



Introduction
Idiopathic pulmonary fibrosis (IPF) is a specific form of chronic, progressive, fibrosing interstitial pneumonia [1] with a median survival rate of 3.8 years after diagnosis [2]. IPF is characterized by excessive scar tissue formation and destruction of lung parenchyma, resulting in gas exchange abnormalities and respiratory failure. The annual cumulative prevalence increased steadily from 202.2 cases per 100000 people in 2001 to 494.5 cases per 100000 people in 2011 in the USA. Currently, drug treatment cannot definitively improve the quality of life for IPF patients or “reverse” fibrosis.
The incidence of IPF increases with age, and it is now widely regarded as a disease of aging [3]. IPF patients under 50 years of age are rare [4]. Two-thirds of IPF patients are older than 60 years at the time of presentation with a mean age of 66 years at the time of diagnosis. Furthermore, the survival rate of IPF patients markedly decreases with age [5]. Aging is considered the main risk factor for IPF [6]. Different mechanisms associated with aging, including stem cell dysfunction [7], aging mediator overexpression [8], and mitochondrial superoxide [9], have been described to participate in the pathogenesis of IPF.
Increasing evidence has implicated that defects in alveolar epithelial cells (AECs) induce lung fibrosis. Histopathologic abnormalities of AEC have been observed in tissue sections from IPF patients and in animal models of pulmonary fibrosis [10]. Mutations in AEC genes, including surfactant proteins A and C, have been linked to familial disease [11]. Transgenic animal experiments have confirmed that targeted injury to AEC is sufficient to initiate lung fibrosis [12, 13].
Mechanisms of AEC injury and dysregulated repair responses have been proposed to be critically involved in the progression of IPF [14]. Senescence of AEC may be detrimental to lung repair. Depletion of senescent epithelial cells in vitro and ex vivo decreases fibrotic markers [15]. However, the pathogenesis of AEC senescence involved in IPF is largely unknown.
Phosphatase and tension homolog deleted on chromosome ten (PTEN) has recently drawn extensive attention for its critical anti-tumor function, as it regulates several fundamental cellular processes, including cell adhesion, growth, migration and apoptosis [16]. Several studies have investigated the effects of PTEN on longevity [17, 18]. Interestingly, while monoallelic loss or mutation of PTEN gene drives cellular proliferation, complete inactivation of PTEN induces a senescence response in tumor cells [19]. Additionally, previous studies have investigated the role of PTEN in aging-related renal impairment [20]. Genetic ablation of PTEN in prostatic epithelial cells induces cell senescence [21]. Thus, we posited that PTEN may be involved in the pathogenesis of the senescence-associated molecular mechanism of IPF.
The protein kinase B (Akt) signaling pathway, which is negatively regulated by PTEN, plays a critical role in the regulation of cell growth and cell survival in various systems. The Akt pathway also constitutes an endogenous negative feedback regulator that reduces oxidative stress and limits pro-inflammatory and apoptotic events in response to injury [22]. There have been many attempts to investigate the involvement of the Akt pathway in the aging process [23–26]. It remains poorly understood if PTEN-regulation of the Akt pathway during AEC senescence is involved in the pathogenesis of pulmonary fibrosis.
In this study, we confirmed that AECs possess senescent properties in IPF patients. Bleomycin-stimulated AECs in vitro and a bleomycin-induced mouse pulmonary fibrosis model demonstrated that the loss of PTEN causes senescence in AECs through activation of the Akt pathway. Knockdown of Akt or inhibition of Akt activation reverses this change.
Results
IPF is a disease of aging
In this study, we used the aging-related markers, P21WAF1 and SA-β-Gal, to detect senescence in IPF lung tissues and AECs. P21WAF1 was detected by methods including western blotting, IHC and immunofluorescence staining. As shown in Figure 1, both senescent markers, P21WAF1 and SA-β-Gal, were over-expressed in the lung tissues from IPF patients compared to age-matched normal controls (Figure 1A–1G). HE and IHC staining showed a destroyed alveolar structure (Figure 1A, 1B), and the positive staining of P21WAF1 was primarily located in AECs of fibrotic areas in the lung tissues of IPF patients. In addition, strongly positive SA-β-Gal staining was primarily located in AECs from lung tissues with IPF, whereas SA-β-Gal staining was barely observed in normal lung tissues (Figure 1C, 1D). Furthermore, immunofluorescence was used to examine the spatial location of P21WAF1 and AEC2 markers (SP-C), in IPF lung and normal tissues (Figure 1E). P21WAF1 was overexpressed in IPF lung tissue but was barely present in normal lung tissue. Western blotting confirmed P21WAF1 expression differences between IPF and normal lung tissues (Figure 1F, 1G). Taken together, these data suggested that IPF is an aging-related disease as previous findings demonstrated and abnormal senescence occurs in AECs from IPF lung tissues.
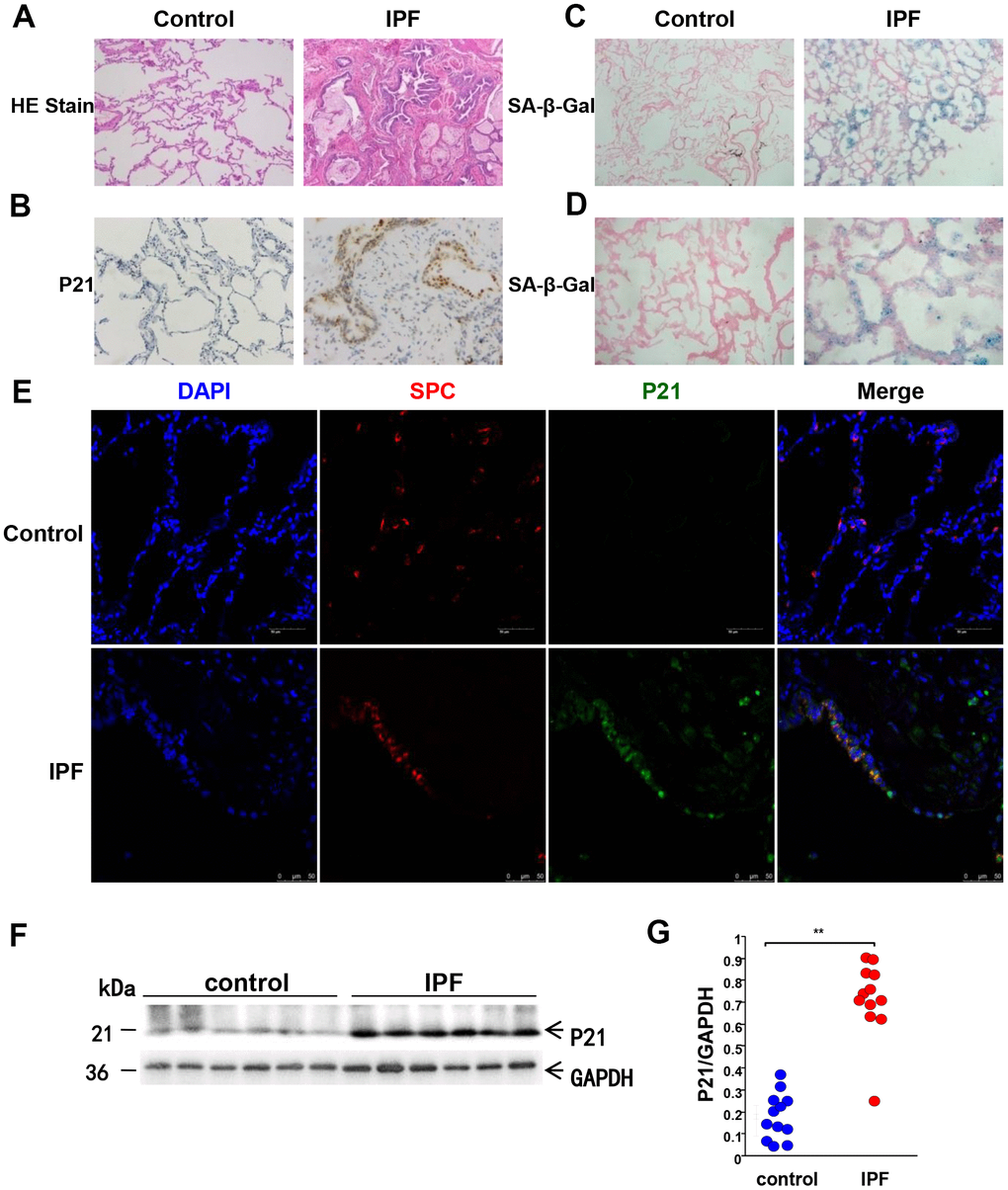
Figure 1. Aging-related markers are significantly overexpressed in the lung tissues of IPF patients. Human lung tissues of IPF (n = 12) and age-matched normal lung tissues (n = 12) were analyzed. (A) Representative images of HE staining of lung tissues (original magnification, 100×). (B) Representative images of the aging-related marker, P21WAF1, via IHC staining of lung tissues (original magnification, 100× in control and 200× in IPF). (C) Representative results of SA-β-Gal staining of lung tissues (original magnification, 100×). (D) Enlarged images of SA-β-Gal staining (200×). (E) Immunofluorescence staining of SP-C (an AEC2-specific marker, red) and P21WAF1(green) was conducted to confirm senescent marker expression in AECs (original magnification, 400×). (F, G) P21WAF1 senescent marker in lung tissues was measured by western blotting. Each dot represents an individual lung tissue. **p < 0.01. Unpaired, two-tailed Student’s t test.
PTEN loss and Akt pathway activation in AECs from IPF
After demonstrating that aging-related markers were overexpressed in AECs from IPF lung tissues, we next investigated if PTEN and the Akt pathway participated in senescence of AECs from IPF lung tissues. As indicated in Figure 2A, the phosphorylation levels of Akt were higher in IPF lung tissues than in normal lung tissues, while the level of PTEN was lower in IPF. Both markers were observed predominantly distributed within AECs in fibrotic areas. western blot analysis was then performed using IPF lung tissues and normal lung tissues. The results of PTEN and p-Akt 473 were consistent with IHC staining (Figure 2C–2E). To further confirm PTEN location, we conducted immunofluorescence staining. As shown in Figure 2B, PTEN was downregulated in IPF lung tissues. In normal lung tissues, PTEN was mainly distributed in AECs, but some PTEN colocalized with SP-C. Therefore, these data suggested that the decreased expression of PTEN may be related to Akt activation in IPF and that this regulation primarily occurs within AECs. To further confirm our findings from clinical samples, in vitro experiments were performed to elucidate the mechanisms underlying AEC senescence.
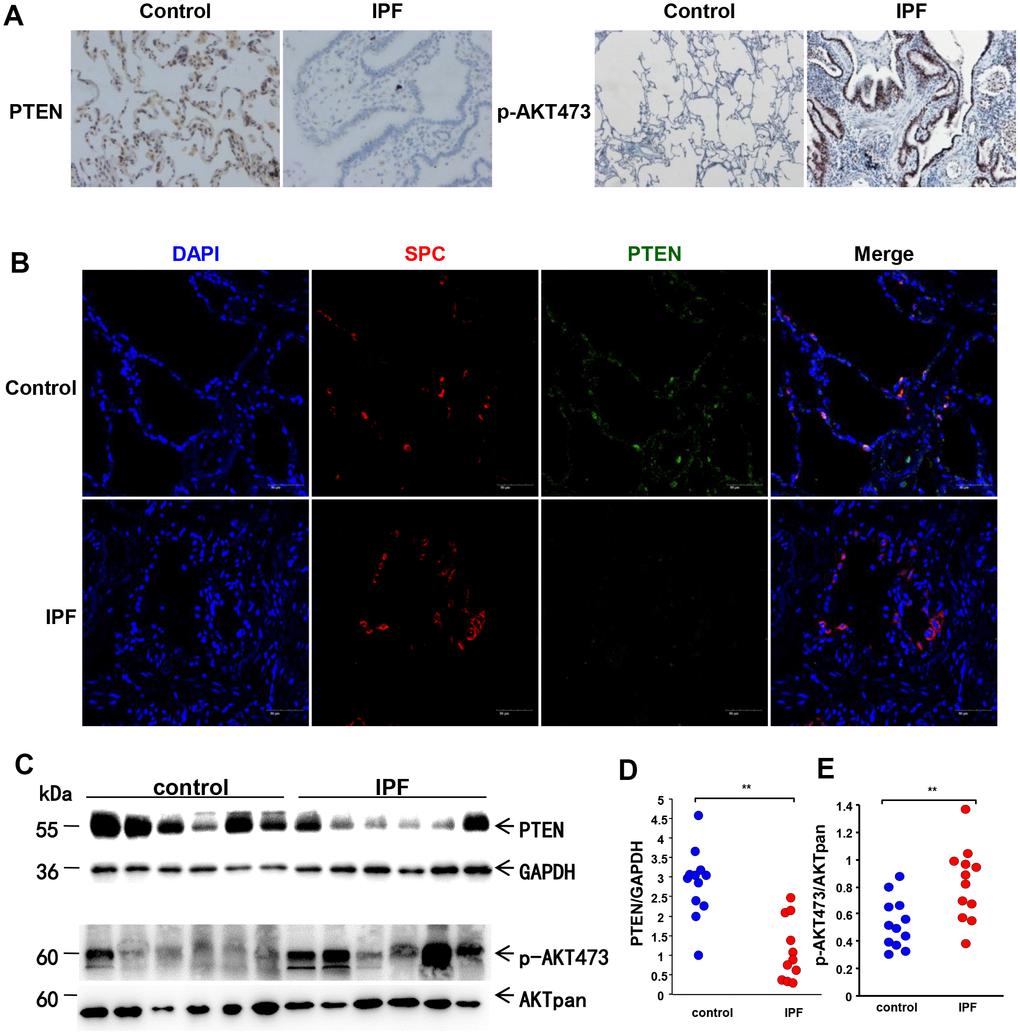
Figure 2. Loss of PTEN and activation of the AKT pathway in lung tissue from IPF patients. (A) Representative results of IHC staining for PTEN and p-AKT 473 in lung tissues (original magnification, 200× for PTEN and 100× for p-AKT 473). (B) Immunofluorescence staining for both SP-C (red) and PTEN (green) were conducted to examine the spatial distribution of PTEN (original magnification, 200×). (C–E) Western blot analysis was applied to detect the expression of PTEN and activation of the AKT pathway. Each dot represents an individual lung tissue. **p < 0.01. Unpaired, two-tailed Student’s t test.
PTEN is decreased in bleomycin-induced AEC senescence and Akt pathway phosphorylation levels are increased
Bleomycin has been used to induce AEC senescence in previous studies. Here, we employed bleomycin to generate a cellular senescence model using A549 cell lines. In many studies, A549 is generally used as a replacement for primary AECs because AECs are difficult to obtain and maintain in culture ex vivo. Gradually increasing concentrations of bleomycin were added to culture medium to stimulate A549 cells for 72 hours, and cells were then transferred to fresh FBS-free medium for another 24 hours to generate the cellular senescence model. SA-β-Gal staining and western blotting were performed. As shown in Figure 3, the intensity of positive SA-β-Gal staining increased with increasing concentrations of bleomycin (Figure 3A, 3B). In addition, the senescence-related marker, P21WAF1, was overexpressed in a dose-dependent manner in stimulated A549 cells (Figure 3C, 3D). To investigate the role of PTEN and the Akt pathway during bleomycin-induced cellular senescence, we examined the expression of PTEN and Akt in bleomycin-induced senescent A549 cells. As shown in Figure 3E–3G, PTEN was significantly reduced with increasing concentrations of bleomycin, and activation of the Akt pathway was detected. These data suggested that loss of PTEN activated the Akt pathway to promote AEC senescence, thus participating in the pathogenesis of IPF. For further examination, we conducted a set of in vitro experiments to elucidate the potential mechanisms underlying cellular senescence in IPF.
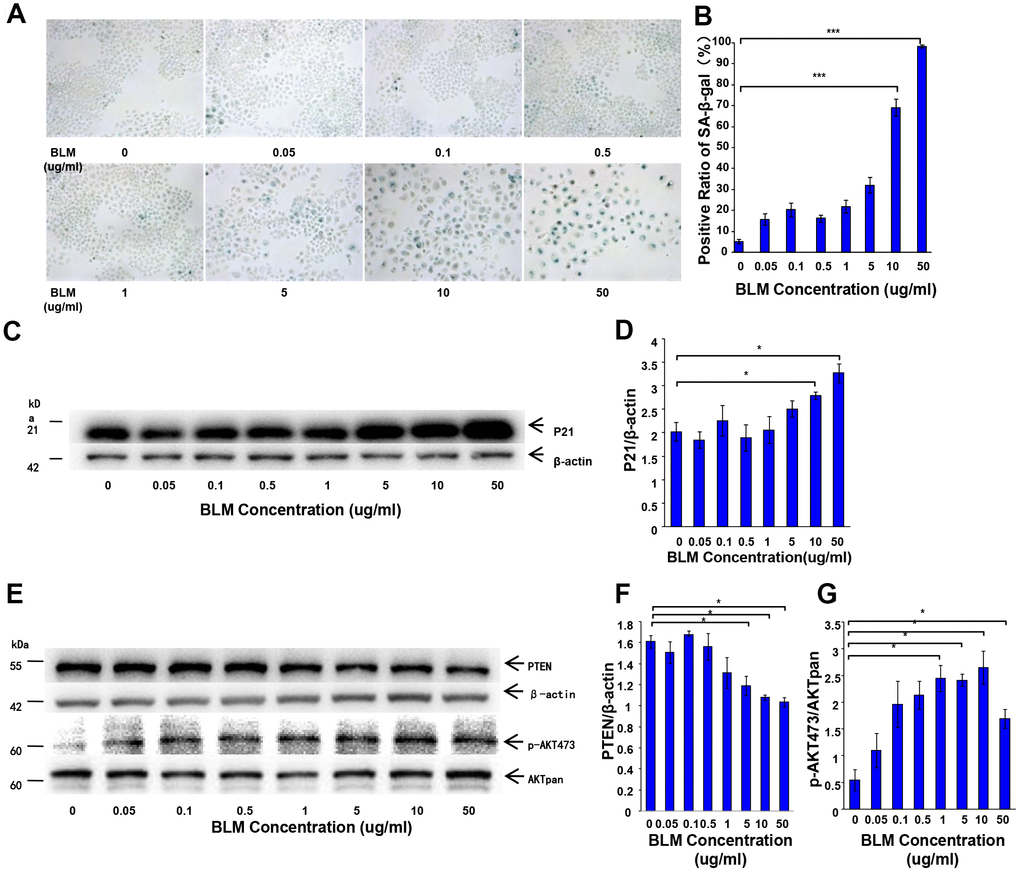
Figure 3. Decrease of PTEN and activation of the AKT pathway in the senescent cell model. Gradually increasing concentrations of bleomycin was added to culture medium to stimulate A549 cells for 72 hours followed by transfer to fresh FBS-free medium for another 24 hours. (A, B) SA-β-Gal staining was performed to detect cellular senescence (original magnification, 200×). (C, D) The expression of the aging-related marker, P21WAF1, was detected by western blot analysis. (E–G) PTEN loss and AKT pathway activation were observed. Data are shown as the mean ± SEM, n ≥ 3 per group. *p < 0.05, ***p < 0.001. One-way ANOVA followed by Dunnett’s Multiple Comparison Test.
Change of PTEN expression affects AEC senescence and Akt activation
Although our initial experiments showed reduced PTEN expression and elevated Akt activation in IPF lung tissues and in the senescence cell model, it remained uncertain if decreased PTEN and increased Akt activation were responsible for the induction of AEC senescence. To determine if the change of PTEN expression affects AEC senescence through the activation of Akt, PTEN was overexpressed or knocked down in A549 cells. Cells were cultured in medium containing bleomycin for 72 hours and then transferred to fresh FBS-free medium for 24 hours. SA-β-Gal staining and western blotting were then performed. As shown in Figure 4, the intensity of positive SA-β-Gal staining decreased when PTEN was overexpressed (Figure 4A, 4B), whereas the intensity of positive SA-β-Gal staining increased after PTEN was knocked down (Figure 4H–4I). The Akt pathway was activated after PTEN knockdown, and the expression levels of the aging-related marker, P21ink4a, were substantially higher in the knockdown cells than in the control cells (Figure 4J–4N). Up-regulation of PTEN caused Akt inactivation in senescence cells (Figure 4C–4G). These results supported our hypothesis that the loss of PTEN accelerates the senescence of AECs via activation of the Akt pathway.
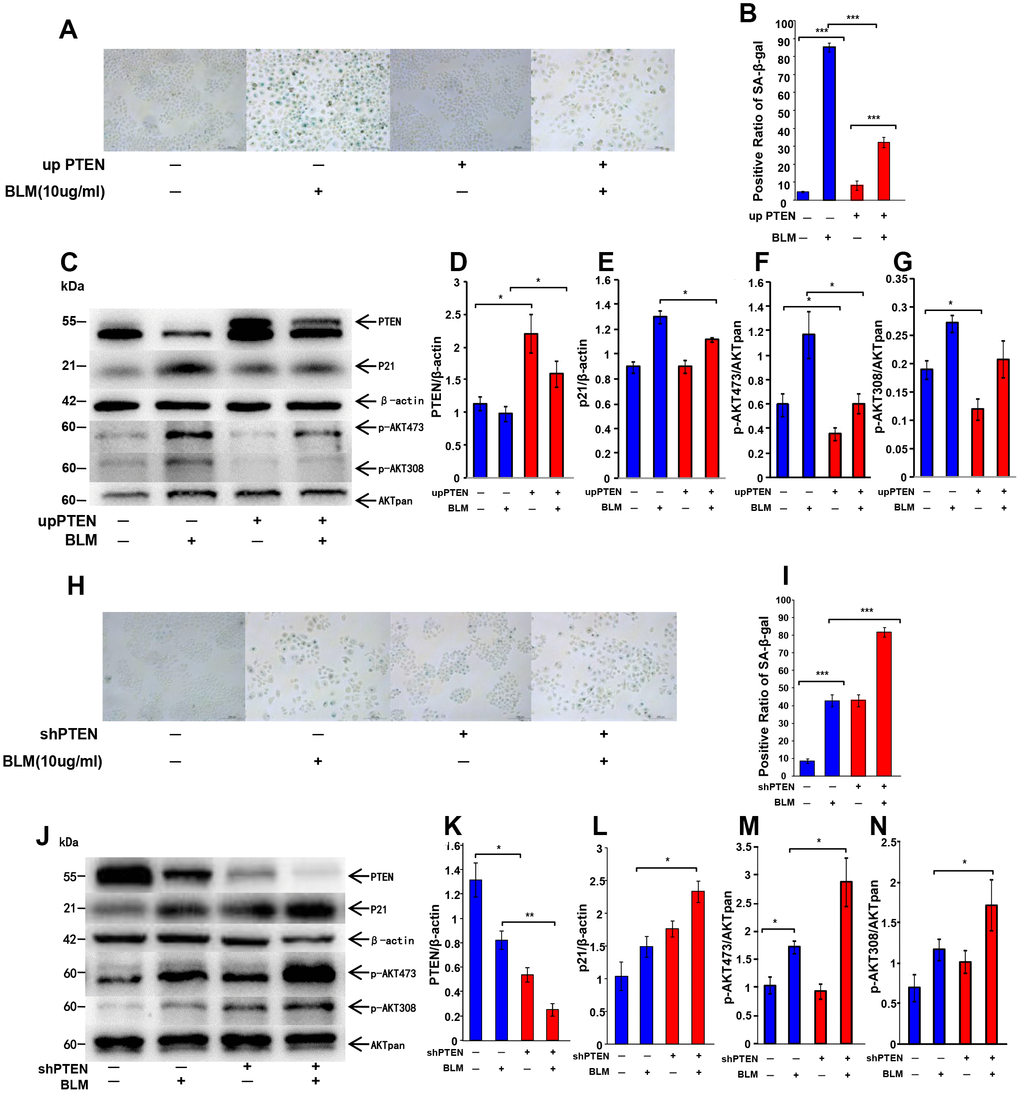
Figure 4. Change of PTEN expression affects cell senescence through AKT pathway activation. Genetic overexpression of PTEN in A549 cells was achieved by a transduction of a lentiviral vector followed by bleomycin (10 μg/ml) stimulation for 72 hours and transfer to fresh medium for 24 hours. (A, B) Cellular senescence was detected by SA-β-Gal staining (original magnification, 200×). (C–G) Western blot analysis was performed to confirm alteration of P21WAF1 and targets of the PTEN/AKT pathway. Lentiviral vector was used to knockdown PTEN expression in A549 cells. Bleomycin was then added to the culture medium to stimulate cells for 72 hours followed by transfer to fresh FBS-free medium for another 24 hours. (H, I) SA-β-Gal staining was applied to detect cellular senescence (original magnification, 200×). (J–N) Change of P21WAF1 and expression of PTEN/AKT targets were determined by western blotting. Data are shown as the mean ± SEM, n ≥ 3 per group. *p < 0.05, **p < 0.01, ***p < 0.001. One-way ANOVA followed by Dunnett’s Multiple Comparison Test.
Knockdown of Akt2, but not Akt1, rescues cell from bleomycin-induced cell senescence
There are three members in the Akt kinase family, namely, Akt1, Akt2, and Akt3, which share a high degree of homology. Akt1 and Akt2 are broadly expressed in many tissues and cell types, whereas Akt3 is predominantly expressed in brain tissue [27, 28]. Each of the Akt isoforms has a distinct but overlapping function in the regulation of cell proliferation, cell apoptosis, protein synthesis, and the cell cycle [29]. Numerous clinical studies have indicated differential expression of Akt isoforms in different types of cancer and cells [30–33]. However, the consequences of suppression of Akt activation by PTEN in AEC senescence and its potential prevention of lung fibrosis are still unknown. Furthermore, it remains unclear if Akt1 and Akt2 both play comparable roles in AEC senescence. To examine if Akt and, particularly, which Akt isoform (Akt1 or Akt2) is involved in the development of AEC senescence, we used lentivirus to knockdown Akt1 or Akt2 in A549 cells to compare the senescence level in bleomycin-stimulated cells. Western blot analysis was used to verify Akt1 and Akt2 silencing. As shown in Figure 5C, 5H, both shRNAs targeting Akt1 and Akt2 (shRNA1 and shRNA2) decreased Akt1 and Akt2 protein levels. SA-β-Gal staining demonstrated that silencing of Akt1 had only a limited impact on cell senescence (Figure 5F, 5G). However, silencing of Akt2 significantly reduced cell senescence rates in bleomycin-stimulated AECs (Figure 5A, 5B) compared to the corresponding control shRNA. A similar result was found with measurement of the expression of the senescence-related marker, P21WAF1 (Figure 5C–5E, 5H–5I). This result indicated that only Akt2 is involved in bleomycin-induced AEC senescence.
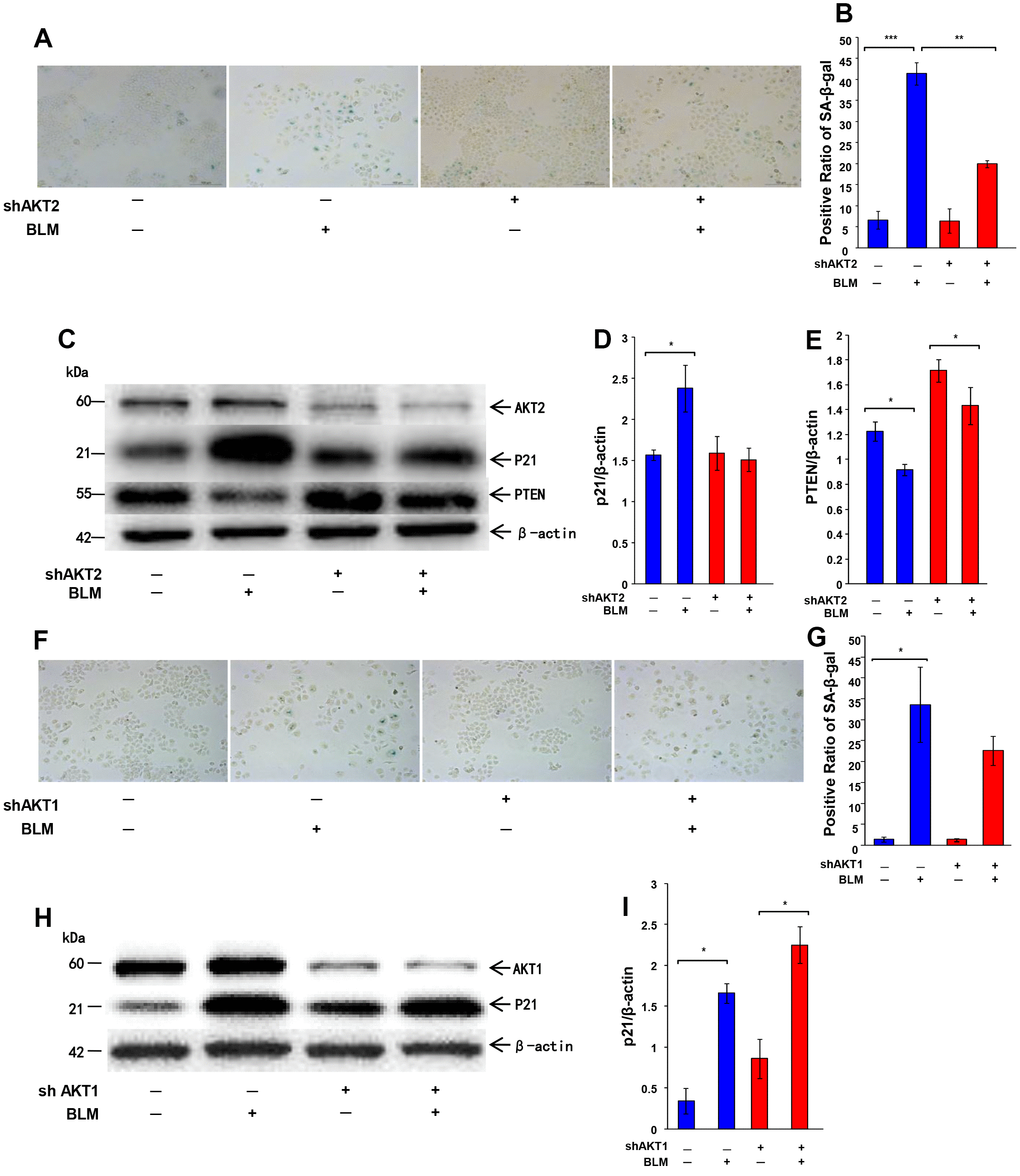
Figure 5. Knockdown AKT2, but not AKT1, rescues cell from bleomycin-induced cell senescence. The expression of AKT2 or AKT1 was knocked down by lentiviral vector in A549 cells followed by bleomycin (10 μg/ml) stimulation for 72 hours and transfer to fresh medium for 24 hours. (A, B) Cellular senescence was detected by SA-β-Gal staining (original magnification, 200×). (C–E) Western blotting was performed to confirm the change of P21WAF1, PTEN and AKT2. (F–G) Cellular senescence after AKT1 knockdown was analyzed by SA-β-Gal staining (original magnification, 200×). (H, I). Expression of P21WAF1 and AKT1 was confirmed by western blotting. Data are shown as the mean ± SEM, n ≥ 3 per group. *p < 0.05, **p < 0.01, ***p < 0.001. One-way ANOVA followed by Dunnett’s Multiple Comparison Test.
Inhibition of Akt activation attenuates AEC senescence
Because the activation of Akt is thought to accelerate cellular senescence, we observed the activation of Akt in senescent AECs, and we then verified if activated Akt also has a regulatory effect on AEC senescence. Two specific inhibitors of the Akt pathway were used. As shown in Figure 6, SA-β-Gal staining and the P21WAF1 senescence marker were significantly reduced after inhibition of Akt activation using LY294002 or MK2206 in bleomycin-stimulated AECs.
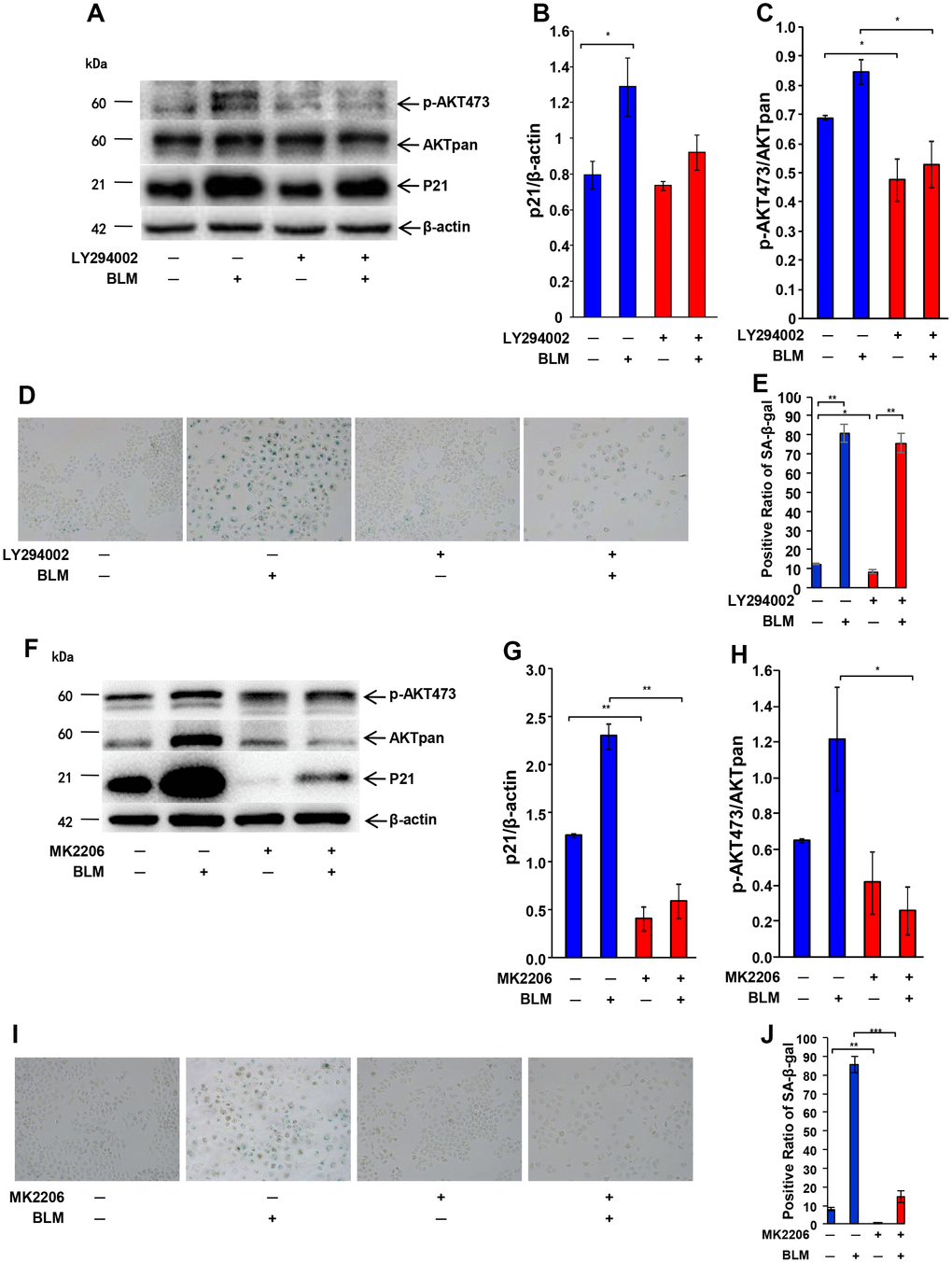
Figure 6. Inhibition of Akt activation attenuates AEC senescence. Inhibitor (20 μm/L LY294002 or 5 μg/ml MK2206) of the AKT pathway was added 1 hour before bleomycin (10 μg/ml). After 72 hours, the medium was changed to fresh medium for 24 hours. AKT inactivation and P21WAF1 expression induced by LY294002 (A–C) or MK2206 (F–H) were detected by western blotting. Cellular senescence after inhibitor LY294002 (D, E) or MK2206 (I, J) pretreatment was analyzed by SA-β-Gal staining. Data are shown as the mean ± SEM, n ≥ 3 per group. *p < 0.05. One-way ANOVA followed by Dunnett’s Multiple Comparison Test.
Akt inactivation rescues AEC senescence in vitro
We next explored if PTEN loss-induced AEC senescence depends on Akt activation. After stable knockdown PTEN, A549 cells were treated with LY294002 or MK2206 for 72 hours, and cells were then cultured in FBS-free medium for another 24 hours. As shown in Figure 7, PTEN loss induced senescence in A549, while inhibition of Akt by LY294002 or MK2206 rescues senescence. Both increased positive SA-β-Gal staining and P21WAF1 expression were decreased when Akt activation was blocked by LY294002 or MK2206 in A549 cells (Figure 7). To further confirm this finding, bleomycin was used to stimulate cells. PTEN-silenced A549 cells were pretreated with LY294002 or MK2206 for 1 hour, and bleomycin was then used to induce cell senescence for 72 hours. Cells were then cultured in fresh medium for an additional 24 hours. SA-β-Gal staining and western blotting showed that LY294002- or MK2206-pretreated cells presented lower intensity of positive SA-β-Gal staining and P21WAF1 protein expression. These results demonstrated that PTEN loss-induced AEC senescence depends on Akt activation.
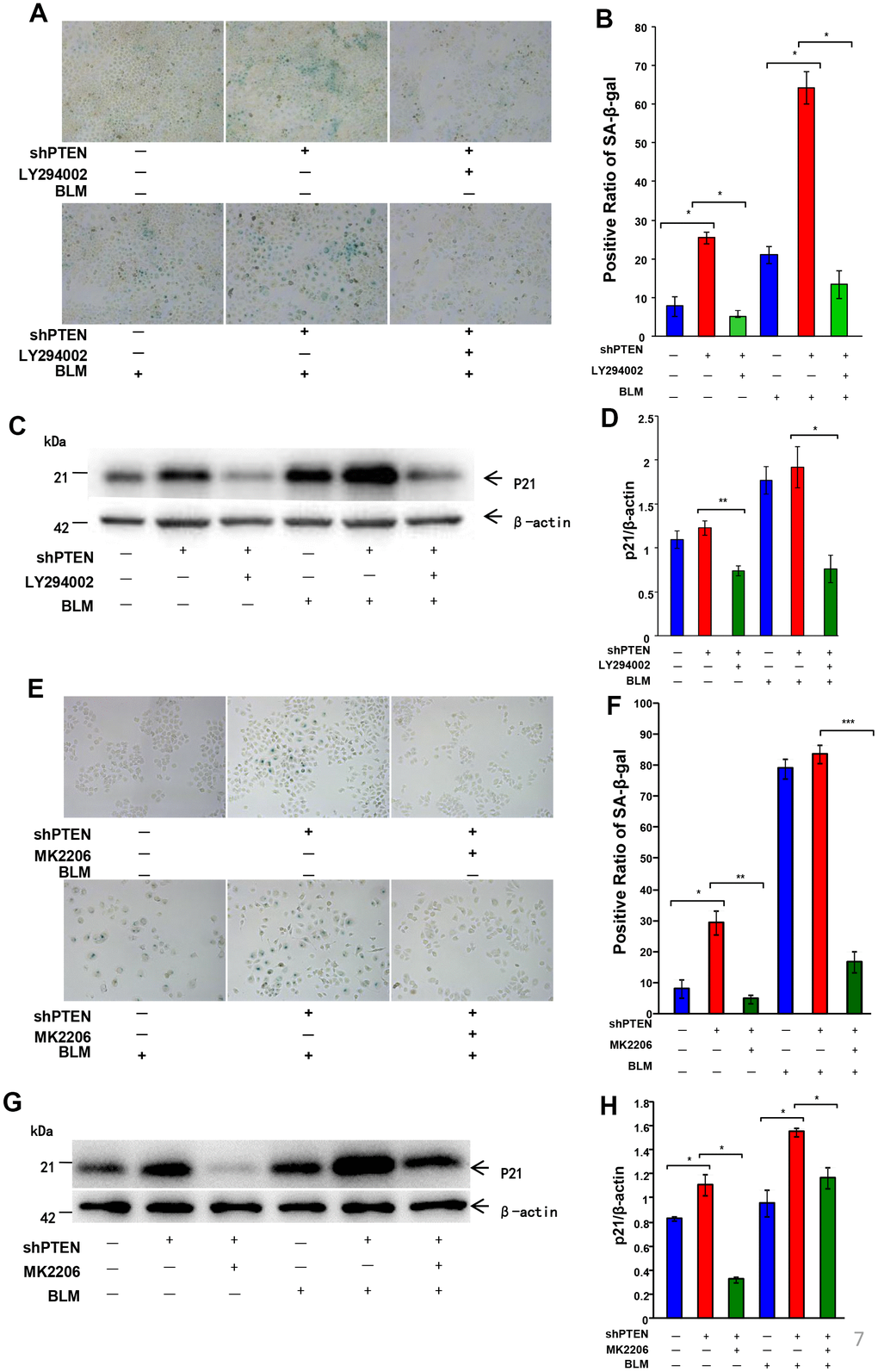
Figure 7. Akt inactivation rescues AEC senescence in vitro. After PTEN was stably knocked down, an inhibitor of the AKT pathway (20 μm/L LY294002 or 5 μg/ml MK2206) was added 1 hour before bleomycin (10 μg/ml) to A549 cells for 72 hours followed by a fresh medium transfer for 24 hours. (A, B) Rescue of cell senescence by LY294002 was determined by SA-β-Gal staining (original magnification, 200×), and decreased expression of P21WAF1 (C, D) was confirmed by western blot. Rescue of cell senescence by MK2206 was determined by SA-β-Gal staining (E, F) and P21WAF1 expression (G, H). Data are shown as the mean ± SEM, n ≥ 3 per group. *p < 0.05, **p < 0.01. One-way ANOVA followed by Dunnett’s Multiple Comparison Test.
Akt inactivation attenuates senescence-related markers and pulmonary fibrosis in vivo
The bleomycin-induced mice pulmonary fibrosis model is a widely used animal model for pulmonary fibrosis. After exposure to bleomycin for 14 days, the expression levels of the senescence-related marker, P21WAF1, was significantly higher in mouse lungs, indicating that cellular senescence occurred in mouse lung tissue and that pulmonary fibrosis developed (Figure 8). Starting on the day of intratracheal injection with bleomycin (5 mg/kg in 100 μl saline), mice were intraperitoneally injected daily with LY294002 (50 mg/kg) for 14 days. Lungs were harvested for western blot analysis. LY294002 attenuated the P21WAF1 senescence marker in mouse lungs, resulting in significantly lower pulmonary fibrosis as assessed by lower collagen1α, hematoxylin and eosin (HE) and Masson Trichrome staining compared to mice treated with vehicle (Figure 8).
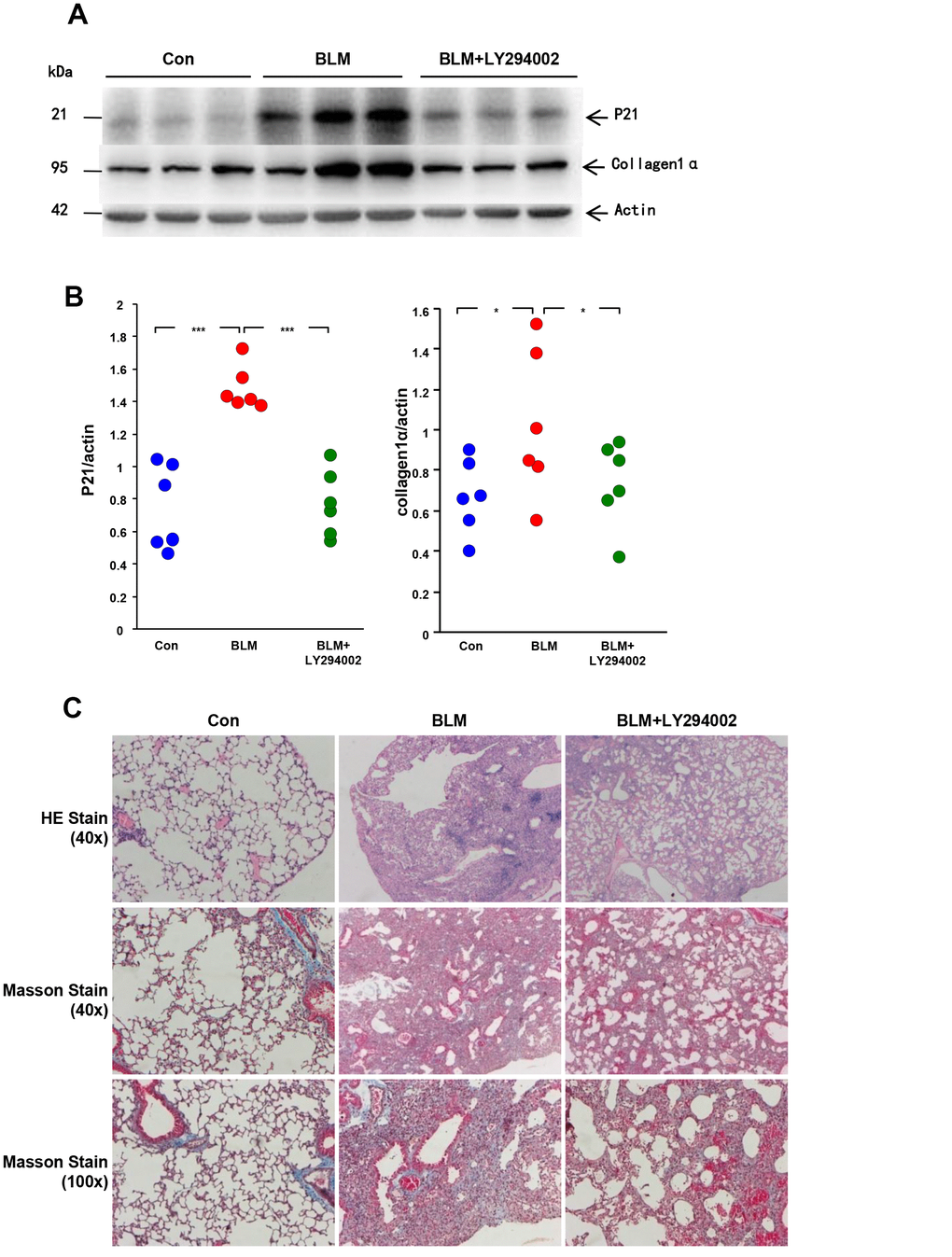
Figure 8. Akt inactivation attenuates AEC senescence and pulmonary fibrosis in vivo. C57BL/6 male mice were randomly divided into vehicle group (n = 6), bleomycin group (n = 6) and inhibitor group (n=6). Mice in the vehicle group were intratracheally injected with 50 μl of 0.9% saline, and mice in the bleomycin group and inhibitor group were injected intratracheally with 50 μl of 5 mg/kg bleomycin. From the day of bleomycin injection, mice in the inhibitor group were treated daily with LY294002 (50 mg/kg) through intraperitoneal injection. On day 14 after bleomycin or saline treatment, lungs were harvested. (A, B) Expression of the P21WAF1 senescent marker and the collagen1α fibrotic marker was detected by western blot analysis. Dots in the graph represent values for each individual mouse. *p < 0.05, ***p < 0.001. One-way ANOVA followed by Dunnett’s Multiple Comparison Test. (C) Representative results of HE and Masson staining of mouse lung tissues.
PTEN physically associates with Akt
To further investigate the interaction between PTEN and Akt, we performed a co-immunoprecipitation (co-IP) assay. As shown in Figure 9, immunoprecipitation of PTEN resulted in the co-IP of Akt in A549 cells. Immunoprecipitation of Akt resulted in the co-IP of PTEN. These results demonstrated that PTEN physically associates with Akt in A549 cells.
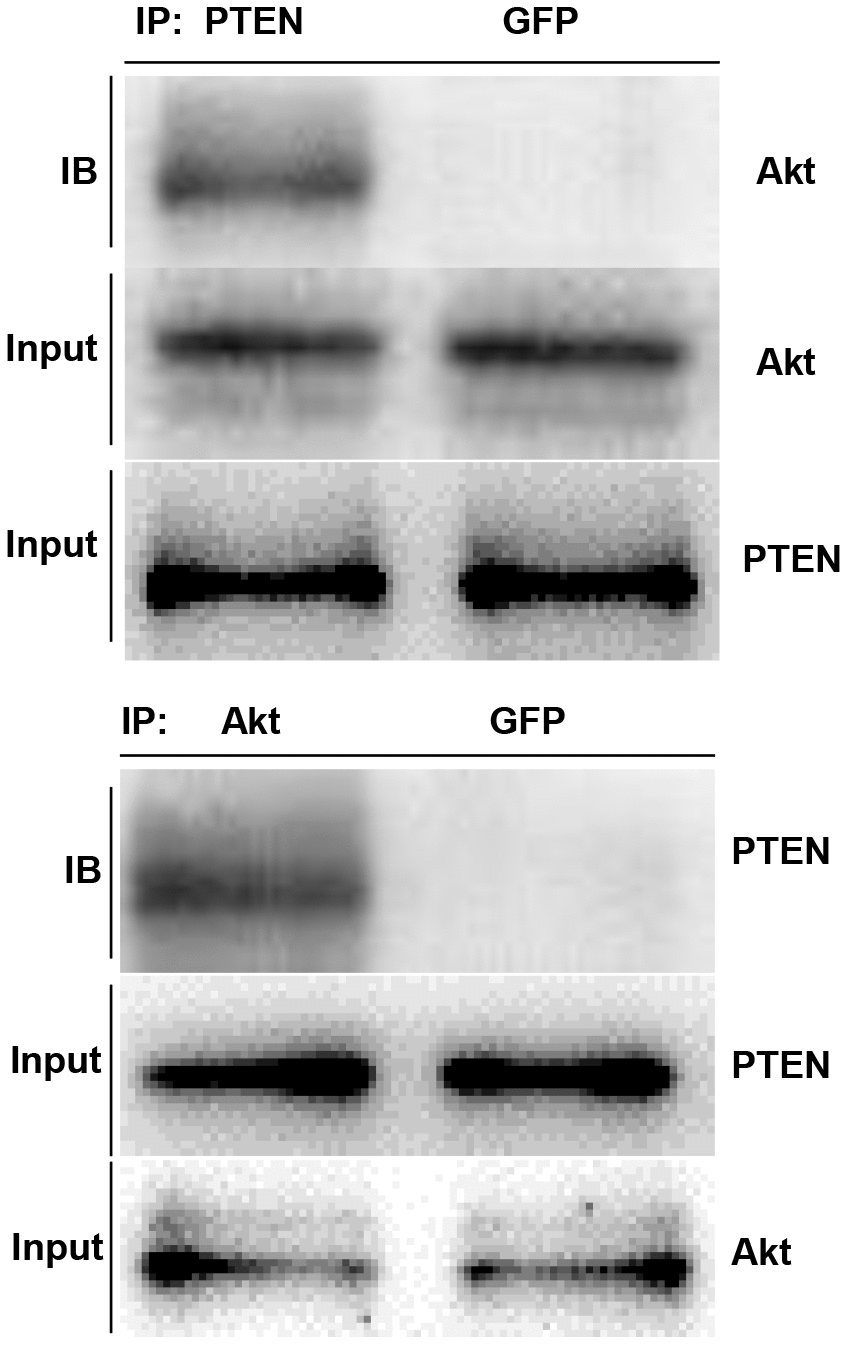
Figure 9. PTEN physically associates with Akt. In A549 cells, immunoprecipitation of PTEN resulted in the co-IP of AKT, and immunoprecipitation of AKT also resulted in the co-IP of PTEN.
Discussion
Because increasing evidence has shown that IPF is an aging-related disease, great effort has been made to identify novel anti-senescence compounds for IPF therapy. Most studies regarding senescence in IPF are mainly focused on fibroblasts, and our recent study demonstrated that lung epithelial cells also play an important role in IPF by triggering collagen deposition in fibroblasts [34]. The present study further investigated the loss of PTEN participation in the pathogenesis of pulmonary fibrosis through regulating the senescence of AECs. We confirmed that PTEN loss-induced AEC senescence is AKT2 dependent.
Both genetic and epigenetic losses of the PTEN tumor suppressor gene are frequently observed in human cancers. Interestingly, while monoallelic loss or mutation of PTEN drives cellular proliferation, complete inactivation of PTEN induces a senescence response, termed PTEN loss-induced cellular senescence (PICS) that opposes tumor development [19]. Low levels of PTEN expression in fibroblasts and epithelial cells from IPF lungs have been detected. Mice with a constitutive deficiency of PTEN develop spontaneous fibrosis via intrinsic defects in fibroblasts and negative regulation of their proliferative responses [35]. A better understanding of the molecular mechanism that loss of PTEN drives senescence in IPF is of great relevance for the clinical development of anti-senescence treatments for IPF patients. However, the mechanism behind PTEN loss in AEC senescence remains elusive thus far.
To address this question, we used stable PTEN-overexpressing and PTEN knockdown cells to establish senescence cell models. We observed increased Akt activation and deteriorated senescence in the PTEN knockdown cell models. However, decreased Akt pathway activation and attenuated senescence were present in the PTEN-overexpressing cell models. These results indicated that the Akt pathway is an important potential barrier to PTEN loss-driven cell senescence.
Akt activation is strongly associated with regulating survival and differentiation of myofibroblasts in the setting of pulmonary fibrosis as it is known to regulate many fibrotic remodeling processes in IPF [36]. Akt signaling has been reported to play a central role in controlling fibroblast proliferation and survival [37–40]. However, the precise role of the Akt pathway in AEC senescence remains unknown.
According to previous studies, Akt1-mediated mitophages contribute to alveolar macrophage apoptosis resistance, which is required for pulmonary fibrosis development [31]. Akt1−/−mice are protected from chronic hypoxia-induced pulmonary vascular and tissue remodeling [41]. The efforts to dissect the molecular mechanisms of Akt isoform-specific signaling will provide new insights for designing more effective and selective therapeutics for IPF treatment. In mammals, the Akt family consists of three isoforms, namely, Akt1, Akt2 and Akt3. Because Akt3 is predominantly expressed in brain tissue, we only studied the Akt1 and Akt2 isoforms. Our study showed that inhibition of Akt pathway activation or knockdown of Akt2 rescues bleomycin-induced cell senescence. Our animal study further demonstrated that inhibition of Akt activation attenuates pulmonary fibrosis. These results were supported by a recent study showing that inhibition of Akt phosphorylation in lungs improves lung function in asthma and fibrosis in mice [42]. Akt2-deficient mice are protected against bleomycin-induced pulmonary fibrosis and inflammation [43].
While fibroblasts were not found to be affected to a large extent in IPF, recent studies have shown that epithelial cells represent a major cell type that is affected by senescence [44]. Senescent epithelial cells secrete several mediators in the SASP, which have been shown to directly influence their surrounding microenvironment, thereby triggering IPF [45]. Studies have revealed that AEC injury is sufficient to initiate lung fibrosis [12, 13]. Senolytic treatment targeting fibrotic lung epithelial cells in vitro and ex vivo attenuates pulmonary fibrosis [15]. These findings indicate that AEC senescence is a promising target for developing novel IPF therapy. The present study also suggested that the senescence of AECs may be a profibrotic initiator in IPF and that the PTEN/Akt pathway may be a new candidate for senolytic drugs, which may advance treatment of IPF.
In conclusion, the present study confirmed that AECs accelerate cellular senescence in IPF. We further showed that PTEN loss-induced AEC senescence occurs in an Akt-dependent manner, which may help identify a potential target for anti-senescent therapy in treatment of lung fibrosis.
Materials and Methods
Patients and specimens
The present study was approved by the Ethics Committee of the Medical School of Nanjing University. Written informed consent was obtained from all the patients enrolled in this study. All human lung tissues of IPF (n = 12) were obtained from the Department of Lung Transplantation, Wuxi People’s Hospital, Wuxi, China. Normal peripheral tissues (n = 12), where were used as controls, were obtained from patients who underwent lobectomy in the Thoracic Surgery Department of Nanjing Drum Tower Hospital of the Affiliated Hospital of Nanjing University Medical School. All diagnoses of IPF were made in accordance with the ATS/ERS criteria for IPF 2011. For the 12 patients who provided normal lung tissues, the average age was 63.16 ± 4.62 years, and 1 female and 11 males were included. For the 12 IPF patients, the average age was 61.83 ± 5.49 years, and 1 female and 11 males were included.
Cell culture
A549 cells were purchased from American Type Culture Collection (Rockville, MD, USA). Cells were cultured in DMEM (Gibco, Australia) supplemented with 10% FBS (Gibco) and 1X penicillin–streptomycin solution (Thermo Fisher Scientific, USA) at 37°C in 5% CO2.
Reagents
Bleomycin was purchased from Nippon Kayaku Co Ltd. (Japan), dissolved in 0.9% saline and stored at −20°C. LY294002, an inhibitor of PI3K/AKT, was purchased from Sigma-Aldrich (USA), dissolved in DMSO (Sigma, USA) and stored at −20°C.Diluted to working concentration of 20μM/L for cell culture. MK2206, an Akt pathway inhibitor, was purchased from Selleckchem, dissolved in DMSO (Sigma, USA) and stored at −20°C. Diluted to 5μg/ml when applied to cells.
Mouse pulmonary fibrosis models
Six- to eight-week old male SPF C57BL/6 mice (Shanghai Laboratory Animal Center, Chinese Academy of Sciences, Shanghai, China) were randomly divided into the vehicle group (n = 6), bleomycin group (n = 6) or inhibitor group (n=6). All protocols were approved by the Ethics Committee for Animal Research of Medical School of Nanjing University. Mice in the vehicle group were intratracheally injected with 50 μl of 0.9% saline, and mice in the bleomycin and inhibitor groups were injected intratracheally with 50 μl of 5 mg/kg bleomycin. From the day of bleomycin injection, mice in the inhibitor group were treated daily with LY294002 (50 mg/kg) through intraperitoneal injection. On day 14 after bleomycin or saline treatment, mice were sacrificed, and lungs were collected for subsequent experiments.
Cellular aging model
A549 cells were stimulated with specific concentrations of bleomycin for 72 hours followed by culture in FBS-free medium for 24 hours to generate the cellular senescence model. Aging-related markers, including p21 and senescence-associated β-galactosidase (SA-β-Gal), were then tested to confirm the senescent state of cells.
Senescence-associated β-galactosidase staining
SA-β-Gal staining was performed according to the manufacturer’s instructions. The Senescence β-Galactosidase Staining Kit was purchased from Beyotime Biotechnology (Shanghai, China). Cells on 6-well chamber slides were fixed with 4% formaldehyde for 10 min at room temperature, rinsed three times with PBS for 5 min and incubated with freshly prepared SA-β-Gal staining solution at 37°C overnight. Slides were washed twice with PBS for 10 min at room temperature. Images were captured using a microscope equipped with a digital camera (Eclipsee800, Nikon). For each slide, at least three fields were obtained to calculate the SA-β-gal intensity.
Western blot
Whole protein from cell lysates or lung tissues was extracted according to manufacturer’s instructions (Keygene, China). Equal amounts of protein were separated on 10% SDS-PAGE gels, blotted onto PVDF membranes (Merck Millipore, Germany), blocked with 5% skim milk in TBST and incubated with primary antibody at room temperature for 4 hours or overnight. Membranes were incubated with HRP-conjugated secondary antibody for one hour, and protein expression was detected using ECL (Merck Millipore, Germany). Primary antibodies against P21, PTEN and Akt were purchased from Cell Signaling Technology (USA).
Immunohistochemical staining
Tissues were fixed in formalin and embedded in paraffin. Immunohistochemical staining for target proteins and hematoxylin–eosin staining (HE) were performed. Four-micron-thick sections were deparaffinized in xylene and rehydrated in graded alcohol. Endogenous peroxidase was quenched with 3% aqueous hydrogen peroxide for 15 min. Antigen retrieval was then performed in a pressure cooker. Sections were incubated with 10% normal goat serum to block any nonspecific reaction. After primary antibody incubation overnight at 4°C, horseradish peroxidase-conjugated secondary antibody was added for 20 min, and 3,3-diaminobenzidine tetrahydrochloride (DAB) was added for 10 min at room temperature. Finally, sections were dehydrated and mounted.
Immunofluorescence assay
Fresh lung tissues were fixed in 4% paraformaldehyde, embedded in paraffin and cut into 4-μm sections. After deparaffinating, rehydration and retrieval, tissues were incubated with primary antibodies overnight at 4°C. After rinsing several times with PBS, sections were incubated with Alexa-488- or Alexa-568-conjugated secondary antibodies (Invitrogen, United Kingdom) for 2 hours. Finally, DAPI was added onto slides for 10 minutes. Images were obtained with a ZEISS Imager A1 fluorescence microscope (Gottingen, Germany). Immunofluorescence was performed using the following primary antibodies: rabbit monoclonal anti-P21 and anti-PTEN antibodies (Abcam, United Kingdom); and mouse monoclonal anti-SPC antibody (Santa Cruz Biotechnology, CA, USA).
Masson’s trichrome staining
Lung tissues were fixed in 4% paraformaldehyde at room temperature overnight followed by dehydration with gradient alcohol and embedding in paraffin. Sections of 5 μm were subjected to Masson’s trichrome staining according to the manufacturer’s instructions (Solarbio, China).
Co-immunoprecipitation
Cells were lysed in buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl,1% NP-40 and protease inhibitor. Cell lysates were placed on ice for 1 hour with mixing every 10 minutes and then centrifuged at 13000 g at 4 °C for 20 minutes. Protein was first incubated with the following primary antibodies at 4 °C for 1 hour: mouse monoclonal anti-PTEN, anti-AKT or anti-GFP (control antibody) antibodies (Santa Cruz Biotechnology, CA, USA). Protein A/G plus-Agarose (20 μl; Santa Cruz Biotechnology, CA, USA) was then added at 4 °C for 4 hours. After washing 4 times with PBS, samples were resuspended in 40 μl of electrophoresis sample buffer and heated at 70 °C for 10 minutes. The supernatant was subjected to SDS-PAGE and western blotting.
Statistical analysis
Data were expressed as the mean values ± SEM. Student’s t-test (2-tailed) was used to determine differences for experiments between two groups, and one-way ANOVA was applied to analyze differences for experiments with more than two groups. p value less than 0.05 was accepted as statistically significant.
Author Contributions
TQ, YT, YG and MM were responsible for performing the experiments and writing the manuscript. HL, JD, YZ and HC were responsible for designing the experiments and providing financial support. XL and HD were primarily responsible for data analysis and creating figures. HW was responsible for immunohistochemical staining. JZ and JC were primarily responsible for designing the experiments and supplying some of the human lung specimens. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare that they have no conflicts of interests.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81670058, 81470253, 81570055, 81570058, 81670059) and Jiangsu Provincial Key Research and Development (BE2016611).
References
- 1. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, Behr J, Cottin V, Danoff SK, Morell F, Flaherty KR, Wells A, Martinez FJ, et al, and American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018; 198:e44–68. https://doi.org/10.1164/rccm.201807-1255ST [PubMed]
- 2. Raghu G, Chen SY, Yeh WS, Maroni B, Li Q, Lee YC, Collard HR. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med. 2014; 2:566–72. https://doi.org/10.1016/S2213-2600(14)70101-8 [PubMed]
- 3. Collard HR. The age of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010; 181:771–72. https://doi.org/10.1164/rccm.201001-0049ED [PubMed]
- 4. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011; 183:788–824. https://doi.org/10.1164/rccm.2009-040GL [PubMed]
- 5. King TE
Jr , Tooze JA, Schwarz MI, Brown KR, Cherniack RM. Predicting survival in idiopathic pulmonary fibrosis: scoring system and survival model. Am J Respir Crit Care Med. 2001; 164:1171–81. https://doi.org/10.1164/ajrccm.164.7.2003140 [PubMed] - 6. Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, Cogan JD, Vulto I, Xie M, Qi X, Tuder RM, Phillips JA
3rd , Lansdorp PM, Loyd JE, Armanios MY. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA. 2008; 105:13051–56. https://doi.org/10.1073/pnas.0804280105 [PubMed] - 7. Cárdenes N, Álvarez D, Sellarés J, Peng Y, Corey C, Wecht S, Nouraie SM, Shanker S, Sembrat J, Bueno M, Shiva S, Mora AL, Rojas M. Senescence of bone marrow-derived mesenchymal stem cells from patients with idiopathic pulmonary fibrosis. Stem Cell Res Ther. 2018; 9:257. https://doi.org/10.1186/s13287-018-0970-6 [PubMed]
- 8. Shaikh SB, Prabhu A, Bhandary YP. Targeting anti-aging protein sirtuin (Sirt) in the diagnosis of idiopathic pulmonary fibrosis. J Cell Biochem. 2018. [Epub ahead of print]. https://doi.org/10.1002/jcb.28033 [PubMed]
- 9. Schuliga M, Pechkovsky DV, Read J, Waters DW, Blokland KE, Reid AT, Hogaboam CM, Khalil N, Burgess JK, Prêle CM, Mutsaers SE, Jaffar J, Westall G, et al. Mitochondrial dysfunction contributes to the senescent phenotype of IPF lung fibroblasts. J Cell Mol Med. 2018; 22:5847–61. https://doi.org/10.1111/jcmm.13855 [PubMed]
- 10. Wheaton AK, Velikoff M, Agarwal M, Loo TT, Horowitz JC, Sisson TH, Kim KK. The vitronectin RGD motif regulates TGF-β-induced alveolar epithelial cell apoptosis. Am J Physiol Lung Cell Mol Physiol. 2016; 310:L1206–17. https://doi.org/10.1152/ajplung.00424.2015 [PubMed]
- 11. Garcia CK. Idiopathic pulmonary fibrosis: update on genetic discoveries. Proc Am Thorac Soc. 2011; 8:158–62. https://doi.org/10.1513/pats.201008-056MS [PubMed]
- 12. Sisson TH, Mendez M, Choi K, Subbotina N, Courey A, Cunningham A, Dave A, Engelhardt JF, Liu X, White ES, Thannickal VJ, Moore BB, Christensen PJ, Simon RH. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med. 2010; 181:254–63. https://doi.org/10.1164/rccm.200810-1615OC [PubMed]
- 13. Kim KK, Dotson MR, Agarwal M, Yang J, Bradley PB, Subbotina N, Osterholzer JJ, Sisson TH. Efferocytosis of apoptotic alveolar epithelial cells is sufficient to initiate lung fibrosis. Cell Death Dis. 2018; 9:1056. https://doi.org/10.1038/s41419-018-1074-z [PubMed]
- 14. Ota C, Ng-Blichfeldt JP, Korfei M, Alsafadi HN, Lehmann M, Skronska-Wasek W, M De Santis M, Guenther A, Wagner DE, Königshoff M. Dynamic expression of HOPX in alveolar epithelial cells reflects injury and repair during the progression of pulmonary fibrosis. Sci Rep. 2018; 8:12983. https://doi.org/10.1038/s41598-018-31214-x [PubMed]
- 15. Lehmann M, Korfei M, Mutze K, Klee S, Skronska-Wasek W, Alsafadi HN, Ota C, Costa R, Schiller HB, Lindner M, Wagner DE, Günther A, Königshoff M. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo.. Eur Respir J. 2017; 50:1602367. https://doi.org/10.1183/13993003.02367-2016 [PubMed]
- 16. Milella M, Falcone I, Conciatori F, Cesta Incani U, Del Curatolo A, Inzerilli N, Nuzzo CM, Vaccaro V, Vari S, Cognetti F, Ciuffreda L. PTEN: multiple functions in human malignant tumors. Front Oncol. 2015; 5:24. https://doi.org/10.3389/fonc.2015.00024 [PubMed]
- 17. Tait IS, Li Y, Lu J. Effects of PTEN on the longevity of cultured human umbilical vein endothelial cells: the role of antioxidants. Int J Mol Med. 2015; 35:277–84. https://doi.org/10.3892/ijmm.2014.1999 [PubMed]
- 18. Ortega-Molina A, Efeyan A, Lopez-Guadamillas E, Muñoz-Martin M, Gómez-López G, Cañamero M, Mulero F, Pastor J, Martinez S, Romanos E, Mar Gonzalez-Barroso M, Rial E, Valverde AM, et al. Pten positively regulates brown adipose function, energy expenditure, and longevity. Cell Metab. 2012; 15:382–94. https://doi.org/10.1016/j.cmet.2012.02.001 [PubMed]
- 19. Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, Salmena L, Sampieri K, Haveman WJ, Brogi E, Richardson AL, Zhang J, Pandolfi PP. Subtle variations in Pten dose determine cancer susceptibility. Nat Genet. 2010; 42:454–58. https://doi.org/10.1038/ng.556 [PubMed]
- 20. El-Horany HE, Gaballah HH, Helal DS. Berberine ameliorates renal injury in a rat model of D-galactose-induced aging through a PTEN/Akt-dependent mechanism. Arch Physiol Biochem. 2018; 1–9. [Epub ahead of print]. https://doi.org/10.1080/13813455.2018.1499117 [PubMed]
- 21. Parisotto M, Grelet E, El Bizri R, Dai Y, Terzic J, Eckert D, Gargowitsch L, Bornert JM, Metzger D. PTEN deletion in luminal cells of mature prostate induces replication stress and senescence in vivo. J Exp Med. 2018; 215:1749–1763. https://doi.org/10.1084/jem.20171207 [PubMed]
- 22. Cao Z, Ren D, Ha T, Liu L, Wang X, Kalbfleisch J, Gao X, Kao R, Williams D, Li C. CpG-ODN, the TLR9 agonist, attenuates myocardial ischemia/reperfusion injury: involving activation of PI3K/Akt signaling. Biochim Biophys Acta. 2013; 1832:96–104. https://doi.org/10.1016/j.bbadis.2012.08.008 [PubMed]
- 23. Jeong H, Liu Y, Kim HS. Dried plum and chokeberry ameliorate d-galactose-induced aging in mice by regulation of Pl3k/Akt-mediated Nrf2 and Nf-kB pathways. Exp Gerontol. 2017; 95:16–25. https://doi.org/10.1016/j.exger.2017.05.004 [PubMed]
- 24. Chang YM, Chang HH, Kuo WW, Lin HJ, Yeh YL, Padma Viswanadha V, Tsai CC, Chen RJ, Chang HN, Huang CY. Anti-apoptotic and pro-survival effect of alpinate oxyphyllae fructus (AOF) in a D-galactose-induced aging heart. Int J Mol Sci. 2016; 17:466. https://doi.org/10.3390/ijms17040466 [PubMed]
- 25. Li HH, Lin SL, Huang CN, Lu FJ, Chiu PY, Huang WN, Lai TJ, Lin CL. MIR-302 attenuates amyloid-β-induced neurotoxicity through activation of Akt signaling. J Alzheimers Dis. 2016; 50:1083–98. https://doi.org/10.3233/JAD-150741 [PubMed]
- 26. Zhang Y, Liu B, Chen X, Zhang N, Li G, Li-Hong Zhang LH, Li-Yan Tan LY. Naringenin Ameliorates Behavioral Dysfunction and Neurological Deficits in a d-Galactose-Induced Aging Mouse Model Through Activation of PI3K/Akt/Nrf2 Pathway. Rejuvenation Res. 2017; 20:462–472. https://doi.org/10.1089/rej.2017.1960 [PubMed]
- 27. Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, Lee VM, Szabolcs M, de Jong R, Oltersdorf T, Ludwig T, Efstratiadis A, Birnbaum MJ. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol. 2005; 25:1869–78. https://doi.org/10.1128/MCB.25.5.1869-1878.2005 [PubMed]
- 28. Xie R, Cheng M, Li M, Xiong X, Daadi M, Sapolsky RM, Zhao H. Akt isoforms differentially protect against stroke-induced neuronal injury by regulating mTOR activities. J Cereb Blood Flow Metab. 2013; 33:1875–85. https://doi.org/10.1038/jcbfm.2013.132 [PubMed]
- 29. Hers I, Vincent EE, Tavaré JM. Akt signalling in health and disease. Cell Signal. 2011; 23:1515–27. https://doi.org/10.1016/j.cellsig.2011.05.004 [PubMed]
- 30. Riggio M, Perrone MC, Polo ML, Rodriguez MJ, May M, Abba M, Lanari C, Novaro V. AKT1 and AKT2 isoforms play distinct roles during breast cancer progression through the regulation of specific downstream proteins. Sci Rep. 2017; 7:44244. https://doi.org/10.1038/srep44244 [PubMed]
- 31. Larson-Casey JL, Deshane JS, Ryan AJ, Thannickal VJ, Carter AB. Macrophage Akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity. 2016; 44:582–96. https://doi.org/10.1016/j.immuni.2016.01.001 [PubMed]
- 32. Tang H, Chen J, Fraidenburg DR, Song S, Sysol JR, Drennan AR, Offermanns S, Ye RD, Bonini MG, Minshall RD, Garcia JG, Machado RF, Makino A, Yuan JX. Deficiency of Akt1, but not Akt2, attenuates the development of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2015; 308:L208–20. https://doi.org/10.1152/ajplung.00242.2014 [PubMed]
- 33. Attoub S, Arafat K, Kamel Hammadi N, Mester J, Gaben AM. Akt2 knock-down reveals its contribution to human lung cancer cell proliferation, growth, motility, invasion and endothelial cell tube formation. Sci Rep. 2015; 5:12759. https://doi.org/10.1038/srep12759 [PubMed]
- 34. Tian Y, Li H, Qiu T, Dai J, Zhang Y, Chen J, Cai H. Loss of PTEN induces lung fibrosis via alveolar epithelial cell senescence depending on NF-κB activation. Aging Cell. 2019; 18:e12858. https://doi.org/10.1111/acel.12858 [PubMed]
- 35. Parapuram SK, Thompson K, Tsang M, Hutchenreuther J, Bekking C, Liu S, Leask A. Loss of PTEN expression by mouse fibroblasts results in lung fibrosis through a CCN2-dependent mechanism. Matrix Biol. 2015; 43:35–41. https://doi.org/10.1016/j.matbio.2015.01.017 [PubMed]
- 36. Xia H, Diebold D, Nho R, Perlman D, Kleidon J, Kahm J, Avdulov S, Peterson M, Nerva J, Bitterman P, Henke C. Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis. J Exp Med. 2008; 205:1659–72. https://doi.org/10.1084/jem.20080001 [PubMed]
- 37. Hsu HS, Liu CC, Lin JH, Hsu TW, Hsu JW, Su K, Hung SC. Involvement of ER stress, PI3K/AKT activation, and lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis. Sci Rep. 2017; 7:14272. https://doi.org/10.1038/s41598-017-14612-5 [PubMed]
- 38. Pereira PR, Oliveira-Junior MC, Mackenzie B, Chiovatto JE, Matos Y, Greiffo FR, Rigonato-Oliveira NC, Brugemman TR, Delle H, Idzko M, Albertini R, Ligeiro Oliveira AP, Damaceno-Rodrigues NR, et al. Exercise Reduces Lung Fibrosis Involving Serotonin/Akt Signaling. Med Sci Sports Exerc. 2016; 48:1276–84. https://doi.org/10.1249/MSS.0000000000000907 [PubMed]
- 39. Kulasekaran P, Scavone CA, Rogers DS, Arenberg DA, Thannickal VJ, Horowitz JC. Endothelin-1 and transforming growth factor-β1 independently induce fibroblast resistance to apoptosis via AKT activation. Am J Respir Cell Mol Biol. 2009; 41:484–93. https://doi.org/10.1165/rcmb.2008-0447OC [PubMed]
- 40. Nho RS, Hergert P. IPF fibroblasts are desensitized to type I collagen matrix-induced cell death by suppressing low autophagy via aberrant Akt/mTOR kinases. PLoS One. 2014; 9:e94616. https://doi.org/10.1371/journal.pone.0094616 [PubMed]
- 41. Abdalla M, Sabbineni H, Prakash R, Ergul A, Fagan SC, Somanath PR. The Akt inhibitor, triciribine, ameliorates chronic hypoxia-induced vascular pruning and TGFβ-induced pulmonary fibrosis. Br J Pharmacol. 2015; 172:4173–88. https://doi.org/10.1111/bph.13203 [PubMed]
- 42. Campa CC, Silva RL, Margaria JP, Pirali T, Mattos MS, Kraemer LR, Reis DC, Grosa G, Copperi F, Dalmarco EM, Lima-Júnior RC, Aprile S, Sala V, et al. Inhalation of the prodrug PI3K inhibitor CL27c improves lung function in asthma and fibrosis. Nat Commun. 2018; 9:5232. https://doi.org/10.1038/s41467-018-07698-6 [PubMed]
- 43. Nie Y, Sun L, Wu Y, Yang Y, Wang J, He H, Hu Y, Chang Y, Liang Q, Zhu J, Ye RD, Christman JW, Qian F. AKT2 regulates pulmonary inflammation and fibrosis via modulating macrophage activation. J Immunol. 2017; 198:4470–80. https://doi.org/10.4049/jimmunol.1601503 [PubMed]
- 44. Minagawa S, Araya J, Numata T, Nojiri S, Hara H, Yumino Y, Kawaishi M, Odaka M, Morikawa T, Nishimura SL, Nakayama K, Kuwano K. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-β-induced senescence of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2011; 300:L391–401. https://doi.org/10.1152/ajplung.00097.2010 [PubMed]
- 45. Disayabutr S, Kim EK, Cha SI, Green G, Naikawadi RP, Jones KD, Golden JA, Schroeder A, Matthay MA, Kukreja J, Erle DJ, Collard HR, Wolters PJ. miR-34 miRNAs regulate cellular senescence in type II alveolar epithelial cells of patients with idiopathic pulmonary fibrosis. PLoS One. 2016; 11:e0158367. https://doi.org/10.1371/journal.pone.0158367 [PubMed]