




Introduction
Age-related macular degeneration (AMD) is the leading cause of vision loss in the elderly in the Western World. It is a complex disease as reflected not only by the ever growing number of genetic, environmental, and systemic risk factors identified to date [1–3], but also our improved understanding of the various clinical phenotypes, through the advent of high resolution imaging modalities used to evaluate both patients and post-mortem tissue pathology [4–8]. Clinically, during the initial stages of AMD development, known as ‘early dry’, patients accumulate extracellular lipid and protein filled deposits below the retinal pigment epithelial (RPE) cell layer [1,5,9], which normally serves as crucial support to the overlying neural retina and forms part of the outer blood retina barrier [10]. These deposits can lead to dysfunction and atrophy of RPE cells, which along with loss of photoreceptors and choroidal endothelial cells, are major steps in AMD progression towards severe vision deterioration associated with the ‘late dry’ sub-type known as geographic atrophy [1,3,11]. Advanced exudative or ‘wet’ AMD is characterized by endothelial invasion through Bruch’s membrane and pathological growth of abnormal new vessels originating from the choroid, below the RPE, resulting in the formation of choroidal neovascular (CNV) lesions, which are responsible for the most severe form of disease-related vision loss [1–3, 12]. Demographically, both the increase in life expectancy of the general population, and prevalence of AMD in the elderly population with age [2,3], ensures that this disease will become an even greater health problem in the near future. Therefore, it is imperative to investigate the pathological pathways that are altered in this complex disease, recognize potential models that demonstrate phenotypic features of AMD, and identify alternative targets, in order to develop treatments and improve the quality of life of patients. Currently there are no therapies available for the ‘dry’ forms of the disease. However, antibody-based treatments targeting vascular endothelial growth factor (VEGF) are offered to patients with ‘wet’ AMD, which are effective to varying degrees in approximately 30-60% of the patients. This leaves more than 30% of the patient population, for which an alternative treatment must be found.
Peroxisome proliferator-activated receptors (PPARs) are ligand activated transcription factors, which belong to the steroid hormone superfamily. Though PPARα, PPARβ/δ and PPARγ are ubiquitously expressed throughout the body [13,14], PPARβ/δ has been shown to be involved in regulation of pathways important in AMD pathogenesis, including lipid metabolism, extracellular matrix remodeling, angiogenesis and inflammation [1,15]. Because of this, we hypothesized that PPARβ/δ is critical in the etiology of AMD. Here we report a novel role of the PPARβ/δ pathway in the pathobiology of AMD. We first established the expression and activity of the pathway in cell culture models of RPE and choroidal endothelial cells, cells vulnerable in AMD. Next we evaluated the ocular phenotype of aged mice harboring the null allele at the PPARβ/δ locus (Pparβ/δ-/-). Finally, we tested the therapeutic potential of targeting the PPARβ/δ pathway in an experimentally induced model of choroidal neovascularization. Our in vitro studies revealed that following PPARβ/δ knockdown there is an increase in expression of select extracellular matrix molecules concomitant with a decrease in expression of growth factors, in both RPE and choroidal endothelial cells. Similarly, PPARβ/δ knockdown impacted the expression of several AMD-related genes in the inflammatory and lipid metabolic pathways. In vivo evaluation of eyes from aged wild-type mice showed accumulation of thin patchy sub-RPE deposits, while genetic ablation of Pparβ/δ in vivo, resulted in increased frequency and severity of continuous sub-RPE deposits along with development of RPE degenerative changes. On the other hand, Pparβ/δ knockout mice develop CNV lesions smaller in volume and area, increased localization of immune cells, and decreased deposition of extracellular matrix molecules, compared to Pparβ/δ+/+ mice. Finally, we observed that treatment with a PPARβ/δ antagonist, GSK0660, resulted in a significant inhibition of neovascular lesion size, and extracellular matrix deposition, in aged mice, while treatment with a PPARβ/δ agonist resulted in a decrease of lipid accumulation in a cell culture model of ‘lipid loaded RPE’ cells. This study establishes a strong basis to consider selectively testing and developing, PPARβ/δ ligands as potential therapies for AMD.
Results
The PPARβ/δ pathway is biologically active in AMD vulnerable cells
The activity of PPARβ/δ in AMD vulnerable cells was examined in human primary RPE cells, the human derived ARPE19, and the macaque derived RF/6A choroidal endothelial cell lines. This was performed by examination of the (i) expression of PPARβ/δ and its binding partners the retinoid X receptor alpha and beta (RXRα and β), (ii) receptor transcriptional activity, by measuring the binding of the receptor-ligand complex to the gene-response element, and (iii) expression of known PPARβ/δ target genes. Freshly isolated human RPE cells and choroid from aged donor eyes, along with primary human RPE cultured cells, ARPE19 and RF/6A cell lines, expressed PPARβ/δ and its binding partners the RXRs (Figure 1A). Additionally, ligand activation of PPARβ/δ with GW0742 (10μM) caused increased transcriptional activity in human primary RPE (Figure 1B), RF/6A cells (Figure 1C) and ARPE19 cells (Figure S1) [16]. These changes were mitigated by the PPARβ/δ antagonist, GSK0660 (10μM), and siRNA-mediated knockdown of PPARβ/δ (Figure 1, B and C). Similarly, ligand activation of PPARβ/δ increased expression of the PPARβ/δ target genes, angiopoietin-like 4 (ANGPTL4) and pyruvate dehydrogenase kinase, isozyme 4 (PDK4), in primary RPE cells (Figure 1, D and E), RF/6A cells and ARPE19 cells (Figure 1, F, G, S1 and S2), an effect that was diminished by treatment with a PPARβ/δ antagonist or PPARβ/δ knockdown.
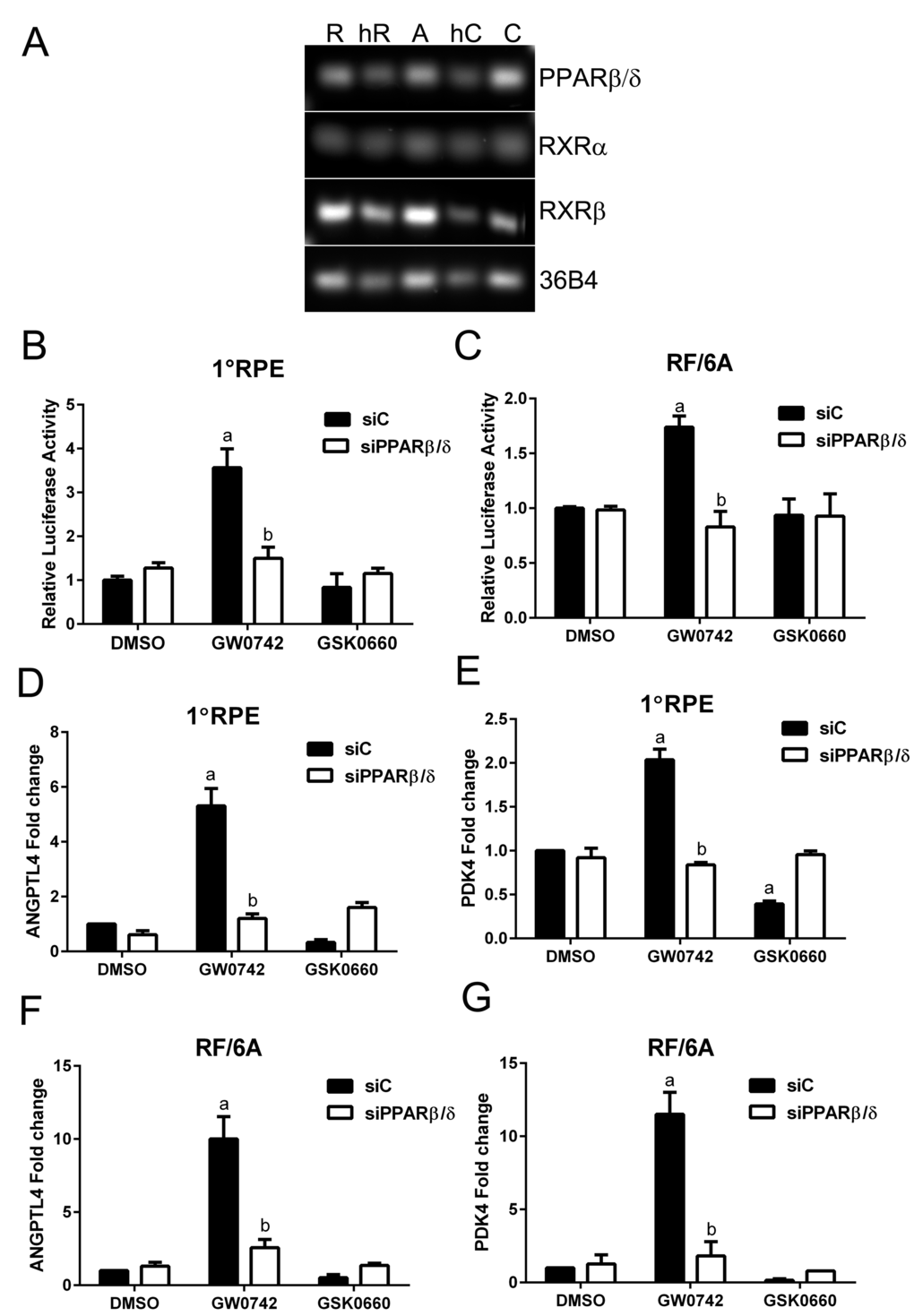
Figure 1. PPARβ/δ signaling pathway is functional in AMD vulnerable cells. (A) Agarose gel image of PCR amplification products of PPARβ/δ and its obligate binding partners RXRα and RXRβ in primary human RPE cells [R], freshly isolated human RPE cells [hR], ARPE19 cells [A], human choroid [hC], and RF/6A cells [C], 36B4 was used as loading control. PPARβ/δ activity in primary RPE (1°RPE) cells (B) and RF/6A cells (C) transfected with the DR1 luciferase reporter and siC or siPPARβ/δ; cells were treated with PPARβ/δ agonist, GW0742 (10μM) or antagonist, GSK0660 (10μM) or DMSO as vehicle control (n = 3): a: p < 0.05 relative to DMSO treated cells; b: p < 0.05 relative to drug+siC treated cells (p < 0.05; two way ANOVA, Sidak’s multiple comparisons test). Expression of ANGPTL4 and PDK4 mRNA in primary RPE (1°RPE) cells (D and E) and RF/6A (F and G) in siC and siPPARβ/δ (100 pmoles/250,000 cells) treated cells in response to GW0742, GSK0660, or DMSO as a control (n = 3); a: p < 0.05 relative to DMSO treated cells; b: p < 0.05 relative to drug+siC treated cells (Two way ANOVA, Sidak’s multiple comparisons test).
Loss of PPARβ/δ results in selective regulation of dry and wet AMD related pathogenic pathways
The discovery of multiple genetic, systemic, and environmental risk factors associated with AMD ontology has resulted in the identification of several AMD-pathogenic pathways. These pathways include, but are not limited to, impairment of extracellular matrix turnover [3,17], increased angiogenesis [18], inflammation [19,20], and dysregulation of lipid processing pathways [21]. Since modulation of the PPARβ/δ pathway has been shown to regulate collagen synthesis in vitro and in vivo [22–24], the effect of PPARβ/δ knockdown (siPPARβ/δ) on the expression of extracellular matrix-related genes was assessed. Deletion of PPARβ/δ expression caused upregulation of collagen type 1A1 (COL1A1) and vitronectin (VTN) in human primary RPE cells, and no significant effect on expression levels of collagen type 4A4 (COL4A4) and fibronectin (FN1) (Figure 2A). In RF/6A cells, deletion of PPARβ/δ resulted in downregulation of the extracellular matrix genes COL1A1, COL4A4 and FN1 (Figure 2B). Increased deposition of collagen type 1A1, collagen 4A4 and vitronectin is characteristic of Bruch’s membrane and human sub-RPE deposits typically observed in dry AMD [25], while endothelial cells require extracellular matrix molecules such as COL4A4, for pericyte recruitment and vessel stabilization during angiogenesis [26,27]. These results demonstrate selective roles for PPARβ/δ in AMD vulnerable cells suggesting it regulates extracellular matrix turnover in RPE cells similar to that reported for dry AMD, yet inhibits an angiogenic phenotype in endothelial cells. Evaluation of the expression of growth factors that regulate vessel stabilization following PPARβ/δ knockdown confirmed this variability in AMD vulnerable cells. A significant decrease in the expression of platelet-derived growth factor receptor beta (PDGFRB), vascular endothelial growth factor A (VEGFA) and transforming growth factor beta 1 (TGFB1) in both primary RPE and RF/6A cells transfected with siPPARβ/δ, as compared to control siRNA (Figure 2, C and D) suggests that disruption of PPARβ/δ expression in both of these AMD-vulnerable cells leads to an anti-angiogenic environment in the RPE and choroid. Interestingly, receptor knockdown resulted in a downregulation of the expression of the neurotrophic agent, pigment epithelial-derived factor (PEDF or SERPINF1) in RPE cells but not in RF/6A cells (Figure 2, C and D). Since modulation of the PPARβ/δ pathway has been shown to regulate inflammation in vitro and in vivo, the effect of PPARβ/δ knockdown on the expression of molecular markers of inflammation was also examined [23,28,29]. Genetic knockdown of PPARβ/δ resulted in the formation of a pro-inflammatory environment in the outer retinal cells, which was evident by the upregulation of inflammatory genes such as, prostaglandin-endoperoxide synthase 2 (PTGS2), interleukin-1 beta (IL1B), chemokine ligand 2 (CCL2) and tumor necrosis factor alpha (TNFA) in RPE cells (Figure 2E); and secreted phosphoprotein 1 (SPP1) and TNFA in RF/6A cells (Figure 2F). Given the role of PPARβ/δ in regulating lipid processing pathways [30], the expression of genes involved in lipid metabolism and previously shown to be altered in AMD was examined. Increased expression of apolipoprotein E (APOE), A (APOA) and low density lipoprotein receptor (LDLR) in human RPE cells (Figure 2G), along with decreased expression of several lipid transfer genes in choroidal endothelial cells (Figure 2H) following PPARβ/δ knockdown was observed. Extracellular and intracellular accumulation of lipids and lipofuscin are characteristics of dry AMD. Good animal models demonstrating significant lipid accumulation in Bruch’s membrane and/or deposits, and not requiring aging mice for long periods of time are currently not available. Therefore, in lieu of that, we examined the effect of activating or antagonizing PPARβ/δ in an in vitro culture model of lipid-loaded RPE cells. Ligand activation of PPARβ/δ resulted in a significant decrease in RPE lipid accumulation (Figure 2I), suggesting a potential therapeutic avenue to pursue in the treatment of early dry AMD, in which removal of extra- and intra-cellular lipids is a goal. Collectively, these data suggest that though PPARβ/δ drives several of the pathogenic pathways associated with AMD development, it may have selective detrimental and beneficial effects in AMD vulnerable cells. To determine the role of PPARβ/δ on the posterior eye, the ocular phenotype of wild-type (Pparβ/δ+/+) and Pparβ/δ-null (Pparβ/δ–/–) mice was examined.
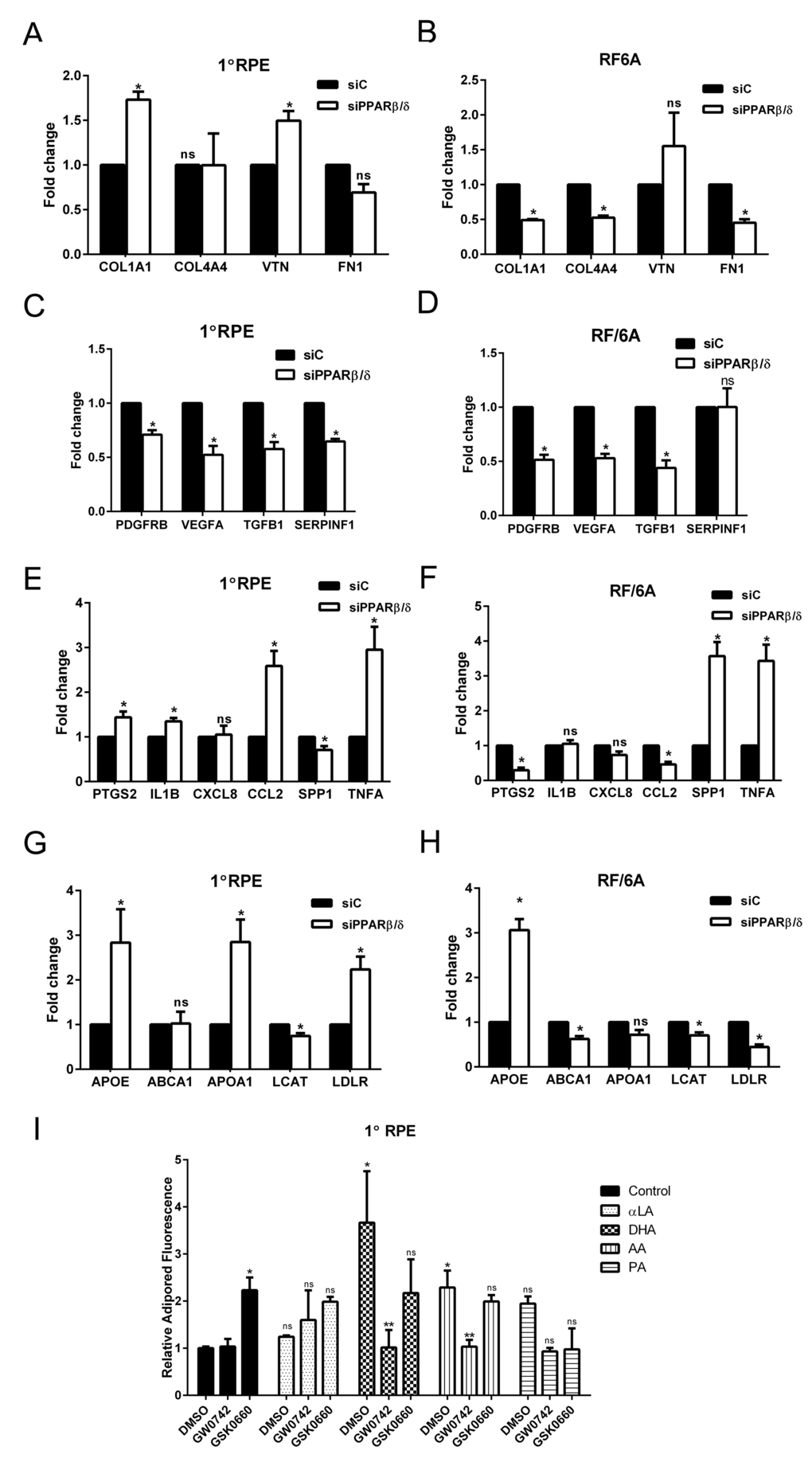
Figure 2. PPARβ/δ regulates both dry- and wet-AMD related pathogenic pathways. Effect of siRNA mediated knockdown of PPARβ/δ on mRNA expression of extracellular matrix genes COL1A1, COL4A4, FN1 and VTN; angiogenesis and fibrosis genes, VEGFA, PDGFB, TGFB1 and SERPINF1; inflammation-related genes, PTGS2, IL1B, CXCL8, CCL2, SPP1, and TNFA; and lipid processing genes APOE, ABCA1, APOA1, LCAT and LDLR in 1°RPE cells (A, C, E, and G) and RF/6A (B, D, F and H) cells. (mean and S.E.M.; n = 3; *p < 0.05, ns: not significant, two way ANOVA, Sidak’s multiple comparisons test); siC, control siRNA; siPPARβ/δ, PPARβ/δ siRNA. Quantification of intracellular lipid accumulation after lipid loading followed by incubation with PPARβ/δ agonist, GW0742 or antagonist, GSK0660 in (I) 1°RPE. (*, p<0.05, compared to DMSO Control; ** p<0.05, compared to DMSO; ns: not significant, n=3, two way ANOVA, Sidak’s multiple comparisons test); Control: DMSO vehicle, αLA: α-linolenic acid, DHA: docosahexaenoic acid, AA: arachidonic acid, and PA: palmitic acid.
Aged Pparβ/δ–/– mice exhibit several phenotypic features of dry AMD
Gene specific differences in the weights of 18-month old Pparβ/δ+/+ and Pparβ/δ–/– mice were not observed (Figure S3). The overall architecture of the retina and RPE/choroid was evaluated and no differences in the morphology of the inner retina and/or thickness of the inner and outer nuclear layers were found (Figure S4). In the outer retina, thin patchy sub-RPE deposits were observed in both 18-month old Pparβ/δ+/+ and Pparβ/δ–/– mice with varying degrees of thickness and length (Figure 3, A, B, C and D). Detailed quantification of the length of deposit relative to length of Bruch’s membrane revealed deposits were present at a higher frequency in Pparβ/δ–/– mice compared to Pparβ/δ+/+ mice (ratio of deposits/BrM length: 0.42
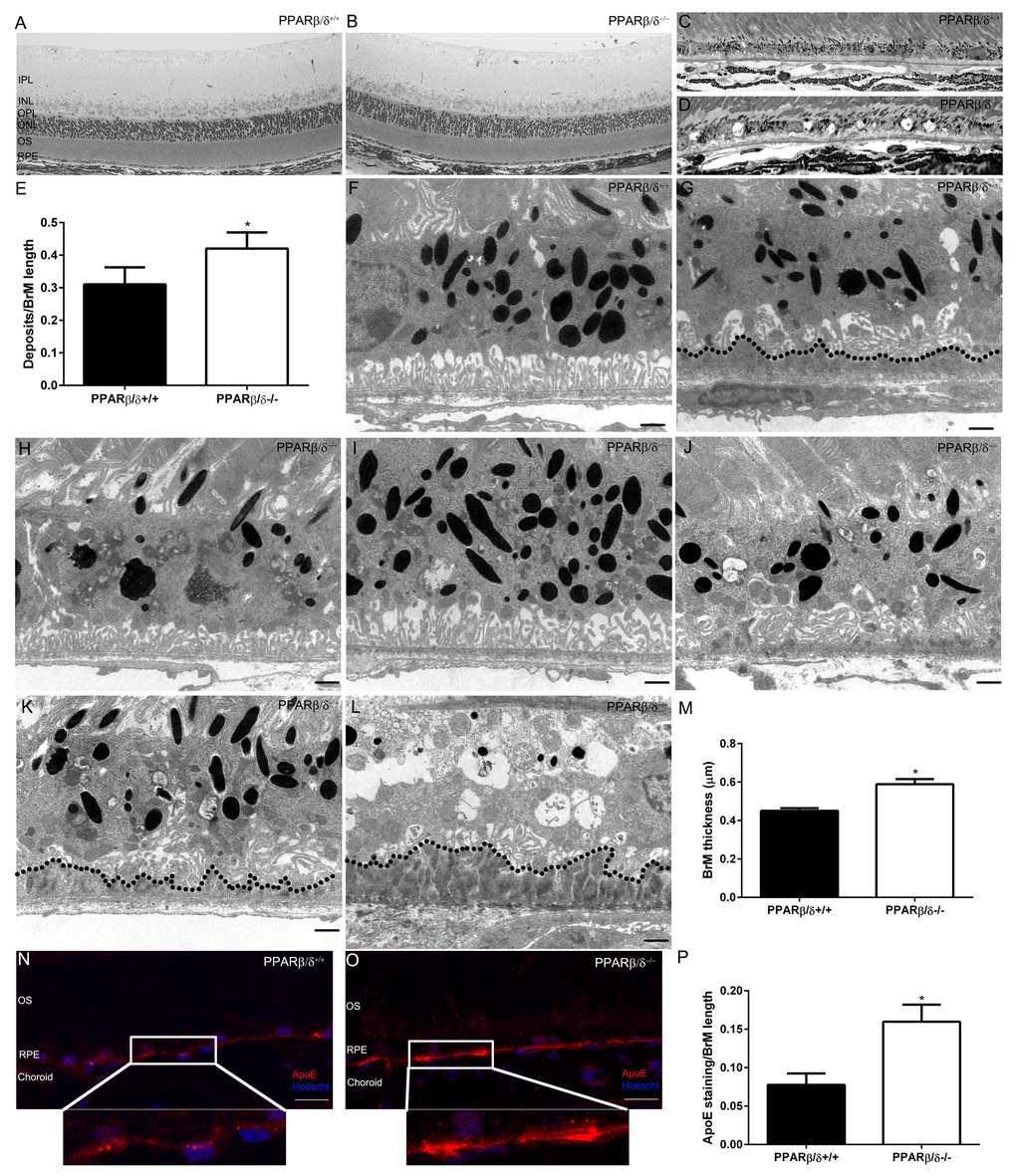
Figure 3. 18-month old Pparβ/δ−/− mice exhibit dry-AMD pathology. Toluidine blue stained images of plastic sections from 18-month old (A)Pparβ/δ+/+ and (B)Pparβ/δ−/− mice displaying retinal layers (IPL: Inner plexiform layer, INL: Inner nuclear layer, OPL: Outer plexiform layer, ONL: Outer nuclear layer, OS: photoreceptor outer segments, RPE: Retinal pigment epithelium). Toluidine blue stained images of the outer retina of (C)Pparβ/δ+/+ and (D)Pparβ/δ−/− mice, which have sub-RPE deposits (Scale bar = 10 μm). (E) Quantification of deposits per Bruch’s membrane (BrM) length in plastic sections (mean and S.E.M., n=10 images/animal, n=4/genotype, * p<0.05). Electron micrographs of RPE/Bruch’s membrane/choroidal junction in 18-mo-old Pparβ/δ+/+ mice display (F) normal RPE morphology (G) with some age related deposits (dotted line), whereas 18-mo-old Pparβ/δ−/− mice show (H) RPE hypo-pigmentation, (I) hyper-pigmentation and (J) abnormal basal infoldings with thin sub-deposits, (K) loss of basal infoldings with thin and (L) thick sub-RPE deposits (dotted line). (M) Quantification of Bruch’s membrane thickness in electron micrographs of Pparβ/δ+/+ and Pparβ/δ−/− mice (n = 10 images per mouse, n = 4 mice per genotype, two tailed t-test). Scale bars in panels F-L: 1 μm. Images of apolipoprotein E (apoE; red) stained sections from (N)Pparβ/δ+/+ and (O)Pparβ/δ−/− mice (Scale bar: 20 μm). Nuclei are stained with Hoescht (blue). (P) Quantification of ratio of apoE stained regions/Bruch’s membrane length (mean and S.E.M., n=4 per group, * p < 0.05, two tailed t-test).
PPARβ/δ promotes laser-induced CNV
Since PPARβ/δ knockdown in both RPE and choroidal endothelial cells in vitro resulted in an anti-angiogenic phenotype, the role of PPARβ/δ in development of CNV in vivo was examined. No evidence of spontaneous CNV or overt vascular changes were observed in aged Pparβ/δ–/– mice. However, laser-induced CNV lesions in 18-20 month old Pparβ/δ–/– were smaller as compared to those observed in Pparβ/δ+/+ mouse eyes (Figure 4, A, B and C). Additionally, examination of the distribution of the lesions in flatmounts of the posterior eye cups revealed a genotype-dependent difference in the number of mouse eyes with single versus merged lesions. Specifically, while Pparβ/δ+/+ mice presented with two or three merged lesions, no eyes from the Pparβ/δ–/– cohort presented with three merged lesions, and only one eye displayed two merged lesions (Figure 4B). Quantitatively, measurement of CNV lesion volume determined from z-stacks and three-dimensional reconstructions confirmed a significantly smaller volume in Pparβ/δ–/– mice compared to Pparβ/δ+/+ mice (Figure 4D). These data demonstrate a functional role for the PPARβ/δ gene in angiogenesis and the development of CNV lesions.
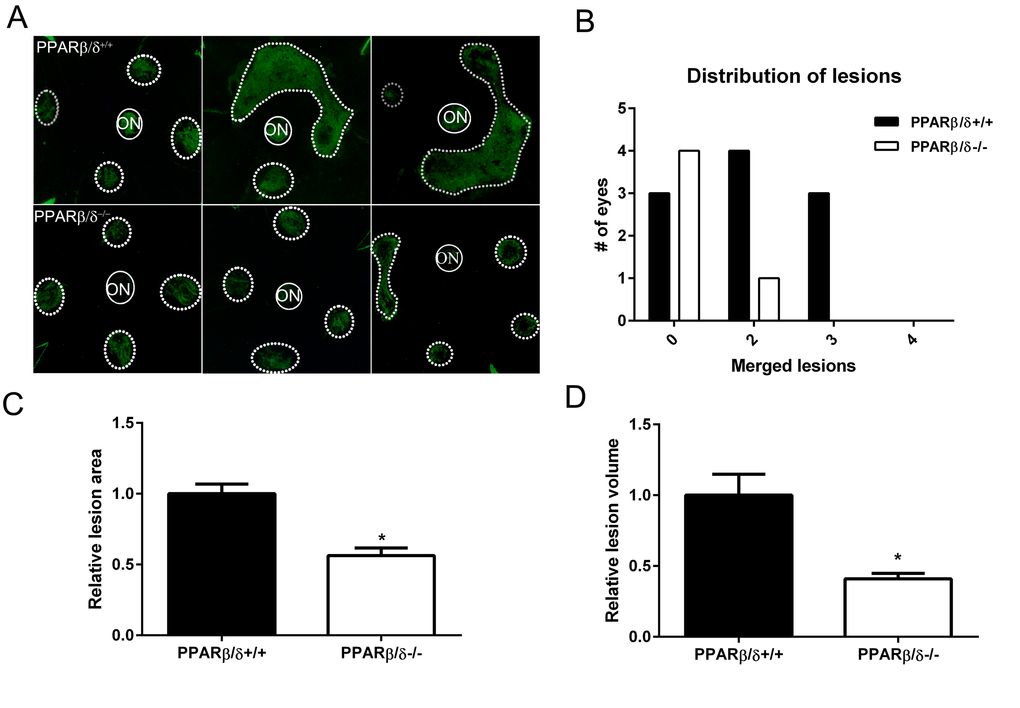
Figure 4. Genetic disruption of PPARβ/δ attenuates laser-induced CNV. (A) Choroidal flat-mounts were prepared from 18–20 month-old laser-induced CNV mice (Pparβ/δ+/+, n = 10 eyes, 40 lesions; Pparβ/δ−/−, n=5 eyes, 20 lesions) and stained with isolectin GS-IB4 (ON, optic nerve). Representative images from three eyes/genotype are shown to demonstrate individual and merged CNV lesions. Dotted and solid line circles demarcate lesions and optic nerves, respectively. (B) Distribution of number of eyes with individual versus merged lesions in Pparβ/δ+/+ and Pparβ/δ−/−. (C) Relative lesion area/animal was measured using ImageJ (mean and S.E.M.; *p < 0.01, two tailed t-test). (D) Relative lesion volume/animal (mean and S.E.M. for each group; *p < 0.05, two tailed t-test).
PPARβ/δ regulates extracellular matrix deposition and immune cell infiltration in CNV lesions
PPARβ/δ has been shown to regulate extracellular matrix and inflammation [31]. With this in mind, extracellular matrix regulation, extracellular matrix deposition and immune cell localization within CNV lesions from aged Pparβ/δ+/+ and Pparβ/δ–/– mice were characterized by examining cross sections of the retina/RPE/choroid. Collagen type 4 (COL4) and fibronectin (FN1) are known components of Bruch’s membrane, sub-RPE deposits [32–34], and associated with CNV lesions [33]. Furthermore, fibronectin is known to be produced in response to vessel injury [35]. Quantitatively, significantly lower staining intensity for both COL4 and FN1 in CNV lesions was measured in aged Pparβ/δ–/– mice compared to age-matched Pparβ/δ+/+ controls (Figure 5, A and B). As mentioned earlier, PPARβ/δ is known to influence inflammation [31], and this is one of the major pathways that regulates the pathogenesis of AMD [29,36]. Thus, cross sections of the retina/RPE/choroid containing CNV lesions were probed with antibodies to ionized calcium binding adaptor molecule 1 (Iba1), which labels microglial cells and macrophages, and adhesion G protein-coupled receptor E1, also known as F4/80, which labels mature macrophages (Figure 5 C and S8). Interestingly, a significant increase in the localization of both F4/80+ and Iba1+ cells within the neovascular lesions of Pparβ/δ–/– mice as compared to age-matched Pparβ/δ+/+ controls was quantified (Figure 5 D and S8). This is a noteworthy observation given the distinct phenotypes that macrophages can differentiate into: 1) the broadly characterized M1 macrophage, which is reported to contribute to pro-fibrotic and pro-inflammatory phenotypes, and 2) M2 macrophages, which also have 4 different subtypes 37]; characterized as having immunosuppressive and tissue remodeling properties [19,38]. This finding warrants future detailed analysis of the polarity of macrophages in the Pparβ/δ–/– CNV lesions. Combined, these results show that the loss of PPARβ/δ expression causes decreased extracellular matrix deposition associated with increased acute immune cell infiltration within small CNV lesions, and may reflect decreased fibrosis modulated by PPARβ/δ.
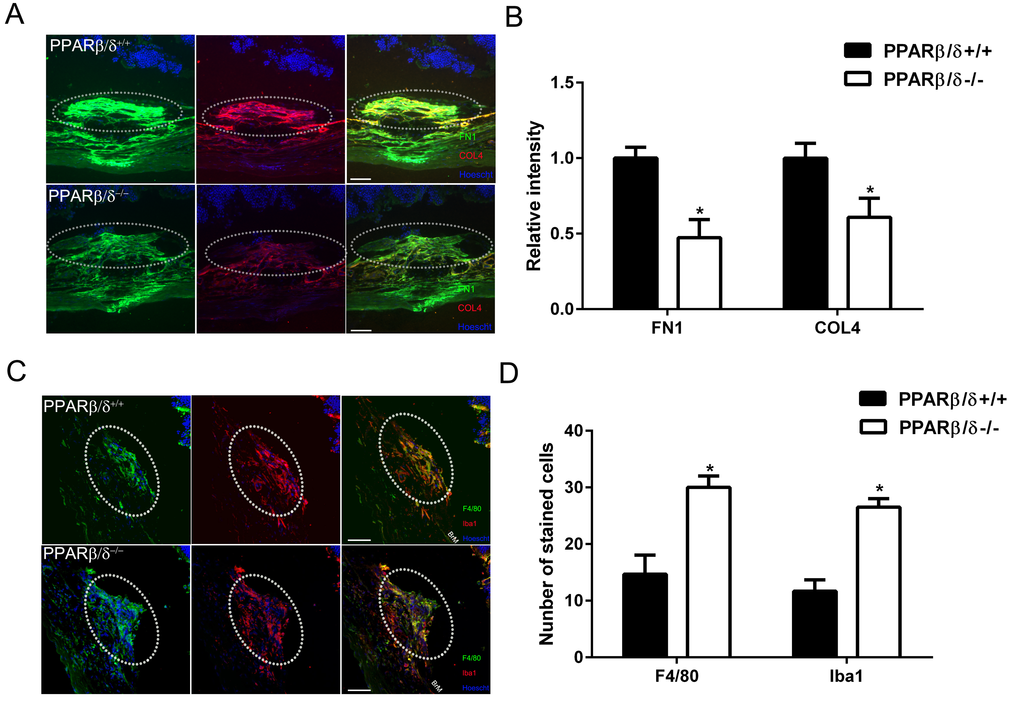
Figure 5. PPARβ/δ regulates extracellular matrix deposition and immune-cell infiltration in CNV lesions. (A) FN1 (green) and COL4 (red) immunolocalization in CNV lesions of Pparβ/δ+/+ and Pparβ/δ−/− mice (dotted oval demarcates the lesion area; nuclei are stained blue with Hoechst; representative images are shown; scale bar = 50 µm). (B) FN1 and COL4 staining intensity was quantified in the CNV lesions of Pparβ/δ+/+ and Pparβ/δ−/− mice using ImageJ (mean and S.E.M.; n = 3/group; **p < 0.01, two tailed t-test). (C) Laser CNV lesions from Pparβ/δ−/− mice display a higher number of F4/80 (green) and Iba1 (red) immunopositive cells (dotted oval demarcates the lesion area; nuclei are stained blue with Hoechst; representative images are shown; scale bar = 50 µm). (D) The numbers of F4/80+ and Iba1+ cells in the CNV lesions of Pparβ/δ+/+ and Pparβ/δ−/− mice were counted using ImageJ (mean and S.E.M.; n = 3/group; *p < 0.01, two tailed t-test).
Pharmacological antagonism of PPARβ/δ inhibits endothelial cell migration and tube formation in vitro
Cellular organization is a central process in neovascularization, which involves endothelial cell migration and tube formation [39,40]. Since disruption of the PPARβ/δ gene caused smaller CNV lesions in vivo, whether pharmacological antagonism of PPARβ/δ could inhibit angiogenesis, using functional assays measuring endothelial migration and tube formation, was determined. Basic fibroblast growth factor (bFGF) was used to induce migration in a scrape wound assay in cultures of choroidal endothelial cells in the presence of the PPARβ/δ agonist, GW0742 or the PPARβ/δ antagonist GSK0660 (Figure 6A). Choroidal endothelial cell migration into the wound was inhibited by antagonism of PPARβ/δ with GSK0660 (Figure 6, A and B), while ligand activation of PPARβ/δ with GW0742 did not influence cell migration. The effect of ligand activation and pharmacological antagonism of PPARβ/δ on the ability of endothelial cells to form a three-dimensional network, indicative of vascular morphogenesis was also assessed (Figure 6C). Similarly, pharmacological antagonism of PPARβ/δ with GSK0660 inhibited tube formation (tube length) in choroidal endothelial cells compared to control and growth factor induced tube formation, whereas ligand activation of PPARβ/δ did not affect tube formation (Figure 6, C and D). In cell viability assays no significant differences were seen between control and PPARβ/δ agonist or antagonist-treated endothelial cells, indicating that the changes in cell migration and tube formation are not due to PPARβ/δ agonist or antagonist-induced cell-death (Figure S9).
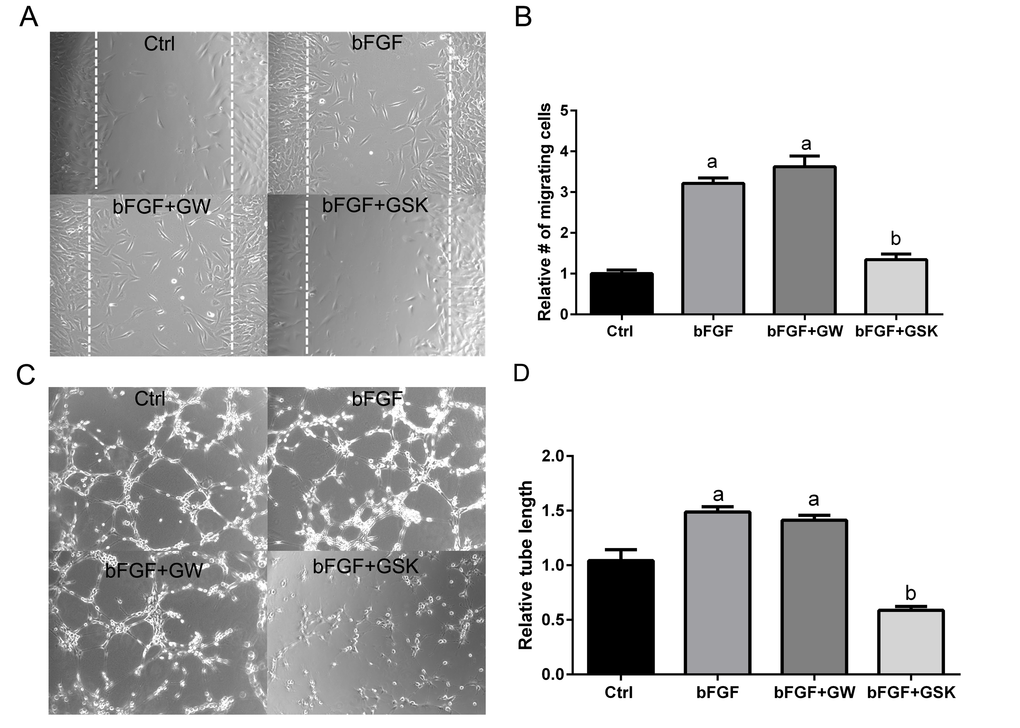
Figure 6. Antagonism of PPARβ/δ blocks endothelial cell migration and tube formation. (A) The effect of ligand activation or antagonism of PPARβ/δ on migration of RF/6A cells was analyzed in a bFGF induced wound-healing assay (n = 3, representative images at t = 36 hours are shown); dotted lines demarcate the boarders of the scrape wound. Ctrl: media only, bFGF: basic fibroblast growth factor (10 μg/ml), bFGF+GW: bFGF plus GW0742 (10μM), bFGF+GSK0660: bFGF plus GSK0660 (10 μM). (B) The cells migrating into the wound were counted using ImageJ (mean and S.E.M.; n = 3; a, p < 0.01 relative to Ctrl; b, p < 0.01 relative to bFGF, one way ANOVA, Tukey’s multiple comparisons test). (C) The effect of ligand activation or antagonism of PPARβ/δ on bFGF-induced tube formation in RF/6A cells was analyzed by an angiogenesis assay in Geltrex™ (n = 3; representative images at t = 3 hours are shown). Ctrl: media only, bFGF: basic fibroblast growth factor (10 μg/ml), bFGF+GW: bFGF plus GW0742 (10 μM), bFGF+GSK0660: bFGF plus GSK0660 (10 μM). Suramin, an inhibitor of tube formation, was used as a negative control (data not shown). (D) Quantification of tube length in Geltrex™ using ImageJ (mean and S.E.M.; n = 3; a, p < 0.01 relative to Ctrl; b, p < 0.01 relative to bFGF, one way ANOVA, Tukey’s multiple comparisons test).
Pharmacological antagonism of PPARβ/δ attenuates CNV lesions in vivo
The therapeutic potential of inhibiting PPARβ/δ activity on CNV formation in 12-13 month old aged Pparβ/δ+/+ mice following experimentally induced laser injury to the back of the eye was examined. Mice were treated with vehicle control, GW0742 (0.5 mg/kg/day) or GSK0660 (1 mg/kg/day) (Figure 7A). The distribution, area, and volume of the lesions in flatmounts of posterior eye-cups stained with isolectin GS-IB4 were assessed. A treatment dependent difference in the distribution of merged lesions was observed. Antagonism of PPARβ/δ with GSK0660 caused a higher proportion of individual neovascular lesions as compared to vehicle or agonist treated mice (Figure 7B). Additionally, the area and volume of the lesions from animals treated with the PPARβ/δ antagonist were significantly lower relative to vehicle control and PPARβ/δ agonist treated mice (Figure 7, C and D). Finally, examination of cross-sections of the retina/RPE/choroid containing CNV lesions revealed decreased deposition of COL4 within the lesions of mice treated with the PPARβ/δ antagonist, while no difference was observed in the distribution of FN1 (Figure 8, A, B and C). Interestingly, this acute treatment with agonist and antagonist did not effect the distribution of Iba1+ and F4/80+ cells within the neovascular lesion of aged Pparβ/δ+/+ mice (data not shown). Noteworthy, this short-term treatment with PPARβ/δ agonist or antagonist, also, did not affect the integrity of the RPE tight junctions (Figure S10). Overall, these results are consistent with the in vitro studies and demonstrate that inhibition of PPARβ/δ activity results in a less severe CNV phenotype and may therefore be beneficial in attenuating lesion formation in vivo.
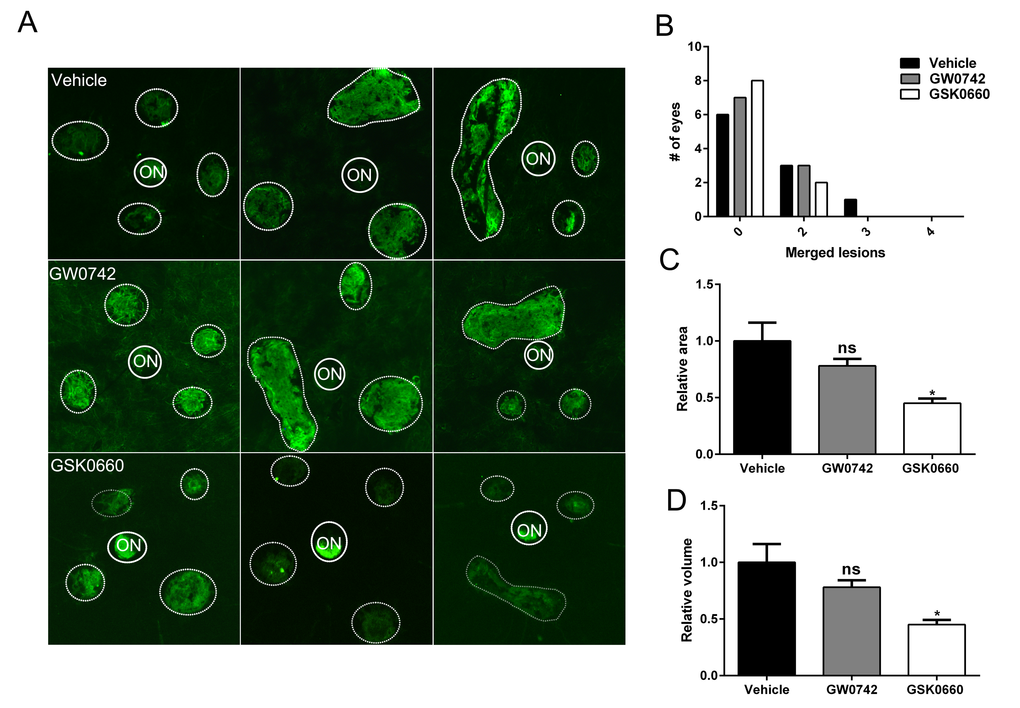
Figure 7. Antagonism of PPARβ/δ pathway attenuates CNV. (A) Choroidal flat-mounts were prepared from 12-13 month-old C57BL/6J (Pparβ/δ+/+) mice subjected to laser CNV and treated with vehicle control (1% DMSO in saline), GW0742 (0.5 mg/kg/day, i.p.), and GSK0660 (1 m/kg/day, i.p.) and stained with isolectin-GS-IB4 (n=10 eyes/group, 40 lesions; ON, optic nerve). Representative images from three eyes/treatment are shown to demonstrate individual and merged CNV lesions (dotted and solid line circles demarcate lesions and ONs, respectively). (B) Distribution of number of eyes with individual versus merged lesions in vehicle control, GW0742 and GSK0660. (C) Relative lesion area/animal was measured using ImageJ (n=10 eyes per group, mean and S.E.M.; *p < 0.01, one way ANOVA, Tukey’s multiple comparisons test). (D) Relative lesion volume/animal (n=10 eyes per group, mean and S.E.M. for each group; *p < 0.05. ns: not significant, one way ANOVA, Tukey’s multiple comparisons test).
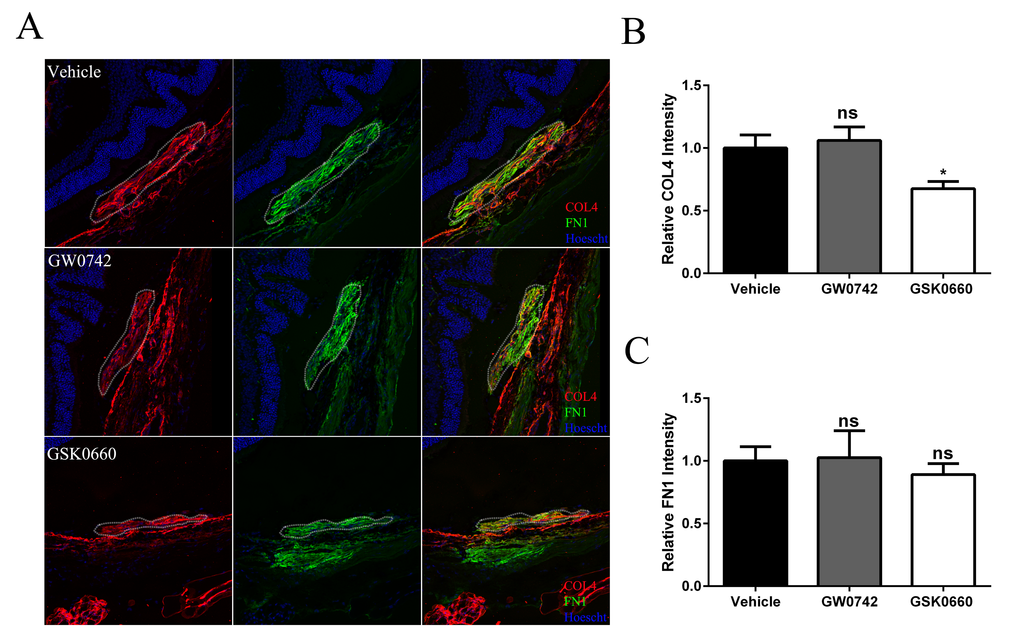
Figure 8. Antagonism of PPARβ/δ decreases accumulation of collagen type 4 in CNV lesions. (A) FN1 (green) and COL4 (red) immunolocalization in CNV lesions of mice treated with vehicle control (1% DMSO in saline), GW0742 (0.5mg/kg/day, i.p.), or GSK0660 (1m/kg/day, i.p.) (dotted oval demarcates the lesion area; nuclei are stained blue with Hoechst; representative images are shown; scale bar = 50 µm). (B) COL4 and (C) FN1 staining intensity was quantified in the CNV lesions using ImageJ (Mean and S.E.M.; n = 3/group; *p < 0.01; ns: not significant, one way ANOVA, Tukey’s multiple comparisons test).
Discussion
These studies demonstrate for the first time a role of PPARβ/δ in regulating different aspects of extracellular matrix turnover, angiogenesis, inflammation, and lipid processing in the eye. PPARβ/δ affects the RPE and choroidal endothelium differentially, selectively impacting the development of several fundamental AMD phenotypes. These data show that PPARβ/δ activity is functionally important in RPE and choroidal endothelial cell models systems; cells that are compromised during the initiation and progression of AMD. Knockdown of PPARβ/δ expression led to upregulation of extracellular matrix gene expression in primary RPE cells but a downregulation in choroidal endothelial cells. Additionally, the observed PPARβ/δ-dependent downregulation of the expression of factors critical for angiogenesis, including VEGFA, PDGFRB and TGFB, in both cell populations, supports the hypothesis that PPARβ/δ regulates signaling pathways important in development of neovascular lesions. Consistent with the in vitro studies, a significant decrease in area and volume of laser induced neovascular lesions was observed in both aged Pparβ/δ-/- mice and in Pparβ/δ+/+ mice following pharmacological antagonism of the receptor. In contrast to these findings, morphological characterization of the ocular phenotype of aged Pparβ/δ–/– mice revealed the exacerbation and development of several features of the early dry AMD phenotype including continuous sub-RPE deposits, increased RPE autofluorescence, Bruch’s membrane thickening, RPE pigmentary changes and disorganized basal infoldings. These in vivo results are supported by in vitro data demonstrating increased dysregulation of extracellular matrix molecules following PPARβ/δ knockdown in human RPE cells. Collectively, these studies illustrate, for the first time, cell-specific effects of PPARβ/δ in two populations of AMD vulnerable cells (Figure 9). Our findings correlate with the concept of selective modulation of PPARs and other nuclear receptors such as the estrogen receptor, in which ligand binding may lead to differential gene expression and biological responses in different cells and tissues [41,42]. Additionally, part of the variability observed in the eye, may be due to differential expression of PPARβ/δ in the RPE versus choroidal endothelial cells (Figure 1A); differential expression of the receptor co-regulatory proteins including co-activators and co-repressors; differential binding affinities of the co-regulatory proteins; and/or varying receptor conformational changes induced by endogenous ligand binding in epithelial versus endothelial cells [43]. This is an area of ongoing investigation.
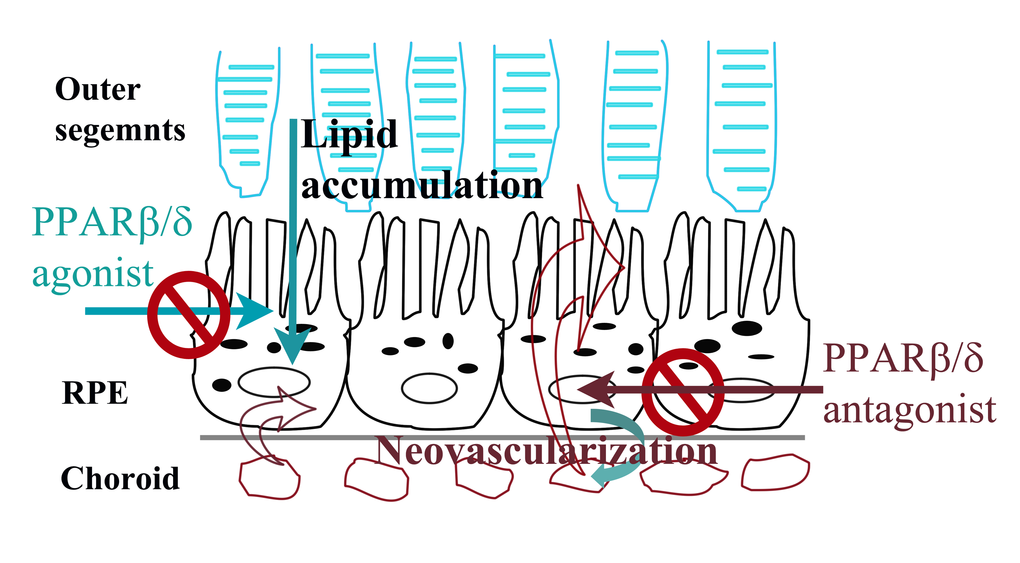
Figure 9. Summary model of selective regulation of dry and wet AMD related pathogenic pathways by PPARβ/δ in AMD vulnerable cells. Ligand activation of PPARβ/δ inhibits lipid accumulation by RPE cells and may have therapeutic effect in dry-AMD. In contrast, antagonism of PPARβ/δ inhibits neovascularization possibly by regulating inflammation and fibrosis in outer-retina.
The increased influx of immune cells within the neovascular lesion in the absence of PPARβ/δ expression is an interesting finding that requires further investigation and may be clarified with full characterization of the polarization status of these immune cells. Recently, PPARβ/δ has been shown to be involved in alternative activation of macrophages in mice [44]. Reportedly, both IL-13 and IL-4 induce expression of PPARβ/δ and modulate the ability of adipose tissue and liver macrophages to transition to the M2 phenotype. It has also been shown that PPARβ/δ coordinates the immune phenotype of alternatively activated macrophages, both in vitro and in vivo [45]. These reports suggest that the PPARβ/δ regulates macrophage polarization in animal models of liver-injury and atherosclerosis. However, it is important to note that to date, no one has critically examined the specific subtype of M2 macrophages actually regulated by PPARβ/δ, and since different subtypes have different functions [37], the precise effect of this particular change requires further investigation. Based on these studies it is plausible that the population of Iba1+ cells observed in the CNV lesions of Pparβ/δ–/– mice are transient and may express markers of the alternatively activated macrophage phenotype, which are involved in immunosuppression and tissue remodeling [19,38]. CNV lesions in Pparβ/δ–/– mice also displayed a reduced deposition of COL4 and FN1 in the lesion supporting a decrease in fibrosis. This is a major factor in responsiveness to anti-VEGF treatment for neovascular AMD [46,47]. To our knowledge this is the first study to report the involvement of the PPARβ/δ pathway in CNV formation.
To date, no differences in PPARβ/δ expression between normal and AMD cohorts have been reported. This may be due to the fact that evaluating the activity of PPARβ/δ along with expression of co-factors and specific target genes is more informative than measuring expression of the receptor in silos. Nevertheless, the purported role of PPARβ/δ in other tissues [13,48–50] in combination with our data from PPARβ/δ knockdown in vitro, and genetic ablation in vivo, gave rise to the hypotheses that pharmacologic targeting of PPARβ/δ may therapeutically improve choroidal neovascularization and lipid accumulation. To test our first hypothesis, the postulate that pharmacologic antagonism of PPARβ/δ could therapeutically improve choroidal neovascularization, the effect of ligand activation and pharmacological antagonism of PPARβ/δ on endothelial migration and tube formation in the choroidal endothelial cell line, RF/6A was determined. Pharmacological antagonism of PPARβ/δ activity inhibited choroidal endothelial cell migration as well as tube formation in a three-dimensional matrix. By contrast, ligand activation of PPARβ/δ had no effect in these angiogenesis assays. These results are consistent with previous studies by others using human retinal microvascular endothelial cells and a rat model of oxygen-induced retinopathy [51]. Modeling retinal neovascularization, it was shown that pharmacological antagonism of PPARβ/δ significantly decreased serum-induced retinal endothelial cell proliferation and tube formation in a dose-dependent manner, and in vivo decreased retinal neovascularization in young rats. Another study from the same group showed that pharmacological antagonism of PPARβ/δ stabilized tight junctions in retinal microvascular endothelial cells treated with VEGF, and reduced VEGFR1/2 expression, suggesting a role in retinal vascular permeability [52]. The present study examined the effect of ligand activation and pharmacological antagonism of PPARβ/δ on choroidal neovascularization, in which the highly fenestrated choriocapillaris; vasculature supporting the outer retina; grow through Bruch’s membrane under the RPE, in vivo, using a laser-induced CNV model. Antagonism of PPARβ/δ provided a therapeutic effect on laser-induced lesion formation, whereas ligand activation of PPARβ/δ had no effect, similar to results obtained from our in vitro analyses. Interestingly, antagonizing PPARβ/δ activity did not negatively impact the integrity of the RPE cell tight junctions. This may be due to the fact that antagonism of PPARβ/δ was acute rather than long term. Equally likely is that genetic ablation of the receptor (as seen in vivo in our mice, but not reported in humans) is detrimental to the RPE cells rather than receptor antagonism itself.
Our second hypothesis supports the notion that it is critical to investigate the therapeutic potential of targeting PPARβ/δ activity in the pathogenesis of dry AMD. Specifically, whether PPARβ/δ activity can be targeted for removal of lipid-rich deposits within Bruch’s membrane and sub-RPE deposits should be examined in more detail. Several studies have found that ligand activation of PPARβ/δ inhibits lipid accumulation as well as expression of pro-inflammatory cytokines in THP-1 macrophages and mouse monocytes [53,54]. Though, the sub-RPE deposits in the aged Pparβ/δ–/– mice contain apoE, they did not stain positively for oil red o or filipin and do not appear to contain neutral lipids or cholesterol (data not shown). As proof of concept, using an in vitro model of lipid-loaded RPE cells, it was found that ligand activation of PPARβ/δ decreased lipid accumulation. Taken together, these results support the need to test the effect of PPARβ/δ agonists in conditions where there are systemic and/or dietary factors contributing to development of early AMD. Though there is selective modulation of the PPARβ/δ in RPE and choroidal endothelial cells, based on the in vitro and in vivo data, ligand activation of PPARβ/δ is beneficial to RPE cells and does not appear to have a negative impact on healthy choroidal endothelial cells. Currently there are no FDA-approved PPARβ/δ drugs, but the recent development of highly specific and selective agonists and antagonists [selective PPARβ/δ modulators (SPPARMs)] such as GW501516, GW0742, GSK0660 and GSK3787 have led to improved approaches to study the PPARβ/δ pathway in disease models including obesity and atherosclerosis [55]. In future studies, it will be imperative to construct PPARβ/δ-selective drug-dose response curves to describe cell-selective responses in a quantitative manner and test the suitability of pharmacological agents in long-term studies using aged animal models of AMD that develop lipid accumulation within Bruch’s membrane.
This is the first study to report on the PPARβ/δ pathway in RPE and choroidal endothelial cells and its potential role in the pathogenesis of AMD. The results indicate, for the first time, selective modulation of a nuclear receptor in the eye. Specifically, that inhibition of PPARβ/δ activity may successfully attenuate neovascular lesion formation concomitant with decreased expression of extracellular matrix molecules and decreased angiogenic factors, in vivo, while activation of PPARβ/δ activity may target lipid accumulation. The results of this treatment may be attributable to PPARβ/δ regulation of several distinct AMD pathogenic pathways: fibrosis, inflammation and lipid metabolism. The Pparβ/δ–/– mice also displayed early dry AMD like pathology, establishing these mice as a model to further study the initiation and progression of the early sub-type of the disease. Additional studies should be conducted to study the contribution of other PPARβ/δ regulatory pathways that may influence disease pathology and the therapeutic potential of selectively targeting PPARβ/δ as a means to inhibit inflammation, fibrosis and lipid accumulation (Figure 9).
Materials and Methods
Cell lines
RF/6A cells, a spontaneously transformed choroidal endothelial cell line derived from the eyes of a rhesus macaque fetus, passages 35–40, and ARPE19 cells, a spontaneously arising human RPE cell line derived from the eyes of a 19 year-old male donor, passages 21–28, were obtained from ATCC (Manassas, VA, USA). Primary cell cultures used included RPE cells (1° RPE) isolated from donor eyes older than 60 years (n=3), collected from the North Carolina Organ Donor and Eye Bank Inc. in less than 6 hours post-mortem and cultured within 24 hours in accordance with the Declaration of Helsinki for research involving human tissue, as previously described [56]. Donor eyes did not reveal any evidence of retinal/RPE changes upon post-mortem evaluation of the posterior eye under a dissecting microscope. Only passages between 4 and 9 of each of the primary RPE cell cultures were used in this study. Final conditions of both ARPE19 and the primary RPE cell cultures were such that cells were post-confluent and demonstrated zonula occludens positive immunoreactivity.
Transcriptional activation assay
The transcriptional activity of PPARβ/δ was measured using a luciferase-based reporter assay and PPARβ/δ target gene expression was quantified using qPCR. Briefly, 50000 human primary RPE cells, ARPE19 cells or RF/6A cells/well were seeded in 24-well plates in phenol red-free medium supplemented with 7.5% charcoal-stripped FBS and cultured overnight. Lipofectin (Invitrogen)-mediated transfection was performed the following day, using plasmids encoding a DR-1 luciferase reporter, CMV-β galactosidase or pBSII, as described previously [57,58]. After overnight culture, cells were transfected with small interfering RNAs (siRNA) (scrambled control siRNA or an siRNA against PPARβ/δ; [100 pmoles per 250,000 cells]); 5 hours after knock-down, the transfected cells were treated with PPARβ/δ agonist and antagonist at doses listed in Supplementary Table S1. The cells were lysed 24 hours later for luminescence reading. Luciferase (reporter) and β-galactosidase [chlorophenol red β-d-galactopyranoside (CPRG) as substrate; transfection normalization] activities were measured using a Perkin-Elmer fusion instrument. Concomitantly, cells were treated with these same compounds for RNA isolation and target gene expression studies. All samples were run in triplicate and experiments were performed a minimum of three times.
siRNA transfection and cell functional assays
siRNAs were used to knock down PPARβ/δ in vitro. A control siRNA (siC) for a non-targeting sequence or 5 different PPARβ/δ siRNAs (siPPARβ/δ, siPPARβ/δ1, siPPARβ/δ2, siPPARβ/δ3, siPPARβ/δ4; GE Dharmacon, Lafayette, CO, USA; Supplementary Table S2) were transfected into cells using Lipofectamine RNAiMAX (Invitrogen, Grand Island, NY, USA) and tested to account for non-specific targeting of the siRNA (Figure S11). Briefly, complexes of siRNA (100 pmoles per 250,000 cells) and RNAiMAX in OPTI-MEM were added to six-well cell-culture plates according to the manufacturer’s protocol. siPPARβ/δ was used in subsequent experiments. ARPE19, 1° RPE or RF/6A cells (250 000 cells/well) were added to each well, along with DMEM/F12 or MEM supplemented with 10% charcoal-stripped fetal bovine serum (CS-FBS) for ARPE19 or 1° RPE, and endothelial cells, respectively. Cells were used 24 hours post-transfection, in cell viability assays, scrape wound migration assays (MEM 1% CS-FBS), tube-formation assays (MEM 1% FBS) and PPAR activity assays (7.5% CS-FBS), as described. RNA was extracted at the indicated time points for quantitative real-time PCR (qPCR). Primer sequences are provided in Supplementary Table S3.
RNA Isolation and qPCR
Total RNA isolation from cultured cells, freshly isolated human RPE cells and choroid; RNA quality assessment; cDNA reverse transcription; and qPCR were completed as previously described [59]. The purities of the freshly isolated human RPE cells and choroid from aged donor eyes were determined by measuring the expression levels of RPE specific markers including BEST1 and RPE65 and a vascular marker, PECAM. Minimal levels of cross contamination were found (Figure S12). qPCR was performed using the Bio-Rad CFX96 Realtime PCR Detection System (Bio-Rad). Melt curves for each pair of primers were inspected to confirm a single amplicon. The Ct values were normalized to a housekeeping gene (acidic ribosomal phosphoprotein P0, 36B4). Gene-expression fold changes were calculated using the ΔΔCT method. Primer sequences used were selected from Primer Bank, http://pga.mgh.harvard.edu/primerbank and are presented in Supplementary Table S3. The amplification products obtained after qPCR using PPARβ/δ, RXRα, RXRβ, BEST1, RPE65, PECAM and 36B4 primers were run on a 1% agarose gel and visualized with ethidium bromide.
Adipored assay
AdipoRed™ (Lonza, Walkersville, MD) assay was used to measure intracellular lipid accumulation according to manufacturer’s protocol. Briefly, human primary RPE cells were plated in a 96-well plate (10000 cells/well) in DMEM/F12 media with 7.5% charcoal stripped FBS. The cells were treated with different lipids (20μM) namely, α-linolenic acid (α-LA), docohexanoic acid (DHA), arachidonic acid (AA), and palmitic acid (PA) for 48 hours, followed by a 24 hour treatment with GW0742 (10 μM) and GSK0660 (10 μM). The cells were then washed with PBS and treated with AdipoRed™ reagent. After 10 minute incubation, the fluorescence was read at 485 nm excitation and 572 nm emission.
Animals
Male and female Pparβ/δ+/+ and Pparβ/δ–/– mice on the C57BL/6 background [60], aged (Pparβ/δ+/+: n=13, 13 females, 3 males; Pparβ/δ–/–: n=10, 7 females, 3 males) were maintained in a temperature (25 °C) and light controlled (12h light/12 h dark) environment and provided standard mouse chow ad libitum. Mice were screened for the confounding retinal degeneration 8 mutation and its absence was confirmed as previously described [56].
Study Approval
The study protocols were approved by the Duke University or Pennsylvania State University Institutional Animal Care and Use Committees. All animal experiments were performed in accordance with the guidelines of the ARVO statement for the Use of Animals in Ophthalmic and Vision Research.
Immunohistochemistry and morphology
For immunohistochemistry, eyes were fixed in 4% paraformaldehyde and cryopreserved. Specimens were cryosectioned from the superior cup through the optic nerve to the inferior cup in 10 µm increments. Cryosections from the nasal, central and peripheral regions of the eye were probed with antibodies (Supplementary Table S4). Non-specific immunostaining in sections was blocked with normal serum (Jackson Immunoresearch, West Grove, PA, USA) appropriate to the secondary antibody species. Secondary antibodies were conjugated to AlexaFluor 568 and 488 (Invitrogen). Control slides containing sequential sections were probed with non-immune serum and buffer without primary antibody. Nuclei were stained with Hoechst 33258 (Invitrogen). Images were collected on a Nikon C1 confocal microscope and visualized and processed using Nikon EZ-C1 Free viewer.
Transmission electron microscopy
For electron microscopy, eyes were fixed in 2% gluteraldehyde, post fixed in 1% osmium tetroxide, and embedded in Spurrs resin. Morphology of the retina/RPE/choroid was studied in 1 μm toluidine blue stained plastic sections. The length of deposits/RPE length was calculated and plotted for Pparβ/δ+/+ and PPARβ/δ−/− mice (10 images per mice, n=4 mice per genotype). Incidence of pathology in PPARβ/δ−/− eyes, including RPE abnormalities (seen in greater than 20% of the eye), presence of sub-RPE deposits, and thickness of Bruch’s membrane, were evaluated in electron microscopy thin sections (10 images per mouse, n = 4 mice per genotype).
Electroretinography (ERG)
ERGs were recorded using the Espion E2 system (Diagnosys LLC) as described previously [56]. Briefly, 14-16-mo-old Pparβ/δ+/+ and PPARβ/δ−/− mice were dark-adapted for four hours and anesthetized by an i.p. injection of a ketamine/xylazine mixture (85/10 mg/kg). Pupils were dilated with 1% cyclopentolate-HCl and 2.5% phenylephrine, and the mouse body temperature was maintained at 37 °C using a water-based warming pad. ERG responses under dark-adapted (“scotopic”) conditions were evoked by a series of nine flashes ranging from 0.0001 cd·s/m2 to 100 cd·s/m2. For flashes up to 0.1 cd·s/m2, responses of 10 trials were averaged. For 0.5 and 1 cd·s/m2 flash responses, three trials were averaged. For brighter stimuli, responses to single flashes were recorded without averaging. Light-adapted (“photopic”) ERGs were evoked by a series of six flashes ranging from 0.2 cd·s/m2 to 2,000 cd·s/m2 whereas rod inputs were suppressed with a steady background illumination of 50 cd/m2. Up to 10 trials were averaged for all flash responses. Analysis of a- and b-wave amplitudes was performed as described [56]
Mouse model of CNV
Laser photocoagulation was performed in cohorts of 18-20 month old Pparβ/δ+/+ (n=7, all females) and PPARβ/δ−/− mice (n=4, 3 females, 1 male), as previously described [59]. Briefly, four thermal burns were induced in each eye around the optic nerve, using a slit lamp delivery system. The mice were euthanized 3 weeks after laser treatment and the eyes were harvested for visualization of laser-induced CNV in posterior pole flat-mounts, or cryopreserved for immunohistochemistry and morphology experiments. To test the efficacy of PPARβ/δ drugs, 12-13-month old Pparβ/δ+/+ mice were divided into three cohorts (n=10/cohort, 5 females and 5 males per cohort), vehicle control (1% DMSO in saline), GW0742 (0.5 mg/kg/day, i.p.) [61,62] and GSK0660 (1 mg/kg/day, i.p.) [63]. Animals were pre-treated with the drugs for 2 days prior to laser CNV induction and euthanized 10 days later. CNV lesion volume, area and size were measured in flat-mounts stained with isolectin GS-IB4 to examine vascularity of the neovascular lesion.
Evaluation of mouse CNV lesions
Following laser CNV, flatmounts of the posterior pole were stained with isolectin GS-IB4 Alexa Fluor® conjugate (Life Technologies, Grand Island, NY) according to manufacturer’s protocol, to examine vascularity and size of the neovascular lesion, and visualized by a Zeiss Axiolan 2 (Carl Zeiss, Thornwood, NY) fluorescent microscope. Horizontal optical section images of the flatmounts were obtained at 1.50 µm intervals. CNV lesion thickness was measured using Nikon EZ-C1 viewer software. Total area of the CNV lesions per lesion per animal was measured using ImageJ software (developed by Wayne Rasband, National Institute of Health, Bethesda, MD) and the volume of the lesions was measured by running the MeasureStacks script in ImageJ. All measurements were normalized to Pparβ/δ+/+ or vehicle control in their respective experiment. Cryosections from the lasered eyes were screened for lesions and stained with F4/80 and Iba1 antibodies (Supplementary Table S4). CNV was demarcated and cells staining positive for F4/80 and Iba1 were counted and plotted. To study extracellular matrix deposition, cryosections were stained with FN1 and COL4 antibodies. CNV lesions were demarcated and mean fluorescence intensity of FN1 and COL4 was measured by ImageJ. Mean intensity was plotted.
Scrape wound migration assay
Following PPARβ/δ knock-down in RF/6A cells by siRNA transfection, as described in the main text, 250000 cells were added to each well in six-well plates; 24 hours after knock-down, the cell monolayer was scraped in a direction perpendicular to a horizontal line, using a 1000 µl pipette tip, to create a wound. bFGF (100 ng/ml)-induced cell motility was observed at t = 0 and 36 hours post-scraping. The total number of cells migrating into the wound at t = 36 were counted using ImageJ and normalized to control siRNA. Similarly RF/6A cells were pretreated with PPARβ/δ agonist, GW0742 or antagonist, GSK0660, at doses listed in Supplementary Table S1 for 24 hours. It was followed by scrape wound assay. Data were generated from four fields of view/experiment in a total of three biological replicates.
Tube-formation assay
Tube formation assay was used as a model for angiogenesis. Geltrex™ (Life Technologies, Grand Island, NY, USA) was thawed overnight at 4°C. Using cold pipette tips, 200 μl/well was added in a 12-well plate. The Geltrex solidified into a thin layer after incubation at 37°C for 1 hour. PPARβ/δ was knocked down in RF/6A endothelial cells by siRNA transfection, trypsinized after 48 hours and plated onto the Geltrex-coated wells (2.5 × 104 cells/well). Network formation was examined after 3 hours, using an inverted phase-contrast microscope, and quantified as total tube length formed by siPPARβ/δ-endothelial cells normalized to siControl, using ImageJ. Similarly RF/6A cells were pretreated with PPARβ/δ agonist, GW0742 or antagonist, GSK0660, at doses listed in Supplementary Table S1 for 24 hours followed by scrape wound assay. Four fields of view/experiment were examined, in a total of three biological replicates.
Cell viability and proliferation assays
Primary human RPE, ARPE19, and RF/6A cells were plated in 96-well plates at different cell densities (5000-, and 10000-cells per well) and treated with DMSO, GW0742 (10μM) and GSK0660 (10μM). Cell viability was measured after four days post-plating using CellTiter-Blue® (Promega, Madison, WI), according to the manufacturer’s protocol. The results obtained from 10000-cells/well are reported.
Lipofuscin Quantification
A Leica Spectral Laser Scanning Confocal Microscope was used to measure autofluorescence throughout the entire RPE layer of 18-mo-old PPARβ/δ-/- and Pparβ/δ+/+ cryosections (n = 3 sections per mouse, n = 3 mice per genotype) as previously described [64]. The three sections were selected randomly from the nasal, central, and temporal regions of the mouse eye. Lambda (λ) scans were performed using a 405-nm laser. Excitation and emission frequencies were measured with a 5-nm-wide band through a spectral range from 422.5 nm to 722.5 nm using serial 30-image scans at ∼10.3-nm intervals. Fluorescent intensities are represented as arbitrary units as defined by the confocal Leica software. The significance of differences in spectra obtained between PPARβ/δ-/- and Pparβ/δ+/+ mice was assessed using a two-tailed t test, with no variance assumptions.
Statistics
Statistical methods for data analysis included two-tailed Student's t-test and two-way ANOVA, with Sidak's multiple comparison test using GraphPad Prism. Values were considered statistically significant at p < 0.05.
Supplementary Materials
Acknowledgements
We thank Mr. Edwin Meade for help with immunohistochemistry, and Mr. Peter Saloupis for assistance with animal experiments, Dr. Donald P. McDonnell and Dr. Ching-Yi Chang for plasmids used in transcriptional activity assays and valuable discussion. Many thanks to Mrs. Ying Hao for assistance with electron microscopy. We thank Dr. Scott Cousins for allowing us to utilize his slit lamp and laser.
Funding
This work was supported by the National Eye Institute grant EY02868 (GM), NEI P30 EY005722 (Duke Eye Center) and Research to Prevent Blindness Core Grant (Duke Eye Center).
Conflicts of Interest
The authors declare that no conflict of interest exists.
References
- 1. Malek G and Lad EM. Emerging roles for nuclear receptors in the pathogenesis of age-related macular degeneration. Cell Mol Life Sci. 2014; 71:4617–36. https://doi.org/10.1007/s00018-014-1709-x [PubMed]
- 2. Ambati J, Ambati BK, Yoo SH, Ianchulev S, Adamis AP. Age-related macular degeneration: etiology, pathogenesis, and therapeutic strategies. Surv Ophthalmol. 2003; 48:257–93. https://doi.org/10.1016/S0039-6257(03)00030-4 [PubMed]
- 3. Ambati J and Fowler BJ. Mechanisms of age-related macular degeneration. Neuron. 2012; 75:26–39. https://doi.org/10.1016/j.neuron.2012.06.018 [PubMed]
- 4. Huang JD, Presley JB, Chimento MF, Curcio CA, Johnson M. Age-related changes in human macular Bruch’s membrane as seen by quick-freeze/deep-etch. Exp Eye Res. 2007; 85:202–18. https://doi.org/10.1016/j.exer.2007.03.011 [PubMed]
- 5. Curcio CA, Johnson M, Huang JD, Rudolf M. Apolipoprotein B-containing lipoproteins in retinal aging and age-related macular degeneration. J Lipid Res. 2010; 51:451–67. https://doi.org/10.1194/jlr.R002238 [PubMed]
- 6. Curcio CA, Johnson M, Huang JD, Rudolf M. Aging, age-related macular degeneration, and the response-to-retention of apolipoprotein B-containing lipoproteins. Prog Retin Eye Res. 2009; 28:393–422. https://doi.org/10.1016/j.preteyeres.2009.08.001 [PubMed]
- 7. Hee MR, Baumal CR, Puliafito CA, Duker JS, Reichel E, Wilkins JR, Coker JG, Schuman JS, Swanson EA, Fujimoto JG. Optical coherence tomography of age-related macular degeneration and choroidal neovascularization. Ophthalmology. 1996; 103:1260–70. https://doi.org/10.1016/S0161-6420(96)30512-5 [PubMed]
- 8. Chen Y, Vuong LN, Liu J, Ho J, Srinivasan VJ, Gorczynska I, Witkin AJ, Duker JS, Schuman J, Fujimoto JG. Three-dimensional ultrahigh resolution optical coherence tomography imaging of age-related macular degeneration. Opt Express. 2009; 17:4046–60. https://doi.org/10.1364/OE.17.004046 [PubMed]
- 9. Li CM, Chung BH, Presley JB, Malek G, Zhang X, Dashti N, Li L, Chen J, Bradley K, Kruth HS, Curcio CA. Lipoprotein-like particles and cholesteryl esters in human Bruch’s membrane: initial characterization. Invest Ophthalmol Vis Sci. 2005; 46:2576–86. https://doi.org/10.1167/iovs.05-0034 [PubMed]
- 10. Strauss O. (1995). The Retinal Pigment Epithelium. In: Kolb H, Fernandez E and Nelson R, eds. Webvision: The Organization of the Retina and Visual System. (Salt Lake City (UT).
- 11. Whitmore SS, Braun TA, Skeie JM, Haas CM, Sohn EH, Stone EM, Scheetz TE, Mullins RF. Altered gene expression in dry age-related macular degeneration suggests early loss of choroidal endothelial cells. Mol Vis. 2013; 19:2274–97. [PubMed]
- 12. Green WR, McDonnell PJ, Yeo JH. Pathologic features of senile macular degeneration. Ophthalmology. 1985; 92:615–27. https://doi.org/10.1016/S0161-6420(85)33993-3 [PubMed]
- 13. Peters JM, Foreman JE, Gonzalez FJ. Dissecting the role of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) in colon, breast, and lung carcinogenesis. Cancer Metastasis Rev. 2011; 30:619–40. https://doi.org/10.1007/s10555-011-9320-1 [PubMed]
- 14. Burdick AD, Kim DJ, Peraza MA, Gonzalez FJ, Peters JM. The role of peroxisome proliferator-activated receptor-beta/delta in epithelial cell growth and differentiation. Cell Signal. 2006; 18:9–20. https://doi.org/10.1016/j.cellsig.2005.07.009 [PubMed]
- 15. Malek G. Nuclear receptors as potential therapeutic targets for age-related macular degeneration. Adv Exp Med Biol. 2014; 801:317–21. https://doi.org/10.1007/978-1-4614-3209-8_40 [PubMed]
- 16. Hollingshead HE, Killins RL, Borland MG, Girroir EE, Billin AN, Willson TM, Sharma AK, Amin S, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) ligands do not potentiate growth of human cancer cell lines. Carcinogenesis. 2007; 28:2641–49. https://doi.org/10.1093/carcin/bgm183 [PubMed]
- 17. Zarbin MA. Current concepts in the pathogenesis of age-related macular degeneration. Arch Ophthalmol. 2004; 122:598–614. https://doi.org/10.1001/archopht.122.4.598 [PubMed]
- 18. Ng EW and Adamis AP. Targeting angiogenesis, the underlying disorder in neovascular age-related macular degeneration. Can J Ophthalmol. 2005; 40:352–68. https://doi.org/10.1016/S0008-4182(05)80078-X [PubMed]
- 19. Ambati J, Atkinson JP, Gelfand BD. Immunology of age-related macular degeneration. Nat Rev Immunol. 2013; 13:438–51. https://doi.org/10.1038/nri3459 [PubMed]
- 20. Anderson DH, Radeke MJ, Gallo NB, Chapin EA, Johnson PT, Curletti CR, Hancox LS, Hu J, Ebright JN, Malek G, Hauser MA, Rickman CB, Bok D, et al. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res. 2010; 29:95–112. https://doi.org/10.1016/j.preteyeres.2009.11.003 [PubMed]
- 21.
- 22.
- 23. Bojic LA, Burke AC, Chhoker SS, Telford DE, Sutherland BG, Edwards JY, Sawyez CG, Tirona RG, Yin H, Pickering JG, Huff MW. Peroxisome proliferator-activated receptor δ agonist GW1516 attenuates diet-induced aortic inflammation, insulin resistance, and atherosclerosis in low-density lipoprotein receptor knockout mice. Arterioscler Thromb Vasc Biol. 2014; 34:52–60. https://doi.org/10.1161/ATVBAHA.113.301830 [PubMed]
- 24. Matsushita Y, Ogawa D, Wada J, Yamamoto N, Shikata K, Sato C, Tachibana H, Toyota N, Makino H. Activation of peroxisome proliferator-activated receptor delta inhibits streptozotocin-induced diabetic nephropathy through anti-inflammatory mechanisms in mice. Diabetes. 2011; 60:960–68. https://doi.org/10.2337/db10-1361 [PubMed]
- 25. Hageman GS, Mullins RF, Russell SR, Johnson LV, Anderson DH. Vitronectin is a constituent of ocular drusen and the vitronectin gene is expressed in human retinal pigmented epithelial cells. FASEB J. 1999; 13:477–84. [PubMed]
- 26. Zhou Z, Pausch F, Schlotzer-Schrehardt U, Brachvogel B, Poschl E. Induction of initial steps of angiogenic differentiation and maturation of endothelial cells by pericytes in vitro and the role of collagen IV. Histochem Cell Biol. 2016.
- 27. Davis GE, Koh W, Stratman AN. Mechanisms controlling human endothelial lumen formation and tube assembly in three-dimensional extracellular matrices. Birth Defects Res C Embryo Today. 2007; 81:270–85. https://doi.org/10.1002/bdrc.20107 [PubMed]
- 28. Schnegg CI and Robbins ME. Neuroprotective Mechanisms of PPAR Modulation of Oxidative Stress and Inflammatory Processes. PPAR Res. 2011; 2011:373560. .
- 29. Choudhary M and Malek G. A Brief Discussion on Lipid Activated Nuclear Receptors and their Potential Role in Regulating Microglia in Age-Related Macular Degeneration (AMD). Adv Exp Med Biol. 2016; 854:45–51. https://doi.org/10.1007/978-3-319-17121-0_7 [PubMed]
- 30. Luquet S, Gaudel C, Holst D, Lopez-Soriano J, Jehl-Pietri C, Fredenrich A, Grimaldi PA. Roles of PPAR delta in lipid absorption and metabolism: a new target for the treatment of type 2 diabetes. Biochim Biophys Acta. 2005; 1740:313–17. https://doi.org/10.1016/j.bbadis.2004.11.011 [PubMed]
- 31. Kilgore KS and Billin AN. PPARbeta/delta ligands as modulators of the inflammatory response. Curr Opin Investig Drugs. 2008; 9:463–69. [PubMed]
- 32. Reale E, Groos S, Eckardt U, Eckardt C, Luciano L. New components of ‘basal laminar deposits’ in age-related macular degeneration. Cells Tissues Organs. 2009; 190:170–81. https://doi.org/10.1159/000187632 [PubMed]
- 33. Grossniklaus HE, Martinez JA, Brown VB, Lambert HM, Sternberg P Jr, Capone A Jr, Aaberg TM, Lopez PF. Immunohistochemical and histochemical properties of surgically excised subretinal neovascular membranes in age-related macular degeneration. Am J Ophthalmol. 1992; 114:464–72. https://doi.org/10.1016/S0002-9394(14)71859-8 [PubMed]
- 34. Newsome DA, Hewitt AT, Huh W, Robey PG, Hassell JR. Detection of specific extracellular matrix molecules in drusen, Bruch’s membrane, and ciliary body. Am J Ophthalmol. 1987; 104:373–81. https://doi.org/10.1016/0002-9394(87)90227-3 [PubMed]
- 35. Clark RA, Quinn JH, Winn HJ, Lanigan JM, Dellepella P, Colvin RB. Fibronectin is produced by blood vessels in response to injury. J Exp Med. 1982; 156:646–51. https://doi.org/10.1084/jem.156.2.646 [PubMed]
- 36. Gupta N, Brown KE, Milam AH. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp Eye Res. 2003; 76:463–71. https://doi.org/10.1016/S0014-4835(02)00332-9 [PubMed]
- 37. Hao NB, Lu MH, Fan YH, Cao YL, Zhang ZR, Yang SM. Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol. 2012; 2012:948098. .
- 38. Murray PJ and Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011; 11:723–37. https://doi.org/10.1038/nri3073 [PubMed]
- 39. Arnaoutova I, George J, Kleinman HK, Benton G. The endothelial cell tube formation assay on basement membrane turns 20: state of the science and the art. Angiogenesis. 2009; 12:267–74. https://doi.org/10.1007/s10456-009-9146-4 [PubMed]
- 40. Guo S, Lok J, Liu Y, Hayakawa K, Leung W, Xing C, Ji X, Lo EH. Assays to examine endothelial cell migration, tube formation, and gene expression profiles. Methods Mol Biol. 2014; 1135:393–402. https://doi.org/10.1007/978-1-4939-0320-7_32 [PubMed]
- 41. Maximov PY, Lee TM, Jordan VC. The discovery and development of selective estrogen receptor modulators (SERMs) for clinical practice. Curr Clin Pharmacol. 2013; 8:135–55. https://doi.org/10.2174/1574884711308020006 [PubMed]
- 42. Feldman PL, Lambert MH, Henke BR. PPAR modulators and PPAR pan agonists for metabolic diseases: the next generation of drugs targeting peroxisome proliferator-activated receptors? Curr Top Med Chem. 2008; 8:728–49. https://doi.org/10.2174/156802608784535084 [PubMed]
- 43. Martinkovich S, Shah D, Planey SL, Arnott JA. Selective estrogen receptor modulators: tissue specificity and clinical utility. Clin Interv Aging. 2014; 9:1437–52. [PubMed]
- 44. Kang K, Reilly SM, Karabacak V, Gangl MR, Fitzgerald K, Hatano B, Lee CH. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008; 7:485–95. https://doi.org/10.1016/j.cmet.2008.04.002 [PubMed]
- 45. Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, Subramanian V, Mukundan L, Ferrante AW, Chawla A. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008; 7:496–507. https://doi.org/10.1016/j.cmet.2008.04.003 [PubMed]
- 46. Channa R, Sophie R, Bagheri S, Shah SM, Wang J, Adeyemo O, Sodhi A, Wenick A, Ying HS, Campochiaro PA. Regression of choroidal neovascularization results in macular atrophy in anti-vascular endothelial growth factor-treated eyes. Am J Ophthalmol.. 2015; 159:9-19. .
- 47. Algvere PV, Steén B, Seregard S, Kvanta A. A prospective study on intravitreal bevacizumab (Avastin) for neovascular age-related macular degeneration of different durations. Acta Ophthalmol. 2008; 86:482–89. https://doi.org/10.1111/j.1600-0420.2007.01113.x [PubMed]
- 48. Wagner KD and Wagner N. Peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) acts as regulator of metabolism linked to multiple cellular functions. Pharmacol Ther. 2010; 125:423–35. https://doi.org/10.1016/j.pharmthera.2009.12.001 [PubMed]
- 49. Vazquez-Carrera M. Unraveling the Effects of PPARbeta/delta on Insulin Resistance and Cardiovascular Disease. Trends in endocrinology and metabolism. TEM. 2016; 27:319–34. [PubMed]
- 50. Peters JM and Gonzalez FJ. Sorting out the functional role(s) of peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) in cell proliferation and cancer. Biochim Biophys Acta. 2009; 1796:230–41. [PubMed]
- 51. Capozzi ME, McCollum GW, Savage SR, Penn JS. Peroxisome proliferator-activated receptor-β/δ regulates angiogenic cell behaviors and oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 2013; 54:4197–207. https://doi.org/10.1167/iovs.13-11608 [PubMed]
- 52. Suarez S, McCollum GW, Bretz CA, Yang R, Capozzi ME, Penn JS. Modulation of VEGF-induced retinal vascular permeability by peroxisome proliferator-activated receptor-β/δ. Invest Ophthalmol Vis Sci. 2014; 55:8232–40. https://doi.org/10.1167/iovs.14-14217 [PubMed]
- 53. Lee CH, Kang K, Mehl IR, Nofsinger R, Alaynick WA, Chong LW, Rosenfeld JM, Evans RM. Peroxisome proliferator-activated receptor delta promotes very low-density lipoprotein-derived fatty acid catabolism in the macrophage. Proc Natl Acad Sci USA. 2006; 103:2434–39. https://doi.org/10.1073/pnas.0510815103 [PubMed]
- 54. Bojic LA, Sawyez CG, Telford DE, Edwards JY, Hegele RA, Huff MW. Activation of peroxisome proliferator-activated receptor δ inhibits human macrophage foam cell formation and the inflammatory response induced by very low-density lipoprotein. Arterioscler Thromb Vasc Biol. 2012; 32:2919–28. https://doi.org/10.1161/ATVBAHA.112.255208 [PubMed]
- 55. Graham TL, Mookherjee C, Suckling KE, Palmer CN, Patel L. The PPARdelta agonist GW0742X reduces atherosclerosis in LDLR(-/-) mice. Atherosclerosis. 2005; 181:29–37. https://doi.org/10.1016/j.atherosclerosis.2004.12.028 [PubMed]
- 56. Hu P, Herrmann R, Bednar A, Saloupis P, Dwyer MA, Yang P, Qi X, Thomas RS, Jaffe GJ, Boulton ME, McDonnell DP, Malek G. Aryl hydrocarbon receptor deficiency causes dysregulated cellular matrix metabolism and age-related macular degeneration-like pathology. Proc Natl Acad Sci USA. 2013; 110:E4069–78. https://doi.org/10.1073/pnas.1307574110 [PubMed]
- 57. Safi R, Kovacic A, Gaillard S, Murata Y, Simpson ER, McDonnell DP, Clyne CD. Coactivation of liver receptor homologue-1 by peroxisome proliferator-activated receptor gamma coactivator-1alpha on aromatase promoter II and its inhibition by activated retinoid X receptor suggest a novel target for breast-specific antiestrogen therapy. Cancer Res. 2005; 65:11762–70. https://doi.org/10.1158/0008-5472.CAN-05-2792 [PubMed]
- 58. Hall JM and McDonnell DP. The molecular mechanisms underlying the proinflammatory actions of thiazolidinediones in human macrophages. Mol Endocrinol. 2007; 21:1756–68. https://doi.org/10.1210/me.2007-0060 [PubMed]
- 59. Choudhary M, Kazmin D, Hu P, Thomas RS, McDonnell DP, Malek G. Aryl hydrocarbon receptor knock-out exacerbates choroidal neovascularization via multiple pathogenic pathways. J Pathol. 2015; 235:101–12. https://doi.org/10.1002/path.4433 [PubMed]
- 60. Peters JM, Lee SS, Li W, Ward JM, Gavrilova O, Everett C, Reitman ML, Hudson LD, Gonzalez FJ. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor beta(delta). Mol Cell Biol. 2000; 20:5119–28. https://doi.org/10.1128/MCB.20.14.5119-5128.2000 [PubMed]
- 61. Galuppo M, Di Paola R, Mazzon E, Esposito E, Paterniti I, Kapoor A, Thiemermann C, Cuzzocrea S. GW0742, a high affinity PPAR-β/δ agonist reduces lung inflammation induced by bleomycin instillation in mice. Int J Immunopathol Pharmacol. 2010; 23:1033–46. [PubMed]
- 62. Paterniti I, Mazzon E, Riccardi L, Galuppo M, Impellizzeri D, Esposito E, Bramanti P, Cappellani A, Cuzzocrea S. Peroxisome proliferator-activated receptor β/δ agonist GW0742 ameliorates cerulein- and taurocholate-induced acute pancreatitis in mice. Surgery. 2012; 152:90–106. https://doi.org/10.1016/j.surg.2012.02.004 [PubMed]
- 63. Toral M, Gómez-Guzmán M, Jiménez R, Romero M, Zarzuelo MJ, Utrilla MP, Hermenegildo C, Cogolludo Á, Pérez-Vizcaíno F, Gálvez J, Duarte J. Chronic peroxisome proliferator-activated receptorβ/δ agonist GW0742 prevents hypertension, vascular inflammatory and oxidative status, and endothelial dysfunction in diet-induced obesity. J Hypertens. 2015; 33:1831–44. https://doi.org/10.1097/HJH.0000000000000634 [PubMed]
- 64. Cai J, Qi X, Kociok N, Skosyrski S, Emilio A, Ruan Q, Han S, Liu L, Chen Z, Bowes Rickman C, Golde T, Grant MB, Saftig P, et al. β-Secretase (BACE1) inhibition causes retinal pathology by vascular dysregulation and accumulation of age pigment. EMBO Mol Med. 2012; 4:980–91. https://doi.org/10.1002/emmm.201101084 [PubMed]