



Introduction
Colorectal cancer (CRC) is one of the most commonly diagnosed malignancies worldwide, and is associated with high morbidity and mortality [1]. Apart from surgical resection, several targeted therapies have been developed against CRC in order to improve prognosis. However, the complex mechanism of CRC genesis considerably limits the therapeutic outcomes in advanced cancer [2, 3]. Therefore, it is essential to determine the mechanisms underlying the development and progression of CRC in order to identity novel therapeutic targets.
Aldo-keto reductase family 1 member B10 (AKR1B10), a member of the AKR1B subfamily, is a 36-kDA cytosolic NADPH-dependent oxidoreductase that catalyzes the reduction of intracellular reactive oxygen species (ROS), retinaldehyde, lipid peroxidation products and xenobiotics [4, 5]. It is commonly expressed in normal epithelial tissues of the digestive tract and presents at very low level in non-gastrointestinal tissues [6, 7]. Aberrant expression of AKR1B10 has been detected in multiple solid tumors such as hepatocellular cancer [8], lung cancer [9], breast cancer [10] and pancreatic cancer [11], and strongly associated with prognosis [12–15], and downregulated in malignancies of the digestive tract, such as gastric cancer and CRC [15, 16]. AKR1B10 normally exerts a gastro-protective effect by metabolizing α, β-unsaturated carbonyl compounds produced by gut microbiota into less toxic hydroxyl compounds [17], promoting the synthesis of fatty acids or lipids in the digestive tract mucosa for the constant renewal of crypt cells [18], and mediating retinoid acid homeostasis and cell differentiation [10]. Thus, it is not surprising that aberrantly low level of AKR1B10 in the gastrointestinal tract is closely linked with the development of cancers [15, 16], as well as inflammatory conditions like diabetic nephropathy [19]. However, little is known regarding the role of AKR1B10 in CRC development, and the molecular mechanisms remain elusive.
Fibroblast growth factor 1 (FGF1) was first identified in brain and pituitary tissues [10], and functions as an insulin sensitizer in type 2 diabetes mellitus along with maintaining adipose tissue and metabolic homeostasis [20, 21]. Studies have also reported anti-inflammatory effects of FGF1 [21, 22], which is significant since metabolic disorders often progress to tumors due to adipose inflammation and systemic circulation of metabolic and inflammatory factors [23]. Therefore, we hypothesized that high level expression of AKR1B10 would suppress CRC development via a non-canonical FGF1-dependent pathway, and our findings demonstrated a novel role of AKR1B10 in CRC and identified its potential diagnostic and therapeutic relevance.
Results
AKR1B10 is downregulated in CRC tissues and related to poor prognosis
The AKR1B10 protein was highly expressed in normal colorectal tissues, and significantly lower in the CRC tissues (Figure 1A–1B, Supplementary Figure 1A). Although in situ AKR1B10 levels were similar between the T1-2 and T3-4 tissues (P > 0.05; Figure 1C), it was significantly decreased in patients with lymph node invasion compared with those without (P < 0.01, Figure 1D). Furthermore, AKR1B10 expression was reduced in CRC tissues with tumor-node-metastasis (TNM) staging I-II compared to III-IV (P < 0.01; Figure 1E). Our results were confirmed with TCGA datasets in the GEPIA platform (Supplementary Figure 1B). In addition, AKR1B10 expression was significantly associated with the depth of invasion (P < 0.05), lymph node invasion (P < 0.001) and TNM staging (P < 0.001, Table 1), while no correlation was observed with other clinicopathological variables such as age, gender, tumor size, tumor location or degree of differentiation (P > 0.05; Table 1). Univariate analysis further revealed that low AKR1B10 expression (P < 0.001), lymph node invasion (P < 0.001), degree of differentiation (P < 0.01), depth of invasion (P < 0.001) and TNM staging (P < 0.001, Table 2) were related to poor prognosis, and low AKR1B10 expression was confirmed as an independent prognostic factor for the survival of CRC patients by multivariate analysis (P < 0.001, Table 2). Therefore, we demarcated the patients according to AKR1B10 expression levels, and found that the survival of AKR1B10neg patients was significantly worse compared to the AKR1B10pos group (P < 0.05; Figure 1F–1G, Supplementary Figure 1C), regardless of age, gender, tumor size, tumor location, venous invasion, neural invasion and lymph node metastasis. In contrast, AKR1B10 expression level had no bearing on the survival of patients with staging T1-T2 invasion (P = 0.355), poor differentiation (P = 0.094) and TNM staging I-II (P = 0.075). Interestingly, elevated AKR1B10 expression was associated with favorable prognosis in patients with TNM staging III-IV but not the staging I-II patients (P = 0.065; P = 0.001; Figure 1H–1I).
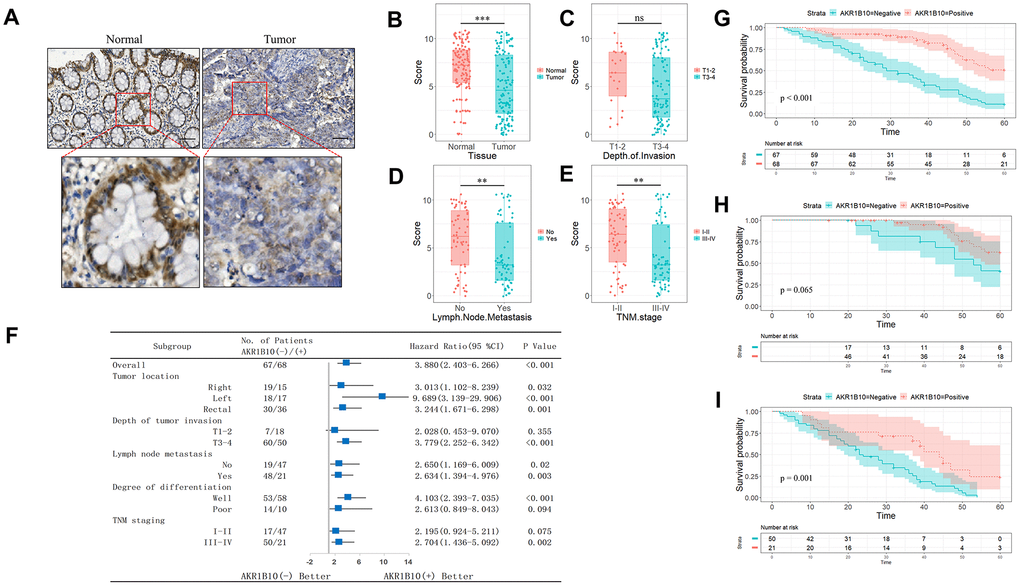
Figure 1. Expression of AKR1B10 in CRC tissues. (A) Representative IHC images showing in situ AKR1B10 expression in CRC and normal tissues (scale bar = 100μm). (B–E) IHC scores of AKR1B10 in (B) CRC vs normal tissues, (C) T I-II vs T III-IV tissues, (D) tumors with or without lymph node invasion, and (E) early vs late TNM staging. (F) OS of AKR1B10pos and AKRiB10neg CRC patients in subgroups demarcated by tumor location, depth of tumor invasion, lymph node metastasis, degree of differentiation and TNM staging. (G–I) OS of (G) AKR1B10pos and AKRiB10neg CRC patients with TNM staging I-II (H) and III-IV (I). CRC, colorectal cancer. OS, overall survival. ns, no significant difference. ** P < 0.01, *** P < 0.001.
Table 1. Relationship between AKR1B10 and clinic-pathological factors in 135 CRC patients.
| Variables | AKR1B10 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Negative | Positive | P value | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age (years) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ≤60 | 30 | 27 | 0.551 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| >60 | 37 | 41 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Gender | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Male | 33 | 24 | 0.863 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Female | 44 | 34 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Size (cm) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| <5 | 26 | 31 | 0.702 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ≥5 | 33 | 45 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Tumor location | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Right | 19 | 15 | 0.595 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Left | 18 | 17 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Rectal | 30 | 36 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Depth of tumor invasion | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| T1-2 | 7 | 18 | 0.017a | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| -4 | 60 | 50 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Lymph node metastasis | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| No | 19 | 47 | <0.001b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Yes | 48 | 21 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Degree of differentiation | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Well | 53 | 58 | 0.347 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Poor | 14 | 10 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TNM staging | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| I-II | 17 | 47 | <0.001b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| III-IV | 50 | 21 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| aP < 0.05, bP < 0.001 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 2. Results of univariate and multivariate analyses of postoperative patients’ survival by Cox’s proportional hazard model.
| Varieties | n | Univariate analysis | Multivariate analysis | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HR | 95% CI | P | HR | 95% CI | P | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age (≤60 or >60 years) | 57/78 | 1.084 | 0.696-1.687 | 0.722 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Gender (Male / Female) | 77/58 | 0.876 | 0.561-1.366 | 0.559 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Size of tumor (≤5 or >5 cm) | 59/76 | 0.654 | 0.418-1.023 | 0.063 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Depth of tumor invasion (T1-2 / T3-4) | 25/110 | 0.223 | 0.102-0.487 | <0.001c | 0.360 | 0.161-0.805 | 0.013a | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Lymph node metastasis (negative / positive) | 66/69 | 0.179 | 0.108-0.298 | <0.001c | 7.731 | 1.656-36.084 | 0.009b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Degree of differentiation (moderate-well/poor) | 111/24 | 0.461 | 0.270-0.787 | 0.005b | 0.799 | 0.457-1.395 | 0.429 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TNM staging (I-II / III-IV) | 64/71 | 0.157 | 0.093-0.264 | <0.001c | 0.033 | 0.006-0.164 | <0.001c | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AKR1B10 expression (negative / positive) | 67/68 | 3.880 | 2.403-6.266 | <0.001c | 2.492 | 1.491-4.164 | <0.001c | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| aP < 0.05, bP < 0.01, cP < 0.001 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ectopic AKR1B10 inhibits proliferation and migration of CRC cells in vitro
Pooled analysis of CRC and normal tissues across 7 Oncomine datasets (Figure 2A) revealed significant downregulation of AKR1B10 mRNA in the CRC tissues, which was also consistent with the findings of Gaedcke et al, Kaiser et al and Hong et al (Supplementary Figure 2A). Furthermore, AKR1B10 expression was also downregulated in the CRC tissues of our cohort compared to the paired normal tissues (Figure 2B, Supplementary Figure 2B–2C), as well as in multiple CRC cell lines (Figure 2C–2D, Supplementary Figure 2D). The HT29 cells expressed the highest levels of AKR1B10, while that in the SW480, HCT116 and RKO cells were relatively low. The biological role of AKR1B10 was further analyzed using knockdown (KD) and overexpression (OE) constructs (Figure 2E). The proliferation rate of AKR1B10-KD cells was significantly higher, and that of AKR1B10-OE cells was inhibited compared to the negative controls (Figure 2F). Consistent with this, the AKR1B10-KD cells also showed enhanced colony-formation ability, which was markedly suppressed in the AKR1B10-OE cells (Figure 2G). Overexpression of AKR1B10 also inhibited in vitro migration of CRC cells, whereas its knockdown had the opposite effect (Figure 2H). Taken together, AKR1B10 acts as a tumor suppressor in CRC, and its ectopic expression promotes the growth of CRC cells in vitro.
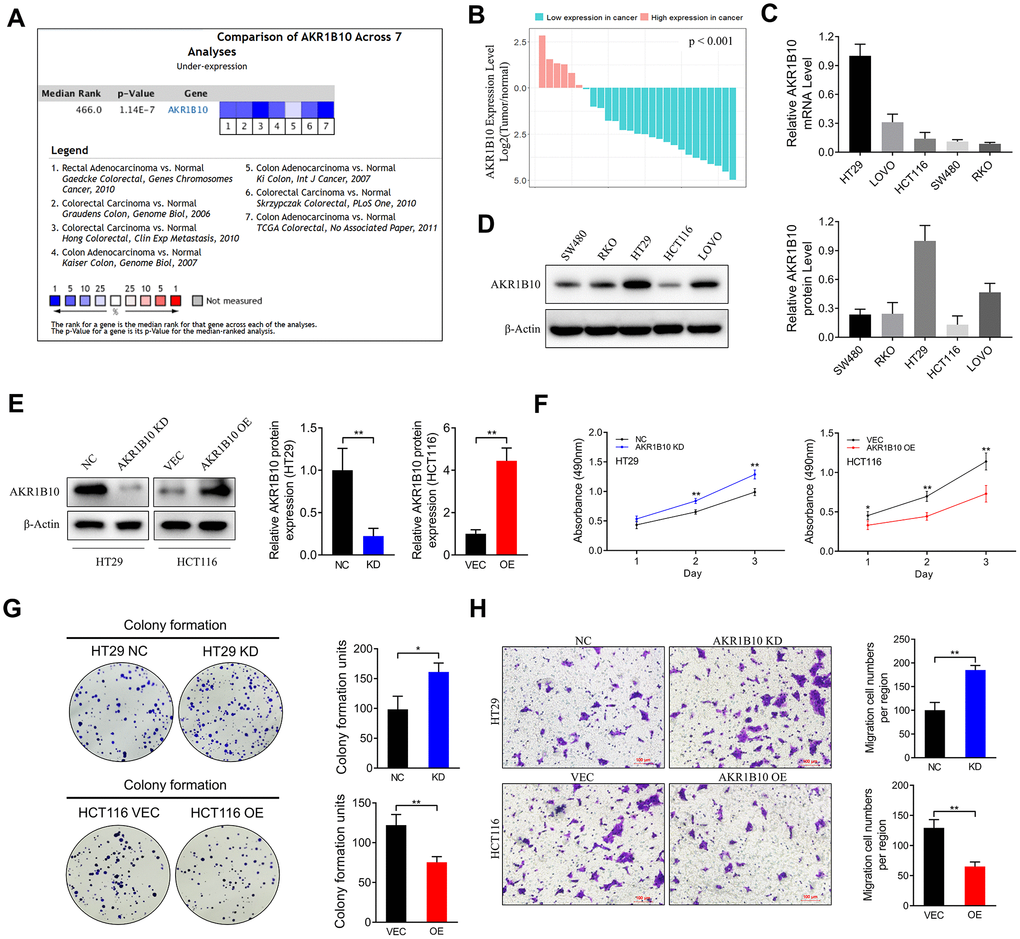
Figure 2. Effect of AKR1B10 on CRC cell proliferation and migration ability. (A) Comparison of AKR1B10 mRNA expression in CRC and normal tissues across 7 Oncomine datasets. (B–C) AKR1B10 mRNA levels in (B) 27 paired CRC and normal tissues and (C) 5 CRC cell lines. (D–E) Immunoblots showing AKR1B10 protein levels in (D) wild type and (E) AKR1B10-KD and AKR1B10-OE CRC cell lines. (F–H) Proliferation rates (F), colony forming capacity (G) and migration rates (H) of AKR1B10-KD and AKR1B10-OE CRC cells. CRC, colorectal cancer. CTL, control; NC, negative control; KD, AKR1B10-shRNA; VEC, vector; OE, AKR1B10 overexpression plasmid. Data are presented as mean ± SD (n=3). *P < 0.05, **P < 0.01, ***P < 0.001.
AKR1B10 is closely related with FGF1 expression levels in CRC tissues
Since FGF1 is associated with inflammation in the tumor microenvironment, we next analyzed the potential correlation between AKR1B10 and FGF1 in TCGA datasets. AKR1B10 expression levels in the CRC tissues were closely related to that of FGF1 (P < 0.001, Figure 3A). Furthermore, FGF1 mRNA levels were also significantly higher in most CRC tissue specimens compared to the paired normal tissues (P < 0.001, Figure 3B, Supplementary Figure 3A, 3B). Interestingly, high AKR1B10 levels were significantly correlated with reduced FGF1 expression in CRC tissues (P = 0.001), while no such correlation was seen in normal tissues (P > 0.05, Figure 3C). Based on both variables, the tumor and normal groups were stratified into two clusters (Supplementary Figure 3C–3D), and most normal specimens belonged to Cluster 1 (71.4%) as opposed to Cluster 2 (28.6%) whereas the tumor samples were concentrated in Cluster 2 (63% compared to 37% in Cluster 1). The FGF1 protein levels were also significantly higher in CRC compared to the normal tissues (Figure 3D–3E), and its reduced expression was predictive of longer survival (Figure 3F). In the cluster analysis as well, the IHC scores of AKR1B10 and FGF1 were significantly different between tumor and normal tissues (Figure 3G), with 23.9% and 76.1% of the normal samples, and 51.9% and 48.1% tumor samples respectively present in Cluster 1 and Cluster 2 (Figure 3H). Taken together, AKR1B10 and FGF1 levels can distinguish between CRC and normal tissues.
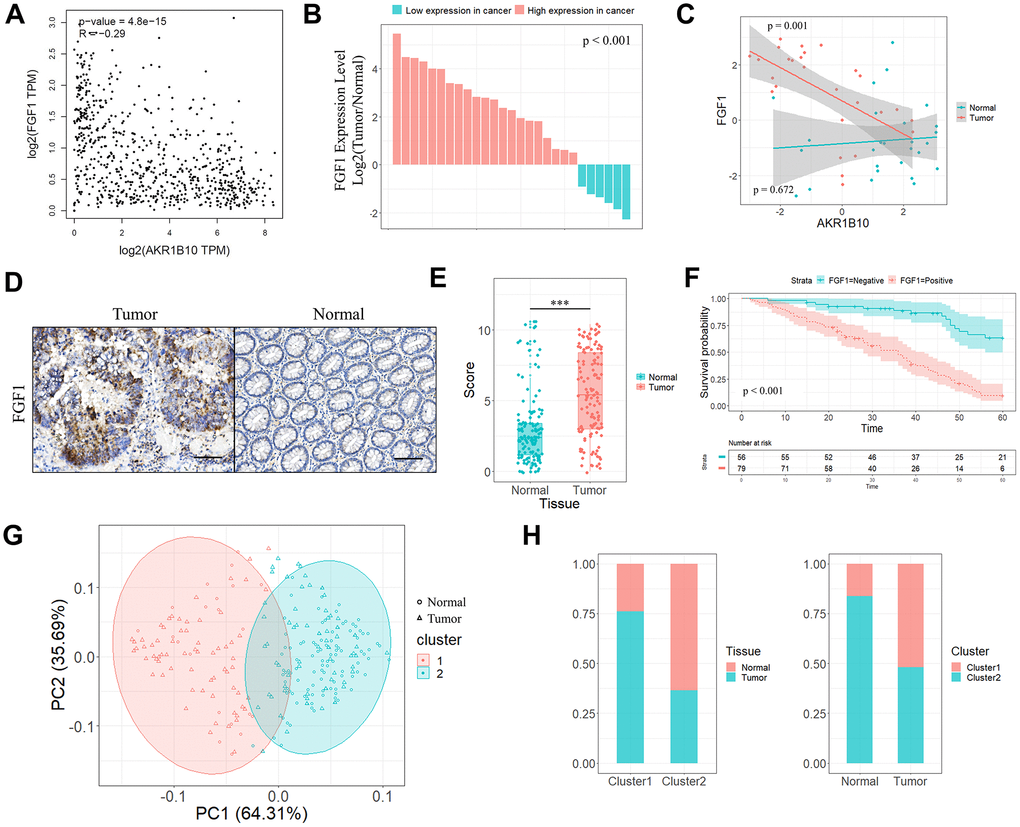
Figure 3. Correlation between AKR1B10 and FGF1 in CRC tissues. (A) Correlation analysis of AKR1B10 and FGF1 levels in CRC tissues from TCGA datasets by GEPIA platform. (B) FGF1 mRNA levels in 27 paired CRC and normal tissues. (C) Correlation between AKR1B10 and FGF1 levels in the above. (D) Representative IHC images showing in situ FGF1 expression in CRC and normal tissues (scale bar = 100μm) and (E) corresponding IHC scores. (F) OS of 135 CRC patients demarcated by FGF1 expression levels. (G) Stratification of 135 pairs of CRC and normal tissues into cluster 1 (red) and cluster 2 (green) according to AKR1B10 and FGF1 IHC scores. (H) Percentage of tumor and normal samples in each cluster. CRC, colorectal cancer. OS, overall survival. *** P < 0.001.
AKR1B10 inhibits colorectal tumorigenesis in vivo by targeting FGF1
The role of AKR1B10 in CRC tumor growth was analyzed by establishing an in vivo xenograft model using wild-type and AKR1B10-KD HT29 cells. Depletion of AKR1B10 had no obvious effect on the body weight of the mice (Figure 4A), but significantly enhanced the proliferative capacity of the CRC cells, which was manifested as increased tumor size (Figure 4B) and weight (Figure 4C–4D) compared to control group. However, the net body weights obtained after subtracting the tumor weights were significantly lower in the mice implanted with AKR1B10-KD CRC cells (Figure 4E). Furthermore, in situ AKR1B10 mRNA levels were markedly lower and that of FGF1 was higher in the AKR1B10-KD tumors (Figure 4F–4G), and showed significant statistical correlation (Figure 4H). We next performed a cluster analysis to consider the combined effects of body weight, tumor volume, tumor weight and AKR1B10/FGF1 levels (Figure 4I), and found that 16.67% of the AKR1B10-KD and 83.33% of the NC group mice were in Cluster 1 (Figure 4J). To gain further mechanism insights, we analyzed the FGF1 levels in CRC cells transfected with AKR1B10-shRNA or AKR1B10 overexpression plasmid, and found that AKR1B10 downregulated FGF1 while knocking it down had the opposite effect (Figure 5A). To further determine the role of FGF1 in AKR1B10-mediated regulation of CRC progression, the HT29 cells were co-transfected with AKR1B10-shRNA and FGF1-shRNA. Interestingly, inhibiting AKR1B10 restored FGF1 expression levels following the latter’s knockdown (Figure 5B) but its overexpression did not rescue the CRC cells from the anti-proliferative effects of FGF1 knockdown (Figure 5C–5E). Taken together, AKR1B10-mediated inhibition of CRC cells is dependent on FGF1.
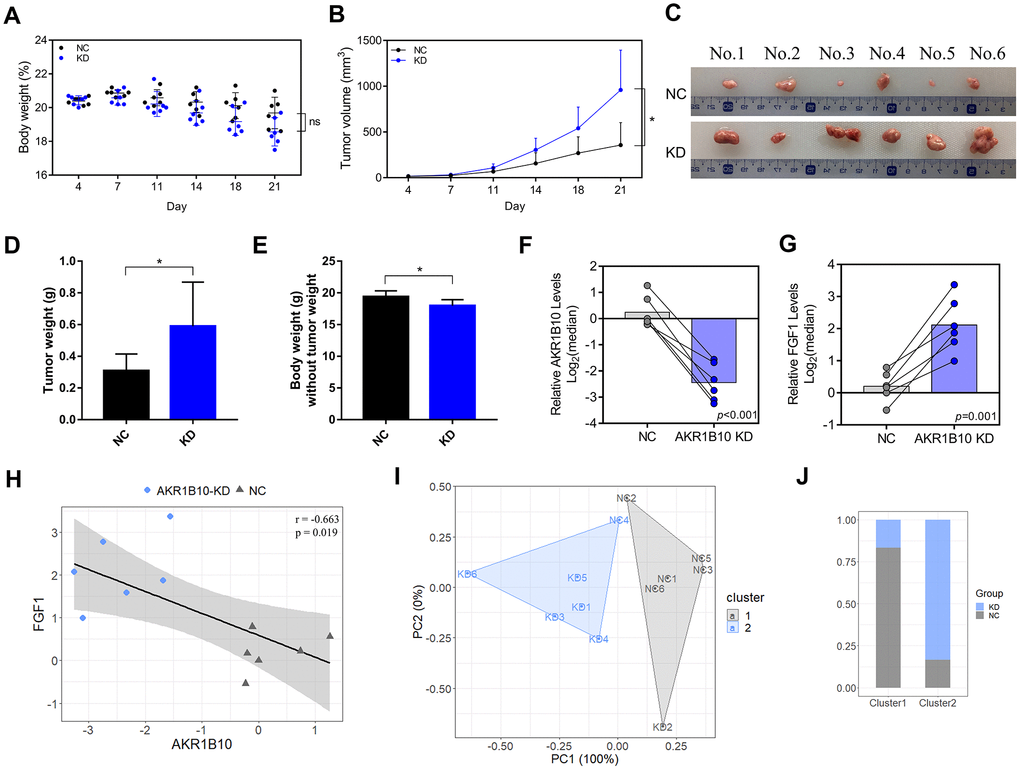
Figure 4. AKR1B10 knockdown suppresses CRC tumor growth in vivo. (A–B) Total body weight (A) and tumor volume (B) of the mice during the experiment. (C) Representative pictures of subcutaneous tumors harvested from NC and AKR1B10-KD group. (D) The weights of tumor masses. (E) Net body weight after subtracting the respective tumor weights. (F–G) Relative AKR1B10 (F) and FGF1 (G) mRNA levels in the tumors and their (H) correlation. (I) Stratification of mice into cluster 1 (grey) and cluster 2 (blue) according to body weight, tumor volume, tumor weight and AKR1B10 and FGF1 mRNA levels. (J) Percentage of NC and AKR1B10-KD mice in each cluster. Data are presented as mean ± SD. CRC, colorectal cancer. NC, negative control; KD, AKR1B10-shRNA. *P < 0.05, **P < 0.01, ***P < 0.001.
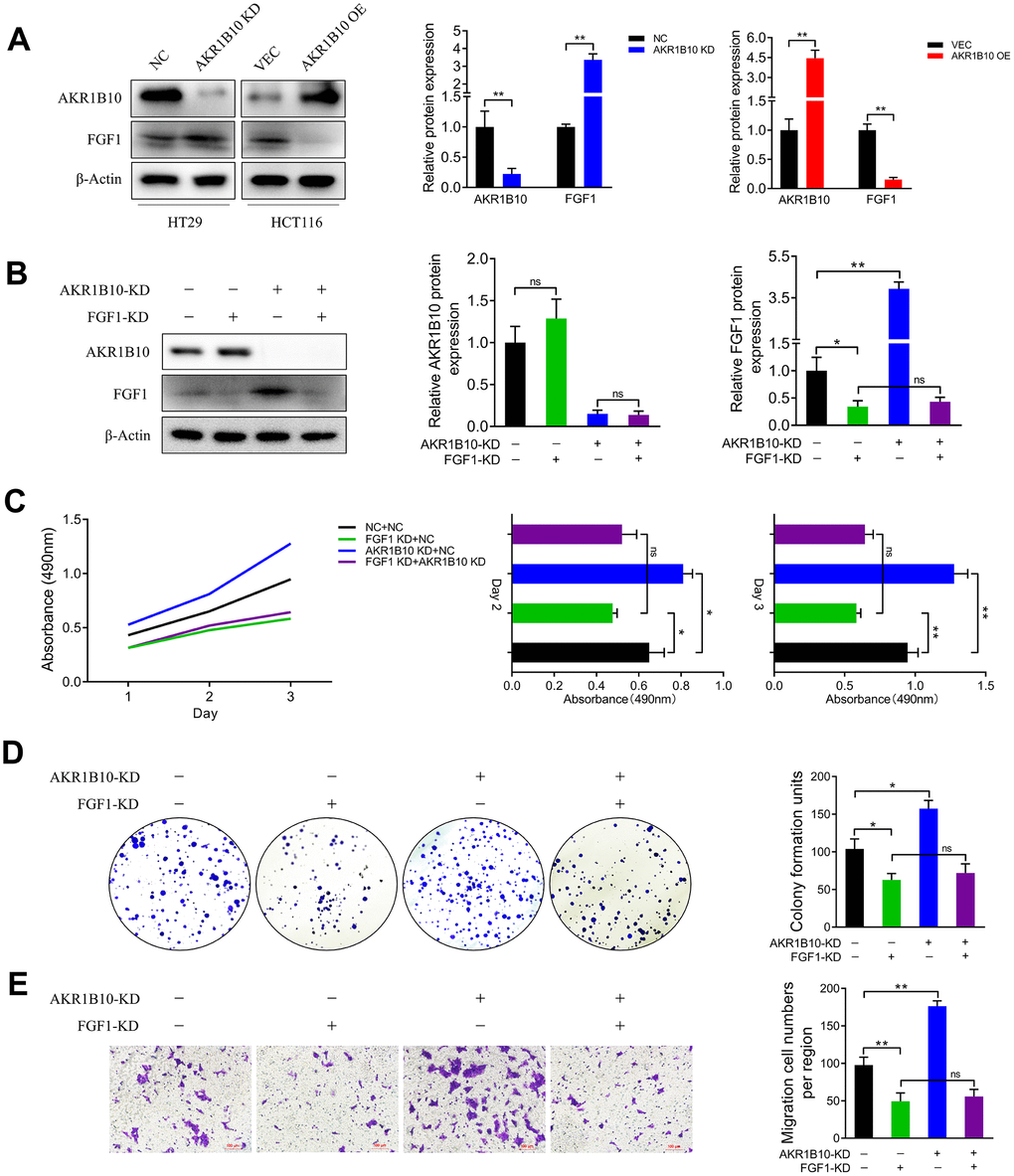
Figure 5. AKR1B10 inhibits CRC cell growth in an FGF1-dependent manner. (A) Immunoblot showing AKR1B10, FGF1 and GAPDH protein levels in HT29 cells transfected with AKR1B10-shRNA and in HCT116 cells transfected with AKR1B10 overexpression plasmid. (B) Immunoblot showing AKR1B10, FGF1 and GAPDH protein levels in HT29 transfected with FGF1-shRNA alone or in combination with AKR1B10-shRNA. (C–E) Proliferation rates (C), colony forming capacity (D) and migration rates (E) of the HT29 cells transfected as above. Data are presented as mean ± SD. NC, negative control; KD, AKR1B10-shRNA; VEC, vector; OE, AKR1B10 overexpression plasmid. “-”, control-shRNA. “+”, AKR1B10 or FGF1 shRNA. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
AKR1B10 metabolizes various substrates such as retinaldehyde, lipid peroxidation products, and xenobiotics [5, 25–27]. It is primarily expressed in normal gastrointestinal epithelial tissues, and usually non-detectable in non-digestive tract tissues [28, 29]. Recent studies have implicated AKR1B10 in tumor growth and metastasis, and reported aberrant expression levels in various cancers [14, 30]. We found that AKR1B10 levels were high in the normal colorectal tissues and decreased significantly in primary CRC tumors compared to the surrounding normal tissues. Furthermore, CRC patients overexpressing AKR1B10 had better OS compared to the low-expressing group, which is consistent with previous studies [16, 31–33]. Nevertheless, the difference in the expression levels of AKR1B10 in the gastrointestinal and other solid tumors [12, 14–16, 34–38] has limited the clinical relevance of AKR1B10 as a therapeutic target. Although a previous study correlated AKR1B10 expression to the prognosis of CRC patients [15], its role in CRC development remains unclear. We found that reduced levels of AKR1B10 in the tumor tissues correlated significantly with advanced stages, greater invasiveness, increased tumor differentiation and poor survival of CRC patients, indicating that AKR1B10 is a potential tumor suppressor in CRC. Consistent with this, ectopic expression of AKR1B10 in the CRC cells significantly inhibited their proliferation, clonal expansion and migration in vitro.
AKR1B10 is a potential biomarker of CRC, although the mechanisms underlying AKR1B10 down-regulation in CRC and AKR1B10-mediated tumorigenesis remain to be clarified. Overexpression of AKR1B10 significantly inhibited the proliferation and migration of CRC cells. Correlation analysis on TGCA datasets showed a significant association between AKR1B10 and FGF1. The latter is a member of the fibroblast growth factor family that is involved in cell proliferation and migration [39–41], and acts as an oncogene in several cancers. FGF1 is aberrantly expressed in pancreatic cancer, lung cancer, glioblastoma and prostate cancer [42–45]. Elevated FGF1 levels are associated with increased angiogenesis and decreased survival in serous ovarian adenocarcinoma [46], and is a potential therapeutic target for ovarian cancer [47, 48]. We found that FGF1 was overexpressed in CRC tissues and predicted poor prognosis. Furthermore, cluster analysis indicated that both FGF1 and AKR1B10 expression levels were able to distinguish between the tumor and adjacent normal tissues, and pointed to a functional relationship as well.
AKR1B10 and AKR1B1 are closely related to inflammation [15, 19], and AKR1B10 in particular regulates inflammatory factors in the tumor microenvironment, which mobilizes the host immune response and promotes tumor suppression [15, 19, 49]. FGF1 activation is mediated via the PI3K-Akt signaling pathway that lies upstream of mTOR [50], which is related to autophagy, apoptosis and metabolism of cancer cells, as well as the NLRP3-mediated inflammatory response [51, 52]. Based on previous evidence and our findings, we hypothesized that AKR1B10 would inhibit the proliferation and migration of CRC cells by regulating FGF1-dependent signaling pathways. Indeed, AKRB110 inhibited FGF1 in CRC cell lines, and elevated FGF1 in response to AKR1B10 depletion promoted xenograft tumor growth in a mouse model. In addition, an inverse correlation between FGF1 and AKR1B10 was also observed in human CRC tumors. The likely mechanism underlying the inhibitory effect of AKR1B10 is the induction of an anti-tumor inflammatory response [15, 53] by targeting FGF1, which is related to the growth and migration of CRC cells [54, 55]. The involvement of an FGF1-dependent pathway is significant in the context of therapeutically targeting AKR1B10 in CRC [56]. Since AKR1B10 was not able to rescue CRC cells after FGF1 knockdown, the latter is possibly a downstream target of AKR1B10. Although the exact regulatory mechanism warrants future investigation, our findings provide a rationale for targeting both as a combination therapy for CRC.
Materials and Methods
Human tissue specimens
A total of 135 pairs of CRC and adjacent normal colon tissues were collected immediately after surgical resection at the Department of General Surgery of the First Affiliated Hospital of Soochow University (Suzhou, China) from 2010 to 2013. None of the patients had received radiotherapy or chemotherapy before radical surgery, and all tissue specimens were verified histo-pathologically. The study was approved by the Independent Ethics Committee of the First Affiliated Hospital of Soochow University (IRB approval number, 2020-076), and all patients provided written informed consent.
Immunohistochemistry (IHC) evaluation
Tissue specimens were fixed with 10% formalin, embedded in paraffin, and cut into 5μm-thick sections. After cleaned in xylene and rehydrated through an ethanol gradient, the sections were treated with 3% hydrogen peroxide to quench endogenous peroxidases, and then boiled in 10mM citrate buffer (pH 6) for antigen retrieval. The processed sections were then blocked with 10% goat serum for 30 min, and incubated overnight with 1:200 diluted polyclonal anti-human AKR1B10 (BOSTER, Wuhan, China) or anti-human FGF1 (BOSTER, Wuhan, China) at 4°C. Color was developed using a tissue staining kit (Zhongshan Biotechnology, Beijing, China). The AKR1B10 or FGF1 staining scores were evaluated in five random fields per slide by two pathologists YuHong Wang (The First Affiliated Hospital of Soochow University) and Zheng Zhi (The Soochow University) in a blinded manner as previously described [24]. The percentage of positively stained cells was scored as follows: 0 - 0-5%; 1 - 6-25%; 2 - 26-50%; 3 - 51-75%; 4 - >75%. The staining intensity was scored as 0 (negative), 1 (weak), 2 (moderate) and 3 (strong). The final score was the average of the percentage score multiplied by intensity score, and graded as follows: – (0), + (1-4), ++ (5-8) and +++ (9-12). Samples with final scores ++ or +++ were graded as positive, and – or + as negative.
Bioinformatics analysis
CRC gene expression datasets were downloaded from the Oncomine (https://www.oncomine.org), CCLE (Cancer Cell Line Encyclopedia, https://portals.broadinstitute.org/ccle) and GEPIA (Gene Expression Profiling Interactive Analysis, http://gepia.cancer-pku.cn) databases, and analyzed by established protocols.
Survival analysis
All patients were followed up by personal or telephonic interviews for 60 months, and the time point was set as the date of CRC-related death or 60 months after surgery. Self-developed R program (version 3.6.1 for Windows, http://cran.r-project.org/) was used for sample classification and prognostic analysis. The patients were classified into two subgroups according to the IHC staining scores, and Kaplan-Meier survival curves were plotted for both groups using the “survminer” package (version 0.4.6, https://cran.r-project.org/web/packages/survminer/index.html). The log-rank test was used for statistical comparison and P < 0.05 was considered significant.
Cell culture and transfection
Five human CRC cell lines (HCT116, HT29, LOVO, SW480 and RKO) were purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, China), and were cultured in RPMI 1640 medium (Hyclone) supplemented with 10% fetal bovine serum (Gibco, USA), penicillin G sodium (100U/ml) and streptomycin (100μg/ml) at 37°C under 5% CO2. The HT29 cells were grown till 70% confluency, and transfected with human AKR1B10 or human FGF1 shRNA according to the manufacturer’s instructions. The transfected cells were selected using 500μg/ml G418 (Roche, Switzerland) for 3-4 weeks, and clones with a stable knockdown of AKR1B10 or FGF1 were selected for further experiments. Control cells were stably transfected with scrambled shRNA. In addition, 70% confluent HCT116 cells were transfected with the AKR1B10 cDNA or empty plasmid using X-tremegene HP at 1:1 ratio, and harvested after 24h. Transient overexpression and silencing were confirmed by RT-PCR and Western blotting. All stable transfectants were used by the 8th passage.
RNA isolation and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from the tissues or cells using TRIzol reagent (Invitrogen, Life Technologies, USA) according to the manufacturer’s protocol. Following DNAse I (Thermo Fisher Scientific, USA) treatment to remove genomic DNA, 1μg RNA was reverse transcribed using a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, USA). The qRT-PCR was performed using Power SYBR® Green PCR Master Mix (ABI, USA) on the 7500 real time PCR system (ABI, USA) according to the manufacturer’s instructions. Fold changes were calculated relative to β-actin (internal control) using the 2-ΔΔCT method. The following primers were used: AKR1B10 forward (5’-CCCAAAGATGATAAAGGTAATGCCATCGGT-3’) and reverse (5’-CGATCTGGAAGTGGCTGAAATTGGAGA-3’); FGF1 forward (5’-GTGGATGGGACAAGGGACAG-3’) and reverse (5’-GGCAGGGGGAGAAACAAGAT-3’); β-actin forward (5’- CCACACTGTGCCCATCTACG-3’) and reverse (5’-AGGATCTTCATGAGGTAGTCAGTCAG-3’). The PCR conditions were: initial denaturation at 95°C for 5 min, followed by 40 cycles of denaturation at 95°C for 30 sec, annealing at 55°C for 30 sec and extension at 72°C for 30 sec, and final extension at 72°C for 7 min.
Protein isolation and Western blotting
Cells were lysed in ice-cold RIPA lysis buffer supplemented with protease and phosphatase inhibitors (KeyGEN Inc., Nanjing, China) according to the manufacturer’s protocol. The extracted proteins were separated by SDS-PAGE and transferred onto PVDF membranes (Millipore, USA). After blocking with 5% non-fat milk for 1h, the membranes were probed overnight with anti-AKR1B1 (1:1000, Cell Signaling Technology), anti-FGF1 (1:1000, Cell Signaling Technology) and anti-β-Actin (1:5000, Cell Signaling Technology) antibodies at 4°C with gentle shaking, followed by horseradish peroxidase-conjugated secondary antibodies. The protein bands were visualized by chemiluminescence and quantified by ImageJ for Windows (NIH, USA).
MTT assay
Cell viability was determined using an MTT assay kit (Amresco, USA) according to the manufacturer’s instructions. Briefly, 2000 transfected cells were seeded in 96-well plates, and cultured for 12, 24, 36, 48, 60 and 72h. The MTT solution was added 4h prior to the termination of each time point, and the supernatants were removed. The formazan crystals were dissolved in 150μl DMSO per well for 10 min with gentle shaking, and the absorbance at 490nm was measured using a microplate reader.
Cell migration assay
Cell migration was assessed using Transwell inserts (pore size 8μm; Corning, New York, USA). The cells were seeded into the upper chambers of the inserts at the density of 50,000 cells/200μl in serum-free RPMI 1640 medium, and the lower chambers were filled with 750μl complete medium per well. After incubating for 24h at 37°C, the cells remaining on the upper surface of the membrane were removed using a cotton swab. The filters were then fixed with 4% paraformaldehyde, and the cells on the lower surface were stained with 0.1% crystal violet and counted in 5 random fields per sample.
Colony formation assay
The suitably transfected cells were seeded in 6-well plates at the density of 1000 cells/well, and cultured for 10 days before being fixed and stained with 0.1% crystal violet. The colonies with more than 100 cells were counted at 40x magnification under an optical microscope (Nikon, Japan) fitted with a digital camera (Nikon, Japan).
Subcutaneous xenograft establishment
SPF male BALB/c nude mice (3-5weeks old and weighing 16-18 g) were purchased from Shanghai SLRC laboratory Animal Co. Ltd. (Shanghai, China). The mice were randomly divided into the AKR1B10 knock down (KD) and negative control (NC) groups (n = 6 per group), and accordingly injected subcutaneously with 5×106 AKR1B10-KD or NC-shRNA HT29 into the left and right dorsal flank on day 0. All animal experiments were approved by the Animal Ethics Committee of Soochow University (Suzhou, China).
Statistical analysis
All data were presented as mean ± SD of three independent experiments. Statistical analyses were performed using SPSS 22.0 software (SPSS Inc., Chicago, IL, USA), GraphPad Prism 8 (San Diego, CA) and R programs. The Student's t-test (unpaired, two-tailed), Mann–Whitney U test or one-way ANOVA were used to compare means between groups. IHC results were analyzed by Chi-squared or Fisher's exact tests. Unsupervised learning cluster analysis was performed using the “cluster” package (version 2.1.0, https://cran.r-project.org/web/packages/cluster/index.html) in R programs. P < 0.05 was considered statistically significant.
Supplementary Materials
Acknowledgments
We gratefully acknowledge the valuable cooperation of YuHong Wang (The First Affiliated Hospital of Soochow University) and Zheng Zhi (The Soochow University) in assessing the IHC score.This work was supported by Project of National Science Foundation of Jiangsu Province, China [grant number BK20161225]. Nanjing Medical University Science and Technology Development Fund (NMUB2018231), Suzhou Special Project of Diagnosis and Treatment for key Clinical Disease (LCZX201814) and Suzhou Science and Technology Development Project (SYSD2018144).
Conflicts of Interest
The authors declare no conflicts of interest.
Funding
This work was supported by Project of National Science Foundation of Jiangsu Province, China [grant number BK20161225]. Nanjing Medical University Science and Technology Development Fund (NMUB2018231), Suzhou Special Project of Diagnosis and Treatment for key Clinical Disease (LCZX201814) and Suzhou Science and Technology Development Project (SYSD2018144).
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019; 69:7–34. https://doi.org/10.3322/caac.21551 [PubMed]
- 2. Punt CJ, Koopman M, Vermeulen L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat Rev Clin Oncol. 2017; 14:235–46. https://doi.org/10.1038/nrclinonc.2016.171 [PubMed]
- 3. Tiwari A, Saraf S, Verma A, Panda PK, Jain SK. Novel targeting approaches and signaling pathways of colorectal cancer: an insight. World J Gastroenterol. 2018; 24:4428–35. https://doi.org/10.3748/wjg.v24.i39.4428 [PubMed]
- 4. Huang L, He R, Luo W, Zhu YS, Li J, Tan T, Zhang X, Hu Z, Luo D. Aldo-keto reductase family 1 member B10 inhibitors: potential drugs for cancer treatment. Recent Pat Anticancer Drug Discov. 2016; 11:184–96. https://doi.org/10.2174/1574892811888160304113346 [PubMed]
- 5. Zhong L, Liu Z, Yan R, Johnson S, Zhao Y, Fang X, Cao D. Aldo-keto reductase family 1 B10 protein detoxifies dietary and lipid-derived alpha, beta-unsaturated carbonyls at physiological levels. Biochem Biophys Res Commun. 2009; 387:245–50. https://doi.org/10.1016/j.bbrc.2009.06.123 [PubMed]
- 6. Hyndman DJ, Flynn TG. Sequence and expression levels in human tissues of a new member of the aldo-keto reductase family. Biochim Biophys Acta. 1998; 1399:198–202. https://doi.org/10.1016/s0167-4781(98)00109-2 [PubMed]
- 7. Cao D, Fan ST, Chung SS. Identification and characterization of a novel human aldose reductase-like gene. J Biol Chem. 1998; 273:11429–35. https://doi.org/10.1074/jbc.273.19.11429 [PubMed]
- 8. Han C, Gao L, Zhao L, Sheng Q, Zhang C, An Z, Xia T, Ding Y, Wang J, Bai H, Dou X. Immunohistochemistry detects increased expression of aldo-keto reductase family 1 member B10 (AKR1B10) in early-stage hepatocellular carcinoma. Med Sci Monit. 2018; 24:7414–23. https://doi.org/10.12659/MSM.910738 [PubMed]
- 9. Hung JJ, Yeh YC, Hsu WH. Prognostic significance of AKR1B10 in patients with resected lung adenocarcinoma. Thorac Cancer. 2018; 9:1492–99. https://doi.org/10.1111/1759-7714.12863 [PubMed]
- 10. Huang C, Cao Z, Ma J, Shen Y, Bu Y, Khoshaba R, Shi G, Huang D, Liao DF, Ji H, Jin J, Cao D. AKR1B10 activates diacylglycerol (DAG) second messenger in breast cancer cells. Mol Carcinog. 2018; 57:1300–10. https://doi.org/10.1002/mc.22844 [PubMed]
- 11. Chung YT, Matkowskyj KA, Li H, Bai H, Zhang W, Tsao MS, Liao J, Yang GY. Overexpression and oncogenic function of aldo-keto reductase family 1B10 (AKR1B10) in pancreatic carcinoma. Mod Pathol. 2012; 25:758–66. https://doi.org/10.1038/modpathol.2011.191 [PubMed]
- 12. Cheng BY, Lau EY, Leung HW, Leung CO, Ho NP, Gurung S, Cheng LK, Lin CH, Lo RC, Ma S, Ng IO, Lee TK. IRAK1 augments cancer stemness and drug resistance via the AP-1/AKR1B10 signaling cascade in hepatocellular carcinoma. Cancer Res. 2018; 78:2332–42. https://doi.org/10.1158/0008-5472.CAN-17-2445 [PubMed]
- 13. Jung YJ, Lee EH, Lee CG, Rhee KJ, Jung WS, Choi Y, Pan CH, Kang K. AKR1B10-inhibitory selaginella tamariscina extract and amentoflavone decrease the growth of A549 human lung cancer cells in vitro and in vivo. J Ethnopharmacol. 2017; 202:78–84. https://doi.org/10.1016/j.jep.2017.03.010 [PubMed]
- 14. Li J, Guo Y, Duan L, Hu X, Zhang X, Hu J, Huang L, He R, Hu Z, Luo W, Tan T, Huang R, Liao D, et al. AKR1B10 promotes breast cancer cell migration and invasion via activation of ERK signaling. Oncotarget. 2017; 8:33694–703. https://doi.org/10.18632/oncotarget.16624 [PubMed]
- 15. Taskoparan B, Seza EG, Demirkol S, Tuncer S, Stefek M, Gure AO, Banerjee S. Opposing roles of the aldo-keto reductases AKR1B1 and AKR1B10 in colorectal cancer. Cell Oncol (Dordr). 2017; 40:563–78. https://doi.org/10.1007/s13402-017-0351-7 [PubMed]
- 16. Yao HB, Xu Y, Chen LG, Guan TP, Ma YY, He XJ, Xia YJ, Tao HQ, Shao QS. AKR1B10, a good prognostic indicator in gastric cancer. Eur J Surg Oncol. 2014; 40:318–24. https://doi.org/10.1016/j.ejso.2013.12.014 [PubMed]
- 17. Ames BN. Dietary carcinogens and anti-carcinogens. J Toxicol Clin Toxicol. 1984; 22:291–301. https://doi.org/10.3109/15563658408992561 [PubMed]
- 18. Laffin B, Petrash JM. Expression of the aldo-ketoreductases AKR1B1 and AKR1B10 in human cancers. Front Pharmacol. 2012; 3:104. https://doi.org/10.3389/fphar.2012.00104 [PubMed]
- 19. Shaw N, Yang B, Millward A, Demaine A, Hodgkinson A. AKR1B10 is induced by hyperglycaemia and lipopolysaccharide in patients with diabetic nephropathy. Cell Stress Chaperones. 2014; 19:281–87. https://doi.org/10.1007/s12192-013-0455-6 [PubMed]
- 20. Gasser E, Moutos CP, Downes M, Evans RM. FGF1 - a new weapon to control type 2 diabetes mellitus. Nat Rev Endocrinol. 2017; 13:599–609. https://doi.org/10.1038/nrendo.2017.78 [PubMed]
- 21. Suh JM, Jonker JW, Ahmadian M, Goetz R, Lackey D, Osborn O, Huang Z, Liu W, Yoshihara E, van Dijk TH, Havinga R, Fan W, Yin YQ, et al. Endocrinization of FGF1 produces a neomorphic and potent insulin sensitizer. Nature. 2014; 513:436–39. https://doi.org/10.1038/nature13540 [PubMed]
- 22. Liang G, Song L, Chen Z, Qian Y, Xie J, Zhao L, Lin Q, Zhu G, Tan Y, Li X, Mohammadi M, Huang Z. Fibroblast growth factor 1 ameliorates diabetic nephropathy by an anti-inflammatory mechanism. Kidney Int. 2018; 93:95–109. https://doi.org/10.1016/j.kint.2017.05.013 [PubMed]
- 23. Iyengar NM, Gucalp A, Dannenberg AJ, Hudis CA. Obesity and cancer mechanisms: tumor microenvironment and inflammation. J Clin Oncol. 2016; 34:4270–76. https://doi.org/10.1200/JCO.2016.67.4283 [PubMed]
- 24. Yao Y, Yang X, Sun L, Sun S, Huang X, Zhou D, Li T, Zhang W, Abumrad NA, Zhu X, He S, Su X. Fatty acid 2-hydroxylation inhibits tumor growth and increases sensitivity to cisplatin in gastric cancer. EBioMedicine. 2019; 41:256–67. https://doi.org/10.1016/j.ebiom.2019.01.066 [PubMed]
- 25. Crosas B, Hyndman DJ, Gallego O, Martras S, Parés X, Flynn TG, Farrés J. Human aldose reductase and human small intestine aldose reductase are efficient retinal reductases: consequences for retinoid metabolism. Biochem J. 2003; 373:973–79. https://doi.org/10.1042/BJ20021818 [PubMed]
- 26. Spite M, Baba SP, Ahmed Y, Barski OA, Nijhawan K, Petrash JM, Bhatnagar A, Srivastava S. Substrate specificity and catalytic efficiency of aldo-keto reductases with phospholipid aldehydes. Biochem J. 2007; 405:95–105. https://doi.org/10.1042/BJ20061743 [PubMed]
- 27. Shen Y, Zhong L, Johnson S, Cao D. Human aldo-keto reductases 1B1 and 1B10: a comparative study on their enzyme activity toward electrophilic carbonyl compounds. Chem Biol Interact. 2011; 191:192–98. https://doi.org/10.1016/j.cbi.2011.02.004 [PubMed]
- 28. Penning TM. The aldo-keto reductases (AKRs): overview. Chem Biol Interact. 2015; 234:236–46. https://doi.org/10.1016/j.cbi.2014.09.024 [PubMed]
- 29. DiStefano JK, Davis B. Diagnostic and prognostic potential of AKR1B10 in human hepatocellular carcinoma. Cancers (Basel). 2019; 11:486. https://doi.org/10.3390/cancers11040486 [PubMed]
- 30. Endo S, Xia S, Suyama M, Morikawa Y, Oguri H, Hu D, Ao Y, Takahara S, Horino Y, Hayakawa Y, Watanabe Y, Gouda H, Hara A, et al. Synthesis of potent and selective inhibitors of aldo-keto reductase 1B10 and their efficacy against proliferation, metastasis, and cisplatin resistance of lung cancer cells. J Med Chem. 2017; 60:8441–55. https://doi.org/10.1021/acs.jmedchem.7b00830 [PubMed]
- 31. Matsunaga T, Suzuki A, Kezuka C, Okumura N, Iguchi K, Inoue I, Soda M, Endo S, El-Kabbani O, Hara A, Ikari A. Aldo-keto reductase 1B10 promotes development of cisplatin resistance in gastrointestinal cancer cells through down-regulating peroxisome proliferator-activated receptor-γ-dependent mechanism. Chem Biol Interact. 2016; 256:142–53. https://doi.org/10.1016/j.cbi.2016.07.008 [PubMed]
- 32. Shen Y, Liao DF, Cao D. AKR1B10 in gastrointestinal diseases. Aging (Albany NY). 2015; 7:221–22. https://doi.org/10.18632/aging.100737 [PubMed]
- 33. Zu X, Yan R, Pan J, Zhong L, Cao Y, Ma J, Cai C, Huang D, Liu J, Chung FL, Liao DF, Cao D. Aldo-keto reductase 1B10 protects human colon cells from DNA damage induced by electrophilic carbonyl compounds. Mol Carcinog. 2017; 56:118–29. https://doi.org/10.1002/mc.22477 [PubMed]
- 34. Hashimoto Y, Imanishi K, Tokui N, Okamoto T, Okamoto A, Hatakeyama S, Yoneyama T, Koie T, Kamimura N, Ohyama C. Carboplatin-gemcitabine combination chemotherapy upregulates AKR1B10 expression in bladder cancer. Int J Clin Oncol. 2013; 18:177–82. https://doi.org/10.1007/s10147-011-0363-7 [PubMed]
- 35. He YC, Shen Y, Cao Y, Tang FQ, Tian DF, Huang CF, Tao H, Zhou FL, Zhang B, Song L, He L, Lin LM, Lu FG, et al. Overexpression of AKR1B10 in nasopharyngeal carcinoma as a potential biomarker. Cancer Biomark. 2016; 16:127–35. https://doi.org/10.3233/CBM-150548 [PubMed]
- 36. Ko HH, Cheng SL, Lee JJ, Chen HM, Kuo MY, Cheng SJ. Expression of AKR1B10 as an independent marker for poor prognosis in human oral squamous cell carcinoma. Head Neck. 2017; 39:1327–32. https://doi.org/10.1002/hed.24759 [PubMed]
- 37. Reddy KA, Kumar PU, Srinivasulu M, Triveni B, Sharada K, Ismail A, Reddy GB. Overexpression and enhanced specific activity of aldoketo reductases (AKR1B1 & AKR1B10) in human breast cancers. Breast. 2017; 31:137–43. https://doi.org/10.1016/j.breast.2016.11.003 [PubMed]
- 38. Sinreih M, Štupar S, Čemažar L, Verdenik I, Frković Grazio S, Smrkolj Š, Rižner TL. STAR and AKR1B10 are down-regulated in high-grade endometrial cancer. J Steroid Biochem Mol Biol. 2017; 171:43–53. https://doi.org/10.1016/j.jsbmb.2017.02.015 [PubMed]
- 39. Sun Y, Fan X, Zhang Q, Shi X, Xu G, Zou C. Cancer-associated fibroblasts secrete FGF-1 to promote ovarian proliferation, migration, and invasion through the activation of FGF-1/FGFR4 signaling. Tumour Biol. 2017; 39:1010428317712592. https://doi.org/10.1177/1010428317712592 [PubMed]
- 40. Wang Z, Li R, Zhong R. Extracellular matrix promotes proliferation, migration and adhesion of airway smooth muscle cells in a rat model of chronic obstructive pulmonary disease via upregulation of the PI3K/AKT signaling pathway. Mol Med Rep. 2018; 18:3143–52. https://doi.org/10.3892/mmr.2018.9320 [PubMed]
- 41. Weaver AN, Burch MB, Cooper TS, Della Manna DL, Wei S, Ojesina AI, Rosenthal EL, Yang ES. Notch signaling activation is associated with patient mortality and increased FGF1-mediated invasion in squamous cell carcinoma of the oral cavity. Mol Cancer Res. 2016; 14:883–91. https://doi.org/10.1158/1541-7786.MCR-16-0114 [PubMed]
- 42. Kawano M, Miura T, Fujita M, Koike S, Imadome K, Ishikawa A, Yasuda T, Imamura T, Imai T, Nakayama F. The FGF1/CPP-C chimera protein protects against intestinal adverse effects of c-ion radiotherapy without exacerbating pancreatic carcinoma. Clin Transl Radiat Oncol. 2018; 14:8–16. https://doi.org/10.1016/j.ctro.2018.10.004 [PubMed]
- 43. Wu D, Yang B, Chen J, Xiong H, Li Y, Pan Z, Cao Y, Chen J, Li T, Zhou S, Ling X, Wei Y, Li G, et al. Upregulation of long non-coding RNA RAB1A-2 induces FGF1 expression worsening lung cancer prognosis. Cancer Lett. 2018; 438:116–25. https://doi.org/10.1016/j.canlet.2018.09.016 [PubMed]
- 44. Hsu YC, Kao CY, Chung YF, Lee DC, Liu JW, Chiu IM. Activation of aurora a kinase through the FGF1/FGFR signaling axis sustains the stem cell characteristics of glioblastoma cells. Exp Cell Res. 2016; 344:153–66. https://doi.org/10.1016/j.yexcr.2016.04.012 [PubMed]
- 45. Shain SA, Sarić T, Ke LD, Nannen D, Yoas S. Endogenous fibroblast growth factor-1 or fibroblast growth factor-2 modulate prostate cancer cell proliferation. Cell Growth Differ. 1996; 7:573–86. [PubMed]
- 46. Birrer MJ, Johnson ME, Hao K, Wong KK, Park DC, Bell A, Welch WR, Berkowitz RS, Mok SC. Whole genome oligonucleotide-based array comparative genomic hybridization analysis identified fibroblast growth factor 1 as a prognostic marker for advanced-stage serous ovarian adenocarcinomas. J Clin Oncol. 2007; 25:2281–87. https://doi.org/10.1200/JCO.2006.09.0795 [PubMed]
- 47. King ML, Lindberg ME, Stodden GR, Okuda H, Ebers SD, Johnson A, Montag A, Lengyel E, MacLean Ii JA, Hayashi K. Wnt7A/β-catenin signaling induces FGF1 and influences sensitivity to niclosamide in ovarian cancer. Oncogene. 2015; 34:3452–62. https://doi.org/10.1038/onc.2014.277 [PubMed]
- 48. Smith G, Ng MT, Shepherd L, Herrington CS, Gourley C, Ferguson MJ, Wolf CR. Individuality in FGF1 expression significantly influences platinum resistance and progression-free survival in ovarian cancer. Br J Cancer. 2012; 107:1327–36. https://doi.org/10.1038/bjc.2012.410 [PubMed]
- 49. Segeritz CP, Rashid ST, de Brito MC, Serra MP, Ordonez A, Morell CM, Kaserman JE, Madrigal P, Hannan NR, Gatto L, Tan L, Wilson AA, Lilley K, et al. hiPSC hepatocyte model demonstrates the role of unfolded protein response and inflammatory networks in α1-antitrypsin deficiency. J Hepatol. 2018; 69:851–60. https://doi.org/10.1016/j.jhep.2018.05.028 [PubMed]
- 50. Mossmann D, Park S, Hall MN. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat Rev Cancer. 2018; 18:744–57. https://doi.org/10.1038/s41568-018-0074-8 [PubMed]
- 51. Cosin-Roger J, Simmen S, Melhem H, Atrott K, Frey-Wagner I, Hausmann M, de Vallière C, Spalinger MR, Spielmann P, Wenger RH, Zeitz J, Vavricka SR, Rogler G, Ruiz PA. Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat Commun. 2017; 8:98. https://doi.org/10.1038/s41467-017-00213-3 [PubMed]
- 52. Li X, Zhang X, Pan Y, Shi G, Ren J, Fan H, Dou H, Hou Y. mTOR regulates NLRP3 inflammasome activation via reactive oxygen species in murine lupus. Acta Biochim Biophys Sin (Shanghai). 2018; 50:888–96. https://doi.org/10.1093/abbs/gmy088 [PubMed]
- 53. Sinreih M, Štupar S, Čemažar L, Verdenik I, Frković Grazio S, Smrkolj Š, Lanišnik Rižner T. Data on expression of genes involved in estrogen and progesterone action, inflammation and differentiation according to demographic, histopathological and clinical characteristics of endometrial cancer patients. Data Brief. 2017; 12:632–43. https://doi.org/10.1016/j.dib.2017.04.050 [PubMed]
- 54. Henriksson ML, Edin S, Dahlin AM, Oldenborg PA, Öberg Å, Van Guelpen B, Rutegård J, Stenling R, Palmqvist R. Colorectal cancer cells activate adjacent fibroblasts resulting in FGF1/FGFR3 signaling and increased invasion. Am J Pathol. 2011; 178:1387–94. https://doi.org/10.1016/j.ajpath.2010.12.008 [PubMed]
- 55. Huang YF, Niu WB, Hu R, Wang LJ, Huang ZY, Ni SH, Wang MQ, Yang Y, Huang YS, Feng WJ, Xiao W, Zhu DJ, Xian SX, Lu L. FIBP knockdown attenuates growth and enhances chemotherapy in colorectal cancer via regulating GSK3β-related pathways. Oncogenesis. 2018; 7:77. https://doi.org/10.1038/s41389-018-0088-9 [PubMed]
- 56. Loeffler-Ragg J, Mueller D, Gamerith G, Auer T, Skvortsov S, Sarg B, Skvortsova I, Schmitz KJ, Martin HJ, Krugmann J, Alakus H, Maser E, Menzel J, et al. Proteomic identification of aldo-keto reductase AKR1B10 induction after treatment of colorectal cancer cells with the proteasome inhibitor bortezomib. Mol Cancer Ther. 2009; 8:1995–2006. https://doi.org/10.1158/1535-7163.MCT-08-0987 [PubMed]