




Introduction
Expansion of gene families with the concomitant acquisition of new functions can be a driving force for the evolutionary differentiation of species. Compared to other mammals, primate and human genomes include many interspersed segmental duplications, which may have been of special relevance for the evolution of the primate lineage [1]. These segmental duplications range between one to several hundred kilobases, and are characterized by a mosaic of repeat structures. They can be associated with rapid structural changes and chromosomal instability. About 430 blocks of the human genome have been identified as having been subject to multiple duplications during hominoid evolution [1]. Clustering analysis of these segmentally duplicated regions in the human genome suggests that a part of the duplication blocks have formed around a “core” or “seed” duplicon [2,3]. Some of the most variable human CNV genes correspond to recently evolved gene families among the human core duplicons (e.g. NPIP and LRRC37A) [4].
The SPATA31 gene family belongs to the core duplicon gene families and it has been shown to be one of the fastest evolving gene families in the human lineage [5]. We have previously characterized in detail the structure, transcriptional pattern, and protein localization of the SPATA31 gene family [6]. It has expanded from a single copy in mouse to at least nine copies in humans, located at seven different sites on both arms of chromosome 9. The coding regions are part of larger segmental duplications and one can distinguish two types, SPATA31A and SPATA31C. Type A is annotated with seven segmental duplications in the human reference genome (genome build hg38), including two pseudogenes, type C is annotated with two copies. The gene lengths and the protein coding regions of SPATA31 genes differ between A and C types, but not much within each type. Compared to the mouse gene, we found that the human SPATA31 genes are broadly expressed and have acquired new functional domains, among them a cryptochrome/photolyase domain, suggesting the acquisition of a function in UV damage repair. Antibody staining showed that the protein is re-localized from the nucleolus to the whole nucleus upon UV irradiation, a pattern known for proteins involved in UV damage sensing and repair. Based on CRISPR/Cas mediated knockouts of members of the gene family in fibroblast cell cultures, we found that the reduction of copy number in cells leads to enhanced sensitivity towards UV-irradiation. Given that increased UV-light resistance of the skin may have played a major role in human evolution, we proposed that the acquisition of an involvement in UV damage sensing or repair has lead to the adaptive evolution of SPATA31 [6].
An interesting side effect of the SPATA31 gene knockouts was that the respective cells survived somewhat longer than normal primary fibroblast cell lines, although this was difficult to quantify. We have therefore used here the alternative approach, namely to over-express a representative member of the SPATA31 gene family, SPATA31A1, and study its effect on cell survival. We find that this over-expression results indeed in premature senescence of the cells, through interference with known aging related pathways. Based on these results, we asked whether natural copy number variation in humans correlates with senescence, in the sense that fewer SPATA31 copies should correlate with longer life span. We can indeed show this effect in a cohort of long-lived individuals. Humans that have reached an age of 95 or higher have on average fewer SPATA31 gene copies than a younger control population.
Results
Over-expression of SPATA31A1 in primary human fibroblast (HFF) cells was achieved using a mammalian expression vector with a CMV enhancer and promoter. Normal expression of SPATA31A1 is low in these cells, while the introduction of the expression vector resulted in a 2.7 fold increase. After the initial transformation, cells were re-cultured every three weeks for up to five additional rounds. We found that SPATA31A1 over-expressing cultures produced relatively fewer cells in each of the replication rounds than the controls transformed with the vector only (Fig. 1A). Based on the ß-galactosidase staining assay for cellular senescence, we observed about twice as many senescent cells in the SPATA31A1 over-expressing cultures than in the controls (Fig. 1B).
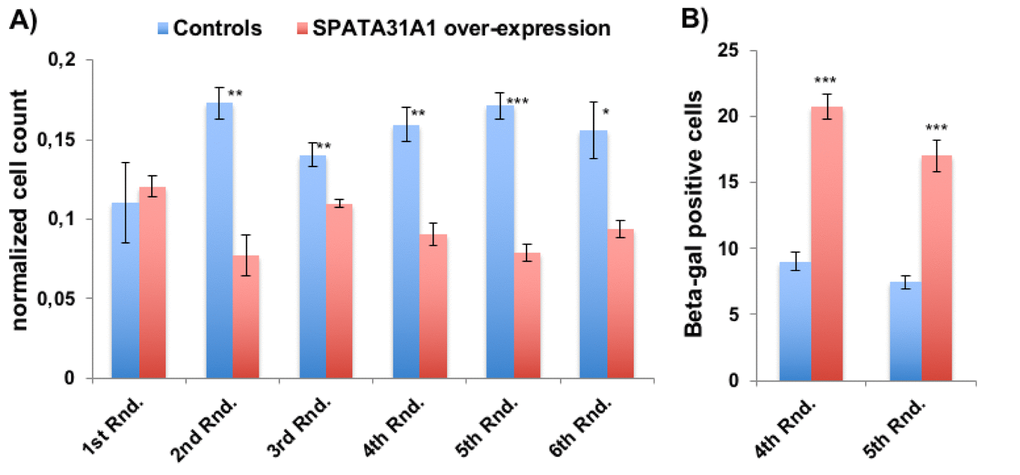
Figure 1. SPATA31A1 over-expression leads to reduced cell growth and increased senescence. (A) Relative number of cells per flask after each round of 3 weeks of culturing. The first round (1st Rnd) represents the normalized cell numbers three weeks after the initial seeding. Differences in normalized cell numbers became significant after the 2nd round, up to the 6th round when the experiment ended. The blue bars represent the averages and standard error of the four replicates of the control cells and the red bars the SPATA31A1 over-expressing cells. The numbers were normalized with respect to total cell counts. See Suppl. Figure S1 for the immunofluorescence images of control-eGFP and SPATA31A1-eGFP expression. (B) Senescence of SPATA31A1 over-expressing cells detected by the ß-galactosidase assay. The fraction of ß- galactosidase (+) staining cells from the 4th and 5th Rnd of re-culturing are shown with standard error between replicates. See suppl. Figure S2 for an example of staining results for senescence-associated ß-galactosidase assay. P-values indicated above the bars (*p< 0.05, **p< 0.01, ***p< 0.001; student t-test).
RNA sequencing of the cells transfected with only the vector (control) or the SPATA31A1 expression plasmid (treatment) after one week of culture (i.e. before differences in cell numbers would become apparent) was used to study the influence of SPATA31A1 on expression networks. Of 23,723 expressed genes in these cells, 8.5% (2,023) showed significant up- or down-regulation (5% false discovery rate) (Suppl. Table S1a,b; Suppl. Figure S3). Functional gene enrichment analysis [7] (Table 1, Suppl. Table S2) was applied to assess the affected pathways. The three most significant KEGG pathways that changed upon over-expression of SPATA31A1, namely cell cycle [8], PI3K-Akt signaling pathway [9] and extracellular matrix (ECM) receptor interaction [10] are known to play a role in aging. ECM homeostasis is centrally involved in UV-damage repair of fibroblasts [11], confirming the previous notion that SPATA31 gene functions are part of this pathway [6]. ECM receptor interaction was also previously shown to be differentially regulated between senescent and young cells [12].
Table 1. Pathway enrichment analysis.
| KEGG Pathway Term1 | ID | Input number | Background number | P-Value | Corrected P-Value | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cell cycle | hsa04110 | 39 | 124 | 1.16e-11 | 3.29e-09 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Pathways in cancer | hsa05200 | 73 | 399 | 2.50e-10 | 3.54e-08 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ECM-receptor interaction | hsa04512 | 27 | 83 | 9.08e-09 | 6.95e-07 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Proteoglycans in cancer | hsa05205 | 45 | 208 | 9.82e-09 | 6.95e-07 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PI3K-Akt signaling pathway | hsa04151 | 61 | 343 | 1.83e-08 | 9.33e-07 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1The corresponding analysis was performed using the web server (http://kobas.cbi.pku.edu.cn/) [7]. Only the five most significant KEGG pathways are listed (full list in Suppl. Table S2). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Among 66 genes that showed a more than 2-fold change (Suppl. Table S1b), we found eleven that had previously been noticed to be differentially regulated or involved in aging or age-related diseases (up-regulated: BEX1 [13], SERPINB2 [13], CORIN [13], CHI3L1 [14], STMN2 (SCG10) [15], [16] and [13], and PTGS2 (COX2) [17,18]; down-regulated: VANGL2 [13], CYTL1 [13], IGF2 [19] and CDH 6 [20]) (Table 2). IGF1, which has been implicated in aging and UV-damage repair [21], is also significantly down-regulated, but less than 2-fold (Table 2).
Table 2. Expression changes for genes discussed in the text.
| Gene ID | Ensembl ID | baseMean1 | log2FoldChange2 | adjusted p | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BDNF | ENSG00000176697 | 3401 | -0.31 | 1.31E-06 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BEX1 | ENSG00000133169 | 166 | 2.07 | 1.53E-40 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CDH6 | ENSG00000113361 | 1514 | -1.02 | 1.91E-44 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CHI3L1 | ENSG00000133048 | 112 | 2.11 | 4.40E-51 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CORIN | ENSG00000145244 | 218 | 1.45 | 3.05E-38 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CYTL1 | ENSG00000170891 | 26 | -1.00 | 2.73E-08 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| DTL | ENSG00000143476 | 258 | 0.77 | 3.91E-10 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IGF1 | ENSG00000017427 | 58 | -0.62 | 1.02E-03 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IGF2 | ENSG00000167244 | 10998 | -1.02 | 2.28E-38 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PCNA | ENSG00000132646 | 2062 | 0.32 | 1.31E-05 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PTGS2 | ENSG00000073756 | 1120 | 1.59 | 1.45E-62 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SERPINB2 | ENSG00000197632 | 120 | 1.55 | 1.22E-30 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SPATA31A1 | ENSG00000204849 | 19 | 1.45 | 6.48E-18 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| STMN2 | ENSG00000104435 | 806 | 1.65 | 1.78E-74 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| VANGL2 | ENSG00000162738 | 131 | -1.12 | 1.75E-16 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1baseMean represents the normalized read counts from all samples | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2log2FoldChange is the calculated fold expression change based on normalized read counts, (full list in Suppl. Table S1b). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
We asked further whether other differentially expressed genes are known to be associated with aging effects. We found that 21% (57 out of 278 expressed in the HFF cells) of age-related genes in the GenAge Database [22] overlap with our list of differentially expressed genes, including PCNA, BDNF, IGF1 and IGF2 (Table 2 and Suppl. Table S1c). Hence, the expression data corroborate the observation of increased senescence of the SPATA31A1 over-expressing cells in culture.
SPATA31A genes carry a PCNA (proliferating cell nuclear antigen) interacting domain [6] and PCNA is significantly up-regulated in SPATA31A1 over-expressing cells (Table 2). PCNA is a cofactor of DNA polymerase delta and was found to be one of the genes involved in translesion DNA synthesis and repair [23]. Therefore, we focused on additional DNA repair genes by extracting the respective GO terms from the Gene Ontology database (Amigo2 accession GO:0006281). Twelve percent (57 out of 472 expressed in the HFF cells) of these DNA repair genes are significantly differentially expressed in our data (44 up and 13 down; Suppl. Table S1d). Some of the up-regulated genes are directly related to PCNA dependent translesion repair genes. This includes DTL (Cdt2-CRL4 complex) which is involved in PCNA-dependent translesion DNA synthesis [24,25].
These results raise the question whether natural expression differences of SPATA31 genes through copy number variation could influence mutational and aging processes in humans, especially since humans have the relatively highest number of SPATA31 gene copies among primates [6]. To obtain an overall pattern of copy number variation in humans, we analyzed the Simons Genome Project (SDGP) [26] data using a read depth approach. Given the differences between the SPATA31 A- and C-types (see introduction), we analyzed them separately. We found that SPATA31A has more copies and is more copy number variable than SPATA31C (Fig. 2), with the largest copy number differences seen for SPATA31A in the African populations. When summed across all SPATA31 copies, averages and variances are similar between the population groups, with the exception of native Americans and Africans having lower averages and Africans also higher variance (Suppl. Figure S4). Given that higher copy numbers are expected to express also more gene product, we use in the following correlations with copy numbers to assess effects of SPATA31 in humans.
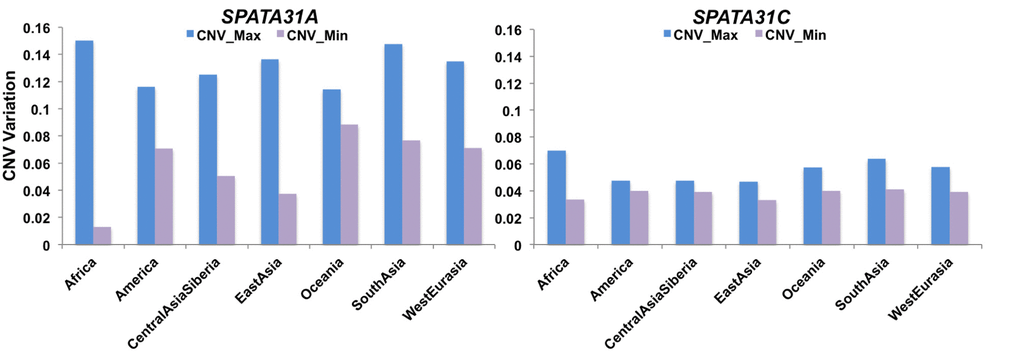
Figure 2. Range of SPATA31 copy numbers in different human population groups. The data are based on normalized read depth from the Simons Genome Project data [26] from which we have also retrieved the classification into population groups. The Y-axis represents normalized read depth measures, minimum and maximum values are provided for each group.
First we asked whether SPATA31 gene copy number could differentially affect the germline mutation rate, especially since SPATA31 is highly expressed in testis [6]. We have tested this by analyzing the data from 250 parent-offspring trios from the Genome of the Netherlands (GoNL) consortium re-sequencing data [27]. We extracted SPATA31 copy numbers of the parents from the genome sequence data and compared these with the numbers of germline mutations detected in this project. We did not find a significant correlation between SPATA31 copy numbers of either parent with de novo mutations in their children (Suppl. Figure S5; Suppl. Table S3), suggesting that the SPATA31 function in the germline does not have an effect on the mutation rate. However, it seems possible that the germline function is anyway different from its somatic function (see Discussion).
There are currently no datasets that would allow testing whether natural somatic mutation rates correlate with SPATA31 copy numbers. But the observation of an increased senescence phenotype in cells with over-expression of SPATA31A1, as well as the expression changes in genes related to aging, prompted us to ask whether copy number variation in human populations could be related to longevity in humans. We analyzed a human DNA sample collection of long-lived individuals (LLI) more than 96 years old and compared it to an average population control of individuals aged 60 to 75 years [28,29]. Since most of the individuals in the control group are still alive, they may include also individuals that become very old. However, by statistical criteria, fewer than 2% are expected to become centenarians [29]. Quantitative droplet PCR was used to measure SPATA31 copy numbers in these samples. We found that LLI had indeed on average a significantly lower copy number than the control group (Fig. 3; Suppl. Table S4).
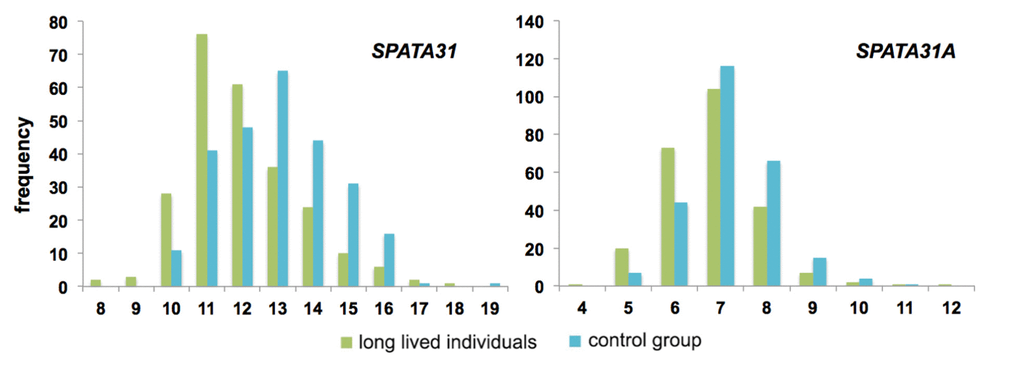
Figure 3. Distribution of copy number classes of SPATA31 genes between long-lived individuals (n=258) and controls (n=249). Long-lived individuals were older than 96 years and control individuals between 60-75 years at the time of sampling [28,29]. Copy number variations were determined by digital PCR using primers that amplify all possible SPATA31 copies (left) or only SPATA31A copies (right). The distributions are significantly different (both p<< 0.0001, Wilcoxon rank sum test).
Given the differences in variability between the SPATA31A and SPATA31C copies, we designed also primers specific for the SPATA31A variants and retested the LLI and control panels. We found the same general pattern as for the full gene set, with LLI having on average significantly fewer copies of SPATA31A (Fig. 3; Suppl. Table S4).
Discussion
The segmentally duplicated genes in primates and humans have received special attention, since they may have significantly contributed to the evolution of these species [1]. However, only few of these genes have been studied in functional detail so far. Together with our previous analysis of the evolution of the SPATA31 gene family [6], the present data suggest that SPATA31 genes are involved in sensing and repairing UV-induced DNA damage, but also in the induction of pathways causing the premature aging of cells. Further, we show that long lived individuals have on average lower numbers of SPATA31 genes in their genome.
The ancestral function of the gene has been in spermatogenesis. Knockouts for SPATA31 in mice lead to spermatogenesis defects and infertility [30]. Given that SPATA31 genes are also highly expressed in the testis of humans, it seems possible that they still have the same function in humans. This would explain why we find no effect on the germline mutation rate, since the testis function would be different from the function in the rest of the body. The specialization for sensing UV-damage and its repair has apparently evolved only together with the acquisition of the new protein domains in primates [6]. The increase in copy number may then have become beneficial as a response to an increased exposure to UV light as consequence of a switch from nocturnal to diurnal lifestyle, as well as increasing loss of body hair in humans. But our results from the over-expression of SPATA31A in epithelial cells suggest that this comes with a cost, namely an increased activation of senescence pathways, coupled with an enhancement of DNA repair processes, which could lead to more somatic mutations.
It has generally been suggested that there is a complex interaction between cellular senescence, tumor incidence due to somatic mutations and aging [31]. Our data imply that SPATA31 genes are part of this process and that their variation in copy number contributes via this effect to longevity in humans. Having more copies may lead to more somatic mutations, including some that cause cancer, while having fewer copies reduces this effect, thus allowing longer life spans. But given that also other senescence pathways are activated by over-expressing SPATA31, the link to life span may also be more complex.
The SPATA31 copy number effect on aging can be seen as an example for antagonistic pleiotropy [32,33]. Higher copy numbers provide a benefit early in life, due to better protection of the skin against sun light, allowing to spend more time during the day for foraging, social life, mate seeking and child care, all factors that should increase reproductive fitness. Hence, there would be positive selection for higher copy numbers. But more copies would also lead to a higher expression of SPATA31 and our cell-culture results show that such a higher expression induces DNA repair pathways. This could lead to a higher incidence of repair-induced damage in the cells and thus to cancer. If this becomes a problem during reproductive age, one would have a potential negative selection against high copy number. Hence, a balance in copy number should be maintained in the population, but with a certain variance. This variance has the effect that total lifetime beyond reproductive age is affected, with individuals with fewer copies having a higher probability to live longer.
Although somatic mutations in humans have long been known to depend on UV-light exposure among other factors [34], the molecular pathways remain to be studied further. SPATA31 copy number variation could turn out to be a risk factor for an increased somatic mutation rate, in particular in combination with UV-exposure [35]. We have not studied this particular aspect here, but it seems possible that humans with a higher sensitivity towards sunlight may have on average fewer SPATA31 copies, implying at the same time that they could have a longer life span.
There have been a series of SNP based genome-wide association studies (GWAS) in humans to detect loci associated human longevity (e.g [29,33,36–38]. and references therein). Several associations were also detected on chromosome 9 (http://genomics.senescence.info/longevity/ [37], which harbors the SPATA31 copies, but none of these regions is close to the location of the mapped functional copies of SPATA31 along the chromosome. In fact, it is unlikely that causative copy number polymorphisms would be picked up in GWAS experiments, since copy number changes tend to evolve faster than their associated SNPs. There have also been two association studies for mortality in long-lived individuals which have focused on copy number variants [39,40]. However, the regions identified in these studies do not overlap with the annotations of the SPATA31 genes. One possible reason may be that the SPATA31 copies occur at multiple places along chromosome 9 and the effect on aging may not be related to any one particular of them. Only screens that would take interaction effects into account might be able to uncover them.
It is currently difficult to estimate how fast copy numbers can change between generations and over time, since this would require phasing of the clusters in family studies, which is currently technically too challenging. We can currently also not rule out the possibility that there is somatic variation in SPATA31 copy number, since we do not have a direct comparison to germline derived copy numbers. Hence, also the modes of inheritance of this copy number variation, for example whether new copy number variation arises frequently between generations or even somatically, remains a question for the future.
Methods
Over-expression of SPATA31A1
SPATA31A1 constitutes a representative member of the SPATA31A genes [6]. A cDNA was obtained from GE Healthcare-Dharmacon (MGC cDNA Clone ID:9056783, MHS6278-213246307) and cloned N-terminally into the mammalian expression vector pEGFPN1 in frame by using EcoR1 (5’) and Kpn1 (3’) restriction enzymes (NEB) (Suppl. Figure S6). Clones were confirmed for integrity by Sanger sequencing. DNA was prepared by using the EndoFree Plasmid Maxi Kit (Qiagen Cat No:12362). Human Foreskin Fibroblast cells (HFF) were purchased from the American Type Culture Collection (ATCC) (HFF2703, CRL-2703). Two rounds of four independent transfections for each of the pSPATA31A1-EGFP (pS) and vector controls pEGFP (pE) were performed using Amaxa Basic Nucleofector Kit Primary Mammalian Epithelial Cell (Lonza Cat No: VPI-1005). Transfected cells were grown in IMDM growth medium (Gibco Cat no: 21980-065) supplemented with 10% FBS and G418 antibiotic (200ng/mL) to ensure that only successfully transfected cells would grow. Suppl. Figure S1 provides examples for the immunofluorescence analysis of the transfected cells. Cells were incubated at 37°C in 5% CO2 atmosphere as a pH regulator. Once the cells reached a sufficient density for splitting, 50,000 cells of each transfection were sub-cultured into 75mm flasks. After 3 weeks of culture, cells were treated with 0.05% Trypsin (Gibco Cat No: 25300-054) to dissociate them from the flask. Equal amount of medium was added and cells were pelleted by centrifugation for 5 min at 300g. Cells were re-suspended in 4 mL of fresh medium and counted with a hematocytometer. Counting was performed always in pairs of each control and treatment group to keep the cells under the same conditions. 50,000 cells were then transferred into a new flask and remaining cells were frozen for long-term preservation. This procedure was repeated every three weeks until the 6th round.
Cellular senescence assay
Senescence associated ß-galactosidase expression was assayed using the ß-gal senescence kit from Sigma (Cat No: CS0030-1KT). The test was performed by seeding 50,000 cells from each culture in the 4th and the 5th round of culturing (84th day and 105th day) on 35mm dishes. After 3 days of growth, cells were analyzed according to the manufacturer’s instruction. To obtain better staining, the staining solution was replaced after 4 hours of initial incubation and then cells were incubated overnight at 37°C. After completion of staining (Suppl. Figure S2 provides examples of stained cells), 10 randomly selected fields were counted from each dish, using a Leica inverted microscope (20x objective), The fraction of stained cells was counted in each field amounting to a total count of about 250 cells per dish.
RNASeq analysis
50,000 cells from preselected culturing at round 0 (before the 1st round) were transferred into 5 cm dishes for each culture. HFF cells were then incubated for an additional week in IMDM growth medium supplemented with 10% FBS and 200ng/ml G418 as selection agent. Total RNA was isolated using the RNeasy Mini Kit (Qiagen Cat No: 74104) and subjected to high throughput sequencing as paired end reads with 2 x 150 cycles (Illumina NextSeq 500). Libraries were prepared with the TruSeq stranded mRNA Library Prep Kit.
The sequencing reads were first quality checked using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and trimmed using Trimmomatic [41]. This yielded on average 30 million paired-end reads that were mapped to the human genome hg38 with tophat2 [42] and read counts per gene annotated in ENSEMBL hg38 were retrieved by featureCounts [43]. The DESeq2 software package 1.8.2 [44] was used to detect significant differential expression between genes of pE1-pE4 cells (control) and pS1-pS4 cells (treatment). Suppl. Table S5 provides the read statistics.
Analysis of GoNL data
We retrieved genome read data for 250 offspring trios (in total 750 individual bam files) with permission of the Genome of Netherlands Project (GoNL) application number 2016126 approved by GoNL Data Access Committee on 12-05-2016 [27]. Total mapped reads of SPATA31 and single copy reference house keeping genes (UBE2, mTOR, TBP, B2M and TERT) (see Suppl. Figure S7 for coordinates) were extracted using samtools. The resulting total reads were mapped against the human reference genome (build hg38) using Bowtie2 [45]. Mapping with bwa-mem yielded similar results, but we chose Bowtie2 because we observed that it better distributes random reads between subtypes of the duplicates. The resulting total mapped reads were counted for each SPATA31 copy and normalized by dividing the total number of mapped reads from the single copy reference house keeping genes. We then performed a linear regression analysis using normalized read depth (CNV) as the explanatory variable and the number of de novo mutations (obtained from GoNL (http://www.nlgenome.nl/?page_id=9)) as the response variable.
DIGITAL PCR for copy number detection
We determined SPATA31 copy numbers in two separate sample sets, in two independent runs. The first included DNA samples from 155 long-lived individuals (LLI - aged at least 96 years at the time of recruitment) and 163 samples of younger controls (60-75 years) from the same population cohort. The second included 96 samples from each of these groups. A detailed description of the study participants and the recruitment procedure is provided in [29] and [28]. Further, we used two different primer sets for each run, the first amplifying both, SPATA31A and SPATA31C variants, the second specific for SPATA31A variants.
All digital PCRs between control and LLI were run using the same master mix and same plate (e.g. 48 controls, 48 LLI for each run). To reduce the sampling error, genomic DNA was first diluted in 99μL by taking 1μL from 50ng/ μL stock concentration and mixed well by pipetting. Primer and probe sequences are provided in Suppl. Figure S8. In detail, the PCR reaction mixture was prepared from 12.5μL of 2x ddPCR Supermix for Probes (Bio-Rad, Hercules, CA, USA) mixed with 1.25μL of 20x primer-probe mix: 18μM PCR primers (each SPATA31 primers, 5μM probe against SPATA31A mixed with 1.25μL of 20X primer-probe mix: 18μM PCR primers (each Albumin primers), 5μM probe against SPATA31 and finally 0.1μL BamH1HF (NEB) added to the reaction. In total 15μL of reaction mixture was prepared in an Eppendorf 96-well twin.tec PCR plate. 10μL of diluted Genomic DNA (in total 5ng) was mixed with 15μL of the reaction mixture. The total 25μL reaction was mixed well by pipetting up and down and loaded into the Automated Droplet Generator (Bio-Rad, Hercules, CA, USA) to generate oil droplets in each well of the plate containing 20μL of the reaction mixture. After droplets were generated, the plate was sealed with a pierceable foil heat seal using PX1™ PCR Plate Sealer (Bio-Rad, Hercules, CA, USA) and then placed on a thermal cycler for amplification. Thermal cycling conditions were as follows: 95°C for 10 min (1 cycle), 94°C for 30 sec (ramp rate 2.5°C/sec) and 56°C for 60 sec (ramp rate 2.5°C/sec) (40 cycles), 98°C for 10 min (1 cycle), and 12°C hold. After PCR, the 96-well PCR plate was loaded on the QX100™ Droplet Reader (Bio-Rad, Hercules, CA, USA) which reads the droplets from each well of the plate. The data obtained were analyzed using QuantaSoft™ analysis software linked with the QX100™ Droplet Reader. We scored the copy numbers by measuring the concentration of the target, SPATA31_All or SPATA31A relative to the concentration of the reference ALBUMIN.
Genome project analysis
We retrieved genome read data for 295 individuals (bam files) from the Simons Genome Project [26]. Reads were extracted and normalized as described above for the GoNL data.
Data availability
RNASeq sequencing reads are available under SRA accession number PRJEB21178.
Supplementary Materials
Author Contributions
Conceived and designed the experiments: CB and DT. Performed the experiment: CB and analysis: CB and CX. Contributed reagents/material/analysis tools: CB, AN. Wrote the manuscript: CB and DT.
Acknowledgements
We thank Guy Reeves for valuable discussion and Sven Künzel for performing sequencing. We thank the GoNL and the Simon project for providing access to their data.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding
This work was supported by institutional funds of the Max-Planck Society to D.T.
References
- 1. Bailey JA, Eichler EE. Primate segmental duplications: crucibles of evolution, diversity and disease. Nat Rev Genet. 2006; 7:552–64. https://doi.org/10.1038/nrg1895 [PubMed]
- 2. Jiang Z, Tang H, Ventura M, Cardone MF, Marques-Bonet T, She X, Pevzner PA, Eichler EE. Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution. Nat Genet. 2007; 39:1361–68. https://doi.org/10.1038/ng.2007.9 [PubMed]
- 3. Marques-Bonet T, Girirajan S, Eichler EE. The origins and impact of primate segmental duplications. Trends Genet. 2009; 25:443–54. https://doi.org/10.1016/j.tig.2009.08.002 [PubMed]
- 4. Alkan C, Kidd JM, Marques-Bonet T, Aksay G, Antonacci F, Hormozdiari F, Kitzman JO, Baker C, Malig M, Mutlu O, Sahinalp SC, Gibbs RA, Eichler EE. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet. 2009; 41:1061–67. https://doi.org/10.1038/ng.437 [PubMed]
- 5. Han MV, Demuth JP, McGrath CL, Casola C, Hahn MW. Adaptive evolution of young gene duplicates in mammals. Genome Res. 2009; 19:859–67. https://doi.org/10.1101/gr.085951.108 [PubMed]
- 6. Bekpen C, Künzel S, Xie C, Eaaswarkhanth M, Lin YL, Gokcumen O, Akdis CA, Tautz D. Segmental duplications and evolutionary acquisition of UV damage response in the SPATA31 gene family of primates and humans. BMC Genomics. 2017; 18:222. https://doi.org/10.1186/s12864-017-3595-8 [PubMed]
- 7. Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li CY, Wei L. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011 (suppl_2); 39:W316-22. https://doi.org/10.1093/nar/gkr483 [PubMed]
- 8. Chandler H, Peters G. Stressing the cell cycle in senescence and aging. Curr Opin Cell Biol. 2013; 25:765–71. https://doi.org/10.1016/j.ceb.2013.07.005 [PubMed]
- 9. Noh EM, Park J, Song HR, Kim JM, Lee M, Song HK, Hong OY, Whang PH, Han MK, Kwon KB, Kim JS, Lee YR. Skin Aging-Dependent Activation of the PI3K Signaling Pathway via Downregulation of PTEN Increases Intracellular ROS in Human Dermal Fibroblasts. Oxid Med Cell Longev. 2016; 2016:6354261. https://doi.org/10.1155/2016/6354261 [PubMed]
- 10. Labat-Robert J. Cell-matrix interactions in aging: role of receptors and matricryptins. Ageing Res Rev. 2004; 3:233–47. https://doi.org/10.1016/j.arr.2003.10.002 [PubMed]
- 11. Panich U, Sittithumcharee G, Rathviboon N, Jirawatnotai S. Ultraviolet Radiation-Induced Skin Aging: The Role of DNA Damage and Oxidative Stress in Epidermal Stem Cell Damage Mediated Skin Aging. Stem Cells Int. 2016; 2016:7370642. https://doi.org/10.1155/2016/7370642 [PubMed]
- 12. Yang KE, Kwon J, Rhim JH, Choi JS, Kim SI, Lee SH, Park J, Jang IS. Differential expression of extracellular matrix proteins in senescent and young human fibroblasts: a comparative proteomics and microarray study. Mol Cells. 2011; 32:99–106. https://doi.org/10.1007/s10059-011-0064-0 [PubMed]
- 13. Glass D, Viñuela A, Davies MN, Ramasamy A, Parts L, Knowles D, Brown AA, Hedman AK, Small KS, Buil A, Grundberg E, Nica AC, Di Meglio P, et al, and MuTHER consortium. Gene expression changes with age in skin, adipose tissue, blood and brain. Genome Biol. 2013; 14:R75. https://doi.org/10.1186/gb-2013-14-7-r75 [PubMed]
- 14. Sanfilippo C, Malaguarnera L, Di Rosa M. Chitinase expression in Alzheimer’s disease and non-demented brains regions. J Neurol Sci. 2016; 369:242–49. https://doi.org/10.1016/j.jns.2016.08.029 [PubMed]
- 15. Liedtke W, Leman EE, Fyffe RE, Raine CS, Schubart UK. Stathmin-deficient mice develop an age-dependent axonopathy of the central and peripheral nervous systems. Am J Pathol. 2002; 160:469–80. https://doi.org/10.1016/S0002-9440(10)64866-3 [PubMed]
- 16. Okazaki T, Wang H, Masliah E, Cao M, Johnson SA, Sundsmo M, Saitoh T, Mori N. SCG10, a neuron-specific growth-associated protein in Alzheimer’s disease. Neurobiol Aging. 1995; 16:883–94. https://doi.org/10.1016/0197-4580(95)02001-2 [PubMed]
- 17. Teismann P, Vila M, Choi DK, Tieu K, Wu DC, Jackson-Lewis V, Przedborski S. COX-2 and neurodegeneration in Parkinson’s disease. Ann N Y Acad Sci. 2003; 991:272–77. https://doi.org/10.1111/j.1749-6632.2003.tb07482.x [PubMed]
- 18. Wu D, Marko M, Claycombe K, Paulson KE, Meydani SN. Ceramide-induced and age-associated increase in macrophage COX-2 expression is mediated through up-regulation of NF-kappa B activity. J Biol Chem. 2003; 278:10983–92. https://doi.org/10.1074/jbc.M207470200 [PubMed]
- 19. Fu VX, Dobosy JR, Desotelle JA, Almassi N, Ewald JA, Srinivasan R, Berres M, Svaren J, Weindruch R, Jarrard DF. Aging and cancer-related loss of insulin-like growth factor 2 imprinting in the mouse and human prostate. Cancer Res. 2008; 68:6797–802. https://doi.org/10.1158/0008-5472.CAN-08-1714 [PubMed]
- 20. Jiang SS, Chen CH, Tseng KY, Tsai FY, Wang MJ, Chang IS, Lin JL, Lin S. Gene expression profiling suggests a pathological role of human bone marrow-derived mesenchymal stem cells in aging-related skeletal diseases. Aging (Albany NY). 2011; 3:672–84. https://doi.org/10.18632/aging.100355 [PubMed]
- 21. Kemp MG, Spandau DF, Travers JB. Impact of Age and Insulin-Like Growth Factor-1 on DNA Damage Responses in UV-Irradiated Human Skin. Molecules. 2017; 22:E356. https://doi.org/10.3390/molecules22030356 [PubMed]
- 22. Tacutu R, Craig T, Budovsky A, Wuttke D, Lehmann G, Taranukha D, Costa J, Fraifeld VE, de Magalhães JP. Human Ageing Genomic Resources: integrated databases and tools for the biology and genetics of ageing. Nucleic Acids Res. 2013; 41:D1027–33. https://doi.org/10.1093/nar/gks1155 [PubMed]
- 23. Mailand N, Gibbs-Seymour I, Bekker-Jensen S. Regulation of PCNA-protein interactions for genome stability. Nat Rev Mol Cell Biol. 2013; 14:269–82. https://doi.org/10.1038/nrm3562 [PubMed]
- 24. Terai K, Abbas T, Jazaeri AA, Dutta A. CRL4(Cdt2) E3 ubiquitin ligase monoubiquitinates PCNA to promote translesion DNA synthesis. Mol Cell. 2010; 37:143–49. https://doi.org/10.1016/j.molcel.2009.12.018 [PubMed]
- 25. Bacquin A, Pouvelle C, Siaud N, Perderiset M, Salomé-Desnoulez S, Tellier-Lebegue C, Lopez B, Charbonnier JB, Kannouche PL. The helicase FBH1 is tightly regulated by PCNA via CRL4(Cdt2)-mediated proteolysis in human cells. Nucleic Acids Res. 2013; 41:6501–13. https://doi.org/10.1093/nar/gkt397 [PubMed]
- 26. Mallick S, Li H, Lipson M, Mathieson I, Gymrek M, Racimo F, Zhao M, Chennagiri N, Nordenfelt S, Tandon A, Skoglund P, Lazaridis I, Sankararaman S, et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature. 2016; 538:201–06. https://doi.org/10.1038/nature18964 [PubMed]
- 27. Francioli LC, Menelaou A, Pulit SL, van Dijk F, Palamara PF, Elbers CC, Neerincx PB, Ye K, Guryev V, Kloosterman WP, Deelen P, Abdellaoui A, van Leeuwen EM, et al, and Genome of the Netherlands Consortium. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet. 2014; 46:818–25. https://doi.org/10.1038/ng.3021 [PubMed]
- 28. Nebel A, Croucher PJ, Stiegeler R, Nikolaus S, Krawczak M, Schreiber S. No association between microsomal triglyceride transfer protein (MTP) haplotype and longevity in humans. Proc Natl Acad Sci USA. 2005; 102:7906–09. https://doi.org/10.1073/pnas.0408670102 [PubMed]
- 29. Flachsbart F, Caliebe A, Kleindorp R, Blanché H, von Eller-Eberstein H, Nikolaus S, Schreiber S, Nebel A. Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc Natl Acad Sci USA. 2009; 106:2700–05. https://doi.org/10.1073/pnas.0809594106 [PubMed]
- 30. Wu YY, Yang Y, Xu YD, Yu HL. Targeted disruption of the spermatid-specific gene Spata31 causes male infertility. Mol Reprod Dev. 2015; 82:432–40. https://doi.org/10.1002/mrd.22491 [PubMed]
- 31. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011; 192:547–56. https://doi.org/10.1083/jcb.201009094 [PubMed]
- 32. Williams G. Pleiotropy, natural selection, and the evolution of senescence. Evolution. 1957; 11:398–411. https://doi.org/10.1111/j.1558-5646.1957.tb02911.x
- 33. Rodríguez JA, Marigorta UM, Hughes DA, Spataro N, Bosch E, Navarro A. Antagonistic pleiotropy and mutation accumulation influence human senescence and disease. Nat Ecol Evol. 2017; 1:55. https://doi.org/10.1038/s41559-016-0055 [PubMed]
- 34. De S. Somatic mosaicism in healthy human tissues. Trends Genet. 2011; 27:217–23. https://doi.org/10.1016/j.tig.2011.03.002 [PubMed]
- 35. Saini N, Roberts SA, Klimczak LJ, Chan K, Grimm SA, Dai S, Fargo DC, Boyer JC, Kaufmann WK, Taylor JA, Lee E, Cortes-Ciriano I, Park PJ, et al. The Impact of Environmental and Endogenous Damage on Somatic Mutation Load in Human Skin Fibroblasts. PLoS Genet. 2016; 12:e1006385. https://doi.org/10.1371/journal.pgen.1006385 [PubMed]
- 36. Pilling LC, Kuo CL, Sicinski K, Tamosauskaite J, Kuchel GA, Harries LW, Herd P, Wallace R, Ferrucci L, Melzer D. Human longevity: 25 genetic loci associated in 389,166 UK biobank participants. Aging (Albany NY). 2017; 9:2504–20. [PubMed]
- 37. Budovsky A, Craig T, Wang J, Tacutu R, Csordas A, Lourenço J, Fraifeld VE, de Magalhães JP. LongevityMap: a database of human genetic variants associated with longevity. Trends Genet. 2013; 29:559–60. https://doi.org/10.1016/j.tig.2013.08.003 [PubMed]
- 38. Nebel A, Kleindorp R, Caliebe A, Nothnagel M, Blanché H, Junge O, Wittig M, Ellinghaus D, Flachsbart F, Wichmann HE, Meitinger T, Nikolaus S, Franke A, et al. A genome-wide association study confirms APOE as the major gene influencing survival in long-lived individuals. Mech Ageing Dev. 2011; 132:324–30. https://doi.org/10.1016/j.mad.2011.06.008 [PubMed]
- 39. Nygaard M, Debrabant B, Tan Q, Deelen J, Andersen-Ranberg K, de Craen AJ, Beekman M, Jeune B, Slagboom PE, Christensen K, Christiansen L. Copy number variation associates with mortality in long-lived individuals: a genome-wide assessment. Aging Cell. 2016; 15:49–55. https://doi.org/10.1111/acel.12407 [PubMed]
- 40. Kuningas M, Estrada K, Hsu YH, Nandakumar K, Uitterlinden AG, Lunetta KL, van Duijn CM, Karasik D, Hofman A, Murabito J, Rivadeneira F, Kiel DP, Tiemeier H. Large common deletions associate with mortality at old age. Hum Mol Genet. 2011; 20:4290–96. https://doi.org/10.1093/hmg/ddr340 [PubMed]
- 41. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30:2114–20. https://doi.org/10.1093/bioinformatics/btu170 [PubMed]
- 42. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013; 14:R36. https://doi.org/10.1186/gb-2013-14-4-r36 [PubMed]
- 43. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014; 30:923–30. https://doi.org/10.1093/bioinformatics/btt656 [PubMed]
- 44. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. https://doi.org/10.1186/s13059-014-0550-8 [PubMed]
- 45. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012; 9:357–59. https://doi.org/10.1038/nmeth.1923 [PubMed]