



Introduction
Adrenal Cushing syndrome (CS), consists of a set of systemic manifestations similar to those found in aging, is caused by glucocorticoid excess due to a cortisol-producing adenoma (CPA) or adrenocortical carcinoma [1]. It is associated with hypertension, physical and cognitive degeneration in aging and accelerated atherogenesis, obesity, and osteoporosis [2–4]. Hypersecretion of cortisol from the adrenal cortex is a character of CPA. Moreover, hypercortisolism might negatively impact telomere maintenance and consequently to premature aging [5,6]. However, the mechanism involved in this hyperfunction of the adrenal cortex in CPA remains unknown.
Synaptophysin (SYP; molecular weight, 38-kDa) is an integral membrane protein and a neuroendocrine marker [7] involved in synaptic vesicle formation [8], and the SYP gene family is involved in neuronal and neuroendocrine differentiation in rats and humans [9,10]. The adrenal cortex is not an intrinsic part of the diffuse neuroendocrine system, but neuroendocrine differentiation appears in some adrenocortical tumours [11]. Previous studies reported that SYP is ubiquitously expressed in adrenocortical tumours and its expression in adrenocortical adenomas may be associated with functions such as transport or secretion of glucocorticoids [12]. Hence, it is possible that SYP may play an important role in adrenocortical adenoma tissues. However, the effect of SYP and the mechanisms of the SYP genomic or genetic alterations in CPA still need to be validated.
One of the most important epigenetic modifications of the genome is DNA methylation, which occurs on cytosine residues at carbon 5 of the pyrimidine ring of simple sequences termed CpG dinucleotides, and subsequently controls gene expression. DNA methylation at CpG islands is strongly related to stable transcriptional repression [13,14]. Thus, DNA methylation regulates expression of many genes and is involved in many human diseases. Recently, aberrant global and gene-specific DNA promoter methylation has been observed in human adrenocortical tumours [15], implicating dysregulation of steroid biosynthesis. Thus, we hypothesized that methylation of cytosine nucleotides in CpG islands of the SYP promoter may regulate SYP gene expression and contribute to the hyperfunction of the adrenal cortex in CPA.
microRNAs are small RNA molecules that regulate gene expression by a posttranslational repression mechanism and are predicted to target up to 30% of the mammalian genome [16,17]. These molecules play important roles in various biological processes, including cell proliferation, differentiation, apoptosis and migration [18,19]. Accordingly, numerous studies have suggested that aberrant expression of certain miRNAs is closely correlated with tumour phenotype, suggesting that miRNAs might function as oncogenes or tumour suppressors [20]. The pleiotropic nature of miRNAs suggests that multiple signalling pathways can be affected by aberrant miRNA expression, thereby significantly directing cancer cell biological behaviour.
Although the role of SYP in adrenocortical tumours has been previously reported, the mechanisms involved in abnormal expression of SYP in adrenocortical tumours had not yet clarified. In the present study, we aimed to investigate the relationship between aberrant SYP methylation status and SYP expression in CPA and the mechanisms involved. Our results showed increased expression of SYP in CPAs, which is a result of hypomethylation of the SYP promoter. Using a miRNA microarray, we found decreased expression of miR-27a-5p in CPAs. By applying gain-of-function and loss-of-function methods and a luciferase reporter assay, we verified that TET3, the demethylation enzyme, was the target of miR-27a-5p. Moreover, we determined that the upregulation of SYP is associated with DNA demethylation induced by TET3 in CPA patients. In addition, we found that a miR-27a-5p-TET3-SYP signal pathway may play a role in the proliferation and cortisol secretion of H295R cells.
Results
Expression of SYP in human adrenocortical adenoma specimens
To identify the expression of SYP in human adrenocortical adenoma, we performed the real time quantitative PCR, western blotting and immunohistochemical staining on 12 adrenocortical adenoma samples from CS and their adjacent normal adrenal cortex tissues.
The qRT-PCR results showed that the expression levels of SYP are significantly higher in the adrenocortical adenoma group than in the control group. Compared to the normal adrenal cortex tissues, the expression level of SYP mRNA was about 12-fold higher in CPAs (Figure 1A). Likewise, the protein expression levels of SYP were also significantly higher in the CPA group compared with the control tissues, as determined by Western blotting (Figure 1B, p<0.05). For the immunostaining, immunoreactivity for SYP was present in both groups. However, the intensity of staining for SYP was significantly stronger in CPAs than in their adjacent normal adrenal cortex tissues (Figure 1C).
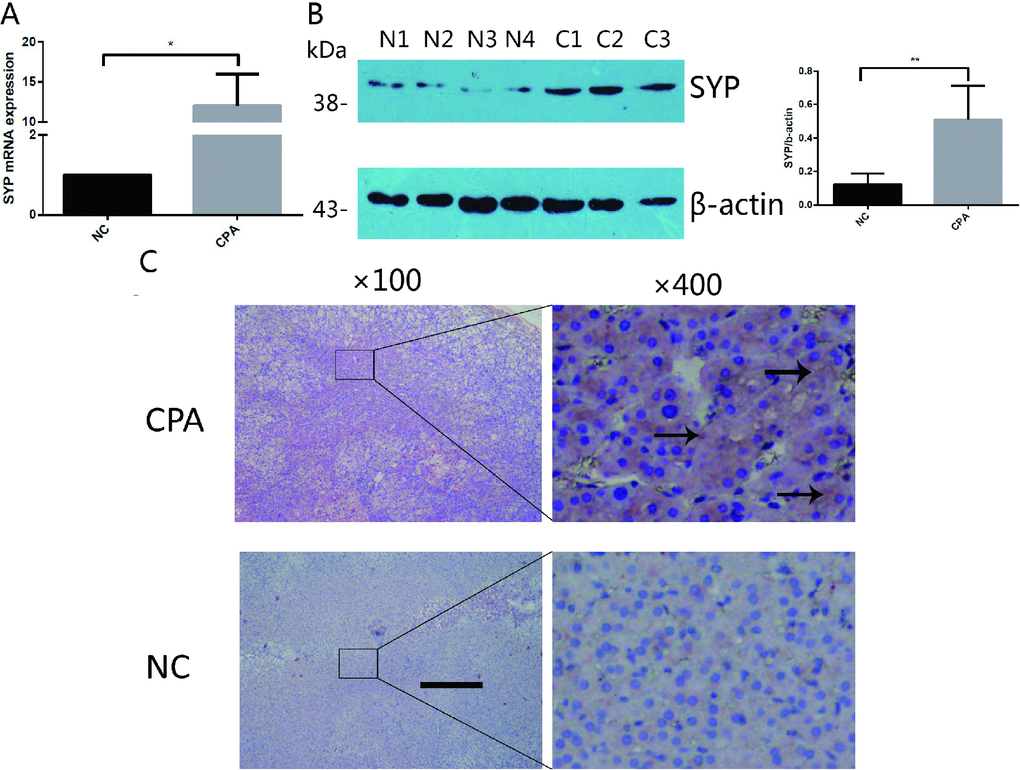
Figure 1. The expression of SYP in CPA. (A and B) The relative mRNA levels of SYP determined by real-time PCR (A) and protein levels of SYP determined by Western blotting (B) in CPAs and normal adrenal tissues (n=12). (C) Immunohistochemisty staining determines the expression of SYP in CPAs and normal adrenal tissues. The brown area indicated by arrow is the positive staining of SYP in CPAs. Three independent experiments were performed, and representative data are shown. The data represent the mean ± SD. NC, normal control. **p<001.
Epigenetic mechanism of SYP up-regulation both in vitro and in vivo
Next, we explored the molecular mechanism that mediates the upregulation of SYP in CPA specimens. Previous studies have demonstrated that there is a CpG island located in the promoter of SYP, and the methylation status of the CpG island is related to SYP expression. Recently study reported that SYP is hypermethylated and decreased in human Alzheimer’s Disease brain tissue [21–23]; therefore, we hypothesised that the upregulation of SYP in CPAs is correlated with the promoter of methylation of SYP. We applied methylation PCR assay to detect the methylation status of SYP in CPA samples and the control samples. The results revealed that in the normal adrenal cortex the promoter of the CpG island was hypermethylated and the methylation rate of CpG sites of SYP reached as high as 60.5% whereas the methylation rate of CpG sites in the CPA group was about 34.0% (Figure 2A). To further confirm the promoter hypermethylation in the regulation of SYP expression, we treated cultured H295R cells (the adrenocortical carcinoma cell line, the most commonly cell line used to study adrenal tumours, which possesses a steroid secretion and regulation pattern similar to that of primary adrenal cell cultures.) with the DNA methyltransferase inhibitor 5-aza after identifying that the H295R cells had a hypermethylation status in the SYP promoter. Our results showed that significantly decreased methylation of the SYP promoter (Figure 2B) is associated with increased expression of SYP upon 5-aza treatment as shown by western blotting (Figure 2C, P<0.05). Thus, we reasoned that DNA methylation of the SYP promoter results in different expression of SYP both in vivo and in vitro.
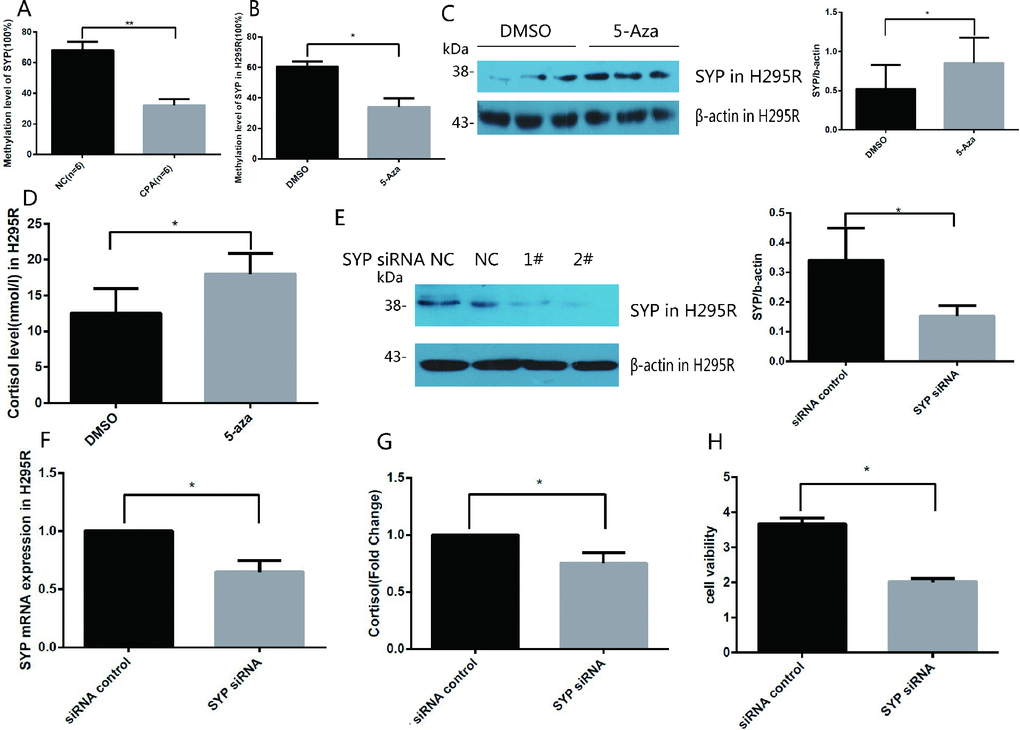
Figure 2. Methylation of the SYP promoter regulates the expression of SYP and cortisol secretion and H295R cells proliferation. (A) SYP methylation levels in CPA and normal adrenal tissues was determined using EpiTect Methyl II PCR Assay (n=12). (B) H295R cells were treated with 5-azaC, SYP methylation levels was determined by EpiTect Methyl II PCR Assay. (C) H295R cells were treated with 5-aza (5 μm) for 48 h the SYP protein level was determined by western blotting. (D) H295R cells were treated with 5-aza (5 μm) for 48 h, then cortisol secretion in H295R cell supernatants were measured in cell supernatants by ELISA. (E and F) Knock-down of SYP by siRNA was confirmed by qRT-PCR (F) and Western blotting (E). (G) The H295R cells was treated with SYP siRNA or control, then cortisol secretion in H295R cell supernatants were measured by ELISA. (H) Knock-down of SYP inhibited H295R cell proliferation measured by CCK assay. Three independent experiments were performed, and representative data are shown. Data are shown as mean ± SD. **p<0.01, compared with normal control. *p<0.05, compared with DMSO control.
DNA methylation of SYP is involved in cortisol secretion and H295R cell proliferation
To determine whether the overexpression of SYP caused by DNA methylation is related to the secretion of cortisol, we measured the secretion of cortisol in cell culture medium after 5-aza treatment in H295R cells. Compared to the control group, the cortisol secretion of the 5-aza treatment group was significantly higher (Figure 2D). To further confirm our speculation that SYP plays a crucial role in regulating the expression of cortisol. We successfully transfected SYP siRNA 2# into H295R cells to knock down the expression of SYP. As shown in Figure 2E, 2FSYP expression was significantly downregulated after SYP siRNA treatment. Not surprisingly, we found that knock-down of SYP results in decreased secretion of cortisol (Figure 2G). Furthermore, the CCK-8 assay showed significant decreases in cell viability with the effective repression of SYP in H295R cells (Figure 2H).
Identification of miRNAs differentially expressed in cortisol-producing adenomas
With the goal of identifying the mechanism involved in dysregulation of SYP through methylation, three adrenocortical adenoma samples associated with CPA and their adjacent normal adrenal cortex tissues were chosen to identify the expression of miRNAs. These tissues were then subjected to miRNA microarray analysis, which identified a large number of miRNAs whose expression changed significantly between cortisol-producing adenomas and normal tissues (Figure 3A). According to the results of the microarray analysis, we chose to further investigate miR-27a-5p. The microarray analysis identified that miR-27a-5p is the most downregulated
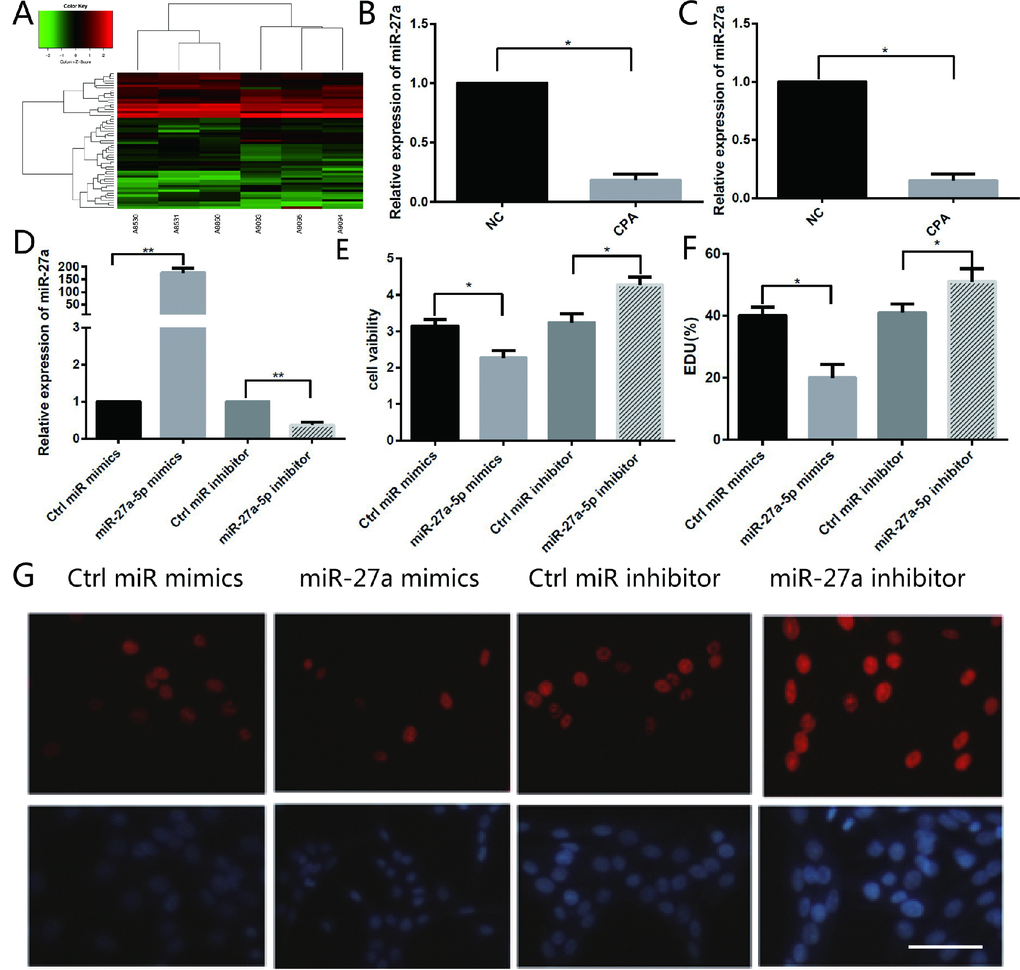
Figure 3. Different miRNA expression in CPA and normal adrenal tissues and the function of miR-27a-5p on the proliferation of H295R cells. (A) Hierarchical clustering of differentially expressed miRNAs in CPA samples and normal adrenal tissues. (B) miR-27a-5p is downregulated about 5.4-fold in CPA in comparison with normal adrenal cortex tissues as indicated by microarray analysis. (C) qRT-PCR analysis of miR-27a-5p in CPA samples and their adjacent normal adrenal cortex tissues. Three independent experiments were performed, and representative data are shown. (D) Upregulation of miR-27a-5p using miR-27a-5p mimics, or downregulation of miR-27a-5p using miR-27a-5p inhibitor in H295R cells were confirmed by qRT-PCR. (E) Cell viability in H295R cells transfected with miR-27a-5p mimics and inhibitor were determined by the CCK-8 assay. (F–G) Analysis of EdU staining on miR-27a-5p mimic-treated H295R cells. The EdU incorporation rate was expressed as the ratio of EdU-positive cells to total DAPI positive cells. Red, EDU; Blue, DAPI. Magnification, 400×. Three independent experiments were performed, and representative data are shown. NC, normal control. **p<0.01. *p<0.05. NC, normal control.
miRNA in CPAs; its expression decreased 5.4-fold in CPAs (Figure 3B). As expected, the qRT-PCR analysis confirmed that the expression of miR-27a-5p decreased significantly by 9.7-fold (Figure 3C). According to the results of the microarray analysis and qRT-PCR result, we chose to further investigate miR-27a-5p for further investigation and hypothesised that miR-27a-5p might be involved in the development of CPA.
miR-27a-5p involved in regulating the proliferation of H295R cells
Sharply decreased expression of miR-27a-5p forced us to ask whether miR-27a-5p has a regulatory effect on the proliferation of H295R cells. To identify the role of miR-27a-5p during the proliferation of H295R cells, we used a gain- and loss-of-function approach to overexpress or inhibit expression of miR-27a-5p. Two days after transfection with miR-27a-5p mimics, miR-27a-5p inhibitors, or control oligos, the number of H295R cells were measured by the CCK-8 assay. qRT-PCR results showed that inhibition of miR-27a-5p inhibited miR-27a-5p expression, whereas over-expression induced endogenous miR-27a-5p levels over 184-fold (Figure 3D). The cell viability was significantly reduced by 28% in miR-27a-5p mimic transfected cells compared with NC in the CCK-8 assay, whereas it increased by 31% in miR-27a-5p inhibitor transfected cells compared with NC (Figure 3E). Moreover, we found that the percentage of EDU-positive cells in miR-27a-5p mimic transfected cells decreased significantly and the percentage of EDU-positive cells in miR-27a-5p inhibitor transfected cells showed the opposite results (Figure 3F, 3G). These results indicate that miR-27a-5p plays a negative role in H295R cell proliferation.
TET3 is the direct target gene of miR-27a-5p
miRNAs exert their function by regulating the expression of their downstream target genes [24]. We utilized several bioinformatic target prediction algorithms to search for targets related to miR-27a-5p. TET3 was revealed to be a potential target of miR-27a-5p (Figure 4A). Accordingly, we evaluated the expression levels of TET3 in the tissue samples used for immunohistochemistry. The intensity of staining for TET3 was significantly stronger in the CPAs than in their adjacent normal adrenal cortex tissues (Figure 4B). Furthermore, we detected downregulation of the TET3 protein in miR-27a-5p mimic transfected cells and upregulation of the TET3 protein in miR-27a-5p inhibitor transfected cells by Western blot analysis (Figure 4C).
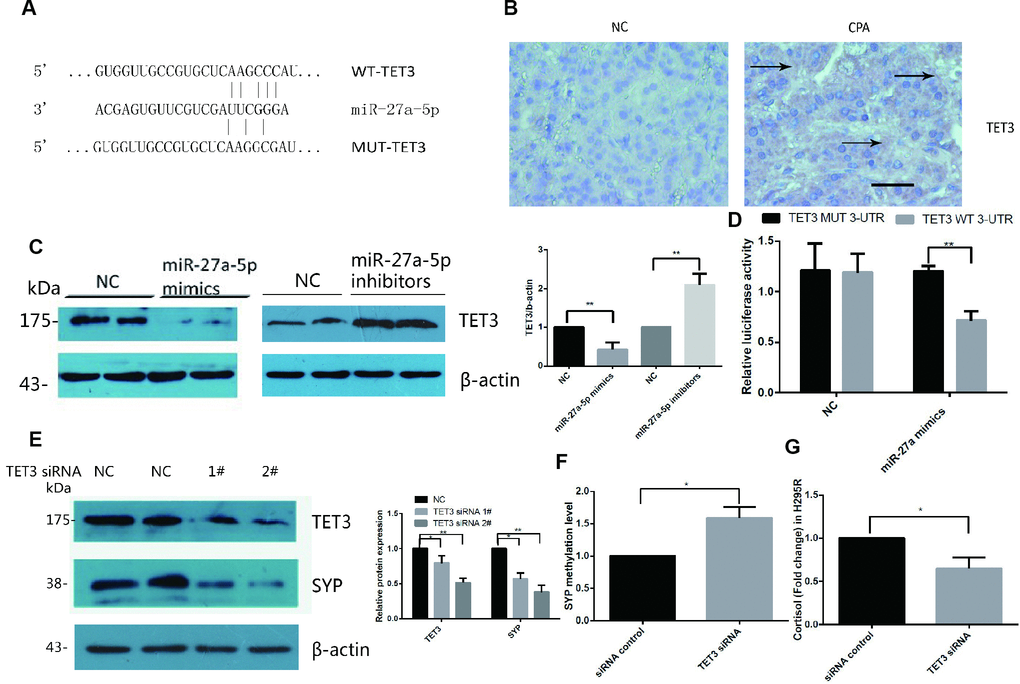
Figure 4. TET3 is a direct target of miR-27a-5p and TET3 regulated SYP expression and cortisol secretion in H295R cells. (A) RNA22 predicts that TET3 is a potential target of miR-27a-5p. (B) Immunohistochemical staining for TET3 in CPA and normal adrenal tissue. DAB staining showed that the intensity of staining for TET3 was significantly stronger in CPAs than in normal adrenal tissues. The brown area indicated by arrow is the positive staining of TET3 in CPAs. Representative data are shown. (C) TET3 protein levels in H295R cells were assessed by Western blotsin control in miR-27a-5p mimics transfected cells and miR-27a-5p inhibitor transfected cells. (D) Luciferase reporter assays were performed using luciferase constructs carrying a WT or mutant TET3 3_-UTR cotransfected into H295R cells with miR-27a-5p mimics compared with an empty vector control. Firefly luciferase activity was normalized to renilla luciferase activity. (E) Western blot analysis of protein levels in H295R cells transfected with TET3 siRNA (siRNA 1#,2#) or scrambled control for 48h. (F) Methylation level of SYP promoter was analysis by EpiTect Methyl II PCR Assay in H295R cell transfected with TET3 siRNA or control. (G) Cortisol secretion were measured in cell supernatants by ELISA from H295R cell transfected with TET3 siRNA or control. Error bars represent SD. Three independent experiments were performed, and representative data are shown. The data represent the mean SD. NC, normal control. **p<0.01. *p<0.05.
To further determine whether miR-27a-5p directly binds TET3 mRNA and regulates its expression, a luciferase reporter construct containing the wild-type or mutant 3′-UTR coding sequences for TET3 was generated and introduced into miR-27a-5p mimics and H295R cells. Overexpression of miR-27a-5p significantly decreased the relative luciferase activity of the WT-3′-UTR of TET3 reporter plasmids, but when the miR-27a-5p seed sequence in TET3 mRNA 3′-UTR was mutated, the inhibitory effect of miR-27a-5p on the relative luciferase activity was abrogated (Figure 4D). Negative control miR-27a-5p mimics did not affect the wild-type or mutant constructs, confirming the specificity of the action. The results confirmed that TET3 is the direct target of miR-27a-5p.
SYP up-regulation is associated with TET3
The DNA methylation/demethylation status was controlled by the TET family of methylcytosine dioxygenases. We hypothesised that TET3 is associated with SYP hypomethylation in CPAs. Thus, we investigated the effect of TET3 knock-down by siRNA in H295R cells. The western blot results confirmed that TET3 was significantly downregulated when transfected with siRNA 2# (Figure 4E). As expected, SYP was significantly downregulated after being transfected with TET3 siRNA in H295R cells for two days (Figure 4E) and the methylation level of it significantly increased (Figure 4F). Secretion of cortisol was decreased at the same time (Figure 4G).
Discussion
In the present study, we analysed the mechanism of the miR-27a-5p-TET3-SYP signal pathway in adrenocortical adenomas causing CPA dysregulation (Figure 5). This analysis resulted in several novel findings that shed light on the molecular mechanism of CPA. Firstly, we used immunohistochemistry, western blotting and qRT-PCR to identify SYP expression signatures in CPA tissues and normal adrenal cortex tissues and found that SYP is significantly upregulated in the CPA tissues compared to the control group. Next, we showed that SYP is regulated by DNA methylation both in vitro and in vivo and the overexpression of SYP is related to the secretion of cortisol and cell proliferation. Then, we used a microRNA array to profile the microRNA expression signatures in CPA and normal cortex tissues and found that miR-27a-5p is one of the most significantly down-regulated microRNAs in the CPA group. We showed that miR-27a-5p plays a pivotal role in the regulation of function of H295R cells. Finally, we found that TET3 is the direct target of miR-27a-5p and confirmed that SYP up-regulation is associated with hypomethylation induced by TET3 in CPA.
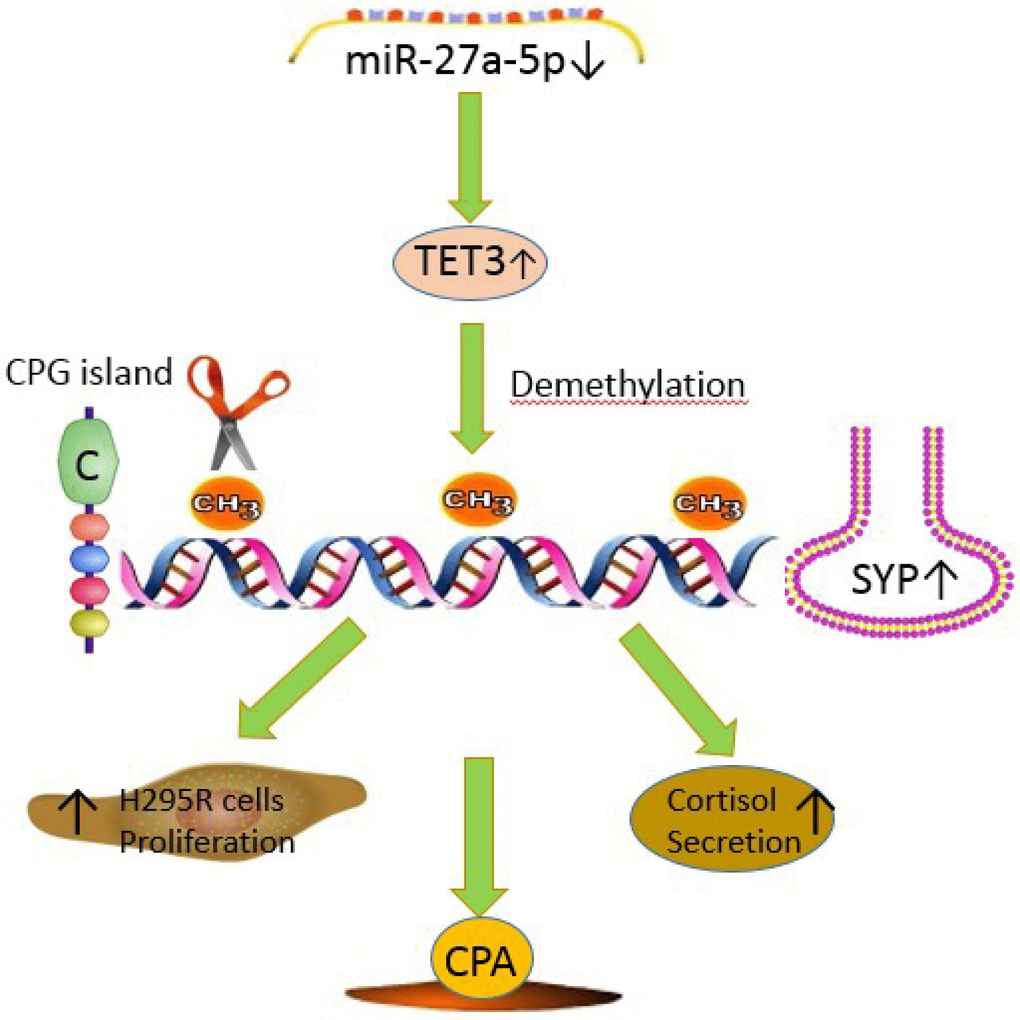
Figure 5. The mechanism diagram about miR-27a-5p-TET3-SYP signal pathway in adrenocortical adenomas causing CPA. miR-27a-5p suppresses SYP through epigenetic repression by targeting TET3 in adrenocortical adenomas causing CPA. miR-27a-5p-TET3-SYP signalling pathway may play a key role in CPA progression, H295R cell proliferation and cortisol secretion.
Recently, increasing evidence has clarified that SYP is ubiquitously expressed in adrenocortical tumours [12]. Nevertheless, there was no study about the the mechanisms involved in SYP aberrantly expressed in adrenocortical tumours. Moreover, further examination of SYP expression among adrenal disorders has not been extensively performed. Hence, we demonstrated that SYP expression was strikingly higher in the CPA than their adjacent normal adrenal cortex tissues by using qRT-PCR and immunohistochemistry. Western blotting, using the same antibodies as in the immunohistochemistry, also demonstrated a significantly increased expression of SYP in CPAs.
Recent studies have shown that a common epigenetic aberration in cancer involves deregulated DNA methylation such as CpG island hypermethylation that leads to gene silencing of specific tumour-suppressor genes [25–27]. Interestingly, a global hypomethylation has recently also been postulated to be an important contributor to tumourigenesis such as hepatocellular carcinoma [28]. Moreover, there are studies that demonstrated that primary and metastatic adrenocortical carcinoma samples have a global hypomethylation pattern compared with normal and benign adrenocortical tissue samples, whereas other studies clarified that some genes involved in cell cycle regulation, and apoptosis in the development of adrenal showed significant and frequent hypermethylation [29–31]. In addition, Brandi et al. found that aldosteronomas are globally hypomethylated and CYP11B2 is overexpressed, which is associated with hypomethylation in these tumours [15,32]. However, only a few reports have addressed DNA methylation in CPA and whether DNA methylation affects SYP expression in CPA has remained unknown. Therefore, we used methylation PCR assay to detect the aberrant methylation of SYP in CPA and the control group. We confirmed the presence of hypomethylation of the SYP promoter in CPA compared with normal adrenal cortex. Moreover, demethylation treatment of the adrenocortical carcinoma cell line H295R using 5-aza resulted in increased expression of SYP, which also supports the finding that SYP expression is regulated by an epigenetic mechanism. These findings provide the first evidence of a mechanism by which SYP is upregulated in CPA through epigenetic regulation of this gene.
A previous study reported that SYP mRNA levels showed a positive correlation with mRNA levels of CYP17A1 encoding 17a-OH [12]. They suggested that SYP expression in adrenocortical cells may be involved in some aspects of adrenal function such as transport or secretion of glucocorticoids. On the other hand, the corticosteroid hormone cortisol is an important determinant of blood pressure and cardiovascular risk [33]. For this reason, we detected whether the over-expression of SYP caused by DNA hypomethylation is related to the secretion of cortisol. After demethylation treatment of the adrenocortical carcinoma cell line H295R with 5-aza, the secretion of cortisol increased compared with the control group. Then, we knocked-down the expression of SYP in H295R cells and demonstrated that the effect of SYP knock-down decreased the secretion of cortisol. Moreover, the finding that SYP inhibits H295R cell proliferation was also confirmed.
Numerous evidences have shown the association of aberrantly expressed miRNAs with tumour development and progression [34–36]. Several studies have reported the different miRNA expression profiles in various types of adrenocortical adenomas compared with normal adrenal gland tissues [37–39]. Nevertheless, there have not been many studies directly comparing CPA and normal tissues using a miRNA microarray. In our present study, we established the specific miRNA expression profile in CPA compared with normal adrenal cortex tissues, and confirmed that the expression levels of miR-27a-5p were dramatically downregulated in CPA samples and involved in regulating the proliferation of H295R cells.
TET1, TET2, and TET3, the ten eleven translocation (TET) family of methylcytosine dioxygenases, can transform 5-methylcytosine (5mC) into 5-hydroxymethylcytosine (5hmC), and eliminate extant methylation labels in cells [40–42]. Specifically, the 5hmC level, which has a close connection with the gene expression of the TET family of methylcytosine dioxygenases, is raised in differentiated cells and sharply decreased in numerous cancer types, suggesting that the 5hmC level is negatively correlated with tumour progression [43,44]. The DNA methylation/demethylation status controlled by the TET family of methylcytosine dioxygenases, may impact the development of tumours. TET3 in mouse is reported to contribute to zygotic epigenetic reprogramming and global demethylation of the male pronucleus. During vertebrate neurogenesis, TET3 also plays an important role in enrichment of 5hmC at neurodevelopmental genes. Recently, a report showed that mutations of the isocitrate dehydrogenase (IDH) genes IDH1 and IDH2 can inactivate methylcytosine dioxygenase activity, resulting in DNA hypermethylation in acute myeloid leukemia cells [45]. This conclusion further enhances the outlook that TET methylcytosine dioxygenase plays a critical role in aberrant epigenetic regulation in cancer progression. Moreover, a study recently found that upregulation of ARL4C, due to DNA hypomethylation induced by TET upregulation, promotes tumourigenesis of lung squamous cell carcinoma [46]. In this study, we demonstrated that miR-27a-5p can directly regulate expression levels of TET3, suggesting that TET3 may serve as a major gene target in mediating the effect of miR-27a-5p in H295R cells proliferation and cortisol secretion. Our results further demonstrated that knock-down of TET3 can reduce the expression level of the SYP gene in H295R cell. Moreover, knock-down of TET3 results in decreased secretion of cortisol.
In this study, we just had twelve CPAs. The number of clinical samples is far from enough to demonstrate the conclusion. Obviously, more clinical samples will be needed to clarify the underlying mechanism.
In summary, our present study revealed that the miR-27a-5p-TET3-SYP signalling pathway may play a key role in CPA progression, H295R cell proliferation and cortisol secretion. SYP acts as a crucial regulator of adrenal hyperfunctions and the mechanism involved was also revealed. In addition, we clarified that TET3 is the direct target gene of miR-27a-5p. The overexpression of SYP is related to its hypomethylation of the CpG sites induced by TET3 in CPA. To the best of our knowledge, our results show for the first time the role of miR-27a-5p and SYP in the expression of cortisol secretion and proliferation of H295R cells. This is also the first time to reveal the mechanism of up-regulation of SYP expression in CPA. Therefore, the study suggested that miR-27a-5p, TET3 and SYP could be potent therapeutic agents. Investigating the role of the miR-27a-5p-TET3-SYP signalling pathway on adrenocortical adenomas will provide new insight into adrenocortical adenomas, and based on this understanding, future drugs targeting specific genes may be developed to treat CPA.
Materials and Methods
Tissue samples
Adrenocortical tissue sample collections were approved by the Ethics Committee of the Second Xiangya Hospital, Central South University, complied with the Declaration of Helsinki and written informed consent was obtained from all participants in our experiments.
Twenty-four adrenal tissue samples (12 adrenocortical adenomas from patients with adrenal Cushing’s syndrome, 12 from paired adjacent normal adrenal cortex) were obtained at surgical resection and immediately snap-frozen and stored at −80°C. The diagnosis of CPA was based on a typical clinical manifestation, increased serum cortisol levels with abnormal diurnal rhythm accompanied by decreased serum ACTH levels, increasing 24 h urinary-free cortisol, an un-suppressible result on the dexamethasone challenge test, and computed tomography results revealing unilateral adrenal tumours in all the patients. Moreover, the pathologic study confirmed the diagnosis. The clinical characteristics are summarized in Table 1.
Table 1. The clinical characteristics of CPA patients.
| Patients | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N | 12 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sex | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Male | 5(41.7%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Female | 7(58.3%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age | 44.92±9.38 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Body mass index(kg/m2) | 26.71±3.38 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8:00 serum cortisol(nmol/l) | 628.98±198.56 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 16:00 serum cortisol(nmol/l) | 571.47±275.78 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 24:00 serum cortisol(nmol/l) 24 h urinary free cortisol(nmol/24h) 8:00 plasma ACTH(ng/l) | 531.24±234.29 1909.39±1059.34 9.04±9.17 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ACTH, Adrenocorticotropic hormone | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Reagents
Dimethyl sulfoxide (DMSO) and 5-Aza-2′-deoxycytidine (5-Aza) were purchased from Sigma-Aldrich (St Louis, Missouri, USA). DMEM and fetal bovine serum were purchased from Gibco BRL Co. (Grand Island, New York). Lipofectamine 2000 was purchased from Invitrogen Co. (Carlsbad, California, USA). The antibody for β-actin was purchased from Abgent Inc. (San Diego, California, USA). Antibodies for SYP and TET3 were purchased from Abcam (Cambridge, England). Maxima SYBR Green/ROX qPCR Master Mix was purchased from Genecopoeia and all of the primers used in this research were purchased from Genecopoeia. The related secondary antibodies and the ECL detection kit were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, California, USA). SYP and TET3 siRNA oligos and control siRNA oligos were purchased from Ribobio (Guangzhou, China). miR-27a-5p mimics, inhibitors and their control oligos were purchased from Ribobio. DAPI was purchased from Solarbia (Beijing, China). The EdU kit was purchased from Ribobio. The CCK8 kit was purchased from 7 sea biotech (Shanghai, China).
Cell culture treatment and transfection
The adrenocortical carcinoma cell line H295R was obtained from the National Platform of Experimental Cell Resources for SciTech (Beijing, China). Cells were maintained in DMEM:F12 medium (Gibco, Life Technologies, Grand Island, New York, USA) supplemented with 2.5% of NuSerum (BD Biosciences, Bedford, Massachusetts, USA), 1% penicillin/ streptomycin (Gibco) and 1% insulin–transferin–selenium culture supplement (BD Biosciences) at 37°C in 5% CO2 infusion and humidified air and the medium was refreshed every 2 days.
After passaging for 12 hours, cells were treated with 5-aza at 5 uM (Sigma-Aldrich) and vehicle for 24 and 48 hours. The medium was replaced every 24 hours. Total protein and DNA was extracted after 48 hours of treatment and analysed for SYP protein and methylation expression level. The experiments were repeated three times. For transient transfection of SYP siRNA oligos, a combination of oligos (50 nM) and Lipofectamine 2000 was mixed following the manufacturer’s instructions and added to cells in 6-well plates at a density of 2 × 105 cells per well.
Measurement of cortisol
Cortisol secreted into the culture media was measured by competitive ELISA using commercial EIA kits (Cayman Chemical) according to the manufacturer’s instructions.
Western blot analysis
Western blot analysis was carried out for detection of SYP, TET3 or β-actin protein levels as previously described. Briefly, 30 μg of protein from each cell layer extract was loaded onto the same SDS PAGE and then transferred to a PVDF membrane. After blocking with 5% non-fat milk, the membrane was incubated with SYP, TET3 and β-actin overnight at 4°C. The next morning, the membrane was washed with PBS three times every ten minutes. The membrane was then incubated with appropriate secondary antibody at 1:2000 dilution in 2% non-fat milk for 1 h. Blots were processed using an ECL kit, exposed to film and then analysed by densitometry.
miRNA microarray analysis
Total RNA was extracted from cortisol-producing adenoma tissue samples and normal adrenal cortex tissue samples. All samples were assessed for enriched miRNA using an agilent 2100 Bioanalyzer. miRNAs were then labelled using the FlashTag Biotin RNA labelling kit (Affymetrix, Santa Clara, CA, USA) following the manufacturer’s protocol and then hybridized to Affymetrix GeneChip miRNA 1.0 microarrays (Affymetrix). Each miRNA was measured in duplicate.
Gene expression estimated using qRT-PCR
Total RNA was extracted from cortisol-producing adenoma tissue samples and normal adrenal cortex tissue samples, and cDNA was prepared. For analysis of miR-27a-5p expression, reverse transcription and quantitative reverse transcription-polymerase chain reaction (qRT–PCR) were carried out using the primer for human miR-27a-5p (Genecopoiea, MmiRQP0538) and U6 snRNA (Genecopoiea, China) according to the manufacturer’s instructions. SYP (HQP017850) and GAPDH (MQP027158) gene primers were purchased from Genecopoiea, and their mRNA expression was also measured by qRT–PCR amplification. Relative quantification was calculated through the 2-△△CT method.
Gene specific DNA methylation determination
Total genomic DNA was isolated from cortisol-producing adenoma tissue samples, normal adrenal cortex tissue samples and H295R cells using QIAamp DNA mini kit (Qiagen, Germany). Genomic DNA was isolated using the binding column according to the manufacturer’s instructions. Gene specific DNA methylation was determined by using an EpiTect Methyl II PCR Assay (Qiagen, Germany) and methyl primer synaptophysin (catalog# EPHS115006-1A). We evaluated the methylation level of SYP according to the manufacturer’s instructions.
Immunohistochemistry
Paraffin-embedded 4-mm-thick specimens were dewaxed in turpentine and rehydrate through decreased concentrations of ethanol. Antigen retrieval was not performed. Endogenous peroxidase activity was blocked by using 3% H2O2 in methnal for 10min and then soaked with phosphate buffered saline (PBS) (pH 7.2–7.4) three times for 5 min. The sections were then pre-incubated with 0.25% trypsin-EDTA for 10 min to block non-specific antigens. The tissue sections were allowed to react overnight at 4°C with anti-SYP (dilution 1/100; Abcam) TET3 (dilution 1/200, Abcam) antibodies. The slides were then incubated at room temperature for 1h, rinsed with PBS three times and incubated with appropriate biotinylated secondary antibodies for 30 min. For the quantification of immunohistochemistry, 10 fields of each section were viewed and analysed with an image analysis program (Bioquant, Nashville, TN).
Plasmid constructs and luciferase reporter assay
For functional analysis of miR-27a-5p, segments of the TET3 3’-UTR, including the predicted miR-27a-5p binding sites, were amplified using PCR and cloned into the PmeI and XbaI restriction sites of the luciferase reporter vector pmirGLO (Promega, Madison, WI, USA), resulting wild-type TET3 3’UTR (WT-TET3-3’UTR). The TET3 mutants for the miR-27a-5p seed regions were prepared using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, San Diego, CA, USA) to get mutant TET3 3’UTR (MUT-TET3-3’UTR). Sequences of the PCR and mutagenic primers are shown in (Table 2). H295R cells were transfected with either WT-TET3-3’UTR or MUT-TET3-3’UTR and miR-27a-5p mimics or control for 48 h. Luciferase activities were detected with the luciferase assay system (Promega).
Table 2. Nucleotide sequences of primers for WT and mutant reporter plasmids.
| Gene | Primer sequence (5′ to 3′) |
| WT TET3 | Forward: 5′ TAGTCTAGAAGAGGTGAGTCAAGAGGCAGTC 3′ |
| Reverse: 5′ GGCCGGCCACGCAACAGGCAGGGAAA 3′ | |
| Mutant TET3 | Forward: GCGGTGTGGTTGCCGTGCTCAAGGCGATGCTGATTTGTAC |
| Reverse: CGCCACACCAACGGCACGAGTTCCGCTACGACTAAACATG |
Statistical analysis
The results of the experiments are presented as means ± SD, and analysis was performed with Statistical Product and Service Solutions (SPSS) software (version 19.0). Comparisons between values of more than two groups were evaluated by an analysis of variance (one-way ANOVA). A level of p<0.05 was considered statistically significant. All experiments were repeated at least three times, and representative experiments are shown in the Figures.
Conflicts of Interest
The authors declare no conflict of interest.
Funding
The work was supported by the National Natural Science Foundation of China (grant numbers 81770881), the National Basic Research Program of China (973 Program) (grant number 2014CB942903), Major R & D projects in Hunan (grant number 217SK2074), the Fundamental Research Funds for the Central Universities of Central South University Grant 2018zzts048.).
References
- 1. Lacroix A, Feelders RA, Stratakis CA, Nieman LK. Cushing’s syndrome. Lancet. 2015; 386:913–27. https://doi.org/10.1016/S0140-6736(14)61375-1 [PubMed]
- 2. Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet. 2006; 367:1605–17. https://doi.org/10.1016/S0140-6736(06)68699-6 [PubMed]
- 3. Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, Montori VM. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008; 93:1526–40. https://doi.org/10.1210/jc.2008-0125 [PubMed]
- 4. Michaud K, Forget H, Cohen H. Chronic glucocorticoid hypersecretion in Cushing’s syndrome exacerbates cognitive aging. Brain Cogn. 2009; 71:1–8. https://doi.org/10.1016/j.bandc.2009.02.013 [PubMed]
- 5. Aulinas A, Ramírez MJ, Barahona MJ, Valassi E, Resmini E, Mato E, Santos A, Crespo I, Bell O, Surrallés J, Webb SM. Telomere length analysis in Cushing’s syndrome. Eur J Endocrinol. 2014; 171:21–29. https://doi.org/10.1530/EJE-14-0098 [PubMed]
- 6. Aulinas A, Ramírez MJ, Barahona MJ, Valassi E, Resmini E, Mato E, Santos A, Crespo I, Bell O, Surrallés J, Webb SM. Dyslipidemia and chronic inflammation markers are correlated with telomere length shortening in Cushing’s syndrome. PLoS One. 2015; 10:e0120185. https://doi.org/10.1371/journal.pone.0120185 [PubMed]
- 7. Wiedenmann B, Franke WW, Kuhn C, Moll R, Gould VE. Synaptophysin: a marker protein for neuroendocrine cells and neoplasms. Proc Natl Acad Sci USA. 1986; 83:3500–04. https://doi.org/10.1073/pnas.83.10.3500 [PubMed]
- 8. Wiedenmann B, Franke WW. Identification and localization of synaptophysin, an integral membrane glycoprotein of Mr 38,000 characteristic of presynaptic vesicles. Cell. 1985; 41:1017–28. https://doi.org/10.1016/S0092-8674(85)80082-9 [PubMed]
- 9. Leube RE. Expression of the synaptophysin gene family is not restricted to neuronal and neuroendocrine differentiation in rat and human. Differentiation. 1994; 56:163–71. https://doi.org/10.1046/j.1432-0436.1994.5630163.x [PubMed]
- 10. Maggiano N, Lauriola L, Serra FG, Ricci R, Capelli A, Ranelletti FO. Detection of synaptophysin-producing cells in human thymus by immunohistochemistry and nonradioactive in situ hybridization. J Histochem Cytochem. 1999; 47:237–43. https://doi.org/10.1177/002215549904700212 [PubMed]
- 11. Haak HR, Fleuren GJ. Neuroendocrine differentiation of adrenocortical tumors. Cancer. 1995; 75:860–64. https://doi.org/10.1002/1097-0142(19950201)75:3<860::AID-CNCR2820750318>3.0.CO;2-G [PubMed]
- 12. Shigematsu K, Nishida N, Sakai H, Igawa T, Toriyama K, Nakatani A, Takahara O, Kawai K. Synaptophysin immunoreactivity in adrenocortical adenomas: a correlation between synaptophysin and CYP17A1 expression. Eur J Endocrinol. 2009; 161:939–45. https://doi.org/10.1530/EJE-09-0596 [PubMed]
- 13. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012; 13:484–92. https://doi.org/10.1038/nrg3230 [PubMed]
- 14. Lin X, Li F, Xu F, Cui RR, Xiong D, Zhong JY, Zhu T, Shan SK, Wu F, Xie XB, Liao XB, Yuan LQ. Aberration methylation of miR-34b was involved in regulating vascular calcification by targeting Notch1. Aging (Albany NY). 2019; 11:3182–97. https://doi.org/10.18632/aging.101973 [PubMed]
- 15. Howard B, Wang Y, Xekouki P, Faucz FR, Jain M, Zhang L, Meltzer PG, Stratakis CA, Kebebew E. Integrated analysis of genome-wide methylation and gene expression shows epigenetic regulation of CYP11B2 in aldosteronomas. J Clin Endocrinol Metab. 2014; 99:E536–43. https://doi.org/10.1210/jc.2013-3495 [PubMed]
- 16. Zhao S, Wang Y, Liang Y, Zhao M, Long H, Ding S, Yin H, Lu Q. MicroRNA-126 regulates DNA methylation in CD4+ T cells and contributes to systemic lupus erythematosus by targeting DNA methyltransferase 1. Arthritis Rheum. 2011; 63:1376–86. https://doi.org/10.1002/art.30196 [PubMed]
- 17. Fang Y, Fang D, Hu J. MicroRNA and its roles in esophageal cancer. Med Sci Monit. 2012; 18:RA22–30. https://doi.org/10.12659/MSM.882509 [PubMed]
- 18. Cherradi N. microRNAs as Potential Biomarkers in Adrenocortical Cancer: progress and Challenges. Front Endocrinol (Lausanne). 2016; 6:195. https://doi.org/10.3389/fendo.2015.00195 [PubMed]
- 19. Li S, Chen T, Zhong Z, Wang Y, Li Y, Zhao X. microRNA-155 silencing inhibits proliferation and migration and induces apoptosis by upregulating BACH1 in renal cancer cells. Mol Med Rep. 2012; 5:949–54. https://doi.org/10.3892/mmr.2012.779 [PubMed]
- 20. Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009; 136:642–55. https://doi.org/10.1016/j.cell.2009.01.035 [PubMed]
- 21. Sandoval-Hernández AG, Hernández HG, Restrepo A, Muñoz JI, Bayon GF, Fernández AF, Fraga MF, Cardona-Gómez GP, Arboleda H, Arboleda GH, Liver X. Liver X Receptor Agonist Modifies the DNA Methylation Profile of Synapse and Neurogenesis-Related Genes in the Triple Transgenic Mouse Model of Alzheimer’s Disease. J Mol Neurosci. 2016; 58:243–53. https://doi.org/10.1007/s12031-015-0665-8 [PubMed]
- 22. Proctor DT, Coulson EJ, Dodd PR. Reduction in post-synaptic scaffolding PSD-95 and SAP-102 protein levels in the Alzheimer inferior temporal cortex is correlated with disease pathology. J Alzheimers Dis. 2010; 21:795–811. https://doi.org/10.3233/JAD-2010-100090 [PubMed]
- 23. Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997; 56:933–44. https://doi.org/10.1097/00005072-199708000-00011 [PubMed]
- 24. Lin X, Zhan JK, Zhong JY, Wang YJ, Wang Y, Li S, He JY, Tan P, Chen YY, Liu XB, Cui XJ, Liu YS. lncRNA-ES3/miR-34c-5p/BMF axis is involved in regulating high-glucose-induced calcification/senescence of VSMCs. Aging (Albany NY). 2019; 11:523–35. https://doi.org/10.18632/aging.101758 [PubMed]
- 25. Kulis M, Esteller M. DNA methylation and cancer. Adv Genet. 2010; 70:27–56. https://doi.org/10.1016/B978-0-12-380866-0.60002-2 [PubMed]
- 26. Taberlay PC, Jones PA. DNA methylation and cancer. Prog Drug Res. 2011; 67:1–23. https://doi.org/10.1007/978-3-7643-8989-5_1 [PubMed]
- 27. Li RJ, Zhang GS, Chen YH, Zhu JF, Lu QJ, Gong FJ, Kuang WY. Down-regulation of mitochondrial ATPase by hypermethylation mechanism in chronic myeloid leukemia is associated with multidrug resistance. Ann Oncol. 2010; 21:1506–14. https://doi.org/10.1093/annonc/mdp569 [PubMed]
- 28. Chuang KH, Whitney-Miller CL, Chu CY, Zhou Z, Dokus MK, Schmit S, Barry CT. MicroRNA-494 is a master epigenetic regulator of multiple invasion-suppressor microRNAs by targeting ten eleven translocation 1 in invasive human hepatocellular carcinoma tumors. Hepatology. 2015; 62:466–80. https://doi.org/10.1002/hep.27816 [PubMed]
- 29. Rechache NS, Wang Y, Stevenson HS, Killian JK, Edelman DC, Merino M, Zhang L, Nilubol N, Stratakis CA, Meltzer PS, Kebebew E. DNA methylation profiling identifies global methylation differences and markers of adrenocortical tumors. J Clin Endocrinol Metab. 2012; 97:E1004–13. https://doi.org/10.1210/jc.2011-3298 [PubMed]
- 30. Fonseca AL, Kugelberg J, Starker LF, Scholl U, Choi M, Hellman P, Åkerström G, Westin G, Lifton RP, Björklund P, Carling T. Comprehensive DNA methylation analysis of benign and malignant adrenocortical tumors. Genes Chromosomes Cancer. 2012; 51:949–60. https://doi.org/10.1002/gcc.21978 [PubMed]
- 31. H’mida Ben-Brahim D, Hammami S, Haddaji Mastouri M, Trabelsi S, Chourabi M, Sassi S, Mougou S, Gribaa M, Zakhama A, Guédiche MN, Saad A. Partial KCNQ1OT1 hypomethylation: A disguised familial Beckwith-Wiedemann syndrome as a sporadic adrenocortical tumor. Appl Transl Genom. 2014; 4:1–3. https://doi.org/10.1016/j.atg.2014.10.001 [PubMed]
- 32. Wang B, Yin BL, He B, Chen C, Zhao M, Zhang W, Xia ZK, Pan Y, Tang J, Zhou X, Yin N. Overexpression of DNA damage-induced 45 α gene contributes to esophageal squamous cell cancer by promoter hypomethylation. J Exp Clin Cancer Res. 2012; 31:11. https://doi.org/10.1186/1756-9966-31-11 [PubMed]
- 33. Whitworth JA, Mangos GJ, Kelly JJ. Cushing, cortisol, and cardiovascular disease. Hypertension. 2000; 36:912–16. https://doi.org/10.1161/01.HYP.36.5.912 [PubMed]
- 34. Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M, Wojcik SE, Iorio MV, Visone R, Sever NI, Fabbri M, Iuliano R, Palumbo T, Pichiorri F, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005; 353:1793–801. https://doi.org/10.1056/NEJMoa050995 [PubMed]
- 35. He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM. A microRNA polycistron as a potential human oncogene. Nature. 2005; 435:828–33. https://doi.org/10.1038/nature03552 [PubMed]
- 36. Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007; 449:682–88. https://doi.org/10.1038/nature06174 [PubMed]
- 37. Robertson S, MacKenzie SM, Alvarez-Madrazo S, Diver LA, Lin J, Stewart PM, Fraser R, Connell JM, Davies E. MicroRNA-24 is a novel regulator of aldosterone and cortisol production in the human adrenal cortex. Hypertension. 2013; 62:572–78. https://doi.org/10.1161/HYPERTENSIONAHA.113.01102 [PubMed]
- 38. He J, Cao Y, Su T, Jiang Y, Jiang L, Zhou W, Zhang C, Wang W, Ning G. Downregulation of miR-375 in aldosterone-producing adenomas promotes tumour cell growth via MTDH. Clin Endocrinol (Oxf). 2015; 83:581–89. https://doi.org/10.1111/cen.12814 [PubMed]
- 39. Patel D, Boufraqech M, Jain M, Zhang L, He M, Gesuwan K, Gulati N, Nilubol N, Fojo T, Kebebew E. MiR-34a and miR-483-5p are candidate serum biomarkers for adrenocortical tumors. Surgery. 2013; 154:1224–28. https://doi.org/10.1016/j.surg.2013.06.022 [PubMed]
- 40. Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010; 466:1129–33. https://doi.org/10.1038/nature09303 [PubMed]
- 41. Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, An J, Lamperti ED, Koh KP, Ganetzky R, Liu XS, Aravind L, Agarwal S, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010; 468:839–43. https://doi.org/10.1038/nature09586 [PubMed]
- 42. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009; 324:930–35. https://doi.org/10.1126/science.1170116 [PubMed]
- 43. Haffner MC, Chaux A, Meeker AK, Esopi DM, Gerber J, Pellakuru LG, Toubaji A, Argani P, Iacobuzio-Donahue C, Nelson WG, Netto GJ, De Marzo AM, Yegnasubramanian S. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget. 2011; 2:627–37. https://doi.org/10.18632/oncotarget.316 [PubMed]
- 44. Yang H, Liu Y, Bai F, Zhang JY, Ma SH, Liu J, Xu ZD, Zhu HG, Ling ZQ, Ye D, Guan KL, Xiong Y. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene. 2013; 32:663–69. https://doi.org/10.1038/onc.2012.67 [PubMed]
- 45. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010; 18:553–67. https://doi.org/10.1016/j.ccr.2010.11.015 [PubMed]
- 46. Fujii S, Shinjo K, Matsumoto S, Harada T, Nojima S, Sato S, Usami Y, Toyosawa S, Morii E, Kondo Y, Kikuchi A. Epigenetic upregulation of ARL4C, due to DNA hypomethylation in the 3′-untranslated region, promotes tumorigenesis of lung squamous cell carcinoma. Oncotarget. 2016; 7:81571–87. https://doi.org/10.18632/oncotarget.13147 [PubMed]