



Introduction
Myocardial ischemia/reperfusion (I/R) injury occurs at the stage of reperfusion due to reactive oxygen species (ROS) overproduction, calcium overload, the inflammatory response and microvascular damage [1, 2]. Mitochondria contribute to myocardial I/R injury by inducing various pathological processes [3–8]. First, most ROS are generated and released by mitochondria when electron transport chain activity is reduced. Second, mitochondria serve as calcium pumps in cardiomyocytes, so they can contribute to intracellular calcium overload when the mitochondrial calcium uniporter is dysregulated. Third, mitochondria-induced oxidative stress and cardiomyocyte death initiate an inflammatory response to repair the damaged myocardium. Fourth, although the content of mitochondria in endothelial cells is relatively low, mitochondrial morphologic disorder has been observed in cardiac microvascular injury following I/R injury. Therefore, several studies have identified mitochondria as the primary targets of strategies to prevent cardiac I/R damage.
Mitochondria are renewable organelles. Damaged mitochondrial fragments can be metabolized by mitophagy and then regenerated through mitochondrial biogenesis [9–11]. Mitophagy determines the degradation rate of old mitochondria, whereas mitochondrial biogenesis sustains mitochondrial population turnover [12–14]. Defective mitophagy has been observed in cardiac I/R injury, and is associated with mitochondrial dysfunction in cardiomyocytes and cardiac microvascular endothelial cells [15, 16]. Similarly, mitochondrial biogenesis was found to be inhibited in a mouse model of cardiac I/R injury; thus, improving mitochondrial biogenesis is considered to be a promising method of alleviating cardiac I/R injury [17, 18]. However, there is not yet an effective drug to promote mitochondrial biogenesis in the heart.
Melatonin, a biological rhythm-related hormone, significantly protects the heart against cardiac I/R injury [19, 20]. The cardioprotective mechanisms of melatonin include the protection of mitochondria, the suppression of inflammation and the inhibition of oxidative stress [21–25]. Mitochondria seem to be the first targets of melatonin treatment [26–28]. Melatonin can normalize mitochondrial oxidative stress, thus stabilizing the mitochondrial membrane potential and promoting adenosine triphosphate (ATP) synthesis. In addition, melatonin regulates mitochondrial morphological alterations such as fission and fusion in the heart during I/R injury. Melatonin also improves mitophagy by inducing optic atrophy 1 (OPA1), FUN14 domain containing 1 and Parkin [7, 21, 29, 30]. Thus, melatonin promotes mitochondrial homeostasis.
Although melatonin has been reported to accelerate mitochondrial biogenesis in early porcine embryos [31], differentiating rat dental papilla cells [32] and Alzheimer’s disease patients [33], its involvement in mitochondrial biogenesis in the setting of cardiac I/R injury has not been investigated. In the present study, we performed cellular experiments to explore the influence of melatonin on mitochondrial biogenesis in cardiac I/R injury.
Results
Mitochondrial biogenesis impaired by hypoxia/reoxygenation (H/R) injury could be improved by melatonin
To assess the alterations in mitochondrial biogenesis following H/R injury, we subjected cardiomyocytes to six-hour hypoxia and six-hour reoxygenation. Then, we performed quantitative real-time PCR (qRT-PCR) and immunoblotting to analyze parameters associated with mitochondrial biogenesis. As shown in Figure 1A–1C, the mRNA levels of peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC1α), transcription factor A mitochondrial (Tfam) and nuclear factor erythroid 2-related factor 2 (Nrf2) were significantly lower in the H/R injury group than in the control group. Melatonin treatment dose-dependently increased PGC1α, Tfam and Nrf2 levels in H/R-injured cells (Figure 1A–1C). We also evaluated the mRNA levels of Sirt3, a novel biomarker of mitochondrial biogenesis. Sirt3 levels in cardiomyocytes were notably reduced after H/R injury, but were restored following melatonin treatment (Figure 1D). Due to the reduced transcription of PGC1α, Tfam and Nrf2, the protein levels of translocase of inner mitochondrial membrane 23 (Tim23) and translocase of outer mitochondrial membrane 20 (Tom20) were also reduced in cardiomyocytes subjected to H/R injury, suggesting that the mitochondrial mass had decreased (Figure 1E and 1F). Melatonin treatment restored the expression of Tim23 and Tom20 (Figure 1E and 1F). These data indicate that melatonin can normalize mitochondrial biogenesis during H/R injury.
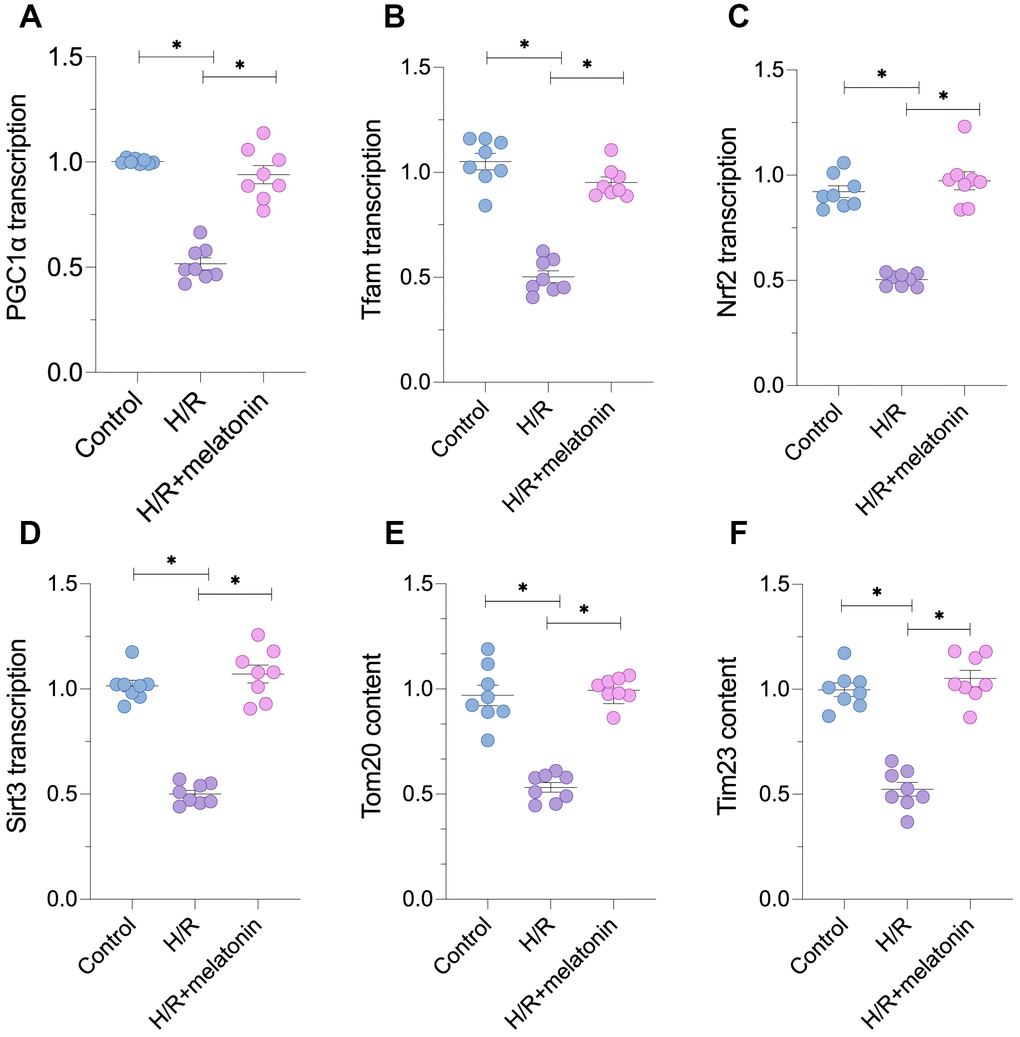
Figure 1. Melatonin treatment restores mitochondrial biogenesis in H/R-treated cardiomyocytes. Cardiomyocytes were subjected to H/R injury, with or without previous melatonin treatment to protect the cardiomyocytes. (A–D) qPCR assays were used to evaluate the transcription of PGC1α, Tfam, Nrf2 and Sirt3. (E, F) Western blots were used to evaluate the alterations in the mitochondria-related proteins Tom20 and Tim23. *p<0.05.
Melatonin promotes mitochondrial biogenesis by inducing the 5’ adenosine monophosphate-activated protein kinase (AMPK) pathway
To determine the molecular basis for the above findings, we analyzed the expression of upstream regulators of PGC1α. Since the AMPK signaling pathway post-transcriptionally modifies PGC1α, we performed an enzyme-linked immunosorbent assay (ELISA) to detect AMPK activity in cardiomyocytes in response to H/R injury. As shown in Figure 2A, AMPK activity was significantly lower in the H/R injury group than in the control group; however, melatonin treatment restored AMPK activity. An immunofluorescence assay confirmed that the expression of phosphorylated (p)-AMPK was reduced in cardiomyocytes exposed to H/R injury, but was restored by melatonin treatment (Figure 2B and 2C).
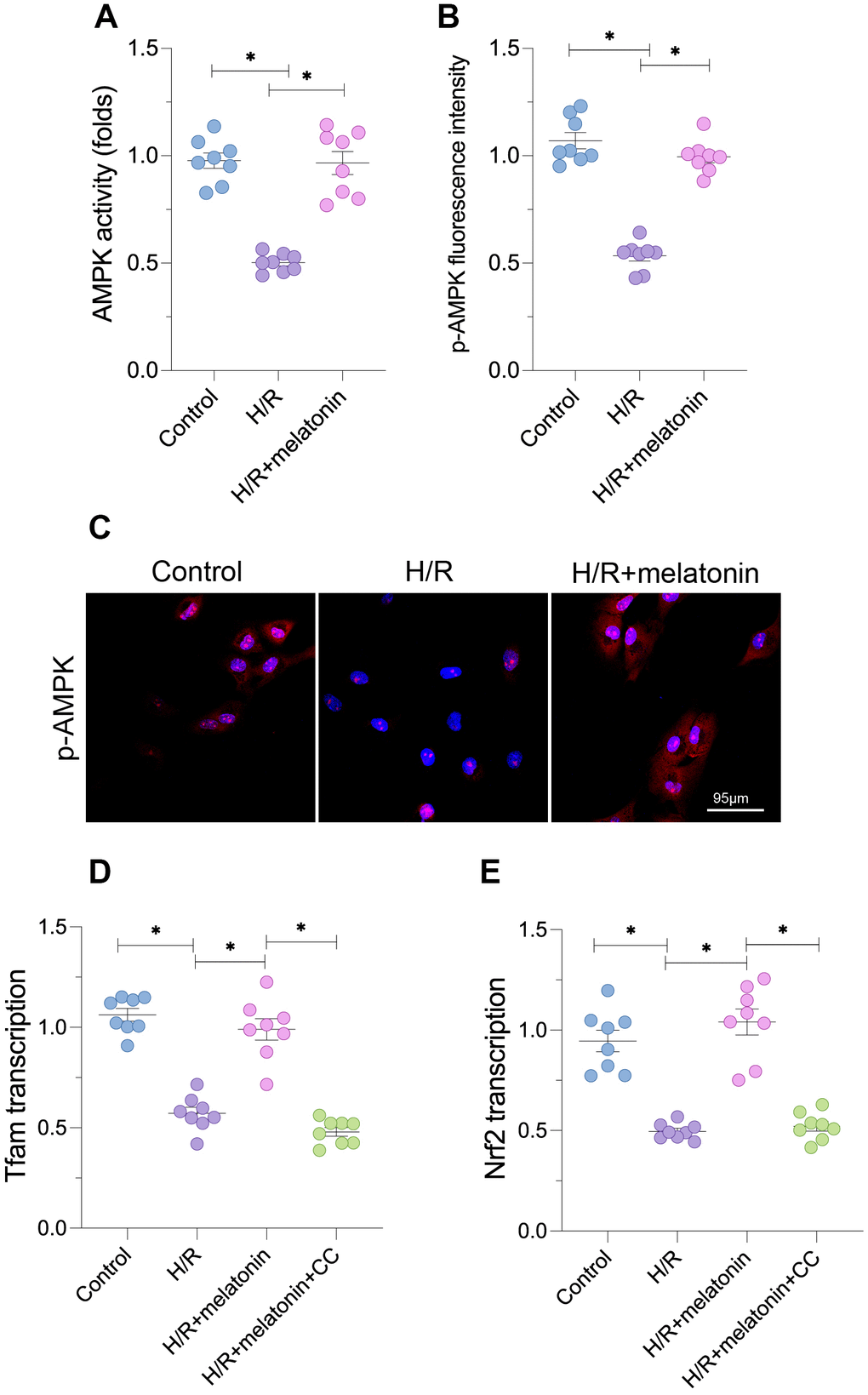
Figure 2. Melatonin activates the AMPK pathway to induce mitochondrial biogenesis. Cardiomyocytes were subjected to H/R injury, with or without previous melatonin treatment to protect the cardiomyocytes. (A) An ELISA was used to evaluate AMPK activity. (B, C) An immunofluorescence assay was used to evaluate the expression of p-AMPK. (D, E) qPCR was used to evaluate the transcription of Tfam and Nrf2. CC was used to inhibit the activity of AMPK. *p<0.05.
To determine whether AMPK was required for melatonin-induced mitochondrial biogenesis, we used compound c (CC) to inhibit AMPK activity in cardiomyocytes. Blocking the AMPK pathway suppressed the melatonin-induced increases in Tfam and Nrf2 mRNA levels (Figure 2D and 2E). CC supplementation also abolished the melatonin-induced increase in the mitochondrial mass (Figure 2D and 2E). Thus, AMPK was required for melatonin-induced mitochondrial biogenesis in H/R-treated cardiomyocytes.
Silencing of PGC1α abolishes the protective effects of melatonin on mitochondrial bioenergetics
Next, we assessed the effects of mitochondrial biogenesis on mitochondrial function in cardiomyocytes damaged by H/R injury. Since our earlier findings indicated that melatonin induced PGC1α, we used small interfering RNA (siRNA) to silence PGC1α in cardiomyocytes. We found that mitochondrial ATP production in cardiomyocytes was reduced by H/R treatment and restored by melatonin treatment; however, the effects of melatonin were nullified when PGC1α was knocked down (Figure 3A). Given that ATP levels were reduced upon H/R injury, we then measured mitochondrial ROS production in cardiomyocytes. As shown in Figure 3B and 3C, mitochondrial ROS fluorescence was greater in the H/R group than in the control group; however, melatonin attenuated mitochondrial ROS production. Notably, when melatonin-treated H/R-injured cardiomyocytes were transfected with siRNA against PGC1α, mitochondrial ROS fluorescence increased again (Figure 3B and 3C). The mitochondrial membrane potential in cardiomyocytes was also reduced in response to H/R injury (Figure 3D and 3E). Melatonin treatment stabilized the mitochondrial membrane potential, but not in PGC1α-silenced cells (Figure 3D and 3E). These data indicate that melatonin enhanced mitochondrial bioenergetics and suppressed oxidative stress by inducing PGC1α.
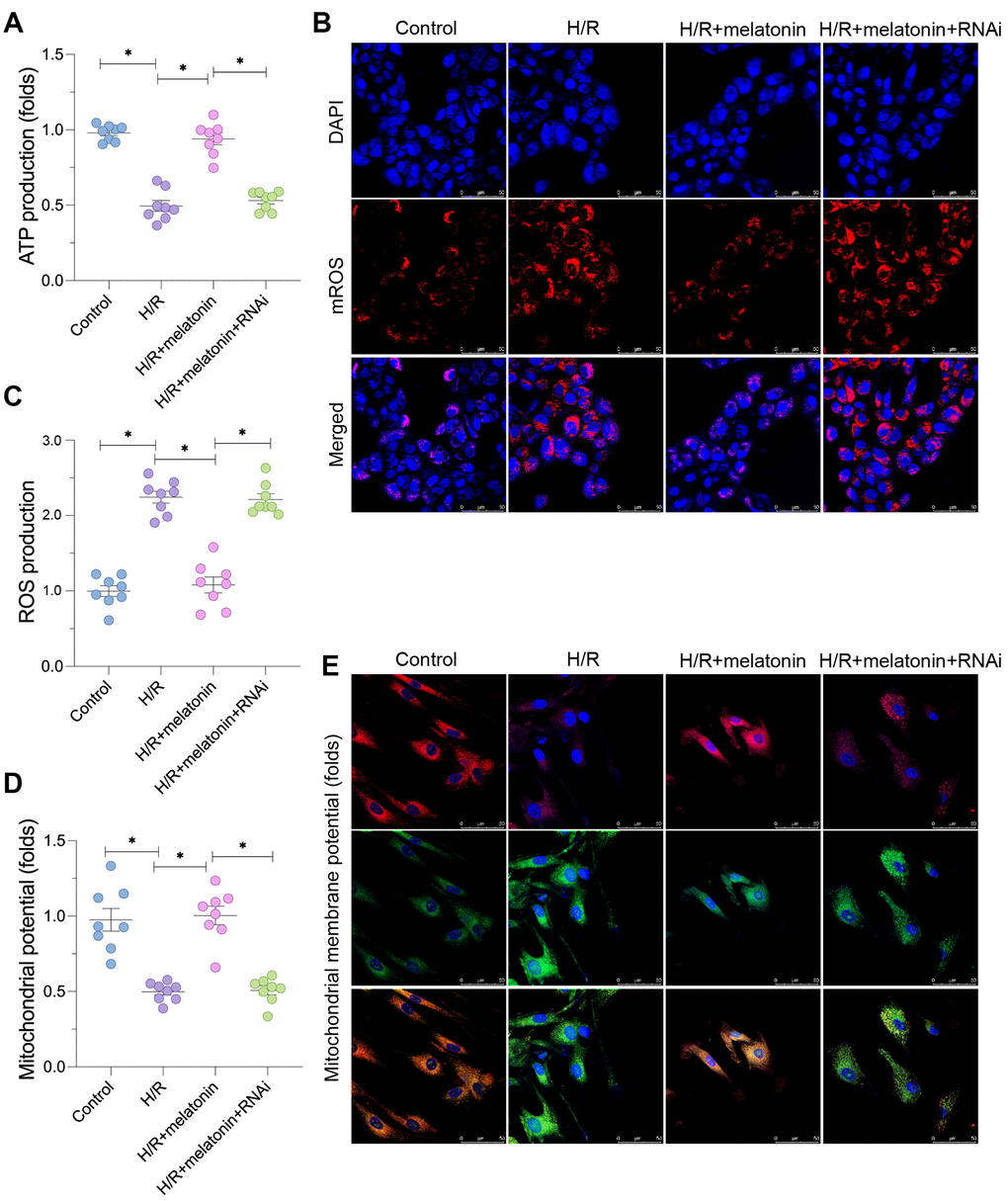
Figure 3. Melatonin preserves mitochondrial function in H/R-treated cardiomyocytes by inducing the AMPK/PGC1α pathway. Cardiomyocytes were subjected to H/R injury, with or without previous melatonin treatment to protect the cardiomyocytes. The cardiomyocytes were transfected with siRNA to knock down PGC1α. (A) An ELISA was used to assess ATP production. (B, C) An immunofluorescence assay was used to measure mitochondrial ROS in cardiomyocytes. (D, E) An immunofluorescence assay was used to measure the mitochondrial membrane potential. *p<0.05.
PGC1α-induced mitochondrial biogenesis also alters the mitochondrial morphology
In addition to monitoring mitochondrial function, we assessed the mitochondrial morphology in cardiomyocytes. As shown in Figure 4A and 4B, H/R injury induced mitochondrial fragmentation, suggesting that H/R injury either increased mitochondrial fission or reduced mitochondrial fusion. The average length of the mitochondrial mass was lower in the H/R group than in the control group (Figure 4A and 4B). Melatonin treatment inhibited the formation of fragmented mitochondria and thus sustained the mitochondrial length; however, this effect depended on the expression of PGC1α (Figure 4A and 4B). At the molecular level, H/R injury significantly repressed the transcription of mitofusin 2 (Mfn2) and Opa1, whereas melatonin upregulated these genes (Figure 4C and 4D). Loss of PGC1α abolished the melatonin-induced upregulation of Mfn2 and Opa1. These data indicate that mitochondrial biogenesis restored mitochondrial fusion in H/R-treated cardiomyocytes.
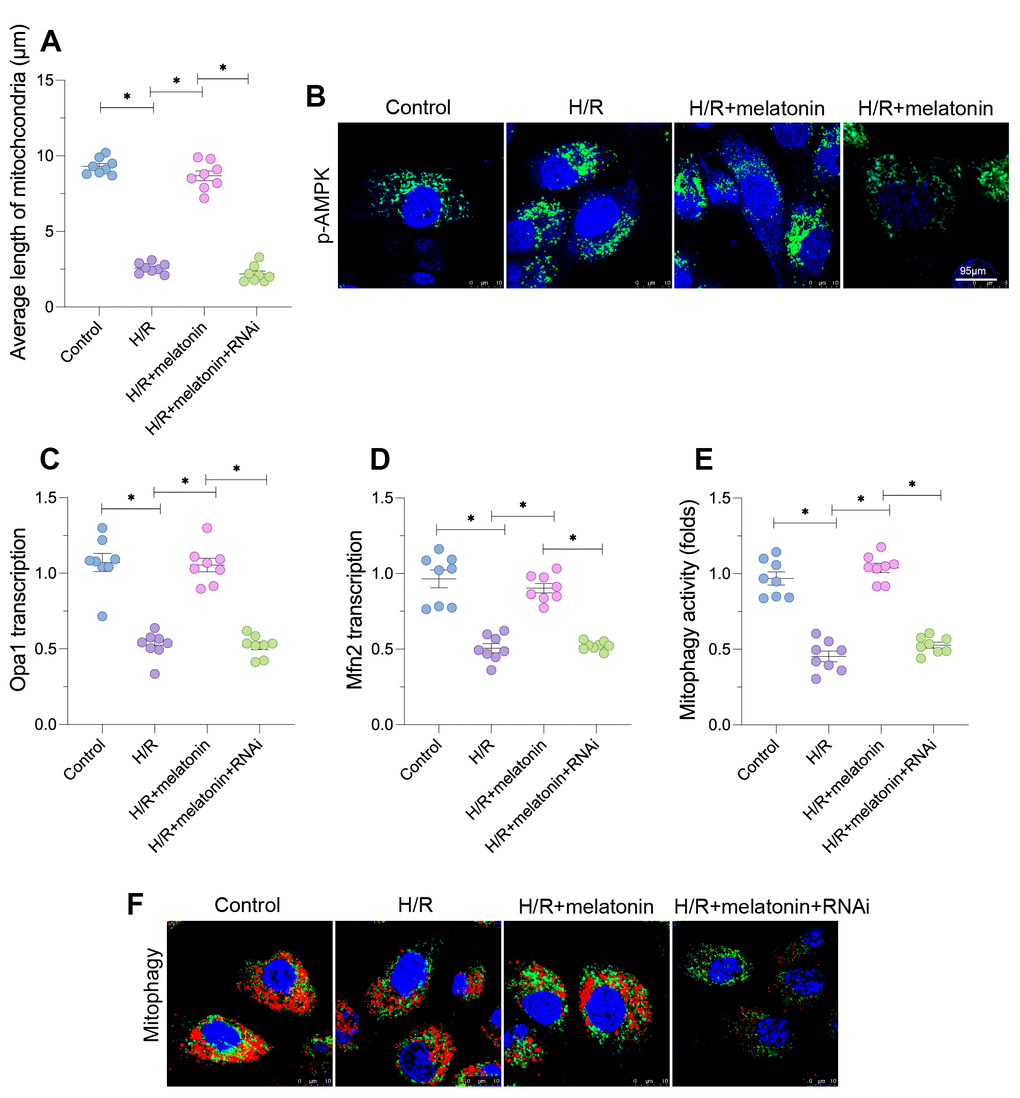
Figure 4. Melatonin alters the mitochondrial morphology in cardiomyocytes by inducing the AMPK/PGC1α pathway. Cardiomyocytes were subjected to H/R injury, with or without previous melatonin treatment to protect the cardiomyocytes. The cardiomyocytes were transfected with siRNA to knock down PGC1α. (A, B) An immunofluorescence assay was used to assess the mitochondrial morphology. (C, D) A qPCR assay was used to evaluate the transcription of Opa1 and Mfn2. (E, F) An mt-kemia assay was used to evaluate mitophagy activity. *p<0.05.
Next, we used the mt-kemia assay to observe the level of mitophagy in H/R-treated cardiomyocytes. As shown in Figure 4E and 4F, mitophagy was downregulated in the H/R injury group compared with the control group, as evidenced by the reduced number of acidic mitochondria in cardiomyocytes. Melatonin restored mitophagy activity, but the loss of PGC1α prevented the melatonin-induced upregulation of mitophagy in cardiomyocytes (Figure 4E and 4F). These results indicate that melatonin normalized the mitochondrial morphology in H/R-treated cardiomyocytes.
Melatonin requires PGC1α-induced mitochondrial biogenesis to inhibit mitochondrial apoptosis in H/R-treated cardiomyocytes
Damaged mitochondria are associated with cardiomyocyte death. Therefore, we evaluated the anti-apoptotic effects of mitochondrial biogenesis. Caspase-9 activity increased rapidly in response to H/R injury, and melatonin prevented this alteration (Figure 5A). However, when PGC1α was silenced, caspase-9 was re-activated in melatonin-treated cardiomyocytes (Figure 5A). The opening rate of the mitochondrial permeability transition pore (mPTP) also increased in response to H/R injury. Melatonin treatment reduced the mPTP opening rate in a manner dependent on PGC1α expression (Figure 5B).
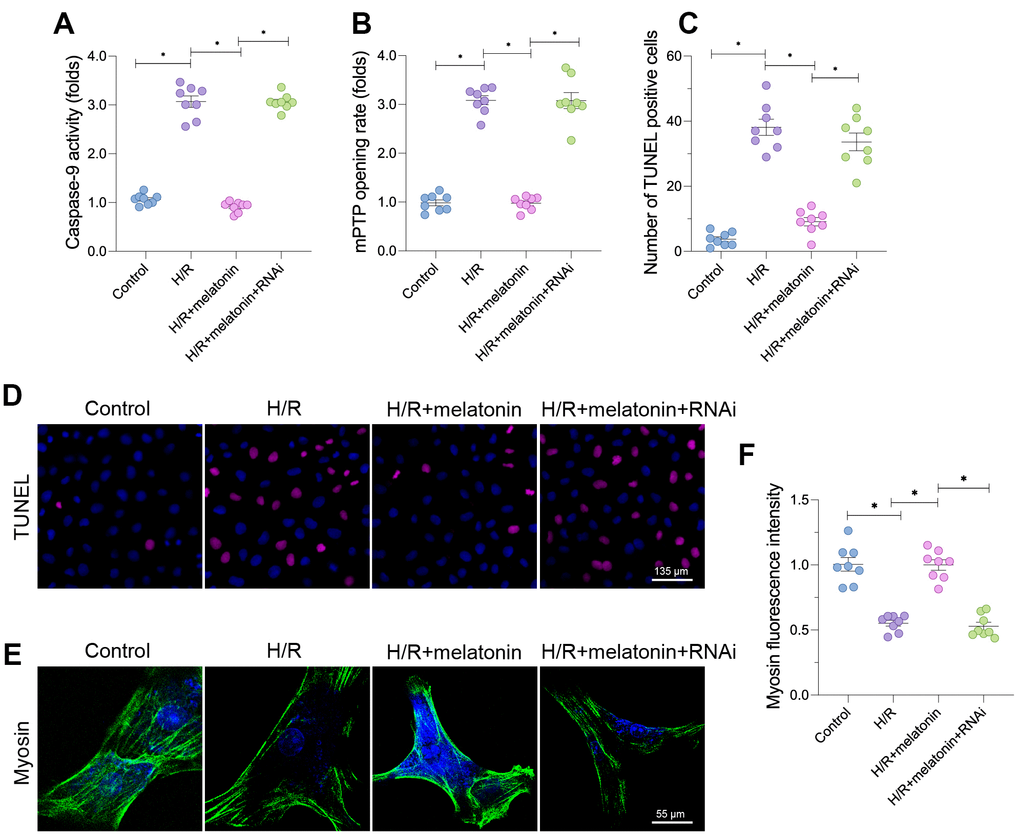
Figure 5. Melatonin-induced mitochondrial biogenesis promotes cardiomyocyte survival. Cardiomyocytes were subjected to H/R injury, with or without previous melatonin treatment to protect the cardiomyocytes. The cardiomyocytes were transfected with siRNA to knock down PGC1α. (A) An ELISA was used to assess caspase-9 activity. (B) The mPTP opening rate was determined in cardiomyocytes. (C, D) TUNEL staining for apoptotic cells in cardiomyocytes. (E, F) An immunofluorescence assay was used to measure the expression of myosin. *p<0.05.
Due to the increased mPTP opening rate and elevated caspase-9 activity, the number of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive cells was greater in the H/R group than in the control group (Figure 5C and 5D). Fewer apoptotic cells were found in the melatonin group, but the loss of PGC1α abolished the inhibitory effect of melatonin on mitochondrial apoptosis. More importantly, H/R injury significantly reduced the fluorescence intensity of myosin (Figure 5E and 5F), indicating that the cardiomyocyte cytoskeleton had been degraded. Melatonin treatment increased the expression of myosin by upregulating PGC1α, confirming that melatonin exerted anti-apoptotic effects on cardiomyocytes during H/R injury. These results indicate that melatonin inhibited mitochondrial apoptosis in H/R-treated cardiomyocytes by stimulating PGC1α-induced mitochondrial biogenesis.
Discussion
In order to pump blood, cardiomyocytes require mitochondria; therefore, mitochondrial homeostasis is vital for cardiomyocyte function. Many studies have demonstrated that enhancing mitochondrial activity is a promising strategy to improve cardiomyocyte viability and cardiac function during myocardial I/R injury. Cardiac I/R injury has been attributed to three molecular mechanisms: cardiomyocyte death, inflammation and oxidative stress [34–38]. However, defective energy metabolism and microvascular damage have also been noted in the reperfused heart [39–41].
Mitochondria seem to be involved in every aspect of the various physiological and pathological events during the development of cardiac I/R injury. For example, mitochondria can induce cardiomyocyte death by releasing pro-apoptotic proteins and activating the endogenous apoptotic pathway. Oxidative stress is primarily caused by ROS overload, and mitochondria are the main producers of ROS through impaired oxidative phosphorylation [42]. Mitochondrial damage-induced oxidative stress also promotes cardiomyocyte death by inducing membrane oxidation and protein post-transcriptional modifications. The inflammatory response may be secondary to cardiomyocyte death and oxidative stress, because the inflammatory response is triggered by inflammatory cell accumulation, which results from cardiomyocyte apoptosis [43–45]. Therefore, mitochondrial homeostasis is linked to the extent of cardiac I/R injury.
Mitochondrial homeostasis is maintained through structural and functional alterations. Mitochondrial morphological adaptions (also known as mitochondrial dynamics) include mitochondrial fission, fusion and mitophagy [46–49], and have been carefully discussed in several articles [50–52]. In this study, we explored the contribution of mitochondrial biogenesis to mitochondrial homeostasis. Mitochondrial biogenesis regenerates mitochondria and ultimately increases the mitochondrial mass, as evidenced by increased levels of mitochondrial DNA and mitochondria-related proteins [53, 54]. Mitochondrial biogenesis rapidly augments the mitochondrial population in a short time, which is necessary for cell metabolism under stress. Although several studies have described the alterations in mitochondrial biogenesis in the setting of cardiac I/R injury [55–57], the protective mechanism of mitochondrial biogenesis has not been fully elucidated. Herein, we found that mitochondrial biogenesis was inhibited in cardiomyocytes subjected to H/R injury. Consequently, mitochondrial bioenergetics were impaired, the mitochondrial morphology was disrupted and mitochondrial function was reduced. Improving mitochondrial biogenesis elevated cardiomyocyte ATP production, prevented mitochondrial fragmentation, enhanced mitochondrial function, stimulated mitophagy and inhibited mitochondrial apoptosis. Therefore, mitochondrial biogenesis may be a critical determinant of mitochondrial structure, function and homeostasis.
Melatonin is an effective drug for the treatment of cardiac I/R injury. Cytoplasmic melatonin can accumulate in the mitochondria and enhance mitochondrial redox balance and bioenergetics. Although melatonin is thought to sustain mitochondrial function through its anti-oxidative actions, melatonin also controls mitochondrial fission, fusion, mitophagy, apoptosis, necroptosis and calcium balance during cardiac I/R injury. In this study, we observed that melatonin stimulated mitochondrial biogenesis. Melatonin activated the AMPK pathway, which thus upregulated PGC1α, Tfam, Nrf2 and Sirt3. Tfam, Nrf2 and Sirt3 have been reported to increase mitochondrial DNA levels and oxidative phosphorylation-related protein translation. Melatonin treatment also increased the mitochondrial mass and improved mitochondrial function. Thus, we have provided a method of restoring mitochondrial biogenesis in the reperfused heart.
There are several limitations to our experiments. First, we did not obtain animal data to support the function of mitochondrial biogenesis in the reperfused heart. Second, we only tested the effects of one concentration of melatonin on mitochondrial biogenesis. Nevertheless, our results indicated that melatonin improved mitochondrial biogenesis and promoted the transmission of protective signals for mitochondria in H/R-treated cardiomyocytes.
Materials and Methods
Cell isolation, culture, treatment and transfection
Primary cultures of neonatal mouse ventricular cardiomyocytes were prepared from the ventricles of three- to five-day-old mice as described previously [58]. Cardiomyocytes were plated on type I collagen-coated cover glasses or culture plates, and were incubated with Dulbecco’s modified Eagle’s medium supplemented with bovine serum albumin or palmitate-bovine serum albumin. Cardiomyocytes were also transfected with siRNA against PGC1α. After 12 hours of transfection, the cardiomyocytes were incubated in Dulbecco’s modified Eagle’s medium with or without palmitate, as described above. H/R injury was induced through six-hour hypoxia and six-hour reoxygenation, as previously described. Melatonin (5 μM) was used 12 hours prior to H/R treatment. CC (10 μM, Selleck Chemicals, Houston, TX, USA) was added to the medium for two hours to prevent AMPK activation [59].
Mitochondrial function
Mitochondrial function was evaluated with a JC-1 Mitochondrial Membrane Potential kit (Cayman Chemical) according to the manufacturer’s instructions. An ELISA was performed, and the signal was detected on a plate reader (EnSpire® Multimode Plate Reader, Perkin Elmer).
The extracellular oxygen consumption rate was evaluated with a fluorescence-based oxygen consumption rate assay kit (Abcam) according to the manufacturer’s instructions [60]. Briefly, cells were treated with 20 μM octyl-α-ketoglutarate or 10 μM AA6. Dimethyl sulfoxide was used as a solvent. The fluorescent signal was detected every minute for one hour on a multi-well plate reader (EnSpire® Multimode Plate Reader) set at 37 °C, excitation/emission=380/650 nm. Signals were normalized to the total DNA content, which was assessed with DRAQ5 (1:1000, Biostatus) [61].
Immunofluorescence and confocal microscopy
Confocal analysis was performed as reported previously. Paraffin-embedded and cryosectioned samples were prepared according to standard histological procedures [62]. Mitochondria were visualized with MitoTracker Red CMXRos (1:2000, Thermo Scientific) and JC-1 (5 μM, AdipoGen). The samples were analyzed on a Leica TCS SP8 confocal microscope [63].
Cell counting kit-8 (CCK-8) assay
A CCK-8 assay was used to assess cell viability [64]. Briefly, adherent cells were detached with trypsin, seeded in 96-well plates at 5×103 cells/well, and allowed to attach to the bottom of the plate for 24 hours at 37 °C. The cells were then subjected to H/R injury with or without melatonin. Lastly, the cell viability was measured at 450 nm using CCK-8 [65].
Western blotting
Western blotting was performed as described previously. Briefly, samples were lysed in complete radioimmunoprecipitation assay buffer (10 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP40, 0.1% sodium dodecyl sulfate, 1 mM phenylmethylsulfonyl fluoride and 1x protease inhibitor cocktail [Roche]) and homogenized with a Sonic Dismembrator 100 (Fisher Scientific) [66]. The protein concentration of the sample homogenate was measured with a Bio-Rad Protein Assay, and equal amounts of soluble proteins were separated on 10% polyacrylamide gels, transferred onto nitrocellulose membranes and subjected to routine Western blot analysis [67].
TUNEL staining
The paraffin-embedded sections obtained from the experiment above were dewaxed with fresh xylene (twice, for 10 min each) and dehydrated using a serial alcohol gradient [68]. Then, the tissue slides were treated with 20 μg/mL DNase-free protease K, incubated at 20-37 °C for 15-30 min, and washed three times with phosphate-buffered saline [69]. Then, 50 μL of TUNEL solution was added to the tissue slides, and the slides were incubated at 37 °C for 60 min in the dark. After being washed three times with phosphate-buffered saline, the slides were treated with an anti-fluorescence quenching agent and observed via fluorescence microscopy at an excitation wavelength of 450-500 nm and an emission wavelength of 515-565 nm [70].
qRT-PCR analysis
An RNAiso Plus Purification Kit (TaKaRa Biotechnology Co., Ltd, 9108) was used to extract total RNA from the cardiomyocytes. The RNA concentration was evaluated based on the optical density of the sample at 260 nm, and the RNA integrity was assessed through 2% agarose gel electrophoresis. The RNA was reverse-transcribed into cDNA using a PrimeScript™ RT Reagent Kit with gDNA Eraser (TaKaRa Biotechnology Co., Ltd, RR047A) [71, 72]. Real-time PCR was performed on a LightCycler machine (Roche) with a commercial SYBR Green reaction reagent (TaKaRa Biotechnology Co., Ltd, RR820A). GAPDH was used as an internal control. The cDNA was denatured for 30 s at 95 °C, followed by 40 cycles of 5 s at 95°C [73].
Statistical analysis
Statistical analyses were performed with GraphPad Prism. The sample sizes (n) are reported in the corresponding figure legends. The present study was exploratory and primarily mechanistic. For all analyses, the observer was blind to the identity of the samples. The variables were analyzed using non-parametric Student’s t-tests or analysis of variance (one-way or two-way). A value of p<0.05 was deemed statistically significant. Results are shown as the mean ± standard error.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1. Davidson SM, Arjun S, Basalay MV, Bell RM, Bromage DI, Bøtker HE, Carr RD, Cunningham J, Ghosh AK, Heusch G, Ibanez B, Kleinbongard P, Lecour S, et al. The 10th Biennial Hatter Cardiovascular Institute workshop: cellular protection-evaluating new directions in the setting of myocardial infarction, ischaemic stroke, and cardio-oncology. Basic Res Cardiol. 2018; 113:43. https://doi.org/10.1007/s00395-018-0704-z [PubMed]
- 2. Eid RA, Alkhateeb MA, Eleawa S, Al-Hashem FH, Al-Shraim M, El-Kott AF, Zaki MS, Dallak MA, Aldera H. Cardioprotective effect of ghrelin against myocardial infarction-induced left ventricular injury via inhibition of SOCS3 and activation of JAK2/STAT3 signaling. Basic Res Cardiol. 2018; 113:13. https://doi.org/10.1007/s00395-018-0671-4 [PubMed]
- 3. Zhou H, Toan S. Pathological Roles of Mitochondrial Oxidative Stress and Mitochondrial Dynamics in Cardiac Microvascular Ischemia/Reperfusion Injury. Biomolecules. 2020; 10:10. https://doi.org/10.3390/biom10010085 [PubMed]
- 4. Kuznetsov AV, Javadov S, Margreiter R, Grimm M, Hagenbuchner J, Ausserlechner MJ. The Role of Mitochondria in the Mechanisms of Cardiac Ischemia-Reperfusion Injury. Antioxidants. 2019; 8:8. https://doi.org/10.3390/antiox8100454 [PubMed]
- 5. Kohlhauer M, Pell VR, Burger N, Spiroski AM, Gruszczyk A, Mulvey JF, Mottahedin A, Costa AS, Frezza C, Ghaleh B, Murphy MP, Tissier R, Krieg T. Protection against cardiac ischemia-reperfusion injury by hypothermia and by inhibition of succinate accumulation and oxidation is additive. Basic Res Cardiol. 2019; 114:18. https://doi.org/10.1007/s00395-019-0727-0 [PubMed]
- 6. Alakoski T, Ulvila J, Yrjölä R, Vainio L, Magga J, Szabo Z, Licht JD, Kerkelä R. Inhibition of cardiomyocyte Sprouty1 protects from cardiac ischemia-reperfusion injury. Basic Res Cardiol. 2019; 114:7. https://doi.org/10.1007/s00395-018-0713-y [PubMed]
- 7. Zhang Y, Wang Y, Xu J, Tian F, Hu S, Chen Y, Fu Z. Melatonin attenuates myocardial ischemia-reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK-OPA1 signaling pathways. J Pineal Res. 2019; 66:e12542. https://doi.org/10.1111/jpi.12542 [PubMed]
- 8. Zhou H, Shi C, Hu S, Zhu H, Ren J, Chen Y. BI1 is associated with microvascular protection in cardiac ischemia reperfusion injury via repressing Syk-Nox2-Drp1-mitochondrial fission pathways. Angiogenesis. 2018; 21:599–615. https://doi.org/10.1007/s10456-018-9611-z [PubMed]
- 9. Carelli V, Maresca A, Caporali L, Trifunov S, Zanna C, Rugolo M. Mitochondria: biogenesis and mitophagy balance in segregation and clonal expansion of mitochondrial DNA mutations. Int J Biochem Cell Biol. 2015; 63:21–24. https://doi.org/10.1016/j.biocel.2015.01.023 [PubMed]
- 10. Zhou H, Wang J, Zhu P, Zhu H, Toan S, Hu S, Ren J, Chen Y. NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1-mediated mitophagy and promoting Mff-required mitochondrial fission by CK2α. Basic Res Cardiol. 2018; 113:23. https://doi.org/10.1007/s00395-018-0682-1 [PubMed]
- 11. Fontecha-Barriuso M, Martin-Sanchez D, Martinez-Moreno JM, Monsalve M, Ramos AM, Sanchez-Niño MD, Ruiz-Ortega M, Ortiz A, Sanz AB. The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules. 2020; 10:10. https://doi.org/10.3390/biom10020347 [PubMed]
- 12. Zhou H, Zhu P, Wang J, Zhu H, Ren J, Chen Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018; 25:1080–93. https://doi.org/10.1038/s41418-018-0086-7 [PubMed]
- 13. Li R, Xin T, Li D, Wang C, Zhu H, Zhou H. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: the role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 2018; 18:229–43. https://doi.org/10.1016/j.redox.2018.07.011 [PubMed]
- 14. He Z, Ning N, Zhou Q, Khoshnam SE, Farzaneh M. Mitochondria as a therapeutic target for ischemic stroke. Free Radic Biol Med. 2020; 146:45–58. https://doi.org/10.1016/j.freeradbiomed.2019.11.005 [PubMed]
- 15. Jin Q, Li R, Hu N, Xin T, Zhu P, Hu S, Ma S, Zhu H, Ren J, Zhou H. DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol. 2018; 14:576–87. https://doi.org/10.1016/j.redox.2017.11.004 [PubMed]
- 16. Zhou H, Zhu P, Guo J, Hu N, Wang S, Li D, Hu S, Ren J, Cao F, Chen Y. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 2017; 13:498–507. https://doi.org/10.1016/j.redox.2017.07.007 [PubMed]
- 17. Yang J, He J, Ismail M, Tweeten S, Zeng F, Gao L, Ballinger S, Young M, Prabhu SD, Rowe GC, Zhang J, Zhou L, Xie M. HDAC inhibition induces autophagy and mitochondrial biogenesis to maintain mitochondrial homeostasis during cardiac ischemia/reperfusion injury. J Mol Cell Cardiol. 2019; 130:36–48. https://doi.org/10.1016/j.yjmcc.2019.03.008 [PubMed]
- 18. Sanz MN, Farine E, Niederberger P, Méndez-Carmona N, Wyss RK, Arnold M, Gulac P, Fiedler GM, Gressette M, Garnier A, Carrel TP, Tevaearai Stahel HT, Longnus SL. Cardioprotective reperfusion strategies differentially affect mitochondria: studies in an isolated rat heart model of donation after circulatory death (DCD). Am J Transplant. 2019; 19:331–44. https://doi.org/10.1111/ajt.15024 [PubMed]
- 19. Singhanat K, Apaijai N, Chattipakorn SC, Chattipakorn N. Roles of melatonin and its receptors in cardiac ischemia-reperfusion injury. Cell Mol Life Sci. 2018; 75:4125–49. https://doi.org/10.1007/s00018-018-2905-x [PubMed]
- 20. Zhou H, Ma Q, Zhu P, Ren J, Reiter RJ, Chen Y. Protective role of melatonin in cardiac ischemia-reperfusion injury: from pathogenesis to targeted therapy. J Pineal Res. 2018; 64:64. https://doi.org/10.1111/jpi.12471 [PubMed]
- 21. Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q, Jin Q, Cao F, Tian F, Chen Y. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J Pineal Res. 2017; 63:63. https://doi.org/10.1111/jpi.12413 [PubMed]
- 22. Zhou H, Li D, Zhu P, Ma Q, Toan S, Wang J, Hu S, Chen Y, Zhang Y. Inhibitory effect of melatonin on necroptosis via repressing the Ripk3-PGAM5-CypD-mPTP pathway attenuates cardiac microvascular ischemia-reperfusion injury. J Pineal Res. 2018; 65:e12503. https://doi.org/10.1111/jpi.12503 [PubMed]
- 23. Wang S, Bian W, Zhen J, Zhao L, Chen W. Melatonin-Mediated Pak2 Activation Reduces Cardiomyocyte Death Through Suppressing Hypoxia Reoxygenation Injury-Induced Endoplasmic Reticulum Stress. J Cardiovasc Pharmacol. 2019; 74:20–29. https://doi.org/10.1097/FJC.0000000000000678 [PubMed]
- 24. Zhang WX, He BM, Wu Y, Qiao JF, Peng ZY. Melatonin protects against sepsis-induced cardiac dysfunction by regulating apoptosis and autophagy via activation of SIRT1 in mice. Life Sci. 2019; 217:8–15. https://doi.org/10.1016/j.lfs.2018.11.055 [PubMed]
- 25. Ma S, Dong Z. Melatonin Attenuates Cardiac Reperfusion Stress by Improving OPA1-Related Mitochondrial Fusion in a Yap-Hippo Pathway-Dependent Manner. J Cardiovasc Pharmacol. 2019; 73:27–39. https://doi.org/10.1097/FJC.0000000000000626 [PubMed]
- 26. Zhong J, Tan Y, Lu J, Liu J, Xiao X, Zhu P, Chen S, Zheng S, Chen Y, Hu Y, Guo Z. Therapeutic contribution of melatonin to the treatment of septic cardiomyopathy: A novel mechanism linking Ripk3-modified mitochondrial performance and endoplasmic reticulum function. Redox Biol. 2019; 26:101287. https://doi.org/10.1016/j.redox.2019.101287 [PubMed]
- 27. Gonzalez-Candia A, Veliz M, Carrasco-Pozo C, Castillo RL, Cárdenas JC, Ebensperger G, Reyes RV, Llanos AJ, Herrera EA. Antenatal melatonin modulates an enhanced antioxidant/pro-oxidant ratio in pulmonary hypertensive newborn sheep. Redox Biol. 2019; 22:101128. https://doi.org/10.1016/j.redox.2019.101128 [PubMed]
- 28. Xia Y, Chen S, Zeng S, Zhao Y, Zhu C, Deng B, Zhu G, Yin Y, Wang W, Hardeland R, Ren W. Melatonin in macrophage biology: current understanding and future perspectives. J Pineal Res. 2019; 66:e12547. https://doi.org/10.1111/jpi.12547 [PubMed]
- 29. Zhou H, Li D, Zhu P, Hu S, Hu N, Ma S, Zhang Y, Han T, Ren J, Cao F, Chen Y. Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARγ/FUNDC1/mitophagy pathways. J Pineal Res. 2017; 63:63. https://doi.org/10.1111/jpi.12438 [PubMed]
- 30. Boga JA, Caballero B, Potes Y, Perez-Martinez Z, Reiter RJ, Vega-Naredo I, Coto-Montes A. Therapeutic potential of melatonin related to its role as an autophagy regulator: A review. J Pineal Res. 2019; 66:e12534. https://doi.org/10.1111/jpi.12534 [PubMed]
- 31. Niu YJ, Zhou W, Nie ZW, Shin KT, Cui XS. Melatonin enhances mitochondrial biogenesis and protects against rotenone-induced mitochondrial deficiency in early porcine embryos. J Pineal Res. 2020; 68:e12627. https://doi.org/10.1111/jpi.12627 [PubMed]
- 32. Jiang LL, Zhang FP, He YF, Fan WG, Zheng MM, Kang J, Huang F, He HW. Melatonin regulates mitochondrial function and biogenesis during rat dental papilla cell differentiation. Eur Rev Med Pharmacol Sci. 2019; 23:5967–79. https://doi.org/10.26355/eurrev_201907_18343 [PubMed]
- 33. Wang CF, Song CY, Wang X, Huang LY, Ding M, Yang H, Wang P, Xu LL, Xie ZH, Bi JZ. Protective effects of melatonin on mitochondrial biogenesis and mitochondrial structure and function in the HEK293-APPswe cell model of Alzheimer’s disease. Eur Rev Med Pharmacol Sci. 2019; 23:3542–50. https://doi.org/10.26355/eurrev_201904_17723 [PubMed]
- 34. Zhou H, Toan S, Zhu P, Wang J, Ren J, Zhang Y. DNA-PKcs promotes cardiac ischemia reperfusion injury through mitigating BI-1-governed mitochondrial homeostasis. Basic Res Cardiol. 2020; 115:11. https://doi.org/10.1007/s00395-019-0773-7 [PubMed]
- 35. Zhou H, Hu S, Jin Q, Shi C, Zhang Y, Zhu P, Ma Q, Tian F, Chen Y. Mff-Dependent Mitochondrial Fission Contributes to the Pathogenesis of Cardiac Microvasculature Ischemia/Reperfusion Injury via Induction of mROS-Mediated Cardiolipin Oxidation and HK2/VDAC1 Disassociation-Involved mPTP Opening. J Am Heart Assoc. 2017; 6:6. https://doi.org/10.1161/JAHA.116.005328 [PubMed]
- 36. Billah M, Ridiandries A, Rayner BS, Allahwala UK, Dona A, Khachigian LM, Bhindi R. Egr-1 functions as a master switch regulator of remote ischemic preconditioning-induced cardioprotection. Basic Res Cardiol. 2019; 115:3. https://doi.org/10.1007/s00395-019-0763-9 [PubMed]
- 37. Bacmeister L, Schwarzl M, Warnke S, Stoffers B, Blankenberg S, Westermann D, Lindner D. Inflammation and fibrosis in murine models of heart failure. Basic Res Cardiol. 2019; 114:19. https://doi.org/10.1007/s00395-019-0722-5 [PubMed]
- 38. Audia JP, Yang XM, Crockett ES, Housley N, Haq EU, O’Donnell K, Cohen MV, Downey JM, Alvarez DF. Caspase-1 inhibition by VX-765 administered at reperfusion in P2Y12 receptor antagonist-treated rats provides long-term reduction in myocardial infarct size and preservation of ventricular function. Basic Res Cardiol. 2018; 113:32. https://doi.org/10.1007/s00395-018-0692-z [PubMed]
- 39. Hadebe N, Cour M, Lecour S. The SAFE pathway for cardioprotection: is this a promising target? Basic Res Cardiol. 2018; 113:9. https://doi.org/10.1007/s00395-018-0670-5 [PubMed]
- 40. Heusch G. Coronary microvascular obstruction: the new frontier in cardioprotection. Basic Res Cardiol. 2019; 114:45. https://doi.org/10.1007/s00395-019-0756-8 [PubMed]
- 41. Heusch G. 25 years of remote ischemic conditioning: from laboratory curiosity to clinical outcome. Basic Res Cardiol. 2018; 113:15. https://doi.org/10.1007/s00395-018-0673-2 [PubMed]
- 42. Rossello X, Yellon DM. The RISK pathway and beyond. Basic Res Cardiol. 2017; 113:2. https://doi.org/10.1007/s00395-017-0662-x [PubMed]
- 43. Liu D, Zeng X, Li X, Mehta JL, Wang X. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res Cardiol. 2017; 113:5. https://doi.org/10.1007/s00395-017-0663-9 [PubMed]
- 44. Jung M, Dodsworth M, Thum T. Inflammatory cells and their non-coding RNAs as targets for treating myocardial infarction. Basic Res Cardiol. 2018; 114:4. https://doi.org/10.1007/s00395-018-0712-z [PubMed]
- 45. Coverstone ED, Bach RG, Chen L, Bierut LJ, Li AY, Lenzini PA, O’Neill HC, Spertus JA, Sucharov CC, Stitzel JA, Schilling JD, Cresci S. A novel genetic marker of decreased inflammation and improved survival after acute myocardial infarction. Basic Res Cardiol. 2018; 113:38. https://doi.org/10.1007/s00395-018-0697-7 [PubMed]
- 46. Wang J, Zhu P, Li R, Ren J, Zhang Y, Zhou H. Bax inhibitor 1 preserves mitochondrial homeostasis in acute kidney injury through promoting mitochondrial retention of PHB2. Theranostics. 2020; 10:384–97. https://doi.org/10.7150/thno.40098 [PubMed]
- 47. Zhou H, Wang J, Hu S, Zhu H, Toanc S, Ren J. BI1 alleviates cardiac microvascular ischemia-reperfusion injury via modifying mitochondrial fission and inhibiting XO/ROS/F-actin pathways. J Cell Physiol. 2019; 234:5056–69. https://doi.org/10.1002/jcp.27308 [PubMed]
- 48. Ding M, Liu C, Shi R, Yu M, Zeng K, Kang J, Fu F, Mi M. Mitochondrial fusion promoter restores mitochondrial dynamics balance and ameliorates diabetic cardiomyopathy in an optic atrophy 1-dependent way. Acta Physiol (Oxf). 2019; 229:e13428. https://doi.org/10.1111/apha.13428 [PubMed]
- 49. Yu F, Abdelwahid E, Xu T, Hu L, Wang M, Li Y, Mogharbel BF, de Carvalho KA, Guarita-Souza LC, An Y, Li P. The role of mitochondrial fusion and fission in the process of cardiac oxidative stress. Histol Histopathol. 2019; 18191. [Epub ahead of print]. https://doi.org/10.14670/HH-18-191 [PubMed]
- 50. Feng ST, Wang ZZ, Yuan YH, Wang XL, Sun HM, Chen NH, Zhang Y. Dynamin-related protein 1: A protein critical for mitochondrial fission, mitophagy, and neuronal death in Parkinson’s disease. Pharmacol Res. 2020; 151:104553. https://doi.org/10.1016/j.phrs.2019.104553 [PubMed]
- 51. Hong Y, Tak H, Kim C, Kang H, Ji E, Ahn S, Jung M, Kim HL, Lee JH, Kim W, Lee EK. RNA binding protein HuD contributes to β-cell dysfunction by impairing mitochondria dynamics. Cell Death Differ. 2019. [Epub ahead of print]. https://doi.org/10.1038/s41418-019-0447-x [PubMed]
- 52. Zhou L, Zhang L, Zhang Y, Yu X, Sun X, Zhu T, Li X, Liang W, Han Y, Qin C. PINK1 Deficiency Ameliorates Cisplatin-Induced Acute Kidney Injury in Rats. Front Physiol. 2019; 10:1225. https://doi.org/10.3389/fphys.2019.01225 [PubMed]
- 53. Dingle AM, Yap KK, Gerrand YW, Taylor CJ, Keramidaris E, Lokmic Z, Kong AM, Peters HL, Morrison WA, Mitchell GM. Characterization of isolated liver sinusoidal endothelial cells for liver bioengineering. Angiogenesis. 2018; 21:581–97. https://doi.org/10.1007/s10456-018-9610-0 [PubMed]
- 54. Chen JJ, Liang X, Wang F, Wen YH, Chen TJ, Liu WC, Gong T, Yang JL, Zhu P. Combinatorial mutation on the β-glycosidase specific to 7-β-xylosyltaxanes and increasing the mutated enzyme production by engineering the recombinant yeast. Acta Pharm Sin B. 2019; 9:626–38. https://doi.org/10.1016/j.apsb.2018.11.003 [PubMed]
- 55. Denton D, Kumar S. Autophagy-dependent cell death. Cell Death Differ. 2019; 26:605–16. https://doi.org/10.1038/s41418-018-0252-y [PubMed]
- 56. Nwadozi E, Ng A, Strömberg A, Liu HY, Olsson K, Gustafsson T, Haas TL. Leptin is a physiological regulator of skeletal muscle angiogenesis and is locally produced by PDGFRα and PDGFRβ expressing perivascular cells. Angiogenesis. 2019; 22:103–15. https://doi.org/10.1007/s10456-018-9641-6 [PubMed]
- 57. Silverblatt JA, Ziff OJ, Dancy L, Daniel A, Carter B, Scott P, Sado DM, Shah A, Bromage DI. Therapies to limit myocardial injury in animal models of myocarditis: a systematic review and meta-analysis. Basic Res Cardiol. 2019; 114:48. https://doi.org/10.1007/s00395-019-0754-x [PubMed]
- 58. Bøtker HE, Hausenloy D, Andreadou I, Antonucci S, Boengler K, Davidson SM, Deshwal S, Devaux Y, Di Lisa F, Di Sante M, Efentakis P, Femminò S, García-Dorado D, et al. Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res Cardiol. 2018; 113:39. https://doi.org/10.1007/s00395-018-0696-8 [PubMed]
- 59. Aanhane E, Schulkens IA, Heusschen R, Castricum K, Leffler H, Griffioen AW, Thijssen VL. Different angioregulatory activity of monovalent galectin-9 isoforms. Angiogenesis. 2018; 21:545–55. https://doi.org/10.1007/s10456-018-9607-8 [PubMed]
- 60. Ansari D, Torén W, Zhou Q, Hu D, Andersson R. Proteomic and genomic profiling of pancreatic cancer. Cell Biol Toxicol. 2019; 35:333–43. https://doi.org/10.1007/s10565-019-09465-9 [PubMed]
- 61. An F, Wang X, Yang M, Luo J, Kong L. Bioactive A-ring rearranged limonoids from the root barks of Walsura robusta. Acta Pharm Sin B. 2019; 9:545–56. https://doi.org/10.1016/j.apsb.2019.02.009 [PubMed]
- 62. Abeysuriya RG, Lockley SW, Robinson PA, Postnova S. A unified model of melatonin, 6-sulfatoxymelatonin, and sleep dynamics. J Pineal Res. 2018; 64:e12474. https://doi.org/10.1111/jpi.12474 [PubMed]
- 63. Aalto AL, Mohan AK, Schwintzer L, Kupka S, Kietz C, Walczak H, Broemer M, Meinander A. M1-linked ubiquitination by LUBEL is required for inflammatory responses to oral infection in Drosophila. Cell Death Differ. 2019; 26:860–76. https://doi.org/10.1038/s41418-018-0164-x [PubMed]
- 64. Agrawal R, Nath V, Kumar H, Kumar V. Deciphering PPARγ activation in cardiometabolic syndrome: studies by in silico and in vivo experimental assessment. J Recept Signal Transduct Res. 2018; 38:122–32. https://doi.org/10.1080/10799893.2018.1436560 [PubMed]
- 65. Bansal R, Gupta S, Rathore AS. Analytical Platform for Monitoring Aggregation of Monoclonal Antibody Therapeutics. Pharm Res. 2019; 36:152. https://doi.org/10.1007/s11095-019-2690-8 [PubMed]
- 66. Alen F, Decara J, Brunori G, You ZB, Bühler KM, López-Moreno JA, Cippitelli A, Pavon FJ, Suárez J, Gardner EL, de la Torre R, Ciccocioppo R, Serrano A, Rodríguez de Fonseca F. PPARα/CB1 receptor dual ligands as a novel therapy for alcohol use disorder: evaluation of a novel oleic acid conjugate in preclinical rat models. Biochem Pharmacol. 2018; 157:235–43. https://doi.org/10.1016/j.bcp.2018.09.008 [PubMed]
- 67. Afonso CB, Spickett CM. Lipoproteins as targets and markers of lipoxidation. Redox Biol. 2019; 23:101066. https://doi.org/10.1016/j.redox.2018.101066 [PubMed]
- 68. Afonso MB, Rodrigues PM, Simão AL, Gaspar MM, Carvalho T, Borralho P, Bañales JM, Castro RE, Rodrigues CM. miRNA-21 ablation protects against liver injury and necroptosis in cholestasis. Cell Death Differ. 2018; 25:857–72. https://doi.org/10.1038/s41418-017-0019-x [PubMed]
- 69. Begum F, Amin S. Investigating the Influence of Polysorbate 20/80 and Polaxomer P188 on the Surface & Interfacial Properties of Bovine Serum Albumin and Lysozyme. Pharm Res. 2019; 36:107. https://doi.org/10.1007/s11095-019-2631-6 [PubMed]
- 70. Cao J, Wang X, Dai T, Wu Y, Zhang M, Cao R, Zhang R, Wang G, Jiang R, Zhou BP, Shi J, Kang T. Twist promotes tumor metastasis in basal-like breast cancer by transcriptionally upregulating ROR1. Theranostics. 2018; 8:2739–51. https://doi.org/10.7150/thno.21477 [PubMed]
- 71. Araki M, Hisamitsu T, Kinugasa-Katayama Y, Tanaka T, Harada Y, Nakao S, Hanada S, Ishii S, Fujita M, Kawamura T, Saito Y, Nishiyama K, Watanabe Y, Nakagawa O. Serum/glucocorticoid-regulated kinase 1 as a novel transcriptional target of bone morphogenetic protein-ALK1 receptor signaling in vascular endothelial cells. Angiogenesis. 2018; 21:415–23. https://doi.org/10.1007/s10456-018-9605-x [PubMed]
- 72. Schreiber T, Salhöfer L, Quinting T, Fandrey J. Things get broken: the hypoxia-inducible factor prolyl hydroxylases in ischemic heart disease. Basic Res Cardiol. 2019; 114:16. https://doi.org/10.1007/s00395-019-0725-2 [PubMed]
- 73. Cui Z, Ni NC, Wu J, Du GQ, He S, Yau TM, Weisel RD, Sung HW, Li RK. Polypyrrole-chitosan conductive biomaterial synchronizes cardiomyocyte contraction and improves myocardial electrical impulse propagation. Theranostics. 2018; 8:2752–64. https://doi.org/10.7150/thno.22599 [PubMed]