



Introduction
Doxorubicin (DOX), a member of the anthracyclines, provided the oncologist with highly effective therapeutic regimen to treat tumors, but the unanticipated side-effects associated with acute or chronic cardiotoxicity greatly limits its clinical use [1]. To maximize the overall survival and minimize the cardiac side-effects, DOX-cardiotoxicity has drawn much attention of both cardiologists and oncologists [2]. The primary mechanism of DOX-induced cardiomyopathy is related to the oxidative injury and mitochondrial dysfunction dose-dependent [1, 3]. The cardiomyocyte is especially susceptible to oxidative damage due to the relative lack of biochemical reserves to mitigate oxidation [2, 4]. Therefore, targeting oxidative stress may be a good therapeutic regimen for preventing and treating DOX-cardiotoxicity.
Pterostilbene (3,5-dimethoxy-4′-hydroxystilbene, PTS) is a natural analog of resveratrol and a known antioxidant mainly exists in blueberries and grapes [5, 6]. A dozen of basic studies reveal that pterostilbene is a potent myocardial protective agent against various cardiac diseases, including hypertrophy, diabetic cardiomyopathy, myocardial infarction and ischemia reperfusion injury [7–10]. These favorable effects of pterostilbene application are largely attributed to the free radical scavenging and anti-inflammatory actions [11, 12]. Interestingly, clinical studies indicate that pterostilbene is generally safe for human use and is good for reducing adult blood pressure [13, 14]. However, whether pterostilbene could protect myocardium against DOX-induced acute cardiotoxicity still remains unknown.
Peroxisome proliferator-activated receptor coactivator 1α (PGC1α) is a vital transcriptional coactivator in regulating mitochondrial functions and maintaining mitochondrial homeostasis through mediating the expression of uncoupling protein 2 (UCP2), nuclear respiratory factor 1 (NRF1), etc [4, 15]. The level and activity of PGC1α are particularly vulnerable to DOX exposure thus leading to the DOX-induced mitochondrial damage [15]. The activity of PGC1α could be posttranslational hindered via acetylation [16]. Moreover, PGC1α’s expression and activity are stimulated by its upstream regulator adenosine monophosphate activated protein kinase (AMPK) and sirtuin1 (SIRT1) [2, 4, 17]. Interestingly, AMPK and SIRT1 are also inhibited after DOX exposure in cardiomyocytes [4, 16, 18]. In present study, we aimed to verify whether pterostilbene could exert cardiac protection against acute DOX-cardiotoxicity via the mechanism of alleviating mitochondrial oxidative stress through activating the AMPK/SIRT1-PGC1α cascades.
Results
Pterostilbene application alleviated DOX-induced H9c2 cell viability inhibition, mitochondrial damage and oxidative stress
As our previous basic research data suggested [4], the dosage of 1 μM DOX was chosen to carry on the present in vitro study. To investigate whether pterostilbene exerts the cardioprotective action against toxicity induced by DOX on H9c2 cells, CCK-8 cell viability assay was conducted to analyze its protective effect. As shown in Figure 1A, pterostilbene treatment (2.5, 5, 7.5 or 10 μM culturing for 24 h) had no effect on H9c2 cell viability, but 1 μM DOX treatment for 24 h significantly inhibited the cell viability. Interestingly, cotreatment of 2.5, 5, 7.5 and 10 μM pterostilbene for 24 h markedly reversed the 1 μM DOX caused decrease of cell viability in a dose-dependent manner. Therefore, this result indicated that pterostilbene exerted cardiac protection against DOX-cardiotoxicity, and 10 μM pterostilbene was chosen to be used for further experimental studies. Generation of ROS thus promoting mitochondrial oxidative stress plays vital actions on the development of cardiac dysfunction. We found 1 μM DOX-exposure for 24 h caused markedly upregulation of ROS level, loss of mitochondrial membrane potential (ΔΨm), and downregulation of ATP content, but pterostilbene cotreatment obviously reversed these DOX-induced mitochondrial oxidative stress by decreasing ROS level and preserving ΔΨm and ATP content (Figure 1B, 1C and Figure 2C). Moreover, the transmission electron microscopic examination on H9c2 cells revealed that 1 μM DOX exposure significantly caused ultrastructural morphology disorder on mitochondria by inducing swelling with cristae disorientation and breakage (Figure 1D). However, pterostilbene cotreatment markedly rescued the myocardial mitochondrion by normalizing the cristae density and architecture (Figure 1D).
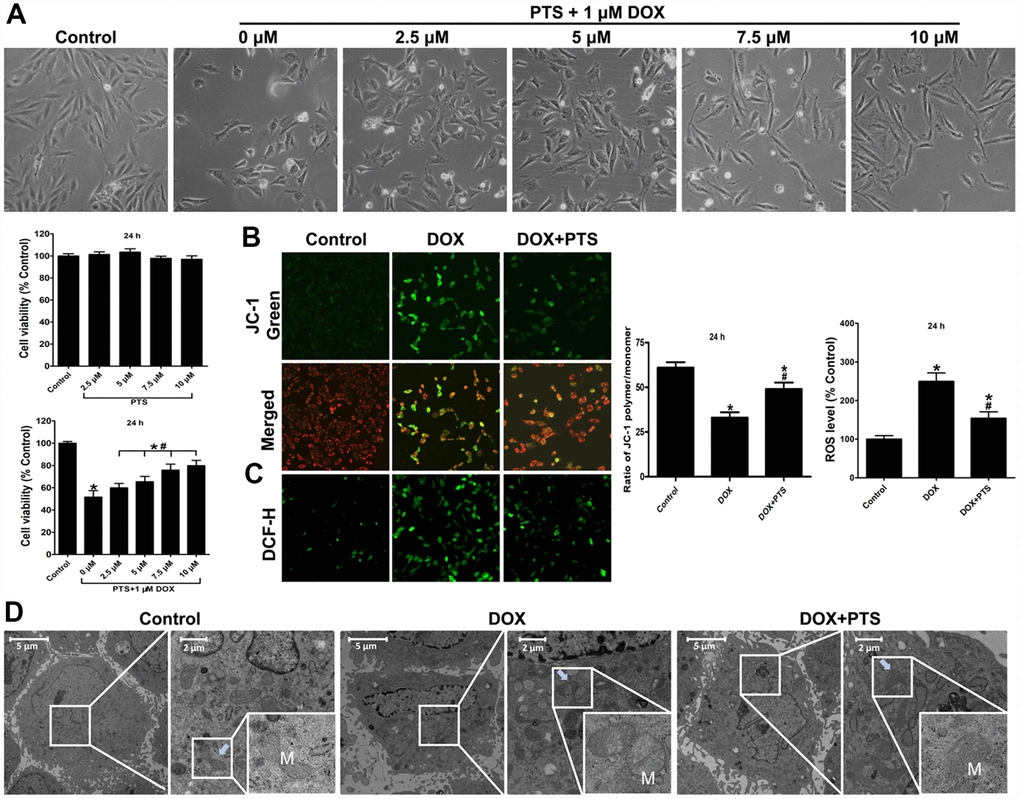
Figure 1. Effect of pterostilbene treatment on cell viability, mitochondrial membrane potential, ROS generation, mitochondrial morphologic changes in DOX-treated H9c2 cells (24 h). (A) single pterostilbene (2.5-10 μM) treatment and cotreatment of 1 μM DOX with increasing concentrations of pterostilbene (2.5-10 μM) on H9c2 cell viability (24 h). (B) mitochondrial membrane potential (ΔΨm) was expressed as the ratio of JC-1 polymer/monomer; red fluorescence represents the mitochondrial JC-1 polymer, and green fluorescence represents the monomeric form of JC-1, indicating ΔΨm depolarization. (C) Representative images and ROS level, and the indexes in the control group are defined as 100%. (D) Representative images of the ultrastructural morphology of mitochondria in each group of H9c2 cells are shown. The results are expressed as the mean ± SEM. *P < 0.05 vs. the control group, #P < 0.05 vs. the 1 μM DOX-treated group.
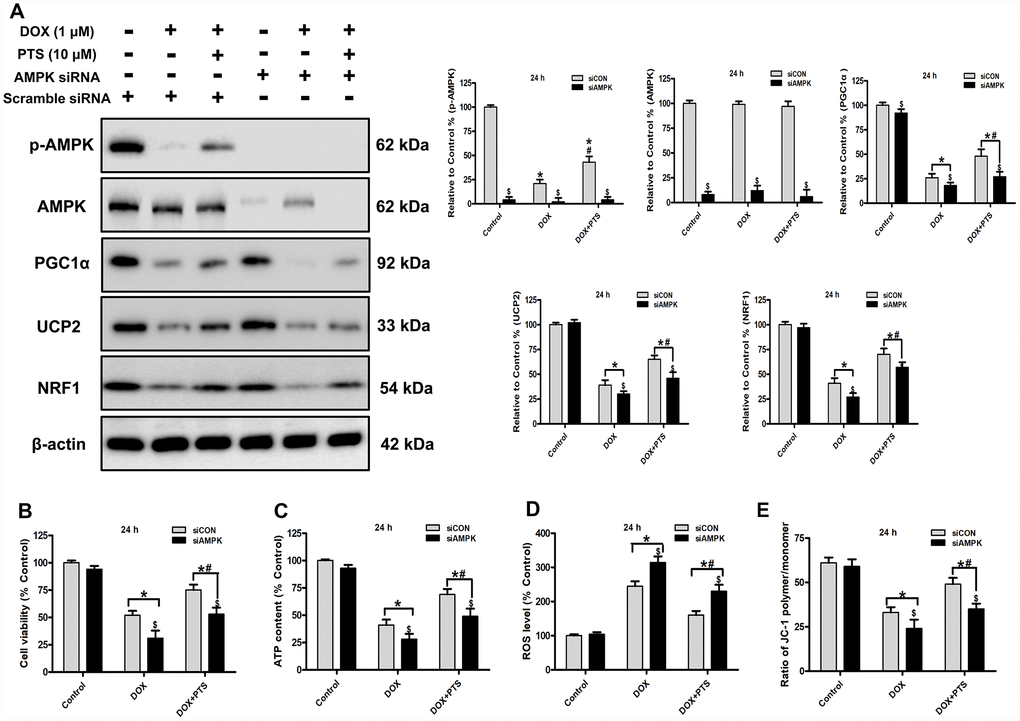
Figure 2. Effect of pterostilbene treatment combined with AMPK siRNA on cell viability, ATP content, ROS generation and ΔΨm, and AMPK and PGC1α signaling in DOX-treated H9c2 cells (24 h). (A) Representative western blot results of p-AMPK, AMPK, PGC1α, UCP2 and NRF1 are shown. Membranes were re-probed for β-actin expression to show that similar amounts of protein were loaded in each lane. (B) Cell viability, (C) cellular ATP content, (D) ROS level and (E) ΔΨm are shown, and (B–D) three indexes in the Control group of siCON are defined as 100%. The results are expressed as the mean ± SEM. *P < 0.05 vs. the Control group, #P < 0.05 vs. the 1 μM DOX-treated group, $P < 0.05 vs. the siCON group with the representative same drug treatments.
Pterostilbene application activated the AMPK, SIRT1 and PGC1α signaling in DOX-treated H9c2 cells
To explore the underlying molecular mechanisms regarding myocardial protective actions of pterostilbene on DOX-cardiotoxicity, we further analyzed the expression of p-AMPK, AMPK, SIRT1, and PGC1α and its downstream signaling proteins (NRF1 and UCP2) in H9c2 cells. Consistent with previously reported studies [4], 1 μM DOX-exposure for 24 h significantly inhibited the AMPK activation (AMPK phosphorylation), and decreased the expression of SIRT1, PGC1α, NRF1 and UCP2 (vs. Control group, P < 0.05, Figure 2A and Figure 3A). Interestingly, cotreatment of 10 μM pterostilbene markedly reversed the above effects caused by DOX treatment compared with DOX group (P < 0.05, Figure 2A and Figure 3A), suggesting activation of AMPK, SIRT1 and PGC1α signaling cascades may be involved in the actions of pterostilbene-exerted protection against DOX-cytotoxicity.
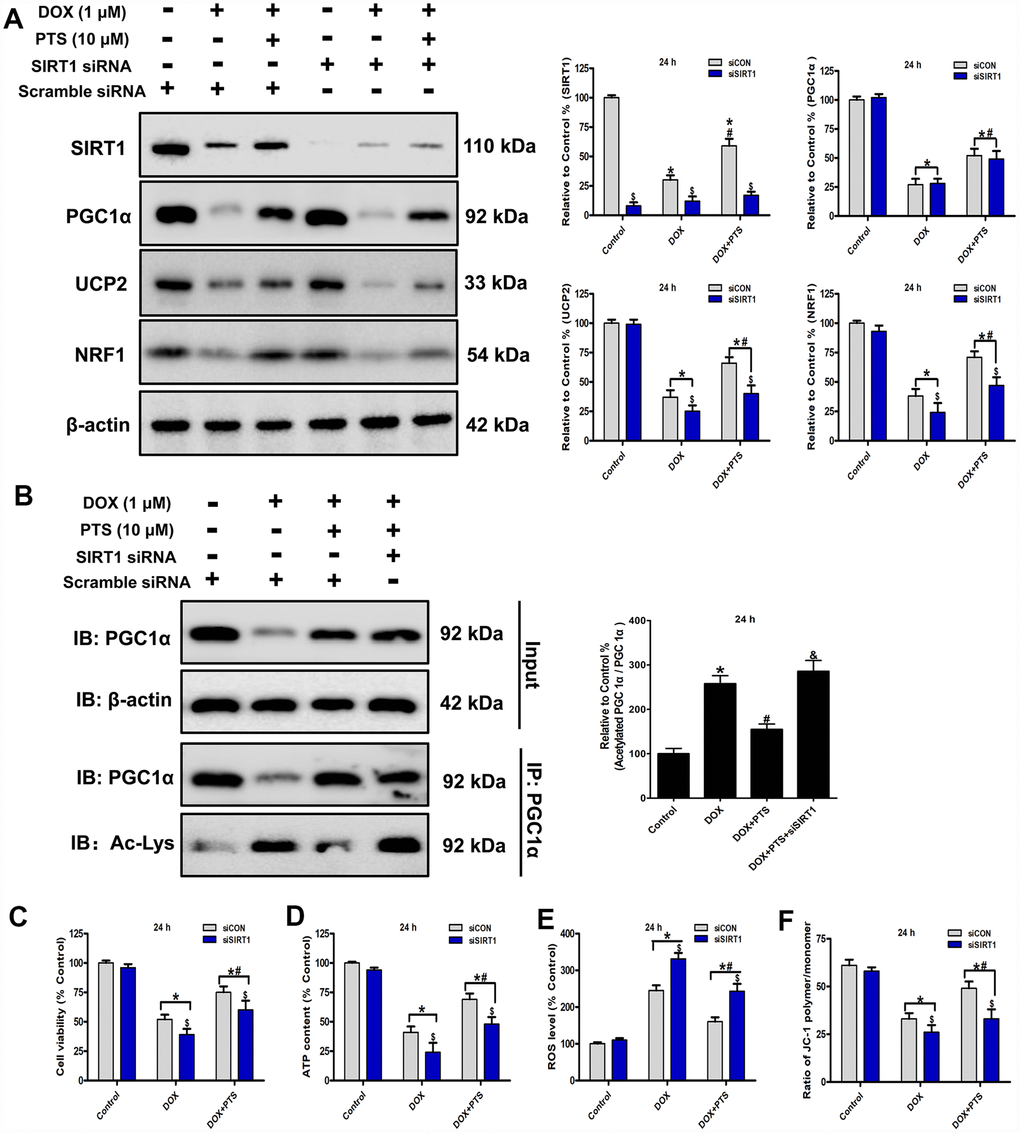
Figure 3. Effect of pterostilbene treatment combined with SIRT1 siRNA on cell viability, ATP content, ROS generation and ΔΨm, and SIRT1 and PGC1α signaling in DOX-treated H9c2 cells (24 h). (A) and (B) Representative western blot results of SIRT1, PGC1α, Ac-PGC1α, UCP2 and NRF1 are shown. Membranes were re-probed for β-actin expression to show that similar amounts of protein were loaded in each lane. IB, immunoblot; IP, immunoprecipitation. (C) Cell viability, (D) cellular ATP content, (E) ROS level and (F) ΔΨm are shown, and (C–E) three indexes in the Control group of siCON are defined as 100%. The results are expressed as the mean ± SEM. *P < 0.05 vs. the Control group, #P < 0.05 vs. the 1 μM DOX-treated group, $P < 0.05 vs. the siCON group with the representative same drug treatments.
Effect of pterostilbene cotreatment with AMPK siRNA on cell viability, oxidative stress parameters, AMPK and PGC1α signaling in DOX-treated H9c2 cells
Our previous study reported that AMPK was a potential upstream regulator on PGC1α signaling, and activation of AMPK-PGC1α cascades was involved in myocardial protection [4]. To investigate the action of AMPK on the myocardial protection of pterostilbene against DOX-cytotoxicity in vitro, a specific AMPK siRNA was applicated to knockdown the AMPK expression in the H9c2 cells. Western blot analyses showed that knockdown of AMPK hindered pterostilbene application-exerted increase of PGC1α and its downstream signaling (NRF1 and UCP2) protein levels in H9c2 cells with DOX exposure (vs. DOX + PTS group, P < 0.05, Figure 2A). Additionally, compared with the pterostilbene and DOX cotreatment group, AMPK knockdown significantly prevented the pterostilbene cotreatment-exerted rise in H9c2 cell viability, preservation of ΔΨm, cellular ATP content and the decrease of ROS generation with DOX-exposure (P < 0.05, Figure 2B–2E). Therefore, AMPK activation was involved in the pterostilbene-induced heart protection against DOX-cytotoxicity via PGC1α upregulation.
Effect of pterostilbene cotreatment with SIRT1 siRNA on cell viability, oxidative stress parameters, SIRT1 and PGC1α signaling in DOX-treated H9c2 cells
SIRT1 is a well-known deacetylase and involved in cardiac protection and mitochondrial biogenesis through its posttranslational deacetylation of PGC1α protein [16]. The activity of PGC1α is regulated by the post-translational level, and acetylation of PGC1α leads to the decrease of its transcriptional ability [19]. In present study, we found cotreatment of pterostilbene markedly increased the SIRT1 expression and decreased the acetylated PGC1α level (vs. DOX group, P < 0.05, Figure 3A and 3B). To explore the action of SIRT1 on the protective effects of pterostilbene against DOX-induced cardiotoxicity in vitro, the SIRT1 was knockdown after SIRT1 specific siRNA application on the H9c2 cells. Western blot analyses revealed that SIRT1 knockdown markedly promoted PGC1α acetylation without influencing PGC1α expression, and downregulated the UCP2 and NRF1 levels in the DOX + PTS + SIRT1 siRNA group compared with the DOX + PTS group (P < 0.05, Figure 3A and 3B). Moreover, compared with the pterostilbene and DOX cotreatment group, SIRT1 knockdown partially reversed the pterostilbene cotreatment-exhibited upregulation of H9c2 cell viability, ΔΨm and cellular ATP content, and the decrease of ROS generation during DOX-exposure (P < 0.05, Figure 3C–3F). These results suggested that SIRT1 participated in the pterostilbene exerted protection against DOX-induced cardiotoxicity via PGC1α deacetylation (activation).
Effect of cotreatment of pterostilbene with Compound C or EX527 on cardiac toxicity and mitochondrial oxidative stress injury on DOX-impaired mice hearts
The mitochondrial damage and concomitant oxidative stress injury were considered to be an important cause of DOX-cardiomyopathy. After DOX exposure for 6 days, compared with sham group, transmission electron microscopy examination on heart tissues showed the obvious disorders in ultrastructural mitochondrial morphology including mitochondrial swelling with cristae disorientation and breakage in the DOX-treated hearts (Figure 4A). Interestingly, pterostilbene cotreatment alleviated the DOX-induced injury on mitochondria through normalizing cristae density and architecture (Figure 4A). Moreover, pterostilbene cotreatment markedly downregulated the ROS generation and promoted the SOD and GPx activities in the DOX-exhibited hearts (vs. DOX group, P < 0.05, Figure 4B–4D). However, when compared with the DOX + PTS group, cotreatment of compound C (a selective AMPK inhibitor) or EX527 (a selective SIRT1 inhibitor) with pterostilbene clearly mitigated the mitochondrial protective and anti-oxidative effects of pterostilbene on DOX-stimulated hearts (Figure 4A–4D). Pterostilbene application combined with DOX and compound C or EX527 showed severe mitochondrial disorders (Figure 4A), as well as increase of ROS level and decreases in GPx and SOD activities (vs. DOX + PTS group, P < 0.05, Figure 4B–4D).
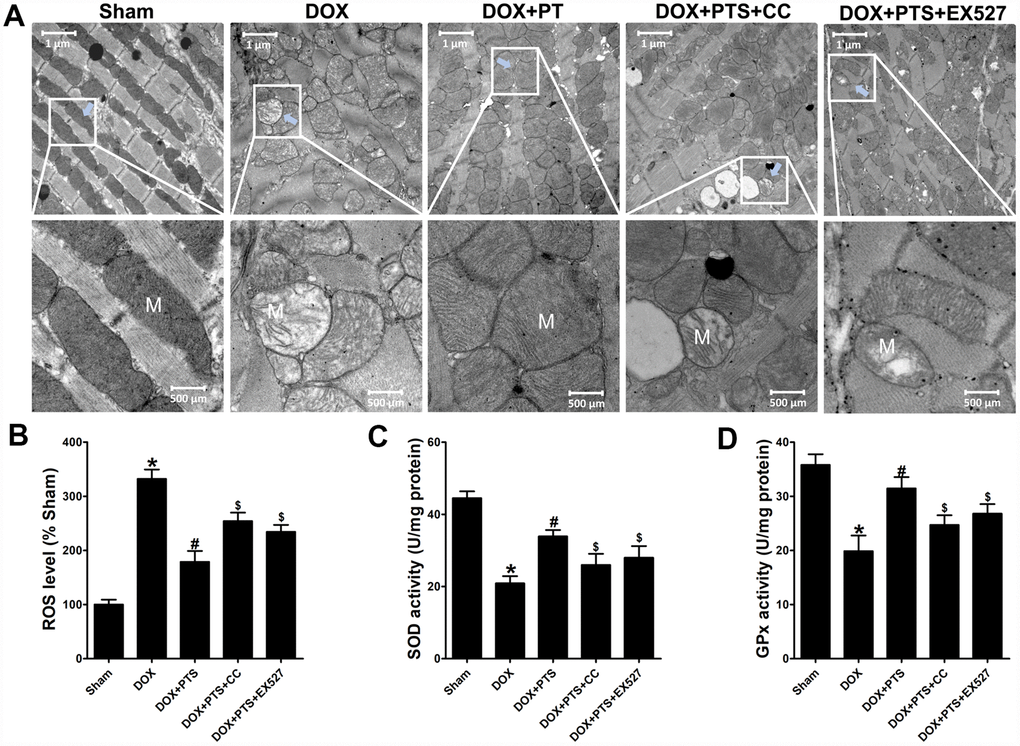
Figure 4. Effect of pterostilbene treatment combined with Compound C or EX527 on cardiac toxicity and oxidative stress in DOX-exposure mice hearts. (A) Representative images of the ultrastructural morphology of mitochondria from the left ventricular myocardium of experimental mice (magnification: upper panel 9900 ×, lower panel 20,500 ×). (B) ROS level, (C) SOD and (D) GPx activity in the myocardial tissues are showed. The results are expressed as the mean ± SEM. *P < 0.05 vs. the Sham group, #P < 0.05 vs. the DOX-treated group, $P < 0.05 vs. the DOX+PTS cotreated group.
Effect of cotreatment of pterostilbene with Compound C or EX527 on AMPK, SIRT1 and PGC1α signaling in the myocardial tissue of DOX-stimulated mice
To further confirm the above in vitro results that AMPK and SIRT1 activation was involved in the protective effects of pterostilbene against DOX-cytotoxicity via stimulating PGC1α signaling.
Western blot analyses were applied in the invivo study, and we found pterostilbene treatment markedly upregulated the p-AMPK and SIRT1 levels, and inhibited the PGC1α acetylation and promoted the expression of PGC1α and its downstream signaling (NRF1 and UCP2) proteins in DOX + PTS group (vs. DOX group, P < 0.05, Figure 5). Interestingly, cotreatment of AMPK inhibitor compound C markedly antagonized pterostilbene induced- upregulation of p-AMPK, PGC1α, NRF1 and UCP2 levels in the DOX + PTS + CC group (vs. DOX + PTS group, P < 0.05, Figure 5). Moreover, SIRT1 activity inhibition by EX527 cotreatment significantly reversed pterostilbene promoted PGC1α deacetylation and the increase of NRF1 and UCP2 expression in the DOX + PTS + EX527 group (vs. DOX + PTS group, P < 0.05, Figure 5). Taken together, above results indicated that AMPK activation-induced PGC1α upregulation and SIRT1 activation-promoted PGC1α deacetylation were involved in the pterostilbene-exerted myocardial protection against DOX-cytotoxicity.
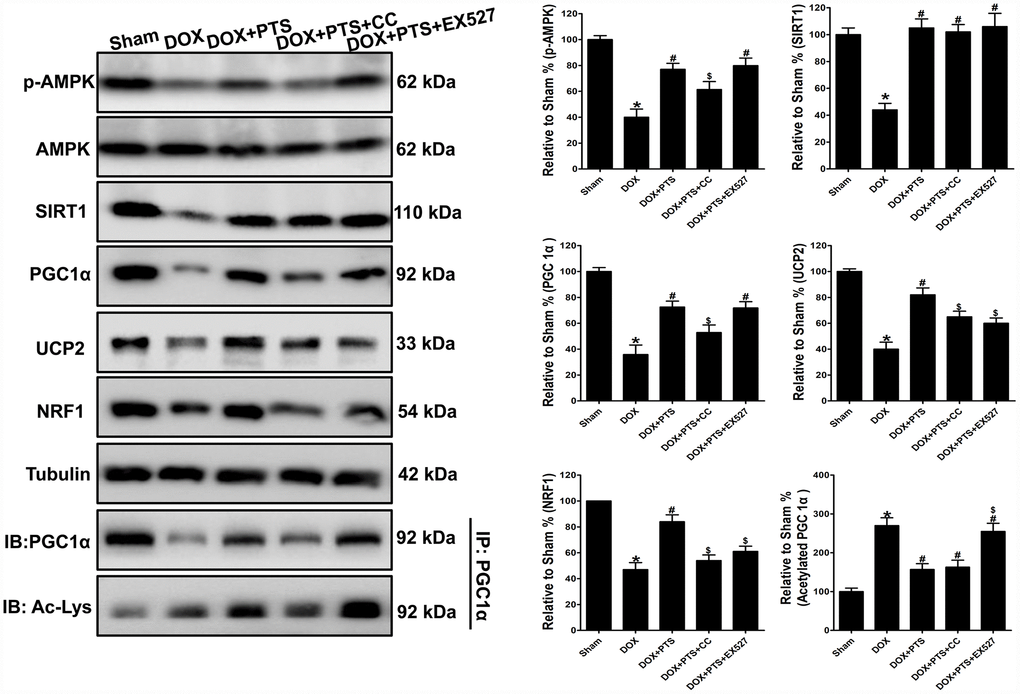
Figure 5. Effect of pterostilbene treatment combined with Compound C or EX527 on AMPK, SIRT1 and PGC1α signaling protein levels in DOX-stimulated mice hearts. Representative western blot results of p-AMPK, AMPK, SIRT1, PGC1α, Ac-PGC1α, UCP2 and NRF1 are shown. Membranes were re-probed for Tubulin expression to show that similar amounts of protein were loaded in each lane. IB, immunoblot; IP, immunoprecipitation. The results are expressed as the mean ± SEM. *P < 0.05 vs. the Sham group, #P < 0.05 vs. the DOX-treated group, $P < 0.05 vs. the DOX+PTS cotreated group.
Discussion
DOX is a potent anticancer agent and has been widely used in the chemotherapeutic treatment regimens for breast, gastric, thyroid, lung, and ovarian cancers [20]. However, DOX dose-dependently causes acute and chronic cardiotoxicity and this severe side-effect greatly limits its use in clinic for cancer treatment [4, 21]. The molecular mechanism of DOX-cardiotoxicity still remains controversial, but a currently major hypothesis of DOX-induced cardiotoxicity is related to oxidative stress stimulation and mitochondrial injury on cardiac myocyte [4, 22, 23]. Therefore, to mitigate the mitochondrial oxidative damage is one of the therapeutic plans for DOX-induced cardiomyopathy [4, 24, 25]. Resveratrol could exert protection against DOX-induced cardiotoxicity via activating SIRT1 and AMPK signaling cascades thus mitigating oxidative stress, mitochondrial injury, cardiomyocyte apoptosis and cardiac fibrosis induced by DOX exposure [19, 26–29]. Pterostilbene is a natural demethylated analogue of resveratrol and riches in blueberries and grapes [30]. Like resveratrol, pterostilbene functions as a potent antioxidant and exerts myocardial protection [7]. Previous studies reported that pterostilbene could preserve cardiac function against hypertrophy, myocardial infarction and ischemia reperfusion injury via oxidative stress inhibition [7, 8, 10]. Interestingly, our present study found that pterostilbene application significantly reduced DOX-induced injury on H9c2 cells. Moreover, pterostilbene markedly ameliorated doxorubicin exposure-caused acute cardiac tissue damages in mice.
Mitochondria play pivotal roles in maintaining cardiac cells homeostasis and exacerbating injury-induced cell death [31]. Mitochondrial dysfunction disrupts the ATP production, increases the ROS generation, and alters redox balance in cardiomyocytes [4, 32, 33]. Moreover, excess ROS generation also causes irreversible mitochondrial damage and exacerbates cardiac diseases [31]. DOX could accumulate in the inner mitochondrial membrane and thus contributes to mitochondrial toxicity and ROS generation [22]. In present study, pterostilbene cotreatment markedly alleviated DOX-induced mitochondrial swelling with cristae disorientation and breakage both in vivo and in vitro. Additionally, pterostilbene application also reversed DOX-induced ROS generation and ATP content decrease in H9c2 cells. To sustain cardiac contractile function, cardiomyocytes contain a large volume of mitochondria to preserve ATP generation [31]. Acute DOX exposure for 6 days significantly disrupted mitochondrial function and induced oxidative stress by promoting ROS generation and downregulating the SOD and GPx activities in mice heart tissues. However, treatment of pterostilbene obviously reversed the above adverse effects induced by DOX, suggesting the mitochondrial protective and anti-oxidant actions of pterostilbene on cardiomyocytes.
The transcriptional coactivator PGC1α is critical for maintaining ATP production and mitochondrial function, and regulates a number of mitochondrial genes (e.g., NRF1 and UCP2) [22, 36]. Loss of PGC1α leads to oxidative stress and metabolic dysfunction by disrupting mitochondrial growth and respiration in cardiomyocytes [37, 38]. Under the regulation of PGC1α, NRF1 transcriptionally regulates the expression of mitochondrial function-related genes encoding the respiratory complexes and mitochondrial enzymes [4]. Moreover, UCP2, a downstream factor of PGC1α, is involved in myocardial protection via inhibiting ROS generation [4]. Previous studies found that DOX application markedly inhibited the expression and activity of PGC1α and its downstream factors in cardiac cells [4]. Consistently, our study revealed that acute DOX exposure significantly downregulated the PGC1α, NRF1 and UCP2 levels both in the H9c2 cells and mice heart tissues. However, pterostilbene cotreatment dramatically preserved the expression of PGC1α, NRF1 and UCP2 in DOX-treated H9c2 cells and cardiac tissues. Therefore, activation of PGC1α and its downstream factors may participate in the pterostilbene-exerted protection against DOX-cardiotoxicity.
AMPK is a serine-threonine kinase that functions as a key metabolic regulator for mitochondrial activity and cardiomyocyte energy homeostasis [36, 37]. Activated AMPK by Thr172 site phosphorylation exerts myocardial protection against various cardiac diseases, including ischemia reperfusion injury, infarction, and DOX-cardiotoxicity [4, 38, 39]. DOX application could markedly inhibit the AMPK activity in cardiomyocytes [38]. Consistently, our study also revealed that the DOX exposure significantly inhibited AMPK phosphorylation both in H9c2 cells and in mice heart tissues. Pterostilbene has been previously reported as a potent AMPK activator [9], and we verified that treatment of pterostilbene markedly reversed the DOX-induced downregulation of AMPK phosphorylation. PGC1α is one of the downstream molecules of AMPK, and its expression could be regulated by the activity of AMPK [40]. Interestingly, AMPK activation by pterostilbene upregulated the PGC1α expression after DOX exposure both in vitro and in vivo. However, knockdown of AMPK by siRNA partially impeded the pterostilbene-exerted PGC1α upregulation, cell viability preservation and antioxidant activities in DOX-treated H9c2 cells. Moreover, AMPK inhibitor Compound C treatment also reversed the pterostilbene-exhibited protective actions of mitochondrial morphology protection, antioxidation and PGC1α preservation against acute DOX-cardiotoxicity. These results indicated that activation of AMPK was involved in the myocardial protection of pterostilbene against DOX exposure.
Resveratrol is a classical natural SIRT1 activator and involved in the cardiac protection against DOX-exerted heart injury via SIRT1 activation [28]. Pterostilbene is an analog of resveratrol, but it exhibits better bioavailability and pharmacological activities than resveratrol due to the presence of two methoxy groups which leads to higher cellular uptake and a longer half-life [30]. Moreover, pterostilbene also is a potent natural SIRT1 activator and exerts protection against injuries on various organs through SIRT1 upregulation [41, 42]. SIRT1 functions as a key NAD+-dependent deacetylase and increases the activity of PGC1α via posttranslational deacetylation [16, 43, 44]. Our present study found that pterostilbene treatment could reverse the SIRT1 reduction and enhanced PGC1α acetylation induced by DOX-exposure both in vitro and in vivo. Interestingly, both SIRT1 knockdown in vitro and SIRT1 activity inhibition by EX527 in vivo markedly hindered the protective activity of pterostilbene and increased the acetylated PGC1α levels in H9c2 cells and mice heart tissues. Thus, above results suggested that the SIRT1-PGC1α cascades activation involved in the protection of pterostilbene against DOX-cardiotoxicity.
Conclusions
The present study exhibited the in vivo and in vitro mechanistic evidence that pterostilbene application could alleviate acute DOX-induced mitochondrial injury and oxidation in cardiomyocytes via promoting AMPK activation and SIRT1 upregulation thus preserving PGC1α activity by upregulated expression and deacetylation (Figure 6). Clinical studies suggest pterostilbene is generally safe at doses up to 250 mg/day for human and is good for cardiovascular system via reducing blood pressure in adults (ClinicalTrials.gov Identifier: NCT01267227) [13, 14]. Moreover, basic researches revealed that pterostilbene could protect heart against hypertrophy, diabetic cardiomyopathy, myocardial infarction and ischemia reperfusion injury [7–10]. To our knowledge, this is the first study reporting the protective actions of pterostilbene against acute DOX-cardiotoxicity. However, further definitive and carefully planned clinical studies are still warranted to verify the cardioprotective effects of pterostilbene against DOX-cardiotoxicity.
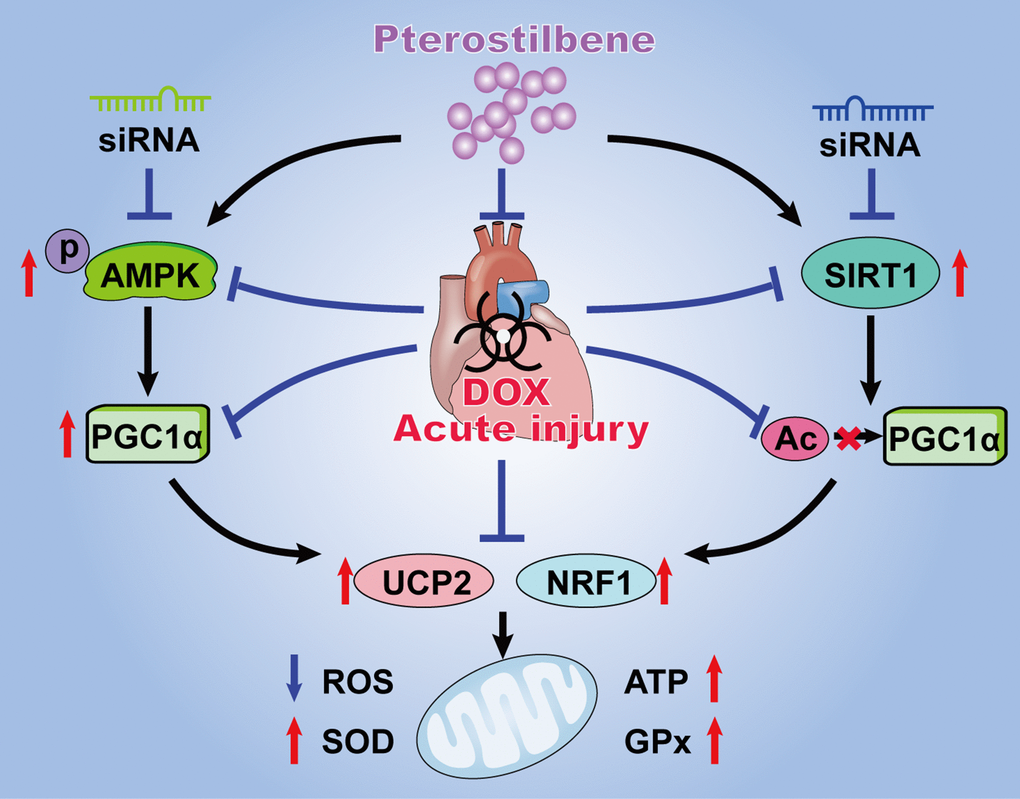
Figure 6. Schematic diagram summarizing the myocardial protective actions of pterostilbene against acute DOX cardiotoxicity via the PGC1α activation through stimulating AMPK and SIRT1 cascades. Pterostilbene treatment enhances the AMPK phosphorylation and SIRT1 upregulation thus increasing the PGC1α expression and inhibiting PGC1α acetylation. These effects markedly increase the levels of UCP2 and NRF1, and inhibits oxidative stress via decreasing ROS generation and increasing ATP content, SOD2 and GPx activities in cardiomyocytes.
Materials and Methods
Cell culture and siRNA transfection
The cardiomyoblast cell line H9c2 (Rattus norvegicus) was purchased from the American Type Culture Collection (ATCC, VA, USA). Cells were cultured in high glucose DMEM medium (Gibco, NY, USA), supplemented with penicillin-streptomycin solution (100 units/ml) (Solarbio, Beijing, China) and 10% fetal bovine serum (FBS, Gibco, USA) according to the providers’ instructions. The commercial rat AMPK siRNA, SIRT1 siRNA and control siRNA were obtained from Santa Cruz Biotechnology (CA, USA). When the H9c2 cells reached 40-60% confluence seeded in the 6-well plates, they were transfected with control or targeted siRNA transfection mixture via using the Lipofectamine 2000 (Invitrogen). Then, cells were cultured in fresh medium after 48 h transfection.
Cell treatments and in vitro experimental design
Pterostilbene, doxorubicin, and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Pterostilbene and doxorubicin stock solutions were maintained in DMSO and diluted in culture medium at certain concentrations for further experiments. The dosage of DOX was chosen based on previously described [4]. The scramble siRNA, AMPK siRNA or SIRT1 siRNA-treated cells were respectively assigned to 3 groups: (i) Control group; (ii) 1 μM DOX treatment group (DOX); (iii) 1μM DOX and 10 μM pterostilbene cotreatment group (DOX + PTS). After those treatments, cells were harvested for further experiments.
Animals and in vivo experimental design
The animal experiments were performed on healthy adult male C57BL/6 mice at 8 weeks of age; the mice were normally kept in a pathogen-free environment with free access to food and water. All experiments on mice were conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (National Institutes of Health Publication No. 85-23, revised 1996). Compound C and EX527 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Pterostilbene, doxorubicin, Compound C and EX527 were dissolved in DMSO and then diluted with normal saline. 75 mice were randomly assigned to five groups (n = 15 for each group): (i) sham-operated control group (Sham); (ii) DOX treatment group (DOX); (iii) DOX and pterostilbene cotreatment group (DOX + PTS); (iv) DOX, pterostilbene and Compound C cotreatment group (DOX + PTS + CC); (v) DOX, pterostilbene and EX527 cotreatment group (DOX + PTS + EX527). DOX was intraperitoneally injected at the dose of 10 mg/kg or the same volume of vehicle (saline) was conducted on day 1 and day 4 for a total of 2 times (20 mg/kg cumulative dose of DOX). Pterostilbene (10 mg/kg/day), Compound C (20 mg/kg/day), EX527 (5 mg/kg/day) or the same volume of vehicle was intraperitoneally injected every day for a total of 7 times (1 day before initial DOX treatment). All mice were euthanized 6 days after the initial injection of DOX. The dosages of DOX, pterostilbene, Compound C and EX527 were chosen based on the previous reports [4, 45, 46].
Cell viability analysis
After the different treatments on H9c2 cells for 24 h, the cell viability was detected via using the CCK-8 kit (7Sea, Shanghai, China) according to the previously described [47]. Optical density (OD) values were measured at 450 nm through the microplate reader (SpectraMax 190, Molecular Device, USA). Cell viability was presented as the ratio of OD values compared with the Control group, and representative images were taken by a 600D camera (Canon Company, Japan).
Cell mitochondrial membrane potential analysis
The H9c2 cells’ mitochondrial membrane potential (ΔΨm) was measured by the fluorescent dye JC-1 staining. After different treatments, cells were incubated with JC-1 working solution for 20 min in the dark at 37 °C. Cells exhibiting red fluorescence are in normal ΔΨm state. Green fluorescence represented the monomeric form of JC-1 and suggested ΔΨm depolarization. Images were obtained using the confocal microscope, and the results were analyzed by the SpectraMax 190 microplate reader, and expressed as the proportion of aggregated JC-1 and monomeric JC-1.
Cellular ATP content analysis
Cellular ATP concentrations were assayed with an ATP detection kit (Beyotime Institute of Biotechnology, China). H9c2 cells were lysed through the ATP assay buffer according to the instructions of the manufacturer. The level of ATP was measured by the emitted light of the mixture of the 50 μl cell lysis supernatant and 50 μl luciferase reagent using the microplate reader.
ROS, SOD and GPx determination
To analyze the oxidative stress parameters, the ROS level and the GPx and SOD activities in cultured cells and heart tissues were detected via using commercially available DCFH-DA (Beyotime Institute of Biotechnology, China), SOD and GPx assay kits (Nanjing Jiancheng Bioengineering Institute, China) according to the manufacturer’s instructions. The data were analyzed by the SpectraMax 190 microplate reader (Molecular Device, USA).
Transmission electron microscopy
The ultrastructure of mitochondria of H9c2 cells and mouse heart samples were observed via using the transmission electron microscopy. After different treatments, cells were collected and fixed in 2% glutaraldehyde for 24 h at 4 °C, and then cells were diced into 2 mm cubes. 6 days after the initial injection of DOX, the mouse hearts were cut into 2 mm cubes placed in 2% glutaraldehyde at 4 °C for 24 h. Later, both the cell and tissue samples were cut into 75 nm thickness sections, then they were stained with uranium acetate and lead citrate; images were observed using a TECNAI G2 Spirit Biotwin 120 kV electron microscope.
Western blot and immunoprecipitation
After various treatments, the cells and heart samples were collected and lysed by the RIPA buffer; a BCA protein assay was used to analyze the protein concentration (Thermo Scientific, Rockford, IL, USA). As for immunoprecipitation, equal amounts of cell or tissue extracts were incubated with 2 ug anti-PGC1α (Santa Cruz, sc-518025) at 4 °C for 12 h on a rotating incubator, and then the samples were further incubated with the 20 μl Protein A/G Agarose (Beyotime Institute of Biotechnology, China) at 4 °C for 2 h rotation. Immunocomplexes were washed four times with NETN buffer before diluted with SDS-PAGE sample loading buffer. Then, the boiled protein samples of each group were fractionated by SDS-PAGE and transferred to PVDF membranes. Later, After blocked with 5% nonfat milk in TBST for 2 h at room temperature, the membranes were incubated with primary antibodies against AMPK (CST, #5831, 1:1000), p-AMPK Thr172 (CST, #50081, 1:1000), PGC1α (Abcam, ab54481, 1:1000), Ac-Lys (CST, #9441, 1:1000), NRF1 (Abcam, ab175932, 1:1000), UCP2 (Abcam, ab97931, 1:500), β-actin (Abcam, ab6276, 1:3000) and Tubulin (CST, #2148, 1:3000) for 12 h at 4 °C. Thereafter, the membranes were washed with TBST and exposed to the corresponding secondary antibodies (1:5000, Zhongshan Company, China) at room temperature for 2 h. The fluorescent signal was detected using a BioRad imaging system (BioRad, Hercules, CA, USA), and the signal was quantified using Image Lab Software (BioRad, Hercules, CA, USA).
Statistical analyses
IBM SPSS 23.0 (SPSS Inc., Chicago, USA) software was used for further data analyses. In this study, between-group comparisons, one-way ANOVA followed by Bonferroni post hoc test. Data are presented as the means ± SEM, and the P value < 0.05 was considered as statistically significant.
Author Contributions
JSY and SBQ designed the experiments and performed the analysis and data interpretation. DL, ZQM, XYZ and LQX performed experiments. DL and ZQM wrote and edited the manuscript. JSY and SBQ discussed data and provided intellectual contributions.
Conflicts of Interest
The authors have no conflicts of interest to declare.
Funding
This work was supported by the CAMS Innovation Fund for Medical Sciences (CIFMS, 2017-I2M-2-005).
References
- 1. Ewer MS, Ewer SM. Cardiotoxicity of anticancer treatments: what the cardiologist needs to know. Nat Rev Cardiol. 2010; 7:564–75. https://doi.org/10.1038/nrcardio.2010.121 [PubMed]
- 2. Vejpongsa P, Yeh ET. Prevention of anthracycline-induced cardiotoxicity: challenges and opportunities. J Am Coll Cardiol. 2014; 64:938–45. https://doi.org/10.1016/j.jacc.2014.06.1167 [PubMed]
- 3. Doroshow JH. Effect of anthracycline antibiotics on oxygen radical formation in rat heart. Cancer Res. 1983; 43:460–72. [PubMed]
- 4. Liu D, Ma Z, Di S, Yang Y, Yang J, Xu L, Reiter RJ, Qiao S, Yuan J. AMPK/PGC1α activation by melatonin attenuates acute doxorubicin cardiotoxicity via alleviating mitochondrial oxidative damage and apoptosis. Free Radic Biol Med. 2018; 129:59–72. https://doi.org/10.1016/j.freeradbiomed.2018.08.032 [PubMed]
- 5. Ma Z, Zhang X, Xu L, Liu D, Di S, Li W, Zhang J, Zhang H, Li X, Han J, Yan X. Pterostilbene: mechanisms of its action as oncostatic agent in cell models and in vivo studies. Pharmacol Res. 2019; 145:104265. https://doi.org/10.1016/j.phrs.2019.104265 [PubMed]
- 6. Yu CL, Yang SF, Hung TW, Lin CL, Hsieh YH, Chiou HL. Inhibition of eIF2α dephosphorylation accelerates pterostilbene-induced cell death in human hepatocellular carcinoma cells in an ER stress and autophagy-dependent manner. Cell Death Dis. 2019; 10:418. https://doi.org/10.1038/s41419-019-1639-5 [PubMed]
- 7. Lacerda D, Ortiz V, Türck P, Campos-Carraro C, Zimmer A, Teixeira R, Bianchi S, de Castro AL, Schenkel PC, Belló-Klein A, Bassani VL, da Rosa Araujo AS. Stilbenoid pterostilbene complexed with cyclodextrin preserves left ventricular function after myocardial infarction in rats: possible involvement of thiol proteins and modulation of phosphorylated GSK-3β. Free Radic Res. 2018; 52:988–99. https://doi.org/10.1080/10715762.2018.1506115 [PubMed]
- 8. Dos Santos Lacerda D, Türck P, Gazzi de Lima-Seolin B, Colombo R, Duarte Ortiz V, Poletto Bonetto JH, Campos-Carraro C, Bianchi SE, Belló-Klein A, Linck Bassani V, Sander da Rosa Araujo A. Pterostilbene reduces oxidative stress, prevents hypertrophy and preserves systolic function of right ventricle in cor pulmonale model. Br J Pharmacol. 2017; 174:3302–14. https://doi.org/10.1111/bph.13948 [PubMed]
- 9. Kosuru R, Kandula V, Rai U, Prakash S, Xia Z, Singh S. Pterostilbene Decreases Cardiac Oxidative Stress and Inflammation via Activation of AMPK/Nrf2/HO-1 Pathway in Fructose-Fed Diabetic Rats. Cardiovasc Drugs Ther. 2018; 32:147–63. https://doi.org/10.1007/s10557-018-6780-3 [PubMed]
- 10. Yu Z, Wang S, Zhang X, Li Y, Zhao Q, Liu T. Pterostilbene protects against myocardial ischemia/reperfusion injury via suppressing oxidative/nitrative stress and inflammatory response. Int Immunopharmacol. 2017; 43:7–15. https://doi.org/10.1016/j.intimp.2016.11.018 [PubMed]
- 11. Kosuru R, Rai U, Prakash S, Singh A, Singh S. Promising therapeutic potential of pterostilbene and its mechanistic insight based on preclinical evidence. Eur J Pharmacol. 2016; 789:229–43. https://doi.org/10.1016/j.ejphar.2016.07.046 [PubMed]
- 12. Li YR, Li S, Lin CC. Effect of resveratrol and pterostilbene on aging and longevity. Biofactors. 2018; 44:69–82. https://doi.org/10.1002/biof.1400 [PubMed]
- 13. Riche DM, Riche KD, Blackshear CT, McEwen CL, Sherman JJ, Wofford MR, Griswold ME. Pterostilbene on metabolic parameters: a randomized, double-blind, and placebo-controlled trial. Evid Based Complement Alternat Med. 2014; 2014:459165. https://doi.org/10.1155/2014/459165 [PubMed]
- 14. Riche DM, McEwen CL, Riche KD, Sherman JJ, Wofford MR, Deschamp D, Griswold M. Analysis of safety from a human clinical trial with pterostilbene. J Toxicol. 2013; 2013:463595. https://doi.org/10.1155/2013/463595 [PubMed]
- 15. Yuan H, Zhang Q, Guo J, Zhang T, Zhao J, Li J, White A, Carmichael PL, Westmoreland C, Peng S. A PGC-1α-Mediated Transcriptional Network Maintains Mitochondrial Redox and Bioenergetic Homeostasis against Doxorubicin-Induced Toxicity in Human Cardiomyocytes: Implementation of TT21C. Toxicol Sci. 2016; 150:400–17. https://doi.org/10.1093/toxsci/kfw006 [PubMed]
- 16. Cui L, Guo J, Zhang Q, Yin J, Li J, Zhou W, Zhang T, Yuan H, Zhao J, Zhang L, Carmichael PL, Peng S. Erythropoietin activates SIRT1 to protect human cardiomyocytes against doxorubicin-induced mitochondrial dysfunction and toxicity. Toxicol Lett. 2017; 275:28–38. https://doi.org/10.1016/j.toxlet.2017.04.018 [PubMed]
- 17. Quan N, Wang L, Chen X, Luckett C, Cates C, Rousselle T, Zheng Y, Li J. Sestrin2 prevents age-related intolerance to post myocardial infarction via AMPK/PGC-1α pathway. J Mol Cell Cardiol. 2018; 115:170–78. https://doi.org/10.1016/j.yjmcc.2018.01.005 [PubMed]
- 18. Yuan YP, Ma ZG, Zhang X, Xu SC, Zeng XF, Yang Z, Deng W, Tang QZ. CTRP3 protected against doxorubicin-induced cardiac dysfunction, inflammation and cell death via activation of Sirt1. J Mol Cell Cardiol. 2018; 114:38–47. https://doi.org/10.1016/j.yjmcc.2017.10.008 [PubMed]
- 19. Schilling J, Kelly DP. The PGC-1 cascade as a therapeutic target for heart failure. J Mol Cell Cardiol. 2011; 51:578–83. https://doi.org/10.1016/j.yjmcc.2010.09.021 [PubMed]
- 20. Luu AZ, Chowdhury B, Al-Omran M, Teoh H, Hess DA, Verma S. Role of Endothelium in Doxorubicin-Induced Cardiomyopathy. JACC Basic Transl Sci. 2018; 3:861–70. https://doi.org/10.1016/j.jacbts.2018.06.005 [PubMed]
- 21. Pakravan G, Foroughmand AM, Peymani M, Ghaedi K, Hashemi MS, Hajjari M, Nasr-Esfahani MH. Downregulation of miR-130a, antagonized doxorubicin-induced cardiotoxicity via increasing the PPARγ expression in mESCs-derived cardiac cells. Cell Death Dis. 2018; 9:758. https://doi.org/10.1038/s41419-018-0797-1 [PubMed]
- 22. Gorini S, De Angelis A, Berrino L, Malara N, Rosano G, Ferraro E. Chemotherapeutic Drugs and Mitochondrial Dysfunction: Focus on Doxorubicin, Trastuzumab, and Sunitinib. Oxid Med Cell Longev. 2018; 2018:7582730. https://doi.org/10.1155/2018/7582730 [PubMed]
- 23. Guo R, Hua Y, Ren J, Bornfeldt KE, Nair S. Cardiomyocyte-specific disruption of Cathepsin K protects against doxorubicin-induced cardiotoxicity. Cell Death Dis. 2018; 9:692. https://doi.org/10.1038/s41419-018-0727-2 [PubMed]
- 24. Wang S, Wang Y, Zhang Z, Liu Q, Gu J. Cardioprotective effects of fibroblast growth factor 21 against doxorubicin-induced toxicity via the SIRT1/LKB1/AMPK pathway. Cell Death Dis. 2017; 8:e3018. https://doi.org/10.1038/cddis.2017.410 [PubMed]
- 25. Ma Z, Xu L, Liu D, Zhang X, Di S, Li W, et al. Utilizing Melatonin to Alleviate Side Effects of Chemotherapy: A Potentially Good Partner for Treating Cancer with Ageing. Oxid Med Cell Longev. 2020. [Epub ahead of print].
- 26. Gu J, Fan YQ, Zhang HL, Pan JA, Yu JY, Zhang JF, Wang CQ. Resveratrol suppresses doxorubicin-induced cardiotoxicity by disrupting E2F1 mediated autophagy inhibition and apoptosis promotion. Biochem Pharmacol. 2018; 150:202–13. https://doi.org/10.1016/j.bcp.2018.02.025 [PubMed]
- 27. Cappetta D, Esposito G, Piegari E, Russo R, Ciuffreda LP, Rivellino A, Berrino L, Rossi F, De Angelis A, Urbanek K. SIRT1 activation attenuates diastolic dysfunction by reducing cardiac fibrosis in a model of anthracycline cardiomyopathy. Int J Cardiol. 2016; 205:99–110. https://doi.org/10.1016/j.ijcard.2015.12.008 [PubMed]
- 28. Zhang C, Feng Y, Qu S, Wei X, Zhu H, Luo Q, Liu M, Chen G, Xiao X. Resveratrol attenuates doxorubicin-induced cardiomyocyte apoptosis in mice through SIRT1-mediated deacetylation of p53. Cardiovasc Res. 2011; 90:538–45. https://doi.org/10.1093/cvr/cvr022 [PubMed]
- 29. Danz ED, Skramsted J, Henry N, Bennett JA, Keller RS. Resveratrol prevents doxorubicin cardiotoxicity through mitochondrial stabilization and the Sirt1 pathway. Free Radic Biol Med. 2009; 46:1589–97. https://doi.org/10.1016/j.freeradbiomed.2009.03.011 [PubMed]
- 30. Ma Z, Yang Y, Di S, Feng X, Liu D, Jiang S, Hu W, Qin Z, Li Y, Lv J, Fan C, Yan X, Li X. Pterostilbene exerts anticancer activity on non-small-cell lung cancer via activating endoplasmic reticulum stress. Sci Rep. 2017; 7:8091. https://doi.org/10.1038/s41598-017-08547-0 [PubMed]
- 31. Sack MN, Fyhrquist FY, Saijonmaa OJ, Fuster V, Kovacic JC. Basic Biology of Oxidative Stress and the Cardiovascular System: Part 1 of a 3-Part Series. J Am Coll Cardiol. 2017; 70:196–211. https://doi.org/10.1016/j.jacc.2017.05.034 [PubMed]
- 32. Ma Z, Xin Z, Di W, Yan X, Li X, Reiter RJ, Yang Y. Melatonin and mitochondrial function during ischemia/reperfusion injury. Cell Mol Life Sci. 2017; 74:3989–98. https://doi.org/10.1007/s00018-017-2618-6 [PubMed]
- 33. Chen LY, Renn TY, Liao WC, Mai FD, Ho YJ, Hsiao G, Lee AW, Chang HM. Melatonin successfully rescues hippocampal bioenergetics and improves cognitive function following drug intoxication by promoting Nrf2-ARE signaling activity. J Pineal Res. 2017; 63:e12417. https://doi.org/10.1111/jpi.12417 [PubMed]
- 34. Kärkkäinen O, Tuomainen T, Mutikainen M, Lehtonen M, Ruas JL, Hanhineva K, Tavi P. Heart specific PGC-1α deletion identifies metabolome of cardiac restricted metabolic heart failure. Cardiovasc Res. 2019; 115:107–18. https://doi.org/10.1093/cvr/cvy155 [PubMed]
- 35. Ding M, Feng N, Tang D, Feng J, Li Z, Jia M, Liu Z, Gu X, Wang Y, Fu F, Pei J. Melatonin prevents Drp1-mediated mitochondrial fission in diabetic hearts through SIRT1-PGC1α pathway. J Pineal Res. 2018; 65:e12491. https://doi.org/10.1111/jpi.12491 [PubMed]
- 36. Bairwa SC, Parajuli N, Dyck JR. The role of AMPK in cardiomyocyte health and survival. Biochim Biophys Acta. 2016; 1862:2199–210. https://doi.org/10.1016/j.bbadis.2016.07.001 [PubMed]
- 37. Ma Z, Fan C, Yang Y, Di S, Hu W, Li T, Zhu Y, Han J, Xin Z, Wu G, Zhao J, Li X, Yan X. Thapsigargin sensitizes human esophageal cancer to TRAIL-induced apoptosis via AMPK activation. Sci Rep. 2016; 6:35196. https://doi.org/10.1038/srep35196 [PubMed]
- 38. Gratia S, Kay L, Potenza L, Seffouh A, Novel-Chaté V, Schnebelen C, Sestili P, Schlattner U, Tokarska-Schlattner M. Inhibition of AMPK signalling by doxorubicin: at the crossroads of the cardiac responses to energetic, oxidative, and genotoxic stress. Cardiovasc Res. 2012; 95:290–99. https://doi.org/10.1093/cvr/cvs134 [PubMed]
- 39. Liu D, Xu L, Zhang X, Shi C, Qiao S, Ma Z, Yuan J. Snapshot: Implications for mTOR in Aging-related Ischemia/Reperfusion Injury. Aging Dis. 2019; 10:116–33. https://doi.org/10.14336/AD.2018.0501 [PubMed]
- 40. Salt IP, Hardie DG. AMP-Activated Protein Kinase: An Ubiquitous Signaling Pathway With Key Roles in the Cardiovascular System. Circ Res. 2017; 120:1825–41. https://doi.org/10.1161/CIRCRESAHA.117.309633 [PubMed]
- 41. Song L, Chen TY, Zhao XJ, Xu Q, Jiao RQ, Li JM, Kong LD. Pterostilbene prevents hepatocyte epithelial-mesenchymal transition in fructose-induced liver fibrosis through suppressing miR-34a/Sirt1/p53 and TGF-β1/Smads signalling. Br J Pharmacol. 2019; 176:1619–1634. https://doi.org/10.1111/bph.14573 [PubMed]
- 42. Liu X, Yang X, Han L, Ye F, Liu M, Fan W, Zhang K, Kong Y, Zhang J, Shi L, Chen Y, Zhang X, Lin S. Pterostilbene alleviates polymicrobial sepsis-induced liver injury: possible role of SIRT1 signaling. Int Immunopharmacol. 2017; 49:50–59. https://doi.org/10.1016/j.intimp.2017.05.022 [PubMed]
- 43. Carloni S, Riparini G, Buonocore G, Balduini W. Rapid modulation of the silent information regulator 1 by melatonin after hypoxia-ischemia in the neonatal rat brain. J Pineal Res. 2017; 63:e12434. https://doi.org/10.1111/jpi.12434 [PubMed]
- 44. Hardeland R. Melatonin and inflammation-Story of a double-edged blade. J Pineal Res. 2018; 65:e12525. https://doi.org/10.1111/jpi.12525 [PubMed]
- 45. Sin TK, Tam BT, Yung BY, Yip SP, Chan LW, Wong CS, Ying M, Rudd JA, Siu PM. Resveratrol protects against doxorubicin-induced cardiotoxicity in aged hearts through the SIRT1-USP7 axis. J Physiol. 2015; 593:1887–99. https://doi.org/10.1113/jphysiol.2014.270101 [PubMed]
- 46. Yang Y, Wang J, Li Y, Fan C, Jiang S, Zhao L, Di S, Xin Z, Wang B, Wu G, Li X, Li Z, Gao X, et al. HO-1 Signaling Activation by Pterostilbene Treatment Attenuates Mitochondrial Oxidative Damage Induced by Cerebral Ischemia Reperfusion Injury. Mol Neurobiol. 2016; 53:2339–53. https://doi.org/10.1007/s12035-015-9194-2 [PubMed]
- 47. Ma Z, Liu D, Di S, Zhang Z, Li W, Zhang J, Xu L, Guo K, Zhu Y, Li X, Han J, Yan X. Histone deacetylase 9 downregulation decreases tumor growth and promotes apoptosis in non-small cell lung cancer after melatonin treatment. J Pineal Res. 2019; 67:e12587. https://doi.org/10.1111/jpi.12587 [PubMed]