peptides in the central nervous system [25, 27, 28].
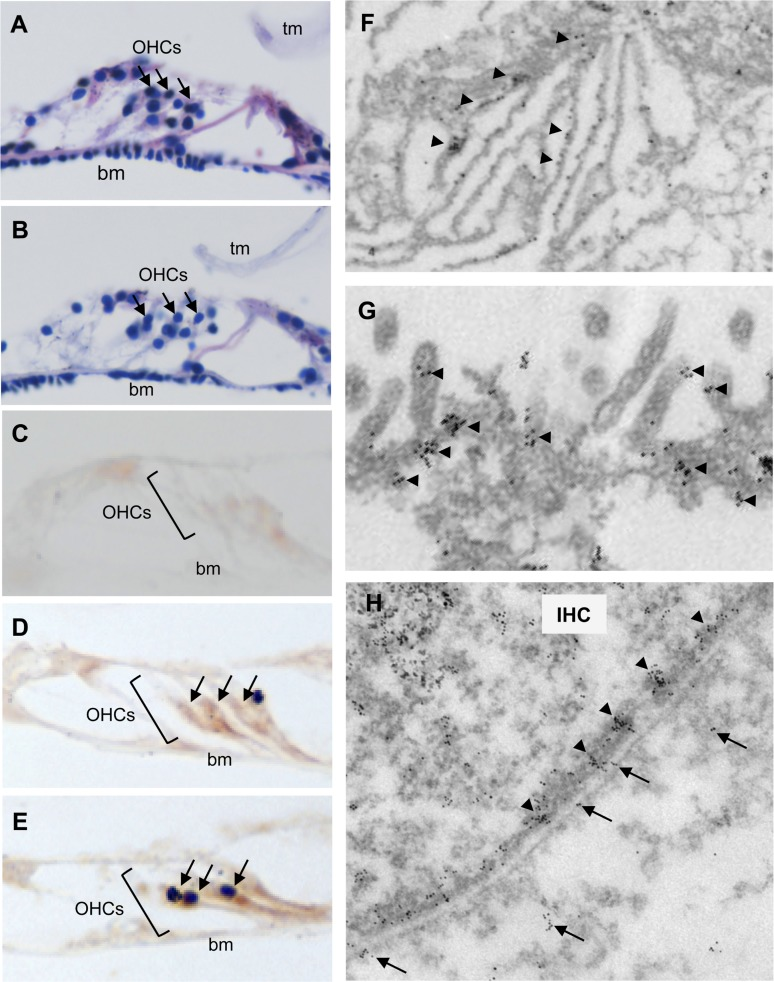
Figure 2. Aβ42Arc is expressed along the hair cells in the cochlea Hematoxylin and eosin staining of sectioned cochleae from 4-month-old non-Tg (A) and Tg(Math1E-Aβ42Arc)1Lt mice (B). OHC, outer hair cell (arrows); tm, tectorial membrane; bm, basilar membrane. (C-E) Immunostaining of cochlear sections at age 4 months using Aβ antibody (6E10). (C) Antibody 6E10 did not stain the cochlea of non-Tg mice. (D) The cochlear OHCs from Tg(Math1E-Aβ42Arc)1Lt were strongly stained by 6E10 (arrows). (E) Possible aggregation of an Aβ-like protein (arrows) in the cochleae of Tg(Math1E-Aβ42Arc)1Lt mice. (F-H) Immunoelectron microscopy of hair cells in Tg(Math1E-Aβ42Arc)1Lt cochlea at age 4 months. An Aβ-like protein accumulated in the endoplasmic reticulum (F), stereo cilia (G), and plasma membrane (H). Arrowheads: clusters of Aβ-like protein were observed around the stereo cilia (G) and plasma membrane (H). IHC: inner hair cell. Arrows: Aβ-like protein were also observed within the neighboring cells (H).
Expression of Aβ derivatives in cochlear hair cells impairs the perception of high-frequency sound
To assess the consequence of Aβ42 or Aβ42Arc expression in cochlear hair cells, we monitored hearing with ABRs. ABRs are electrical action potentials in the cochlea and brain induced by an auditory stimulus and can be detected via electrodes placed on the scalp [29]. We monitored threshold for the first peak of the ABR response (1–2 ms after sound stimulation) because the first peak is generated exclusively by the activity of sensory hair cells and auditory nerves [19]. We first examined the response to a click sound, which is not frequency tunable but reflects the activity in a broad region of the cochlear hair cells, and we compared Tg mice and non-Tg mice that were wild-type littermates of the Tg mice. We found that the sound threshold for the non-Tg, Tg(Math1E-Aβ42)1Lt, and Tg(Math1E-Aβ42Arc)1Lt mice were not significantly different from age 2 to at least 8 months, suggesting that sound perception itself was not affected by the expression of Aβ in the hair cells (Fig. 3A, D). Next, we monitored the response to a high-frequency tone (32 kHz) and found that the thresholds for each of the Tg(Math1E-Aβ42)1Lt and Tg(Math1E-Aβ42Arc)1Lt mice was slightly increased from age 3 to 5 months (Fig. 3B, E, arrows), whereas the mid-range frequency (8 kHz) threshold of the Tg(Math1E-Aβ42)1Lt and Tg(Math1E-Aβ42Arc)1Lt mice was comparable to that of the non-Tg siblings during this period (Fig. 3C, F).
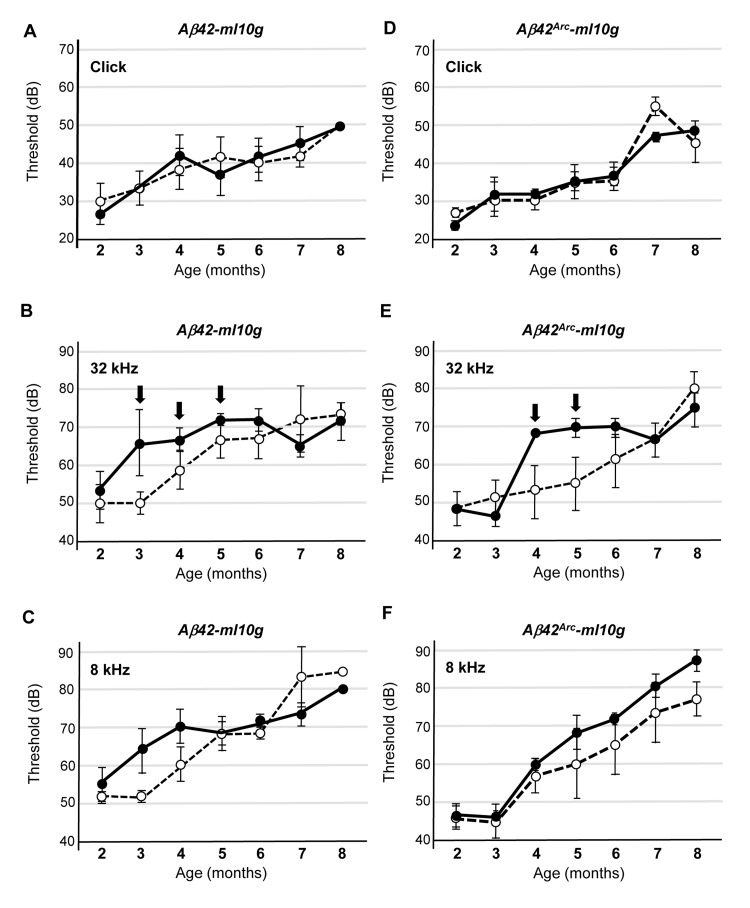
Figure 3. Tg(Math1E-Aβ42)1Lt and Tg(Math1E-Aβ42Arc)1Lt mice show slight hearing defects in high-frequency sound perception (A-C) ABR thresholds of Tg(Math1E-Aβ42-ml10g)1Lt (filled circles) and non-Tg (open circles) mice from age 2 to 8 months for the click sound (A), the 32 kHz tone (B), and the 8 kHz tone (C). ABR thresholds were slightly increased from age 3 to 5 months for the 32 kHz tone (B) (arrows). Non-Tg: n = 8 (5 male, 3 female), Tg: n = 10 (6 male, 4 female). (D-F) ABR thresholds of Tg(Math1E-Aβ42Arc-ml10g)1Lt (filled circles) and non-Tg (open circles) mice from age 2 to 8 months for the click sound (D), the 32 kHz tone (E), and the 8 kHz tone (F). ABR thresholds were slightly increased from age 4 to 5 months for the 32 kHz tone (E) (arrows). Non-Tg: n = 6 (5 male, 1 female), Tg: n = 10 (5 male, 5 female).
Although there were apparent differences in the ability of Tg mice to hear high-frequency sound, the ABR threshold increased with age for all mouse strains, and after the mice were 6 months old, we could no longer detect a difference between non-Tg and Tg mice.
The confluence of threshold might be a consequence of the genetic background of the Tg mice, which is C57BL/6 (B6). B6 mice have a point mutation in Cadherin 23, which induces an age-related hearing defect that leads to an increased dB threshold after age 6 months (Supplemental Fig. S1) [30, 31]. It has been shown that B6 and 129/SvJ (129) heterozygotes do not suffer from this age-related hearing defect [32]. Thus, we crossed Tg(Math1E-Aβ42Arc)1Lt mice with the 129 line and measured ABR of the heterozygotes (Tg(Math1E-Aβ42Arc)1Lt/129). We first compared the response to high-frequency (32 kHz) tone sound stimulation at 2 and 4 months of age and found that hearing ability was improved in the B6/129 background; the heterozygotes responded to a 40 dB sound stimulation, whereas the B6 mice no longer responded to this stimulation at age 4 months (Fig. 4A, B, compare to Fig. 3). We found that both non-Tg (B6/129) and Tg(Math1E-Aβ42Arc)1Lt/129 mice showed a similar response at age 2 months, whereas the first peak could no longer be detected in Tg(Math1E-Aβ42Arc)1Lt/129 mice at 4 months (Fig. 4A, B, arrow).
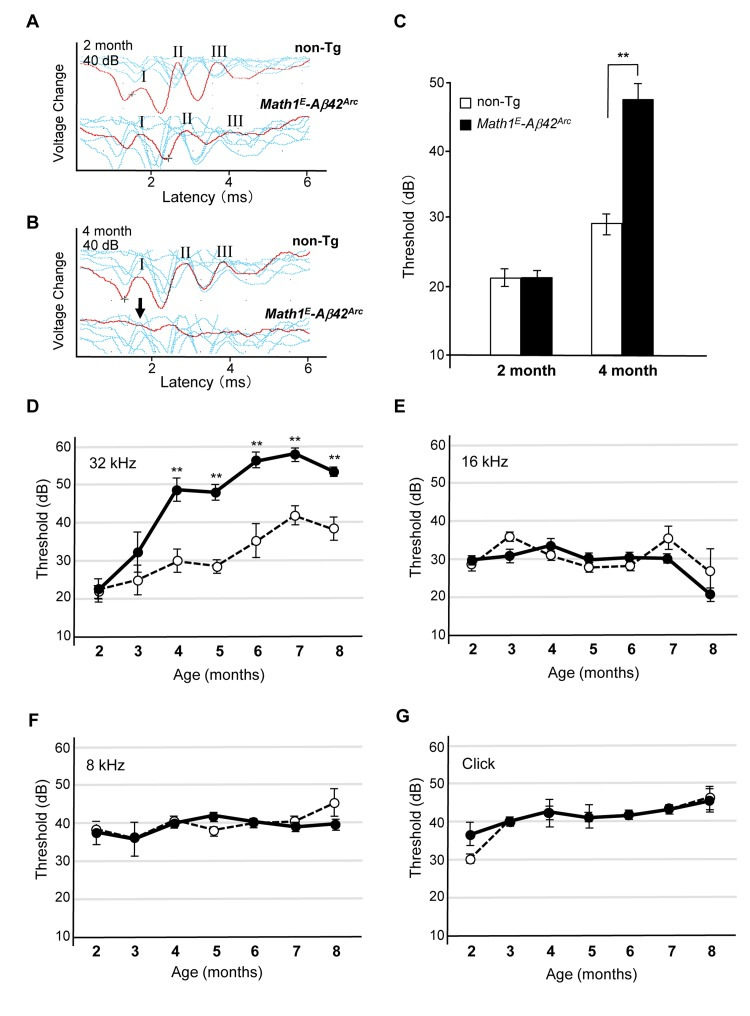
Figure 4. Tg(Math1E-Aβ42Arc)1Lt/129 mice showed dramatic hearing impairment for high-frequency sound over time (A) Representative ABR data (Red line) of non-Tg (B6/129) and Math1E-Aβ42Arc (Tg(Math1E-Aβ42Arc)1Lt/129) mice in age 2 month for the 32 kHz at 40 dB. There were at least three peaks (I–III) in both genotypes (see METHODS). (B) Representative ABR data (Red line) of non-Tg (B6/129) and Math1E-Aβ42Arc (Tg(Math1E-Aβ42Arc)1Lt/129) mice in age 4 month for the 32 kHz at 40 dB. No first peak was observed in Math1E-Aβ42Arc mice (arrow). (C) ABR threshold of non-Tg (B6/129) (white bar) and Math1E-Aβ42Arc (Tg(Math1E-Aβ42Arc)1Lt/129) (black bar) mice in age 2 and 4 months for the 32 kHz at 40dB. (D-G) ABR threshold of Tg(Math1E-Aβ42Arc)1Lt/129 and non-Tg (B6/129) mice from age 2 to 8 months for the 32 kHz tone (D), 16 kHz tone (E), and 8 kHz (F), and click sound (G). Tg(Math1E-Aβ42Arc)1Lt/129 mice (filled circles) showed increasing ABR threshold compared with non-Tg (open circles) (D). No increase in ABR threshold was observed for the 16 kHz tone (E), 8 kHz tone (F), or click sound (G). Data represent the mean ± SEM. non-Tg: n = 4 (3 male, 1 female); Tg: n = 4 (2 male, 2 female). **P < 0.01.
We then determined threshold levels at age 2 and 4 months and found that the Tg(Math1E-Aβ42Arc)1Lt/129 mice showed a greatly increased threshold relative to the non-Tg (B6/12 9) mice at age 4 months (Fig. 4C).
Next, we monitored changes in threshold level from age 2 to 8 months in Tg(Math1E-Aβ42Arc)1Lt/129 mice and confirmed that the mice showed an increasing threshold for high-frequency (32 kHz) sound stimulation from age 4 to at least 8 months (Fig. 4D). The hearing impairment of Tg(Math1E-Aβ42Arc)1Lt/129 mice seemed to be exclusively in high-frequency sound perception; the threshold of the Tg(Math1E-Aβ42Arc)1Lt/129 mice did not change for perception of lower-frequency sound (16 and 8 kHz) or click sound (Fig. 4E–G). These results suggeste d that Aβ42Arc expression in the hair cells exclusively affected the ability of the mice to respond to high-frequency sound (>32 kHz). To confirm that the defect in Tg(Math1E-Aβ42Arc)1Lt/129 mice was exclusively in high-frequency sound perception, we analyzed the threshold response to a 40 kHz tone at age 8 months and found that the threshold was still significantly higher for Tg mice (Supplemental Fig. S2).
Expression of Aβ derivatives in the hair cells causes loss of hair cells in the basal region of cochlea
High-frequency sound is perceived by the hair cells in the basal region of the cochlea [19]. Previous studies have suggested that the hair cells in the basal region of cochleae are lost during aging [4]. To elucidate the mechanism behind the defect in the ability to perceive high-frequency sound at age 8 months, we stained surface preparations of the organ of Corti derived from 8-month-old non-Tg and Tg(Math1E-Aβ42Arc)1Lt/129 mice with Alexa Fluor 488–phalloidin and DAPI and visualized the hair cells (Fig. 5A). Non-Tg epithelia had one row of inner hair cells and three rows of outer hair cells at the basal, mid, and apical levels of the cochlear duct [1, 2]. We compared the hair cells in each region of the cochlea and found that the number of hair cells appeared to be comparable between non-Tg and Tg mice in the apical and middle regions of cochlea (the region responsible for perceiving frequencies <20 kHz) (Fig. 5A–C, F). However, the number of hair cells in the basal region of the cochlea (region responsible for perceiving frequencies >20 kHz) was dramatically reduced in the Tg mice (Fig. 5D–F). These results suggested that expression of Aβ42Arc specifically affects the survival of hair cells in the basal region of cochlea. This is correlated with the fact that neuronal cell death in the central nervous system is typical of AD pathology [33].
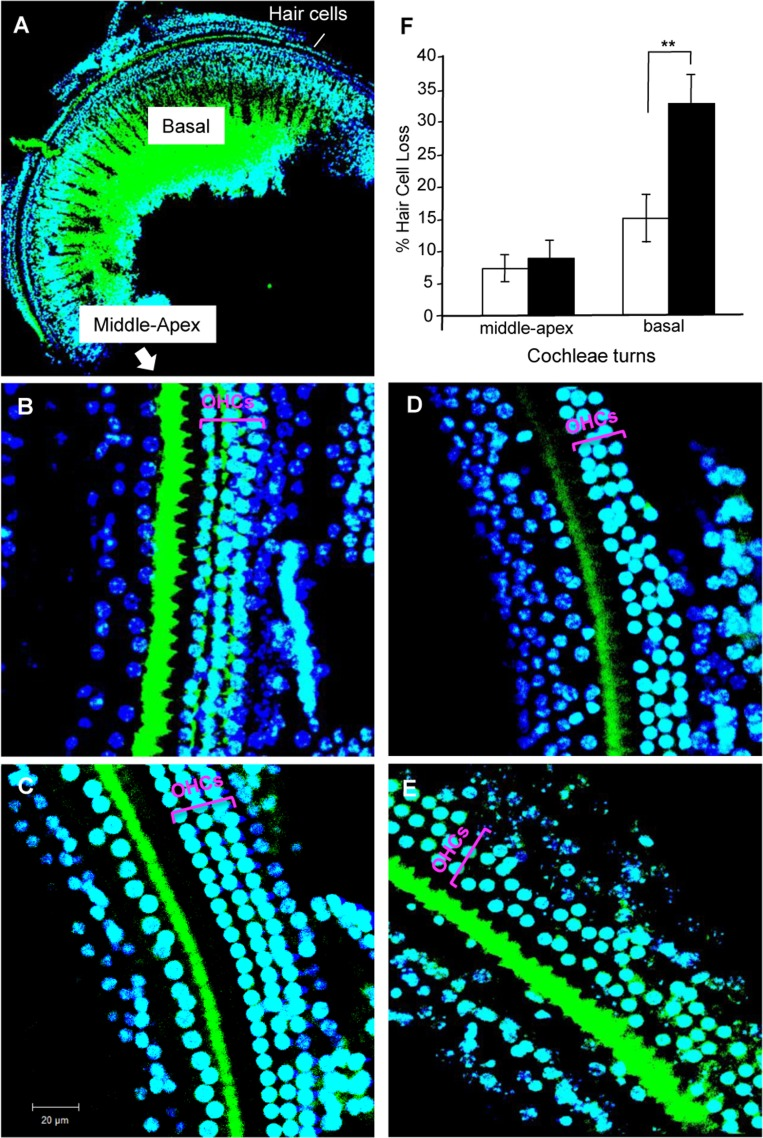
Figure 5. Hair cell loss in cochleae of Tg(Math1E-Aβ42Arc)1Lt/129 mice and control non-Tg siblings at age 8 months (A) Cochleae of basal region staining with Alexa Fluor 488–phalloidin and DAPI. (B) Middle region of cochleae in non-Tg sibling. (C) Middle region of the cochlea in Tg(Math1E-Aβ42Arc)1Lt/129. (D) Basal region of the cochlea in non-Tg sibling. (E) Basal region of the cochlea in Tg(Math1E-Aβ42Arc)1Lt/129. OHCs, outer hair cells. The results of the OHCs count at each cochlea turns (% of hair cells loss) are shown in (F) (number of cochleae = 5, in each group). Middle-apex; middle to apex region. Basal; basal region. Data are the mean ± SEM. **P < 0.01.
Expression of the human microtubule-associated protein tau (MAPT) in cochlear hair cells exacerbates the hearing defects caused by Aβ42Arc
MAPT is thought to be another major player in the development of AD [34]. Many studies indicate that there are synergistic effects between Aβ and tau in the neuronal toxicity of AD in the brain [35, 36]. To elucidate if Aβ42Arc-dependent high-frequency hearing defect reflects other aspects of AD in the brain, we looked for genetic interaction between Aβ42Arc and tau in terms of hearing defects in the cochlear hair cells. We established another Tg mouse line, Tg(MathE-MAPT)1Lt, that expressed human tau (2N4R) in cochlear hair cells, and we monitored auditory function of these mice over time (Supplemental Fig. 3A, B). No significant defects in hearing were seen in these mice when exposed to sound frequencies of 32 or 8 kHz or to click sounds (Fig. 6A–C). To determine whether there is interaction between Aβ42Arc and tau, we established a double Tg mouse line (Tau/Aβ42Arc) harboring both Tg(Math1E-Aβ42Arc)1Lt and Tg(MathE-MAPT)1Lt. We found that these mice had severe hearing defects; the ABR threshold was significantly increased at age 2 and 3 months for both the 32 and 8 kHz tones (Fig. 6D, E). However, we did not detect an increase in the threshold for the click sound, suggesting that the hearing defect was only in the middle- to high-frequency sound perception (>8 kHz; Fig. 6F). The significant shift in the onset of the hearing defect from age 4 months in the Aβ42Arc mice to age 2 months in the tau/Aβ42Arc double Tg-mice strongly suggested that there is a synergistic interaction between Aβ and tau in this system.
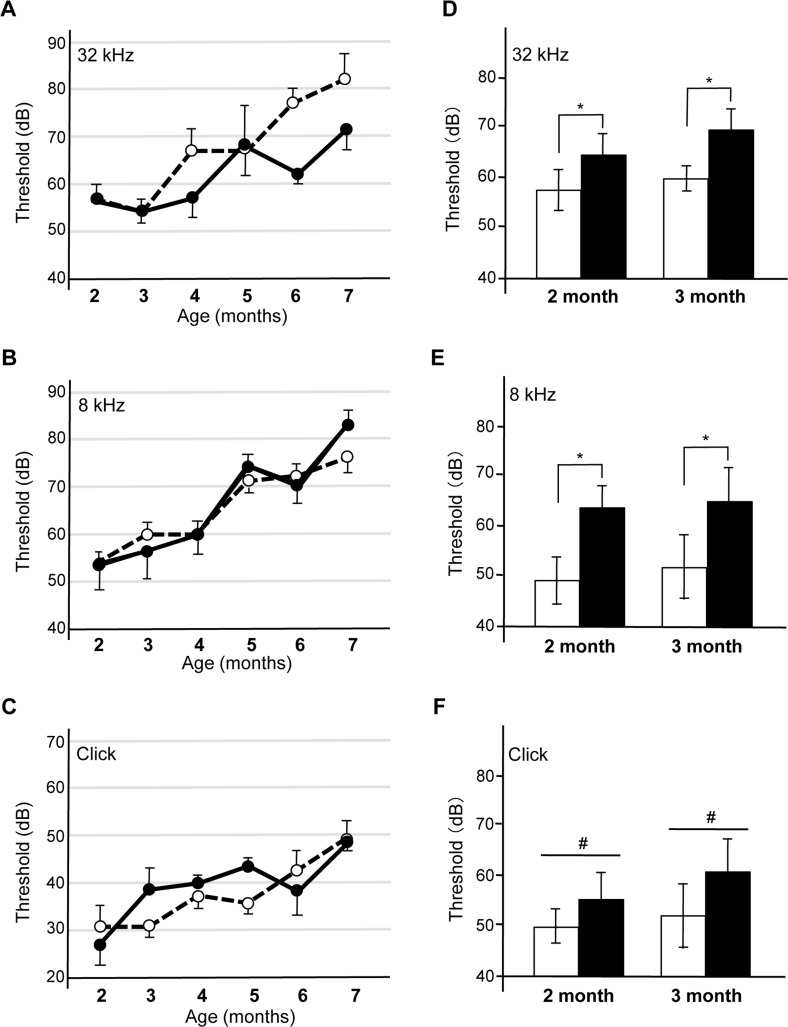
Figure 6. The expression of human tau in the cochleae of Tg(Math1E-Aβ42Arc)1Lt mice induces a synergistic auditory deficit (A-C) ABR threshold of Tg(MathE-MAPT)1Lt (filled circles) and non-Tg (open circles) mice from age 2 to 7 months for the 32 kHz (A) and 8 kHz (B) tones and the click sound (C). (D-F) ABR threshold for the non-Tg (white bars) and tau/Aβ42Arc (black bars) mice at age 2 and 3 months. tau/Aβ42Arc mice showed the threshold-increase onset at age 2 and 3 months for the 32 kHz (D) and 8 kHz (E) tones. The threshold for the click sound did not increase for any of the mice at any age (F). The data are expressed as the mean ± SEM. *p < 0.01; #, not significant. Non-Tg: n = 9 (6 male, 3 female); Tg (tau): n = 9 (4 male, 5 female); Tg (tau/Aβ42Arc): n = 5 (3 male, 2 female).
Discussion
Relationship between Aβ-induced hearing defects and ARHL
During aging, the survival of auditory hair cells decreases, especially in the basal region of the cochlea, and these cells are thought to be very sensitive to stressors such as excessive noise exposure [4]. This vulnerability of hair cells is likely the basis of the development of ARHL, as many animals with ARHL-related mutations show auditory defects after several hours of exposure to extreme noise [5]. We examined whether vulnerability of hair cells is involved in the hearing defects of Tg(Math1E-Aβ42Arc)1Lt mice by investigating if exposure to white noise at 100 dB for 2 h would affect the hearing ability of 3-month-old mice. One week after exposure, we measured the ABR threshold, however, no significant defects in hearing were seen in Tg(Math1E-Aβ42Arc)1Lt mice when exposed to sound frequencies of 32, 8 kHz or to click sounds (data not shown). Therefore, we think that auditory defects caused by Aβ differ from those associated with ARHL.
Tg(Math1E-Aβ42Arc)1Lt mice are a potential new assay system for the study of AD
To investigate the toxic effects of Aβ during AD development, several different types of mouse models for AD have been established, many of which express human APP in the brain [46]. These APP-expressing mice are very useful as an assay system for senile plaque formation in the brain and for learning/memory impairment [46]. However, these APP-expressing mice have several disadvantages as a model system for the development of drugs against AD. First, it takes approximately one year to obtain the robust phenotype caused by Aβ toxicity. Second, the progression of disease in these systems is based on behavioral symptoms, which are assessed by a behavioral test such as the water maze or Barnes test [46]. Thus, these systems are very useful for measuring higher-order brain functions, such as learning and memory, but they are not well suited for quantitative analysis [46]. Third, APP-expressing mice do not show neuronal cell loss, which is a typical pathological feature found in AD patients [33, 34].
Herein, we describe a new Tg system to explore AD based on Tg(Math1E-Aβ42Arc)1Lt mice. Our Tg-mouse system can easily monitor the effect of Aβ toxicities using electrophysiological methods. In other words, we can assess the impact of Aβ in live animals quantitatively over time. In particular, this system can also detect cellular defects like hair cell loss when Aβ is expressed. Therefore, we expect that this system will be a powerful tool when it is complementary used with traditional AD mouse models for evaluating the effects of Aβ toxicity and developing therapeutic drugs against AD.
Methods
Animals
Animal care and treatment were performed in accordance with institutional guidelines following approval by the Animal Care and Use Committee of National Center for Geriatrics and Gerontology (NCGG) (Obu, Japan). Before treatments, animals were anesthetized with isoflurane (Wako).
Mice were obtained from breeding colonies of Tg(Math1E-Aβ42)1Lt, Tg(Math1E-Aβ42Arc)1Lt, and Tg(MathE-MAPT)1Lt, which were heterozygous for the transgene, and were maintained at the Animal Center of NCGG. The Tg mice were maintained in a mixed C57BL/6 background and back-crossed to generate Tg(Math1E-Aβ42)1Lt/B6, Tg(Math1E-Aβ42Arc)1Lt/B6 or Tg(MathE-MAPT)1Lt/B6. We refer to the resulting lines as Tg(Math1E-Aβ42)1Lt, Tg(Math1E-Aβ42Arc)1Lt, and Tg(MathE-MAPT)1Lt, respectively. Wild-type littermates of the Tg mice served as controls (non-Tg mice). The Tg(Math1E-Aβ42Arc)1Lt mice were also crossed with 129 to generate Tg(Math1E-Aβ42Arc)1Lt/129.
Vector construction
We cloned the mouse Math1 minimal enhancer (Math1E) sequence into a pBluescript plasmid (Stratagene) containing the cytomegalovirus promoter and IRES-EGFP::L10-β-globin heavy chain-poly(A) sequence [24]. Aβ42 and Aβ42Arc constructs containing an upstream rat prepro-enkephalin signaling sequence, which had previously been constructed from cDNA of APP by PCR, or MAPT were placed downstream of the cytomegalovirus promoter [47, 48]. The mouse Math1E was kindly provided by Dr. J. E. Johnson (University of Texas Southwestern Medical Center, Dallas, TX).
Real-time qPCR
The qPCR was performed as described [49]. Briefly, RNA was extracted from cochleae using Isogen (Wako) and the RNeasy Mini kit (Qiagen), and the different cDNAs were individually synthesized with Primescript RT reagent (Takara). The mRNA expression levels were quantified using a Thermal Cycler Dice Real-Time System and SYBR Premix Ex Taq (Takara). Data were normalized to β-actin mRNA expression level. Myo7a, unconventional myosins playing an important role in morphogenesis and organization of cochlear hair cell bundles, was used as a positive control. The thermal cycling parameters were 40 cycles at 95°C for 10 s and 60°C for 30 s.
Oligonucleotides for real-time PCR analysis were as follows.
β-actin:
sense, 5′-CTAAGGCCAACCGTGAAAAG-3′
antisense, 5′-ACCAGAGGCATACAGGGACA-3′
Aβ:
sense, 5′-TCCGACATGACTCAGGATATGA-3′
antisense, 5′-CCCACCATGAGTCCAATGA-3′
myo7a:
sense, 5′-GCCATTGCTGACAACTGCTA-3′
antisense, 5′-TTGTGCTCTCTGTCTTGCCA-3′
ELISA
Soluble and insoluble (formic acid extractable) fractions were prepared from extracts of cochleae from Tg(Math1E-Aβ42Arc)1Lt and non-Tg mice. Briefly, frozen cochleae were homogenized with a metal pestle in Tris-buffered saline containing protease inhibitor cocktail (Complete Mini, Roche). After centrifugation at 100,000 × g for 1 h at 4°C, the soluble fraction was subjected to ELISA using the High Sensitivity Human β-Amyloid ELISA kit (Wako) to detect Aβ-like protein. The pellet was treated with 2% (w/v) sodium dodecyl sulfate and then 70% (v/v) formic acid and centrifuged at 100,000 × g for 1 h at 4°C. The supernatant, which represented the insoluble fraction, was subjected to ELISA.
Histochemistry
Cochleae were isolated from Tg(Math1E-Aβ42Arc)1Lt and non-Tg mice at age 4 months and fixed in paraffin. Paraffin sections were stained with hematoxylin and eosin or immunostained using Aβ antibody 6E10 (BioLegend).
Epon-embedded tissue sections were prepared as described [47]. Briefly, dissected cochleae were fixed in 2.5% (v/v) glutaraldehyde and decalcified for 3 days at 4°C in 5 mM EDTA, 0.1 M sodium phosphate (pH 7.4). Tissues were then fixed with 1% (w/v) OsO4 (Wako) in PBS (pH 7.4) for 1 h at room temperature. Tissues were then washed with PBS for 2 h with the wash solution changed every 15 min and then dehydrated as follows: once for 15 min in 50% (v/v) ethanol, twice for 30 min in 70% (v/v) ethanol, and once for 30 min in 85% (v/v) ethanol. After treatment with propylene oxide (Nisshin EM Corporation.), tissues were embedded in Epon 812 resin (TAAB Laboratories Equipment.) and incubated at 60±C for 48 h.
Phalloidin staining was performed by the standard protocol [1]. Briefly, dissected cochleae were fixed overnight with 4% paraformaldehyde in PBS. After cochleae were decalcified for 3 days at 4°C in 5 mM EDTA, 0.1 M sodium phosphate, they were incubated with phalloidin antibody coupled to Alexa Fluor 488 (Thermo). Dissected cochleae were mounted with Vectashield Mounting Medium that contained DAPI (Vector laboratories).
ABR measurement
ABRs were recorded in anesthetized mice. Electro-brainstem responses to the click (100 ms, 0–90 dB sound pressure level) and pure tonal sounds (8–32 kHz in half-octave steps, 20–100 dB sound pressure level in 5-dB steps, 3 ms, 1-ms cosine-squared rise-fall envelope) were recorded with subdermal silver wire electrodes at the ear (positive, active), the vertex (negative, reference), and the back of the mice (ground). ABR recordings were analyzed for consecutive amplitude deflections (waves). The first peak (wave I; latency, 1.2–1.9 ms) was monitored, and the hearing threshold for a given frequency or click stimulus was calculated for the lowest sound pressure leading to reliable ABR signals. Each ABR threshold was obtained by comparing the amplitude levels 5 dB above the hearing threshold for each group. Recordings were made for 12.5 ms with stimulus presentations of alternating polarity to eliminate electrical artifacts. In each case, stimulus presentation was at 0 ms. Signals were amplified (1000-fold, 40–60 dB), band pass filtered (0.2–5 kHz Butterworth, −20 dB/decade), averaged across 64–256 repetitions (dependent on the signal-to-noise ratio) at each sound pressure (usually 0–100 dB sound pressure level in steps of 5 dB), and recorded at 40 kHz sample frequency. Stimuli were delivered to the ear in a calibrated open system by a loudspeaker (model ES-1, TDT) placed 5 mm lateral to the pinna by an ear coupler. Sound pressure was calibrated online prior to each measurement with a microphone (NA42S, RION) placed under the scalp near the ear.
Mice were exposed to inhaled isoflurane anesthesia during ABR recording using a vaporizer.
Data analysis
For statistical analyses, we used the Microsoft Excel Macro program (Microsoft) and JMP (SAS Institute).
Supplementary Materials
Acknowledgments
We thank Drs. Shigeo Hayashi (Riken Center for Developmental Biology, Japan) and Katsuhiko Yanagisawa (NCGG, Japan) for helpful discussions and Drs. Hasegawa (Tokyo Metropolitan Institute of Medical Science, Japan) for kindly providing the human tau cDNA clone. We also thank Dr. K. Watanabe (NCGG, Japan) for critical reading of the manuscript. We also acknowledge Dr. M. Yoshikawa (NCGG, Japan) for useful information about mouse strain.
Funding
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Conflicts of Interest
The authors have no conflict of interests to declare.
References
- 1. Liu Z, Fang J, Dearman J, Zhang L, Zuo J. In vivo generation of immature inner hair cells in neonatal mouse cochleae by ectopic Atoh1 expression. PLOS ONE. 2014; 9: e89377 [PubMed] .
- 2. Oghalai JS. The cochlear amplifier: augmentation of the traveling wave within the inner ear. Curr Opin Otolaryngol Head Neck Surg. 2004; 12: 431 -438. [PubMed] .
- 3. Fettiplace R and Hackney CM. The sensory and motor roles of auditory hair cells. Nat Rev Neurosci. 2006; 7: 19 -29. [PubMed] .
- 4. Friedman LM, Dror AA, Avraham KB. Mouse models to study inner ear development and hereditary hearing loss. Int J Dev Biol. 2007; 51: 609 -631. [PubMed] .
- 5. Yang CH, Schrepfer T, Schacht J. Age-related hearing impairment and the triad of acquired hearing loss. Front Cell Neurosci. 2015; 9: 276 [PubMed] .
- 6. Ahmed HO, Dennis JH, Badran O, Ismail M, Ballal SG, Ashoor A, Jerwood D. High-frequency (10-18 kHz) hearing thresholds: reliability, and effects of age and occupational noise exposure. Occup Med. 2001; 51: 245 -258. .
- 7. Flint Beal M. Aging, energy, and oxidative stress in neurodegenerative diseases. Annals of Neurology. 1995; 38: 357 -366. [PubMed] .
- 8. Butterfield DA and Kanski J. Brain protein oxidation in age-related neurodegenerative disorders that are associated with aggregated proteins. Mechan Aging Dev. 2001; 122: 945 -962. .
- 9. Benilova I, Karran E, De Strooper B. The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci. 2012; 15: 349 -357. [PubMed] .
- 10. Cescato R, Dumermuth E, Spiess M, Paganetti PA. Increased generation of alternatively cleaved beta-amyloid peptides in cells expressing mutants of the amyloid precursor protein defective in endocytosis. J Neurochem. 2000; 74: 1131 -1139. [PubMed] .
- 11. DeArmond SJ. Alzheimer's disease and Creutzfeldt-Jakob disease: overlap of pathogenic mechanism. Curr Opin Neurol. 1993; 6: 872 -881. [PubMed] .
- 12. Lee HG, Zhu X, O'Neill MJ, Webber K, Casadesus G, Marlatt M, Raina AK, Perry G, Smith MA. The role of metabotropic glutamate receptors in Alzheimer's disease. Acta Neurobiol Exp. 2004; 64: 89 -98. .
- 13. Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005; 8: 1051 -1058. [PubMed] .
- 14. Attems J, Walker L, Jellinger KA. Olfaction and aging: A mini-review. Gerontology. 2015; 61: 485 -490. [PubMed] .
- 15. Attems J, Walker L, Jellinger KA. Olfactory bulb involvement in neurodegenerative diseases. Acta Neuropathol. 2014; 127: 459 -475. [PubMed] .
- 16. Benarroch EE. Olfactory system: functional organization and involvement in neurodegenerative disease. Neurology. 2010; 75: 1104 -1109. [PubMed] .
- 17. Doty RI. Olfaction in Parkinson's disease and related disorders. Neurobiol Dis. 2012; 46: 527 -552. [PubMed] .
- 18. Ruan Y, Zheng XY, Zhang HL, Zhu W, Zhu J. Olfactory dysfunctions in neurodegenerative disorders. J Neurosci Res. 2012; 90: 1693 -1700. [PubMed] .
- 19. Knipper M, Dijk PV, Nunes I, Ruttiger L, Zimmermann U. Advances in the neurobiology of hearing disorders: Recent developments regarding the basis of tinnitus and hyperacusis. Progress in Neurobiology. 2013; 111: 17 -33. [PubMed] .
- 20. Fitzsch B, Beisel KW, Pauley S, Soukup G. Molecular evolution of the vertebrate mechanosensory cell and ear. Int Dev Biol. 2007; 51: 663 -678. .
- 21. Oliver D, Klocker N, Schuck J, Baukrowitz T, Ruppersberg JP, Fakler B. Gating of Ca2+-activated K+ channels controls fast inhibitory synaptic transmission at auditory outer hair cells. Neuron. 2000; 25: 595 -601. [PubMed] .
- 22. Men Y, Zhang A, Li H, Zhang T, Jin Y, Li H, Zhang J, Gao J. LKB1 is required for the development and maintenance of stereocilia in inner ear hair cells in mice. PLOS ONE. 2015; DOI: 10.1371 .
- 23. Cai Z, Yan LJ, Li K, Quazi SH, Zhao B. Roles of AMP-activated protein kinase in Alzheimer's disease. Neuromolecular Med. 2012; 14: 1 -14. [PubMed] .
- 24. Helms AW, Abney AL, Ben-Arie N, Zoghbi HY, Johnson JE. Autoregulation and multiple enhancers control Math1 expression in the developing nervous system. Development. 2000; 127: 1185 -1196. [PubMed] .
- 25. Cescato R, Dumermuth E, Spiess M, Paganetti PA. Increased generation of alternatively cleaved beta-amyloid peptides in cells expressing mutants of the amyloid precursor protein defective in endocytosis. J Neurochem. 2000; 74: 1131 -1139. [PubMed] .
- 26. Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, Condron MM, Bitan G, Teplow DB, Shea LE, Ruotolo BT, Robinson CV, Bowers MT. Amyloid-b protein oligomerization and the importance of tetramers and dodecamers in the etiology of Alzheimer's disease. Nat Chem. 2009; 1: 326 -331. [PubMed] .
- 27. LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer's disease. Net Rev Neurosci. 2007; 8: 499 -509. .
- 28. Iijima K, Chiang HC, Hearn SA, Hakker I, Gatt A, Shenton C, et al. Aβ42 mutants with different aggregation profiles induce distinct pathologies in Drosophila. PLOS ONE. 2008; 3: e1703 [PubMed] .
- 29. Willott JF. Overview of methods for assessing the mouse auditory system. Curr Protoc Neurosci. 2006; Chapter 8: Unit8.21A .
- 30. Johnson KR, Zheng QY, Erway LC. A major gene affecting age-related hearing loss is common to at least ten inbred strains of mice. Genomics. 2000; 70: 171 -180. [PubMed] .
- 31. Di Palma F, Holme RH, Bryda EC, Belyantseva IA, Pellegrino R, Kachar B, Steel KP, Noben-Trauth K. Mutations on Cadh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat Genet. 2001; 27: 103 -107. [PubMed] .
- 32. Ouagazzal AM, Reiss D, Romand R. Effects of age-related hearing loss on startle reflex and prepulse inhibition in mice on pure and mixed C57BL and 129 genetic background. Behav Brain Res. 2006; 172: 307 -315. [PubMed] .
- 33. Terry RD, Masliah E, Salmon DP, Butters N, De Teresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991; 30: 572 -580. [PubMed] .
- 34. Ittner LM and Gotz J. Amyloid-β and tau—a toxic pas de deux in Alzheimer's disease. Nat Rev Neurosci. 2011; 12: 65 -72. [PubMed] .
- 35. Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301ltau transgenic mice induced by Abeta 42 fibrils. Science. 2001; 293: 1491 -1495. [PubMed] .
- 36. Chabrier MA, Cheng D, Castello NA, Green KN, LaFeria FM. Synergistic effects of amyloid-beta and wild-type human tau on dendritic spine loss in a floxed double transgenic model of Alzheimer's disease. Neurobiol Dis. 2014; 64: 107 -117. [PubMed] .
- 37. Gago MF, Fernandes V, Ferreira J, Yelshyna D, Silva HD, Rodrigues ML, Rocha L, Bicho E, Sousa N. Role of the visual and auditory systems in postural stability in Alzheimer's disease. J Alz Dis. 2015; https://doi.org/10.3233/JAD-150131 .
- 38. Lin FR, Metter J, O'Brein RJ, Resnick S, Zonderman AB, Ferrucci L. Hearing Loss and Incident Dementia. Arch Neurol. 2011; 68: 214 -220. [PubMed] .
- 39. Perez SE, Lumayag S, Kovacs B, Mufson EJ, Xu S. Beta-amyloid deposition and functional impairment in the retina of the APPswe/PS1DeltaE9 transgenic mouse model of Alzheimer's disease. Invest Ophthalmol Vis Sci. 2009; 50: 793 -800. [PubMed] .
- 40. Casas-Tinto S, Zhang Y, Sanchez-Garcia J, Gomez-Velazquez M, Rincon-Limas DE, Fernandez-Funez P. The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum Mol Genet. 2011; 20: 2144 -2160. [PubMed] .
- 41. Haywood AF and Staveley BE. Parkin counteracts symptoms in a Drosophila model of Parkinson's disease. BMC Neurosci. 2004; 5: 14 [PubMed] .
- 42. Jackson GR, Salecker I, Dong X, Yao X, Arnheim N, Faber PW, MacDonald ME, Zipursky SL. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron. 1998; 21: 633 -642. [PubMed] .
- 43. Cao W, Song HJ, Gangi T, Kelkar A, Antani I, Garza D, Konsolaki M. Identification of novel genes that modify phenotypes induced by Alzheimer's beta-amyloid overexpression in Drosophila. Genetics. 2008; 178: 1457 -1471. [PubMed] .
- 44. Venderova K, Kabbach G, Abdel-Messih E, Zhang Y, Parks RJ, Imai Y, Gehrke S, Ngsee J, Lavoie MJ, Slack RS, Rao Y, Zhang Z, Lu B, Haque ME, Parks DS. Leucine-Rich Repeat Kinase 2 interacts with Parkin, DJ-1 and PINK-1 in a Drosophila melanogaster model of Parkinson's disease. Hum Mol Genet. 2009; 18: 4390 -4404. [PubMed] .
- 45. Kaltenbach LS, Romero E, Becklin RR, Chettier R, Bell R, Phansalkar A, Strand A, Torcassi C, Savage J, Hurlburt A, Cha GH, Ukani L, Chepanoske CL, et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLOS Genet. 2007; 3: e82 [PubMed] .
- 46. Webster SJ, Bachstetter AD, Nelson PT, Schmitt FA, Van Eldik LJ. Using mice to model Alzheimer's dementia: an overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front Genet. 2014; 5: Article 88 1 -23. [PubMed] .
- 47. Omata Y, Lim YM, Akao Y, Tsuda L. Age-induced reduction of autophagy-related gene expression is associated with onset of Alzheimer's disease. Am J Neurodegen. 2014; 3: 134 -142. .
- 48. Jakes R, Noval M, Davison M, Wischik CM. Identification of 3- and 4-repeat tau isoforms within the PHF in Alzheimer's disease. EMBO J. 1991; 10: 2725 -2729. [PubMed] .
- 49. Lim YM, Hayashi S, Tsuda L. Ebi/AP-1 suppresses pro-apoptotic gene expression and permits long-term survival of Drosophila sensory neurons. PLOS ONE. 2012; 7: e37028 [PubMed] .