




Introduction
Cigarette smoking is a significant and well-known risk factor for many diseases of aging such as cardiovascular diseases, atherosclerosis and cancer. Smoking compromises both the life expectancy and the quality of life [1]. Previous studies have demonstrated that the development of kidney diseases could be associated with cigarette smoking [2, 3]. Particularly, in patients suffering from hypertension, diabetes induced polycystic kidney disease and kidney transplantation, cigarette smoking further worsens the progression of chronic kidney disease (CKD) [4–6].
Cigarette smoke contain thousands of compounds and among them nicotine is the key additive component [7]. Apart from its additive role, nicotine is shown to be involved in the pathogenesis of many diseases like aging, atherosclerosis, pulmonary fibrosis, obesity, hypertension and cancer [8, 9]. Recent studies have established the role of nicotine in renal diseases. Nicotine is associated with oxidative stress, enhanced mesangial proliferation, renal fibrosis and extracellular matrix deposition [10–12]. Nicotine was shown to promote glomerular injury in rodent models of acute nephritis and diabetic nephropathy [13]. In diabetic nephropathic mouse model, nicotine promotes extracellular matrix deposition resulting in renal injury [14]. Nicotine illicit its effects via nicotinic acetylcholine receptors (nAChRs) which are Ca2+ activated channels [14]. Nicotine also mediates its action by increasing oxidative stress in the kidney [6, 15] and renal proximal tubule cells in in vitro [16]. Previous reports indicate that α7nAChR is activated by nicotine in the proximal tubule. The active α7nAChR initiates the biosynthesis of profibrotic and proinflammatory cytokines [12]. However, the exact mechanism of how cigarette smoking accelerates the progression of CKD is still uncertain.
NLRP3 inflammasome is known to act as a sensor and is shown to be involved in inflammatory as well as non-inflammatory responses [17–21]. The pathogenic role of NLRP3 inflammasome has been established in several diseases like diabetes, silicosis, obesity, gout, acetaminophen-induced liver toxicity [22–29], unilateral ureteral obstruction [30, 31], acute ischemia/reperfusion-induced kidney injury [32], non-diabetic kidney disease [31] and obesity-induced glomerular injury [33]. The NLRP3 inflammasome is also activated by other triggers like bacterial toxins [34], monosodium urate crystals [23], cholesterol crystals [35], ATP, β-amyloid [36], visfatin [37], muramyl dipeptide [38] and other stimuli [22]. Recently, nicotine has been shown to be involved in development of inflammatory atherosclerotic plaques via NLRP3 inflammasome activation [39]. Hence, in the current study we tested whether nicotine activates NLRP3 inflammasomes in podocytes and contributes to podocyte damage.
Results
Nicotine causes podocyte damage
To delineate the in vitro effects of nicotine on podocyte injury, cultured podocytes were treated with different concentrations of nicotine (2 μM, 4 μM and 8 μM) for overnight. Our results demonstrate that nicotine dose dependently decreased the expression of both podocin and nephrin which are markers of podocyte damage (Figure 1A, 1B). So, for further studies we selected 8 μM as an effective dose for remaining experiments.
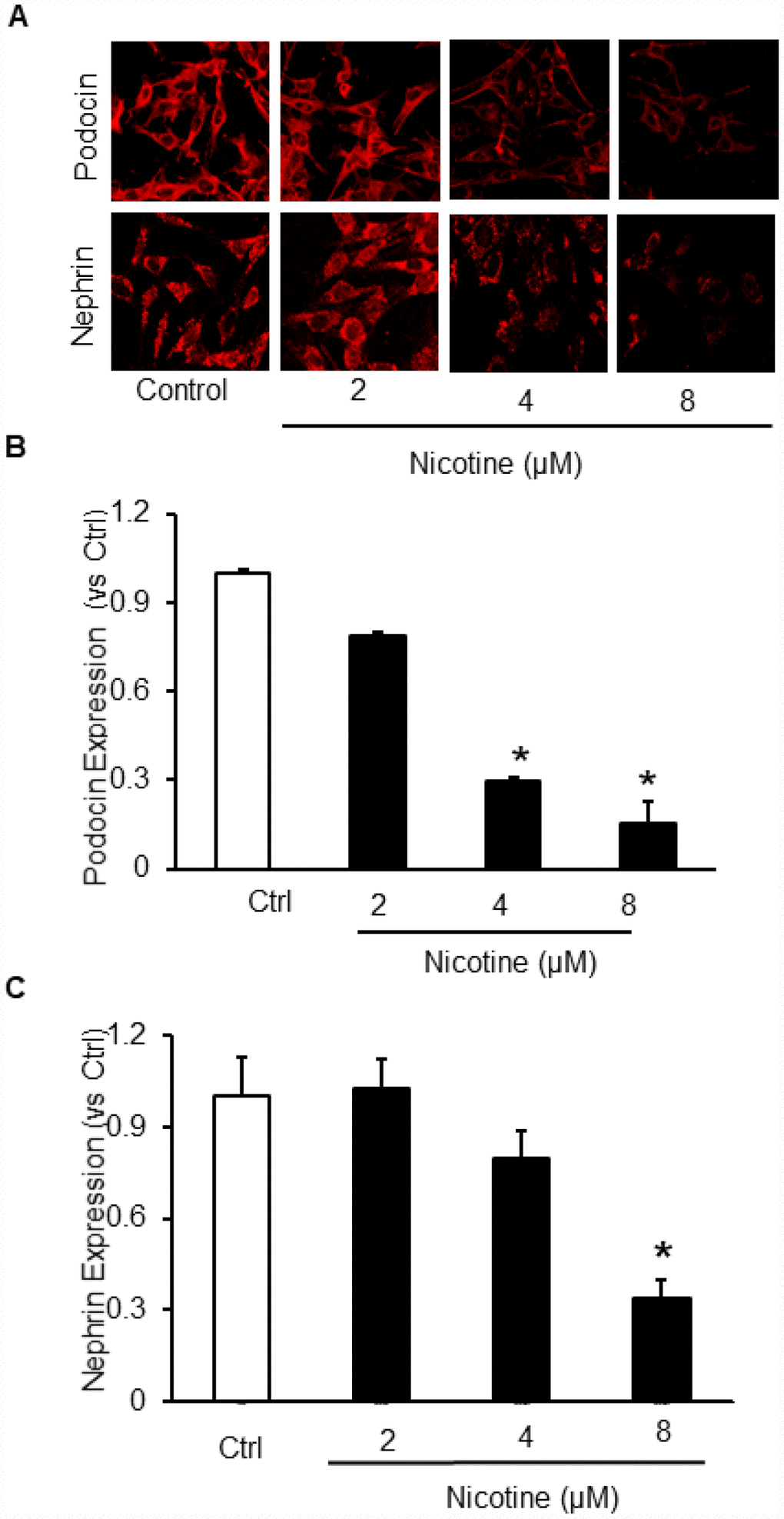
Figure 1. Effect of Nicotine on podocyte injury. Representative immunofluorescence images (A) and summarized quantification data shows Podocin (B) and Nephrin (C) expression in podocytes treated with different concentrations of Nicotine (2μM, 4μM, 8μM). Images were quantified using Image J software. N=5. * Significant difference from control.
Activation of NLRP3 inflammasome by nicotine in cultured mouse podocytes
We hypothesized that nicotine induces inflammasome activation leading to podocyte damage. As shown in Figure 2, nicotine induced co-localization of Nlrp3 (green) with ASC (red) as shown by increased yellow staining in podocytes. However, such nicotine-induced colocalization of Nlrp3 with Asc was blocked by WEHD, a caspase-1 inhibitor (Figure 2A). The summarized quantitative analysis of co-localization of Nlrp3 with Asc in podocytes is shown in Figure 2B. Furthermore, we have found that nicotine treatment enhanced the caspase-1 activity (Figure 3A) and increased production of IL-1β (Figure 3B) compared to control cells. Prior treatment of podocytes with caspase-1 inhibitor, WEHD attenuated nicotine-induced caspase-1 activity and IL-1β production (Figure 3A and 3B).
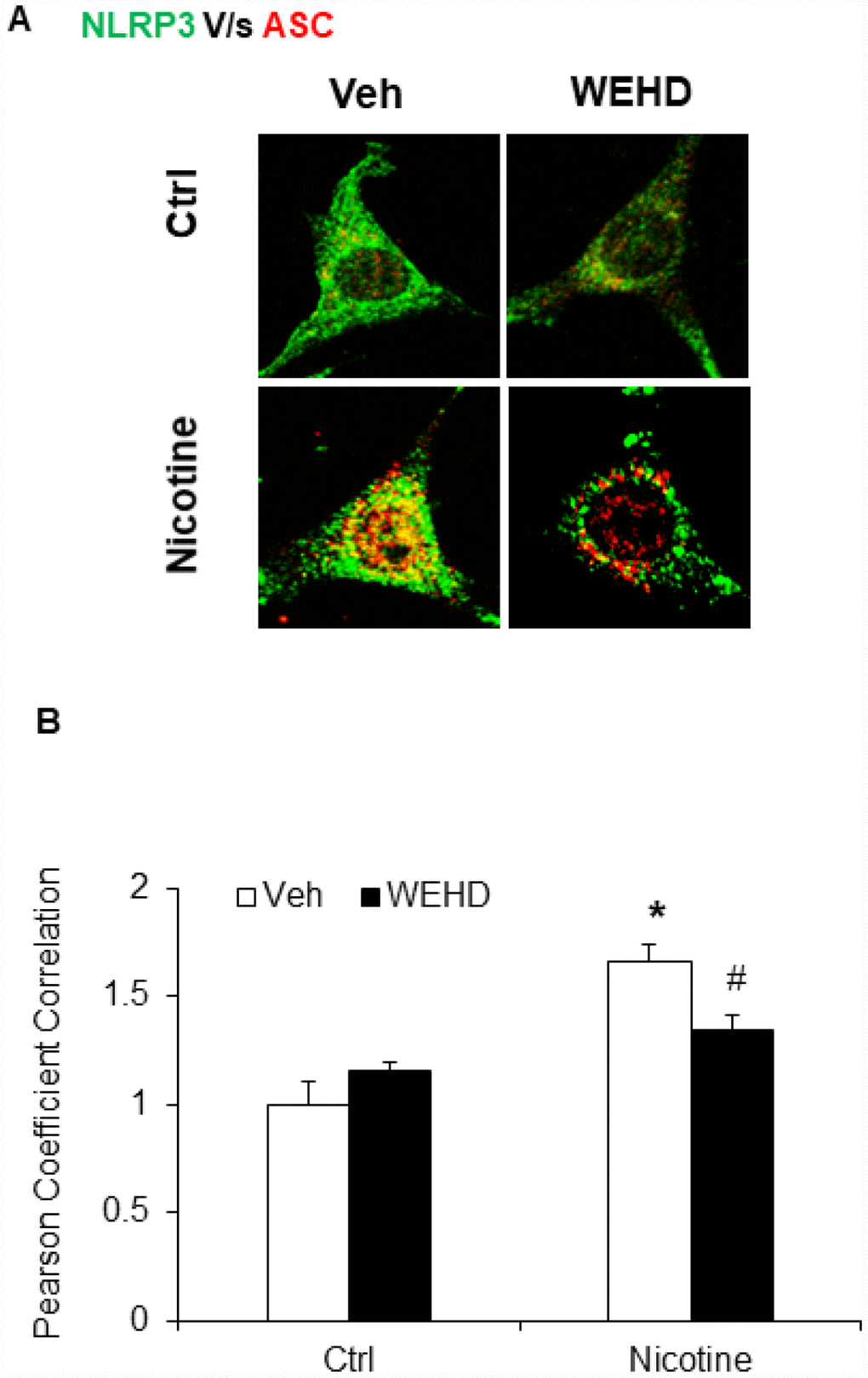
Figure 2. Nicotine induced NLRP3 inflammasome formation in podocytes. (A) Representative confocal fluorescence images show the colocalization of NLRP3 with ASC. (B) Summarized data shows the fold changes of pearson coefficient correlation (PCC) for the colo-calization of NLRP3 with ASC with or without stimulation of nicotine and/or caspase-1 inhibition by WEHD. N=5. Veh: Vehicle. *significant difference from control, # significant difference from nicotine treated group.
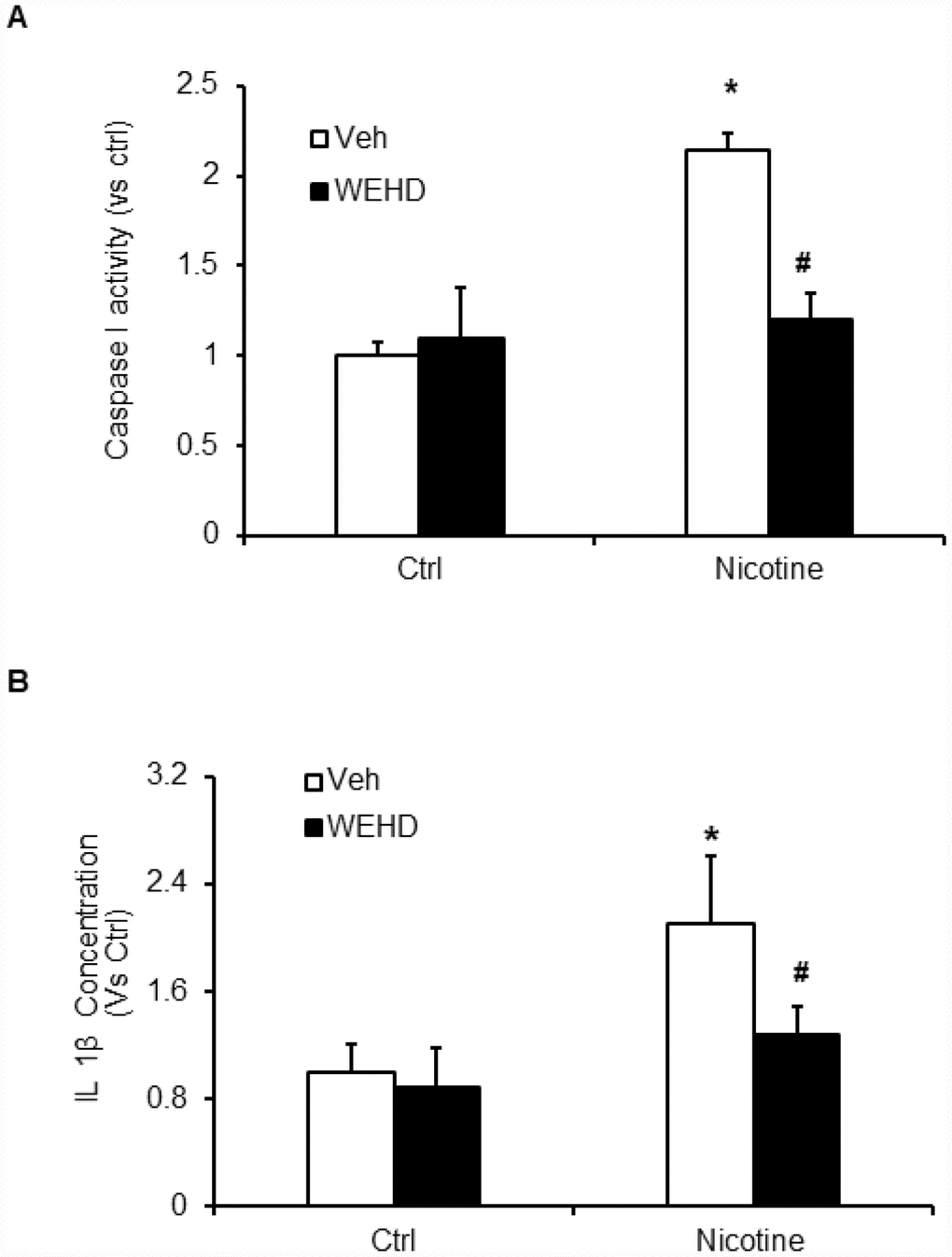
Figure 3. Inflammasome activation by nicotine in podocytes. Values are arithmetic means ± SEM (n=6 each group) of caspase-1 activity (A) and IL-β production (B) in podocytes with or without stimulation of nicotine and/or WEHD. *significant difference from control, #significant difference from nicotine treated group.
Inhibition of caspase-1 protects the podocytes from nicotine-induced damage
Podocyte-specific markers such as podocin and neprhin are down regulated during podocyte injury [33, 40]. Hence, the expression of podocin and desmin were monitored to assess the podocyte damage. Nicotine treatment resulted in an intense reduction of podocin and nephrin expression following immunofluorescence analysis demonstrating significant podocyte damage (Figure 4A–4D). Conversely, prior treatment with caspase-1 inhibitor, WEHD protected the podocytes from damage as shown by normalized protein expression of podocin and nephrin compared to control levels (Figure 4A–4D). These results confirm that nicotine is involved in podocyte dysfunction through the activation of NLRP3 inflammasomes in podocytes.
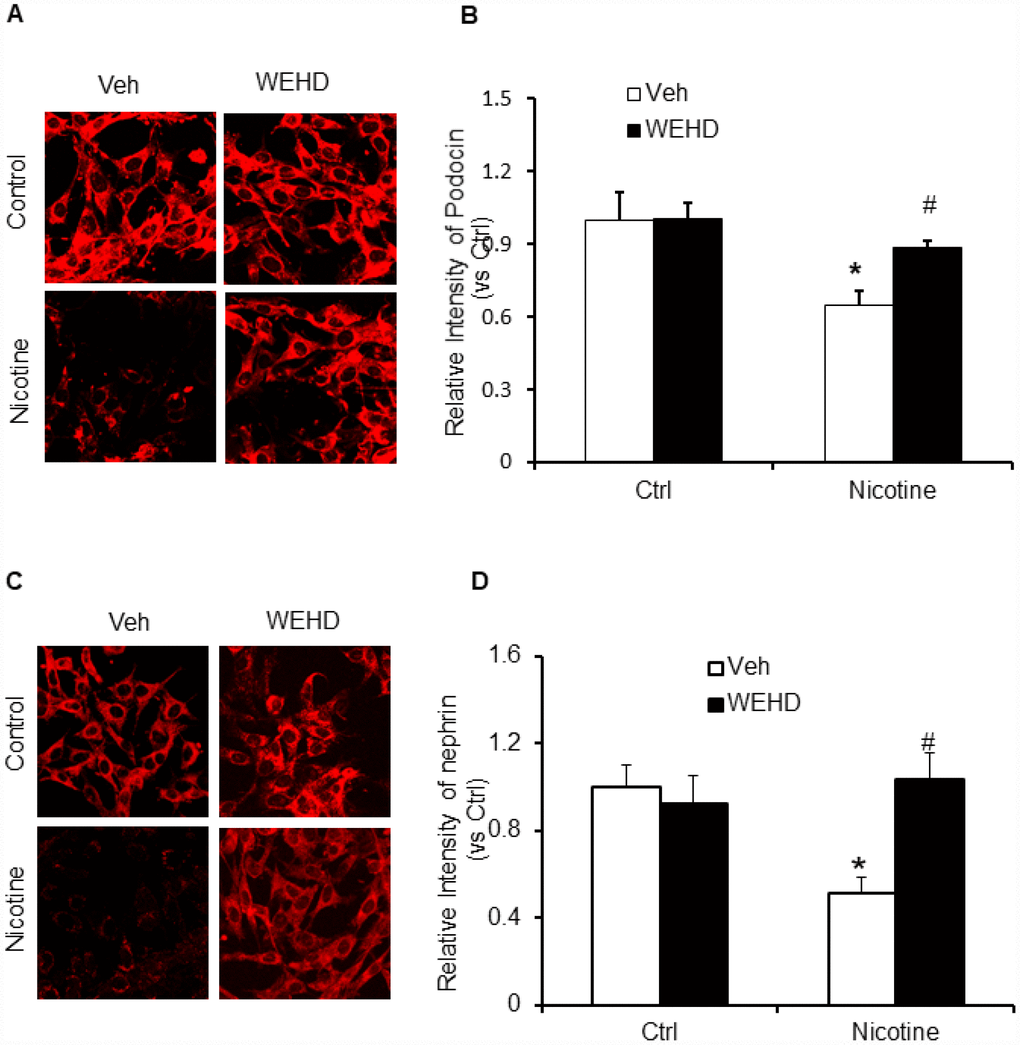
Figure 4. Inhibition of inflammasome abolishes nicotine-induced podocyte injury. Representative immunofluorescence images and summarized quantification data shows Podocin (A) and Nephrin (B) expression in podocytes treated with or without stimulation of nicotine and/or WEHD. Images were quantified using Image J software. N=5. *significant difference from control, #significant difference from nicotine treated group.
Nicotine-induced podocyte monolayer permeability and dysfunction
Next, we tested the role of nicotine in mediating podocyte dysfunction by examining its effect on the permeability of podocyte monolayers to FITC-dextran. As shown in Figure 5, nicotine significantly increased podocyte monolayer permeability as compared to untreated podocytes. Conversely, prior treatment with WEHD significantly attenuated the nicotine-induced increase in the podocyte monolayer permeability (Figure 5). This result suggests that nicotine is associated with podocyte monolayer disruption via NLRP3 inflammasomes.
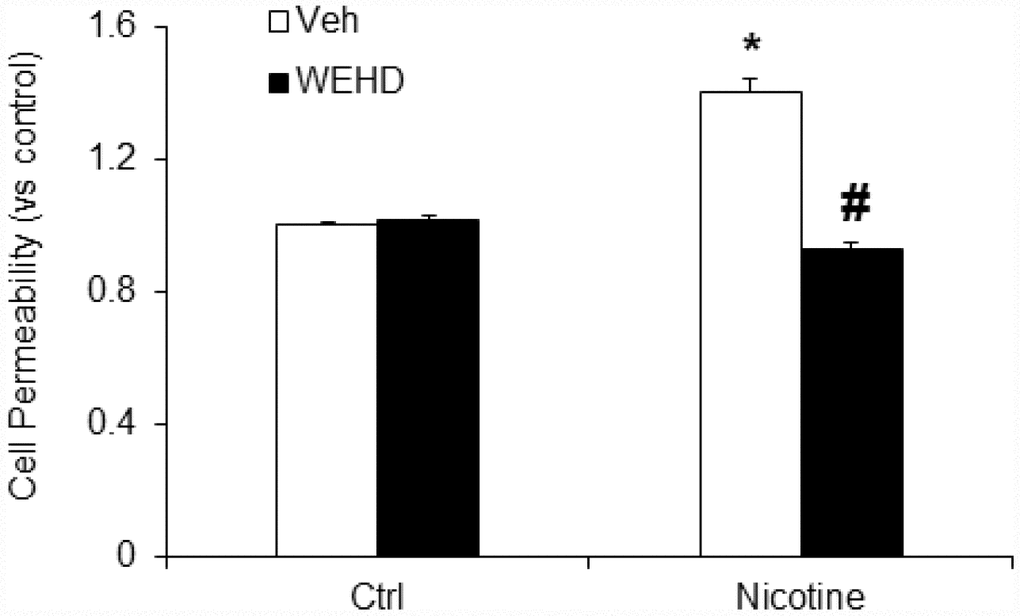
Figure 5. Inhibition of inflammasome abolishes nicotine-induced cell permeability in podocytes. Values are arithmetic means ± SEM (n=6 each group) of cell permeability in podocytes with or without stimulation of nicotine and/or WEHD. *significant difference from control, #significant difference from nicotine treated group.
Nicotine-induced podocyte injury requires ROS
The role of reactive oxygen species (ROS) in NLRP3 inflammasomes activation in different cell types is well established [20, 33]. Hence, we tested whether nicotine-induced O2.- production in podocytes. We found that nicotine treatment significantly increased the O2.- production compared to control cells. On the other hand, prior treatment with caspase-1 inhibitor, WEHD did not alter the nicotine-induced O2.- production (Figure 6). This data suggests that nicotine-induced O2.- production is upstream of NLRP3 inflammasomes activation.
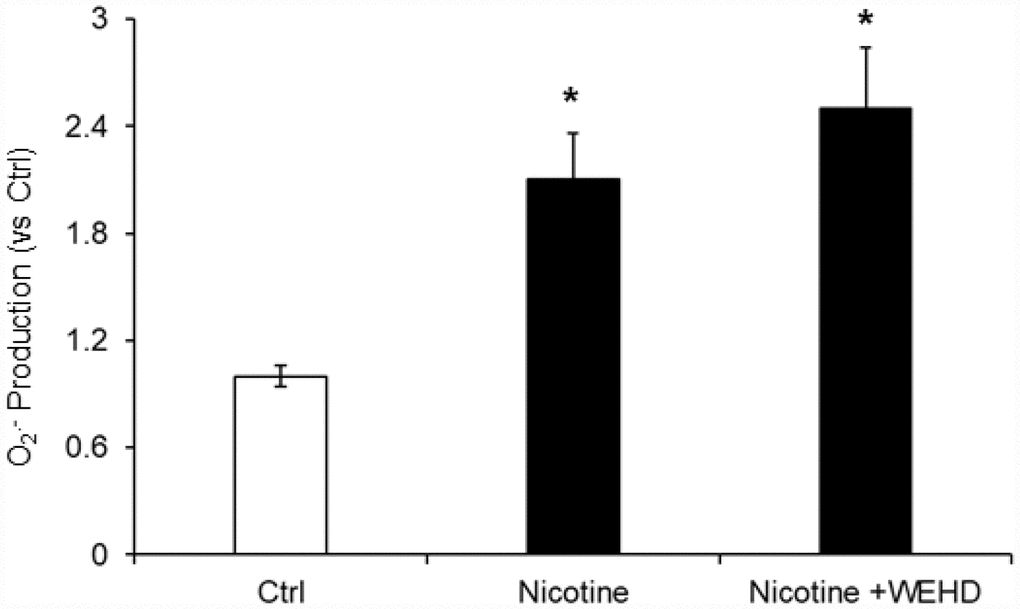
Figure 6. O2. – Production in podocytes with or without nicotine and/or WEHD treatment. Values are arithmetic means ± SE (n=4 each group) of O2. - production in podocytes with or without nicotine and/or WEHD treatment. Ctrl: Control, * Significant difference (P<0.05) compared to the control group.
Subsequently, we examined whether, N-acetyl- cysteine (NAC), a scavenger of ROS attenuates nicotine induced inflammasome activation and podocyte damage. As shown in Figure 7, the activity of caspase-1 and production of IL-1β was significantly increased in podocytes upon nicotine treatment. However, prior treatment with ROS scavenger NAC attenuated the activity of caspase-1 activity and production of IL-1β induced by nicotine (Figure 7). Furthermore, we also found that NAC rescued podocyte injury as evident from restored podocin and nephrin expression in podocytes treated with nicotine along with NAC (Figure 8).
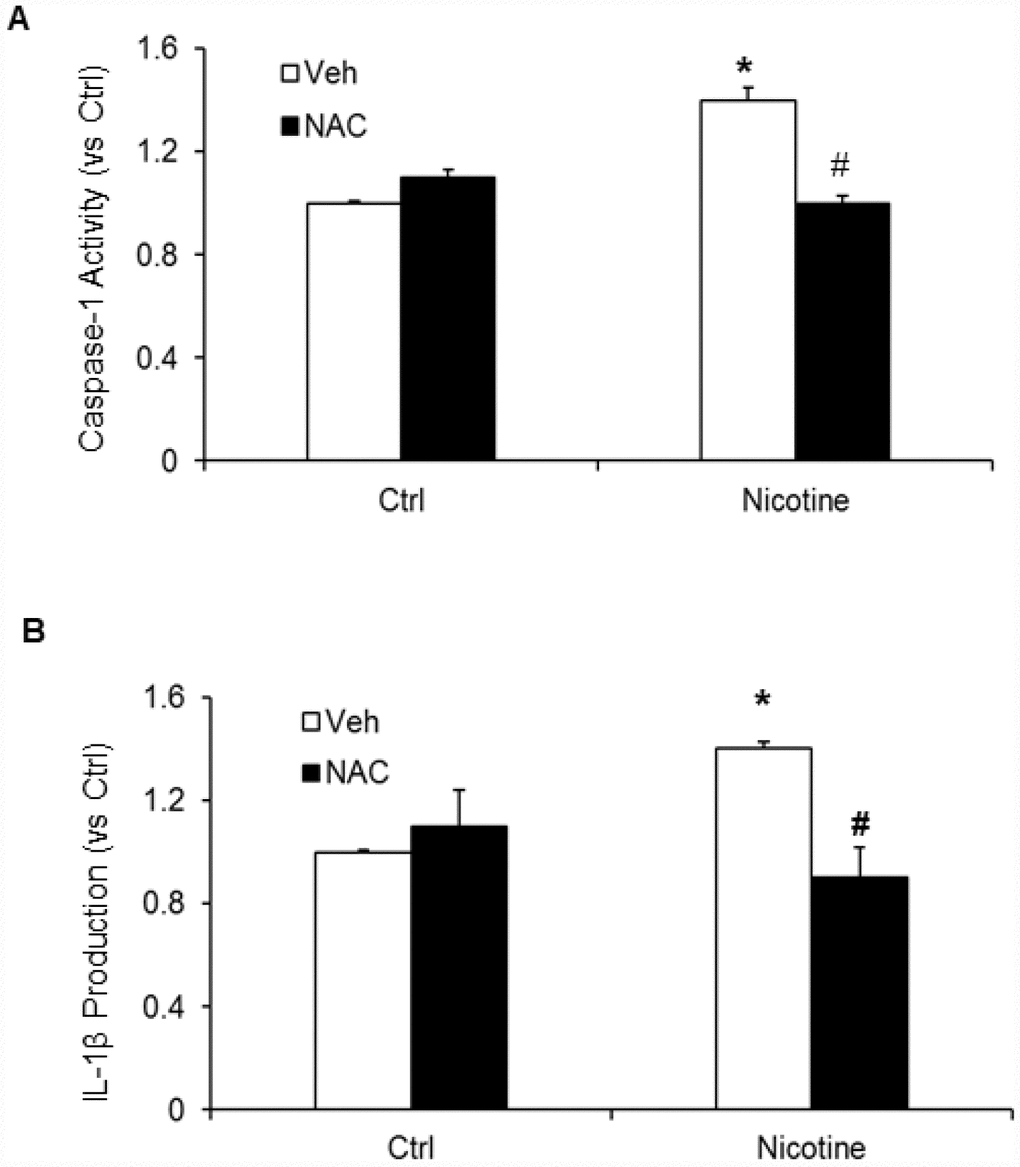
Figure 7. Effect of NAC (N-acetyl-cysteine), ROS scavenger on nicotine-induced inflammasome activation. Values are arithmetic means ± SEM (n=6 each group) of caspase-1 activity (A) and IL-β production (B) in podocytes with or without stimulation of nicotine and/or NAC. *significant difference from control, # significant difference from nicotine treated group.
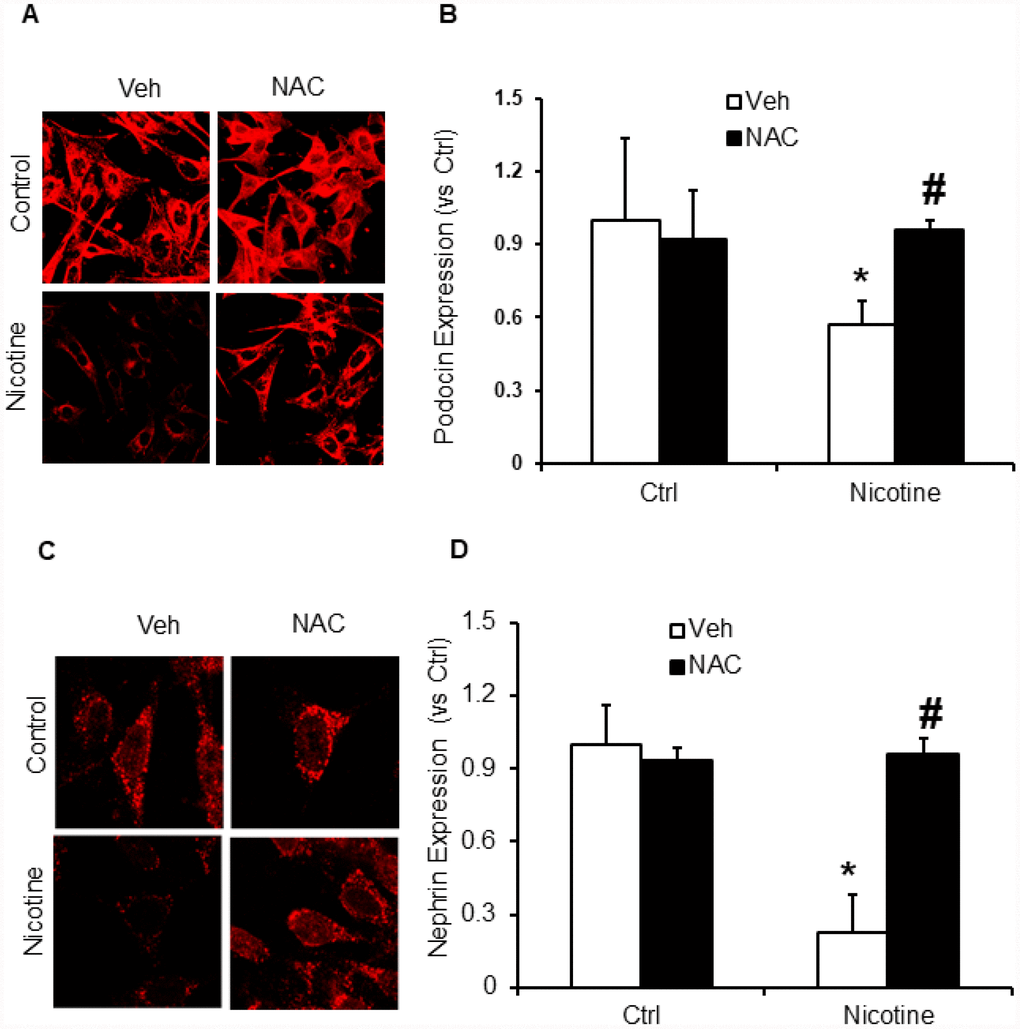
Figure 8. ROS scavenger N-acetyl-cysteine (NAC) protected podocyte from nicotine-induced podocyte injury. Representative immunofluorescence images and summarized quantification data (n=6 each group) shows podocin (A) and nephrin (B) expression in podocytes treated with or without stimulation of nicotine and/or NAC. Images were quantified using Image J software. *significant difference from control, #significant difference from nicotine treated group.
Discussion
The major goal of our current study was to identify whether activation of inflammasomes contributes to nicotine induced podocyte damage and consequent glomerular sclerosis. Our results demonstrate that nicotine induced aggregation of NLRP3 inflammasome components, caspase-1 activation and IL-1β production leading to injury of the cultured podocytes. However, such activation of inflammasome and podocyte injury by nicotine was attenuated in WEHD treated cells. In addition, we also confirmed the role of ROS in nicotine associated podocyte damage via activation of NLRP3 inflammasomes. Our results demonstrate the critical role of nicotine in turning on inflammatory response via activating NLRP3 inflammasomes leading to renal inflammation and renal dysfunction during smoking and may help in understanding the pathophysiology of nicotine associated chronic kidney diseases.
Cigarette smoking is known to accelerate the rate of development of chronic kidney diseases, polycystic kidney disease, diabetes mellitus [2, 41], hypertension [4, 5], IgA nephropathy, lupus nephritis [42], and post kidney transplantation [3, 6]. Likewise, investigational studies revealed that the exposure to environmental tobacco smoke worsens renal injury in diabetic nephropathy and aging mouse models [12]. In another study, it was shown that nicotine causes the podocyte damage through JNK, ERK1/2, and p38 pathways that are regulated ROS generation [43]. However, despite the existing experimental and clinical data, the exact mechanisms responsible for the deleterious effects of nicotine have not been identified. Nevertheless, NLRP3 inflammasome activation has been recently associated with various pathological conditions such as gout, diabetes, chronic kidney diseases including podocyte damage and glomerular injury [17, 23–25, 27–30, 32, 33] and to a number of other diseases like acute lung injury, silicosis, Alzheimer’s disease, liver toxicity, and cystic fibrosis [22–25, 27, 28]. Yet, it is not known whether nicotine activates NLRP3 inflammasome and contributes to podocyte damage. In the present study, we have shown that colocalization of NLRP3 with ASC, caspase-1 activity and IL-1β production was increased by nicotine stimulation suggesting the formation and activation of NLRP3 inflammasomes. This nicotine-induced inflammasome formation and activation was abolished in podocytes with prior treatment with caspase-1 inhibitor, WEHD (Figure 2 and 3). Thus, our results indicate that nicotine enhanced activation of Nlpr3 inflammasome in podocytes, which may contribute to the development of podocyte damage. These results are consistent with earlier reports that nicotine treatment increased the expression of NLRP3, ASC, caspase-1 activation and IL-1β production in human aortic endothelial cells (HAECs) and silencing NLRP3 and ASC in endothelial cells by RNA interference (siRNA) significantly attenuated the nicotine-induced activation of NLRP3, ASC, caspase-1, IL-1β and IL-18 production [39]. To our knowledge, the current study provides the first experimental evidence indicating that nicotine induced NLRP3 inflammasome formation and activation in podocytes. Our results clearly indicate that nicotine induced activation of NLRP3 inflammasomes in podocytes contributes to the development of kidney diseases.
During physiological and pathological processes, podocytes play a critical role in the prevention of glomerular protein leakage through the formation of slit diaphragm [43]. Smoking worsens the kidney diseases and clinical studies have demonstrated that smoking enhances proteinuria [11, 43]. Hence, it could be possible that the tobacco smoke contents directly affect the podocytes in the kidney [43]. In this context, we demonstrated that nicotine reduced the nephrin expression in podocytes, signifying that it triggered podocyte injury [43]. However, it remains unknown whether inhibition of inflammasome by WEHD protects against the nicotine-induced podocyte injury. In the present study, we found that nicotine treatment significantly reduced podocin and nephrin expression in podocytes, which was abolished by prior treatment with WEHD, a caspase-1 inhibitor suggesting the critical role of NLRP3 inflammasomes in nicotine associated podocyte injury (Figure 4). Functional significance of nicotine associated NLRP3 activation was further explored by studying nicotine-induced enhancement of podocyte monolayer permeability. Increased vascular permeability resulting in increased glomerular permeability is known to contribute to the development and advancement of glomerular injury [44, 45]. The present study demonstrates that nicotine increased the permeability of podocyte monolayer and this nicotine-induced increase in podocyte permeability was significantly attenuated by treatment with a caspase-1 inhibitor, WEHD, suggesting the role of NLRP3 inflammasomes in nicotine associated increase in podocyte permeability (Figure 5).
Next, we confirmed the mechanisms underlying nicotine induced NLRP3 inflammasome activation in podocytes. Inflammasome activation is known to sense changes in cellular oxidative stress and activation of the NLRP3 inflammasome by increased ROS is the most widely accepted and reasonably known mechanism [20, 46, 47]. Consistent with these reports, our study demonstrated that treatment with ROS scavenger, N-acetyl-cysteine (NAC) attenuated nicotine-induced caspase-1 activity as well as IL-1β production in podocytes. NAC treatment protected podocytes from ROS induced injury as demonstrated by the preserved expression of podocin and nephrin in nicotine treated cells, suggesting the role of ROS in nicotine induced NLRP3 inflammasome activation (Figures 7 and 8). However, further studies are required to reveal which nicotinic receptors are mediating the inflammasome activation in podocytes. In this regard, there were reports that nicotine mediates its effects through the activation of different nicotinic acetylcholine receptors (nAChRs) in different cell types including α4, α5, α7, β2, β3 and β4 which are expressed in human mesangial cells [14, 43]. α3, α5, and β1 are expressed in renal proximal tubular epithelial cells (HK-2), α5, α6, α7 and β3 are expressed in podocytes [43, 48]. Recently Lu et al reported that α7 nAChR activation contributed to the NLRP3 inflammasome inhibition in peritoneal mouse macrophages and dendritic cells [49]. On-going studies in our laboratory will further clarify the role of these nicotine subunits on nicotine-induced activation of inflammasome activation and podocyte damage. In conclusion, the current study confirmed for the first time that nicotine-induced NLRP3 inflammasome activation in podocytes and thereby results in podocyte injury. Hence, targeting Nlrp3 inflammasome might be a promising therapeutic approach to inhibit inflammasome activation and protect podocytes against nicotine-induced injury.
Materials and Methods
Cell culture
A conditionally immortalized mouse podocyte cell line was cultured undifferentiated with 10 U/ml recombinant mouse interferon-γ at 33°C on collagen I-coated flasks in serum containing RPMI 1640 medium containing 10% fetal bovine serum, 100 U/ml penicillin and 100 mg/ml streptomycin. After differentiation at 37°C for 10–14 days without interferon–γ, podocytes were used for the following experiments. Podocytes were pretreated with WEHD or N-acetyl cysteine (NAC) (10μM) for 30 min prior to nicotine treatment (8μM) for overnight.
Immunofluorescence staining of cells
Cells were fixed in 4% PFA, and then blocked with 1% BSA for 30 min. After incubated with primary antibody (podocin, nephrin, NLRP3 and ASC) 1:200 dilution in 0.1% BSA overnight, the cells were stained with secondary antibodies. After washing, the cells were mounted with DAPI-containing mounting solution. Then the slides were observed under a fluorescent microscope and images were taken (Nikon, Japan). For, confocal analysis, the slides were visualized through sequentially scanning on Leica laser scanning confocal microscope (Leica SP-8, Germany). Colocalization was analyzed by Image J, and the co-localization coefficient was represented by Pearson's correlation coefficient [40].
Caspase-1 activity and IL-1β production assay
After nicotine treatment, cells were harvested and homogenized to extract proteins for caspase-1 activity assay by using a commercially available kit (Biovision). These data were expressed as the fold change compared with control cells. In addition, the cell supernatant was also collected to measure the IL-1β production by a mouse IL-1β ELISA kit (Bender Medsystems, Burlingame, CA) according to the protocol described by the manufacturer [21, 50].
Electronic spin resonance (ESR) analysis of O2.- production
For detection of O2.- production, proteins from podocytes were extracted using sucrose buffer and resuspended with modified Kreb’s–Hepes buffer containing deferoximine (100 mM, Sigma) and diethyldithiocarbamate (5 mM, Sigma). The podocytes (20 μg protein) were incubated for 10 min at 37 °C in the presence or absence of SOD (200 U/ml), and then supplied with 1mM O2 .- specific spin trap 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH, Noxygen, Elzach, Germany). The mixture was loaded in glass capillaries and immediately analyzed for O2 .- production kinetically for 7 min in a Miniscope MS5000 electromagnetic spin resonance [33, 37, 45] spectrometer (Rotunda Scientific Technologies, Stow, Ohio). The results were expressed as the fold changes of control.
Cell permeability assay
The permeability of podocyte layer in culture was measured according to the methods as described previously [19]. In brief, podocyte were seeded in the upper chambers of 0.4 μm polycarbonate transwell filters of a 12-well filtration microplate (Whatman Inc.). After reaching confluence, the culture medium was replaced with fresh serum free RPMI 1640 media in presence of nicotine (8μM) with or without WEHD and incubated overnight. Following day fresh phenol red-free RPMI 1640 with 70 kDa FITC-dextran (2.5 μmol/l) in the upper chambers was added and incubated for 2.5 hrs. The filtration microplate was removed and the medium from the lower compartment was collected, and then fluorescence was measured in a spectrofluorimeter at 494 nm excitation and 521 nm emissions. The relative permeable fluorescence intensity was used to represent the cell permeability [45].
Statistical analysis
Data are provided as arithmetic means ± SEM; n represents the number of independent experiments. All data were tested for significance using ANOVA or paired and unpaired Student’s t-test as applicable. Only results with p<0.05 were considered statistically significant.
Acknowledgments
This work was supported by grants from National Institutes of Health (R01DK104031 to K.B and R56HL143809 to S.K) and American Heart Association (19AIREA34380223 to S.K).
Conflicts of Interest
The authors of this manuscript declare that they have no conflicts of interest.
References
- 1. Boyle P. Cancer, cigarette smoking and premature death in Europe: a review including the Recommendations of European Cancer Experts Consensus Meeting, Helsinki, October 1996. Lung Cancer. 1997; 17:1–60. https://doi.org/10.1016/S0169-5002(97)00648-X [PubMed]
- 2. Orth SR. Smoking—a renal risk factor. Nephron. 2000; 86:12–26. https://doi.org/10.1159/000045708 [PubMed]
- 3. Orth SR, Hallan SI. Smoking: a risk factor for progression of chronic kidney disease and for cardiovascular morbidity and mortality in renal patients—absence of evidence or evidence of absence? Clin J Am Soc Nephrol. 2008; 3:226–36. https://doi.org/10.2215/CJN.03740907 [PubMed]
- 4. Bleyer AJ, Shemanski LR, Burke GL, Hansen KJ, Appel RG. Tobacco, hypertension, and vascular disease: risk factors for renal functional decline in an older population. Kidney Int. 2000; 57:2072–79. https://doi.org/10.1046/j.1523-1755.2000.00056.x [PubMed]
- 5. Hörner D, Fliser D, Klimm HP, Ritz E. Albuminuria in normotensive and hypertensive individuals attending offices of general practitioners. J Hypertens. 1996; 14:655–60. https://doi.org/10.1097/00004872-199605000-00016 [PubMed]
- 6. Orth SR, Viedt C, Ritz E. Adverse effects of smoking in the renal patient. Tohoku J Exp Med. 2001; 194:1–15. https://doi.org/10.1620/tjem.194.1 [PubMed]
- 7. Stedman RL. The chemical composition of tobacco and tobacco smoke. Chem Rev. 1968; 68:153–207. https://doi.org/10.1021/cr60252a002 [PubMed]
- 8. Heeschen C, Jang JJ, Weis M, Pathak A, Kaji S, Hu RS, Tsao PS, Johnson FL, Cooke JP. Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat Med. 2001; 7:833–39. https://doi.org/10.1038/89961 [PubMed]
- 9. Roman J, Ritzenthaler JD, Gil-Acosta A, Rivera HN, Roser-Page S. Nicotine and fibronectin expression in lung fibroblasts: implications for tobacco-related lung tissue remodeling. FASEB J. 2004; 18:1436–38. https://doi.org/10.1096/fj.03-0826fje [PubMed]
- 10. Haass M, Kübler W. Nicotine and sympathetic neurotransmission. Cardiovasc Drugs Ther. 1997; 10:657–65. https://doi.org/10.1007/BF00053022 [PubMed]
- 11. Jain G, Jaimes EA. Nicotine signaling and progression of chronic kidney disease in smokers. Biochem Pharmacol. 2013; 86:1215–23. https://doi.org/10.1016/j.bcp.2013.07.014 [PubMed]
- 12. Rezonzew G, Chumley P, Feng W, Hua P, Siegal GP, Jaimes EA. Nicotine exposure and the progression of chronic kidney disease: role of the α7-nicotinic acetylcholine receptor. Am J Physiol Renal Physiol. 2012; 303:F304–12. https://doi.org/10.1152/ajprenal.00661.2011 [PubMed]
- 13. Jaimes EA, Tian RX, Joshi MS, Raij L. Nicotine augments glomerular injury in a rat model of acute nephritis. Am J Nephrol. 2009; 29:319–26. https://doi.org/10.1159/000163593 [PubMed]
- 14. Jaimes EA, Tian RX, Raij L. Nicotine: the link between cigarette smoking and the progression of renal injury? Am J Physiol Heart Circ Physiol. 2007; 292:H76–82. https://doi.org/10.1152/ajpheart.00693.2006 [PubMed]
- 15. Arany I, Grifoni S, Clark JS, Csongradi E, Maric C, Juncos LA. Chronic nicotine exposure exacerbates acute renal ischemic injury. Am J Physiol Renal Physiol. 2011; 301:F125–33. https://doi.org/10.1152/ajprenal.00041.2011 [PubMed]
- 16. Arany I, Clark J, Reed DK, Juncos LA. Chronic nicotine exposure augments renal oxidative stress and injury through transcriptional activation of p66shc. Nephrol Dial Transplant. 2013; 28:1417–25. https://doi.org/10.1093/ndt/gfs596 [PubMed]
- 17. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nuñez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010; 464:1357–61. https://doi.org/10.1038/nature08938 [PubMed]
- 18. Rajamäki K, Lappalainen J, Oörni K, Välimäki E, Matikainen S, Kovanen PT, Eklund KK. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010; 5:e11765. https://doi.org/10.1371/journal.pone.0011765 [PubMed]
- 19. Boini KM, Hussain T, Li PL, Koka S. Trimethylamine-N-Oxide Instigates NLRP3 Inflammasome Activation and Endothelial Dysfunction. Cell Physiol Biochem. 2017; 44:152–62. https://doi.org/10.1159/000484623 [PubMed]
- 20. Schroder K, Tschopp J. The inflammasomes. Cell. 2010; 140:821–32. https://doi.org/10.1016/j.cell.2010.01.040 [PubMed]
- 21. Zhang C, Boini KM, Xia M, Abais JM, Li X, Liu Q, Li PL. Activation of Nod-like receptor protein 3 inflammasomes turns on podocyte injury and glomerular sclerosis in hyperhomocysteinemia. Hypertension. 2012; 60:154–62. https://doi.org/10.1161/HYPERTENSIONAHA.111.189688 [PubMed]
- 22. Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, Flavell RA, Mehal WZ. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest. 2009; 119:305–14. https://doi.org/10.1172/JCI35958 [PubMed]
- 23. Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006; 440:237–41. https://doi.org/10.1038/nature04516 [PubMed]
- 24. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010; 11:136–40. https://doi.org/10.1038/ni.1831 [PubMed]
- 25. De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011; 32:373–79. https://doi.org/10.1016/j.it.2011.05.004 [PubMed]
- 26. Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008; 320:674–77. https://doi.org/10.1126/science.1156995 [PubMed]
- 27. Horng T, Hotamisligil GS. Linking the inflammasome to obesity-related disease. Nat Med. 2011; 17:164–65. https://doi.org/10.1038/nm0211-164 [PubMed]
- 28. Stienstra R, Joosten LA, Koenen T, van Tits B, van Diepen JA, van den Berg SA, Rensen PC, Voshol PJ, Fantuzzi G, Hijmans A, Kersten S, Müller M, van den Berg WB, et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010; 12:593–605. https://doi.org/10.1016/j.cmet.2010.11.011 [PubMed]
- 29. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011; 17:179–88. https://doi.org/10.1038/nm.2279 [PubMed]
- 30. Anders HJ, Muruve DA. The inflammasomes in kidney disease. J Am Soc Nephrol. 2011; 22:1007–18. https://doi.org/10.1681/ASN.2010080798 [PubMed]
- 31. Vilaysane A, Chun J, Seamone ME, Wang W, Chin R, Hirota S, Li Y, Clark SA, Tschopp J, Trpkov K, Hemmelgarn BR, Beck PL, Muruve DA. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J Am Soc Nephrol. 2010; 21:1732–44. https://doi.org/10.1681/ASN.2010020143 [PubMed]
- 32. Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, Eisenbarth SC, Florquin S, Flavell RA, Leemans JC, Sutterwala FS. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci USA. 2009; 106:20388–93. https://doi.org/10.1073/pnas.0908698106 [PubMed]
- 33. Boini KM, Xia M, Koka S, Gehr TW, Li PL. Instigation of NLRP3 inflammasome activation and glomerular injury in mice on the high fat diet: role of acid sphingomyelinase gene. Oncotarget. 2016; 7:19031–44. https://doi.org/10.18632/oncotarget.8023 [PubMed]
- 34. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004; 430:213–18. https://doi.org/10.1038/nature02664 [PubMed]
- 35. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, Becker CE, Ediriweera HN, Mullick AE, Golenbock DT, Stuart LM, Latz E, Fitzgerald KA, Moore KJ. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. 2013; 14:812–20. https://doi.org/10.1038/ni.2639 [PubMed]
- 36. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008; 9:857–65. https://doi.org/10.1038/ni.1636 [PubMed]
- 37. Koka S, Xia M, Zhang C, Zhang Y, Li PL, Boini KM. Podocyte NLRP3 Inflammasome Activation and Formation by Adipokine Visfatin. Cell Physiol Biochem. 2019; 53:355–65. https://doi.org/10.33594/000000143 [PubMed]
- 38. Martinon F, Agostini L, Meylan E, Tschopp J. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol. 2004; 14:1929–34. https://doi.org/10.1016/j.cub.2004.10.027 [PubMed]
- 39. Wu X, Zhang H, Qi W, Zhang Y, Li J, Li Z, Lin Y, Bai X, Liu X, Chen X, Yang H, Xu C, Zhang Y, Yang B. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018; 9:171. https://doi.org/10.1038/s41419-017-0257-3 [PubMed]
- 40. Boini KM, Xia M, Xiong J, Li C, Payne LP, Li PL. Implication of CD38 gene in podocyte epithelial-to-mesenchymal transition and glomerular sclerosis. J Cell Mol Med. 2012; 16:1674–85. https://doi.org/10.1111/j.1582-4934.2011.01462.x [PubMed]
- 41. Rossing P, Hougaard P, Parving HH. Risk factors for development of incipient and overt diabetic nephropathy in type 1 diabetic patients: a 10-year prospective observational study. Diabetes Care. 2002; 25:859–64. https://doi.org/10.2337/diacare.25.5.859 [PubMed]
- 42. Ward MM, Studenski S. Clinical prognostic factors in lupus nephritis. The importance of hypertension and smoking. Arch Intern Med. 1992; 152:2082–88. https://doi.org/10.1001/archinte.1992.00400220098017 [PubMed]
- 43. Lan X, Lederman R, Eng JM, Shoshtari SS, Saleem MA, Malhotra A, Singhal PC. Nicotine Induces Podocyte Apoptosis through Increasing Oxidative Stress. PLoS One. 2016; 11:e0167071. https://doi.org/10.1371/journal.pone.0167071 [PubMed]
- 44. Bernhard D, Csordas A, Henderson B, Rossmann A, Kind M, Wick G. Cigarette smoke metal-catalyzed protein oxidation leads to vascular endothelial cell contraction by depolymerization of microtubules. FASEB J. 2005; 19:1096–107. https://doi.org/10.1096/fj.04-3192com [PubMed]
- 45. Boini KM, Zhang C, Xia M, Han WQ, Brimson C, Poklis JL, Li PL. Visfatin-induced lipid raft redox signaling platforms and dysfunction in glomerular endothelial cells. Biochim Biophys Acta. 2010; 1801:1294–304. https://doi.org/10.1016/j.bbalip.2010.09.001 [PubMed]
- 46. Latz E. The inflammasomes: mechanisms of activation and function. Curr Opin Immunol. 2010; 22:28–33. https://doi.org/10.1016/j.coi.2009.12.004 [PubMed]
- 47. Koka S, Xia M, Chen Y, Bhat OM, Yuan X, Boini KM, Li PL. Endothelial NLRP3 inflammasome activation and arterial neointima formation associated with acid sphingomyelinase during hypercholesterolemia. Redox Biol. 2017; 13:336–44. https://doi.org/10.1016/j.redox.2017.06.004 [PubMed]
- 48. Kim CS, Choi JS, Joo SY, Bae EH, Ma SK, Lee J, Kim SW. Nicotine-Induced Apoptosis in Human Renal Proximal Tubular Epithelial Cells. PLoS One. 2016; 11:e0152591. https://doi.org/10.1371/journal.pone.0152591 [PubMed]
- 49. Lu B, Kwan K, Levine YA, Olofsson PS, Yang H, Li J, Joshi S, Wang H, Andersson U, Chavan SS, Tracey KJ. α7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol Med. 2014; 20:350–58. https://doi.org/10.2119/molmed.2013.00117 [PubMed]
- 50. Chen Y, Pitzer AL, Li X, Li PL, Wang L, Zhang Y. Instigation of endothelial Nlrp3 inflammasome by adipokine visfatin promotes inter-endothelial junction disruption: role of HMGB1. J Cell Mol Med. 2015; 19:2715–27. https://doi.org/10.1111/jcmm.12657 [PubMed]