Introduction
According to the International Guidelines of the Surviving Sepsis Campaign, septic shock is characterized by sepsis-induced persistent hypotension, in spite of sufficient fluid resuscitation [1]. As a chief source of infection, severe sepsis and septic shock with sepsis-related multiple organ failure account for a significant proportion of mortality in intensive care units (ICUs) even in developed countries [2], with the published mortality rate among hospitals ranging between 25% and 70% on a global scale. Based on the widely-accepted concepts involving sepsis and septic shock, its clinical manifestations occur as a result of interactions between inflammatory, coagulation pathways and infectious pathogens [3]. Although the rates of septic shock-related mortality have exhibited a gradual downtrend largely owing to increased awareness, careful attention, improved early antibiotic treatment and the discovery of hemodynamic indicators, there are still no specific drug treatment regimens for patients who develop the condition [4]. Genetic epidemiology suggests that there is an element of susceptibility in relation to an individual’s genetic make-up, resulting in significant differences in responses to infection, thus posing an added degree of complexity for clinicians [5]. Therefore, the aim of this study was to explore the genetic influence of nucleotide oligomerization domain (NOD)-like receptor (NLR) family CARD domain containing protein 4 (NLRC4) on the treatment of septic shock by mediating the NLR pathway.
Genetic factors play a major role in the mortality of patients with septic shock caused by infections [2]. Reports have documented multiple over-expressed genes in innate immunity, including chemokine receptor, cytokine, and toll-receptor pathways [6]. The gene silencing paradigm is embedded in a larger framework of genetic recombination, and is implicated in the generation of functional and clinical phenotypes in certain inflammatory diseases [7]. Over a decade ago, NLRC4 was identified and regarded as a member of the NLR family of intracellular sensors [8]. Furthermore, another member of the NLR family, the NLRP3 inflammasome, has been regarded as a chief component of the innate immune system in the identification of viral infections [9]. The dysregulation of the innate immune system is well-known to serve as a crucial factor in the onset of sepsis, combined with genetic variability as a potential targeted therapy for sepsis and septic shock [5]. The innate immune system is also characterized by dendritic cell (DC) loss and contributes to dismal outcomes or nosocomial infections [10]. Therefore, this study aimed at investigating the effects of NLRC4 gene silencing on inflammatory reactions, shock-induced lung tissue injuries, and DC-mediated immune responses in septic shock mice models by mediating the NLR pathway.
Results
In silico analysis of NLRC4 in septic shock
Datasets regarding septic shock obtained from the gene expression omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) were differentially analyzed, with adj.P.Val < 0.05 and |LogFoldChange| > 2 as the screening threshold. A total of 1123, 864, 416 and 585 differentially expressed genes (DEGs) were retrieved from the GSE95233, GSE57065, GSE8121 and GSE26378 datasets (Table 1) respectively, among which the top 70 DEGs with the largest fold change were selected from each microarray for intersectional analysis with a Venn diagram (http://bioinformatics.psb.ugent.be/webtools/Venn/) (Figure 1A). The diagram highlighted 6 intersected genes (S100A12, UPP1, GYG1, NLRC4, METTL9, and CYSTM1) as candidates for further investigation. Additionally, disease genes associated with septic shock were retrieved from the DisGeNET database (http://www.disgenet.org/web/DisGeNET/menu/search?4), with the top 20 genes (TNF, C5AR1, GC, AQP1, NOS2, SELL, TBXA2R, A2M, ELANE, IL6, IL10, TLR4, CASP1, ADM, IL1B, LTA, CD14, SAA1, SAA2 and SLC5A1) considered to be septic shock-related genes. The interaction between septic shock-related genes and DEGs was analyzed using the String database (https://string-db.org/). The PPI net was produced via Cytoscape 3.6.0 software [11] (Figure 1B), wherein NLRC4 was discovered to be closely associated with several septic shock-related genes, signifying association with the occurrence of septic shock. Supplementary Figure 1A, 1B illustrate the heat maps of the top 70 DEGs in the microarrays of GSE95233 (Figure 1C) and GSE57065 (Figure 1D). Moreover, higher expression of NLRC4 was observed among patients with septic shock compared to healthy individuals. NLRC4 is a member of the NLR family that is involved in the activation of the NLR pathway associated with inflammatory reactions [12, 13]. Previous reports have demonstrated that NOD1 and NOD2 exert significant roles in septic shock [14, 15], yet the exact extent of the influence and mechanism of NLRC4 in septic shock still remains unclear. Given the above bioinformatics analysis and preliminary findings, we suspected that abnormal expressions of NLRC4 might influence the NLR pathway in septic shock.
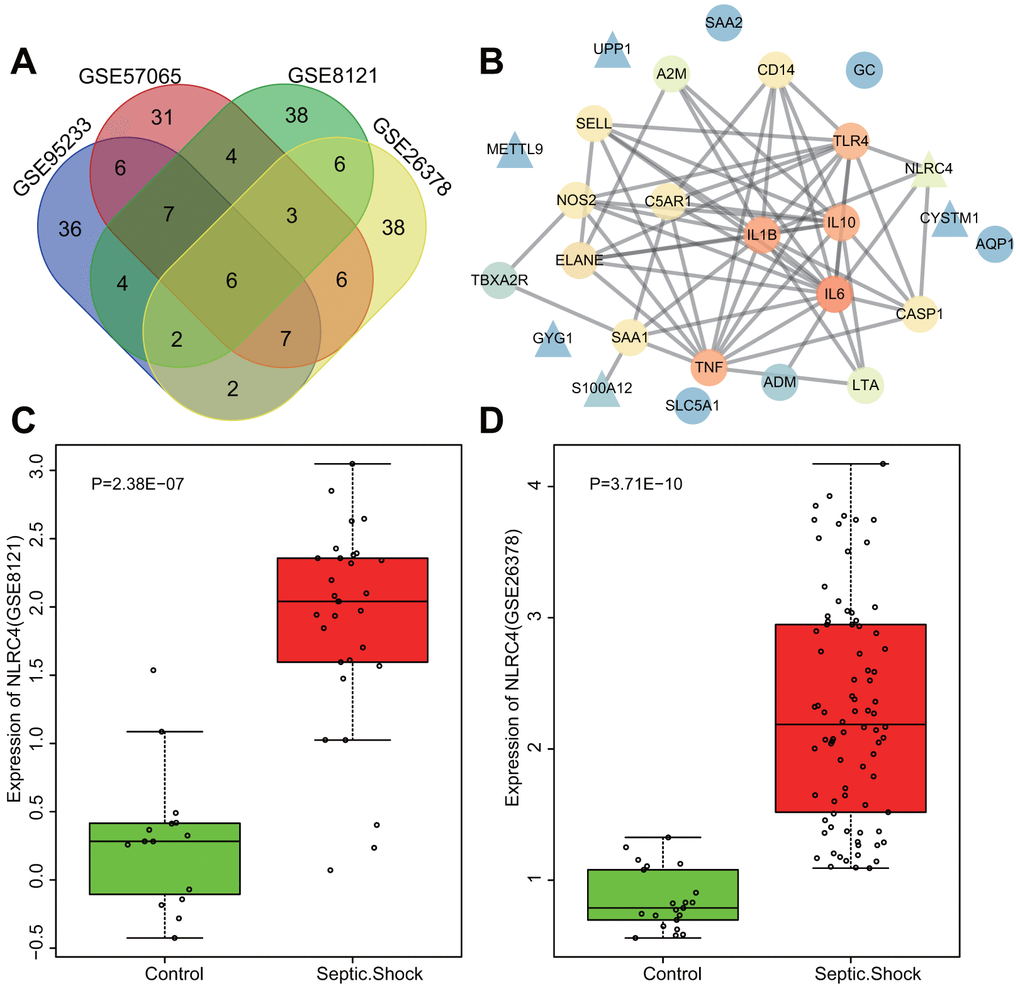
Figure 1. Highly expressed NLRC4 was found in septic shock. (A) 6 intersected genes detected among the top 70 DEGs from the microarray expression profiles of GSE95233, GSE57065, GSE8121 and GSE26378. (B) PPI network of the DEGs and septic shock-related genes (the triangle signifies the DEGs, the circle indicates the septic shock-related genes; the color of the genes shows the correlation degree with other genes, with orange coloration indicative of a high correlation degree and a blue coloration signifying a low correlation degree). (C, D) The expression of NLRC4 in septic microarray expression profiles GSE8121 and GSE26378.
Table 1. Baseline information of septic shock expression profiles GSE95233, GSE57065, GSE8121 and GSE26378.
| Accession | Platform | Organism | Tissue | Sample |
| GSE95233 | GPL570 | Homo sapiens | Blood | 51 septic shock patients and 22 healthy volunteers |
| GSE57065 | GPL570 | Homo sapiens | Whole blood | 28 septic shock patients and 25 healthy volunteers |
| GSE8121 | GPL570 | Homo sapiens | Whole blood | 15 normal children and 30 children with septic shock |
| GSE26378 | GPL570 | Homo sapiens | Whole blood | 82 children with septic shock and 21 normal controls |
NLRC4 silencing alleviates lung tissue injury induced by septic shock
Lung tissue injury induced by septic shock was assessed after DCs were transfected with NLRC4-siRNA. Hematoxylin and eosin (HE) staining results (Figure 2A) revealed that in the sham group, the lobule-alveolar structure in the mouse lung tissue was intact with mild widened-gaps without fiber tissue hyperplasia or inflammatory cell exudation in the alveolar spaces. The mice in the cecal ligation and puncture (CLP) and negative control (NC)-siRNA groups presented with symptoms and signs of alveolar congestion, hemorrhage, alveolar wall thickening, fracture and collapse in most of the mouse lung tissue. Large amounts of inflammatory cells were also detected in the alveoli lumen and interstitial tissues of lungs. In the NLRC4-siRNA group, mice displayed intact alveolar structures in the lungs, with the presence of mild widened-gaps but without hemorrhage, well-preserved fibrous structure, or inflammatory exudate in the alveolar spaces. Lung tissue injury scoring revealed that the CLP and NC-siRNA groups received significantly higher lung tissue injury scores compared to that of the sham group. In comparison, significantly lower lung tissue injury scores were observed in the NLRC4-siRNA group relative to that of the CLP and NC-siRNA groups (Figure 2B). Based on the aforementioned observations, we concluded that NLRC4 gene silencing could alleviate lung tissue injury induced by septic shock.
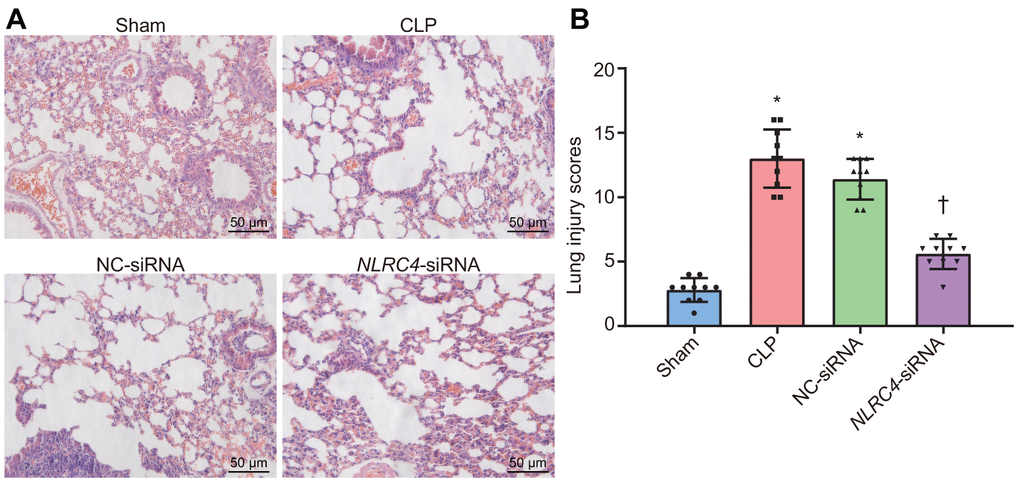
Figure 2. HE staining results illustrate the amelioration of lung tissue injury induced by septic shock in mice after DCs transfected with NLRC4-siRNA. (A) The lung tissue injury detected by HE staining (scale bar = 50 μm). (B) The lung tissue injury scores. n = 10. * p < 0.05 vs. the sham group. † p < 0.05 vs. the CLP and NC-siRNA groups. Data comparison among multiple groups was analyzed by one-way analysis of variance (ANOVA), followed by Tukey’s post hoc test. The experiment was repeated 3 times independently.
Silencing of NLRC4 inhibits the NLR pathway by down-regulating the expressions of NOD1, NOD2, RIP2 and NF-κB in mice lung tissues
After HE staining, the mRNA and protein expression of NLR pathway-related genes in mice’s lung tissues were quantified. Reverse transcription quantitative polymerase chain reaction (RT-qPCR) results revealed that the CLP and NC-siRNA groups presented with significantly elevated mRNA expressions of NLRC4, NOD1, NOD2, RIP2, and NF-κB when compared to the sham group (all psham vs. CLP < 0.001; psham vs. NC-siRNA < 0.001), with no notable changes detected regarding expressions of NLRC4 (psham vs. NLRC4-siRNA = 0.334), NOD1 (psham vs. NLRC4-siRNA = 0.425), NOD2 (psham vs. NLRC4-siRNA = 0.964), RIP2 (psham vs.NLRC4-siRNA = 0.367) or NF-κB (psham vs. NLRC4-siRNA = 0.414) in the NLRC4-siRNA group. In comparison to the CLP and NC-siRNA groups, the NLRC4-siRNA group displayed markedly diminished mRNA expressions of NLRC4, NOD1, NOD2, RIP2, and NF-κB (all pCLP vs. NLRC4-siRNA < 0.001; pNC-siRNA vs. NLRC4-siRNA < 0.001). These results indicated that NLRC4 gene silencing functioned to block the NLR pathway.
Western blot analysis results elaborated that the CLP and NC-siRNA groups exhibited significantly increased protein expressions of NLRC4, NOD1, NOD2, RIP2, and NF-κB compared to that of the sham group (all psham vs. CLP < 0.001; psham vs. NC-siRNA < 0.001), while no changes were detected regarding protein expressions of NLRC4 (psham vs. NLRC4-siRNA = 0.636), NOD1 (psham vs. NLRC4-siRNA = 0.124), NOD2 (psham vs. NLRC4-siRNA = 0.398), RIP2 (psham vs. NLRC4-siRNA = 0.389), or NF-κB (psham vs. NLRC4-siRNA = 0.574)in the NLRC4-siRNA group. In contrast with the CLP and NC-siRNA groups, remarkably declined protein levels of NLRC4, NOD1, NOD2, RIP2, and NF-κB were noted in the NLRC4-siRNA group (pCLP vs. NLRC4-siRNA < 0.001; pNC-siRNA vs. NLRC4-siRNA < 0.001). These results further verified that NLRC4 gene silencing blocked the NLR pathway (Figure 3A–3C).
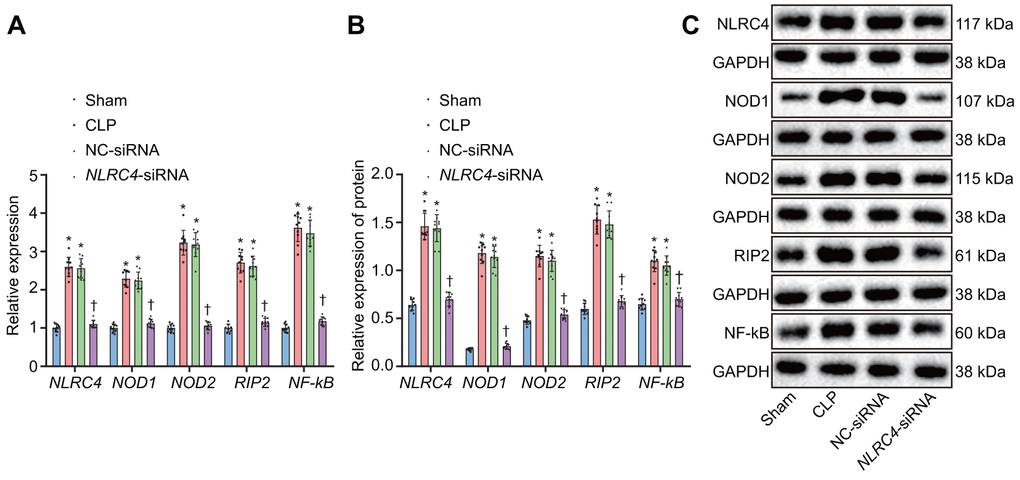
Figure 3. Silencing of NLRC4 inactivates the NLR pathway in mouse lung tissues. (A) The mRNA expression of NLRC4, NOD1, NOD2, RIP2, and NF-κB in DCs determined by RT-qPCR. (B) The protein expression of NLRC4, NOD1, NOD2, RIP2, and NF-κB normalized to GAPDH in DCs measured by Western blot analysis. (C) Western blot bands of NLRC4, NOD1, NOD2, RIP2, and NF-κB in different transfection groups. N = 10. * p < 0.05 vs. the sham group. † p < 0.05 vs. the CLP and NC-siRNA groups. Data comparison among multiple groups was analyzed by one-way ANOVA, followed by Tukey’s post hoc test. The experiment was repeated 3 times independently.
NLRC4 gene silencing alleviates inflammatory reaction and reduces inflammatory cell infiltration
Enzyme-linked immunosorbent assay (ELISA) was performed on septic mice after their DCs were transduced with different vectors to evaluate the effects of NLRC4 gene silencing on the inflammatory reaction of septic shock. Results (Figure 4A) demonstrated that the CLP and NC-siRNA groups exhibited markedly elevated levels of Interleukin (IL)-1β, tumor necrosis factor α (TNF-α) and IL-6 compared to that of the sham group (all psham vs. CLP < 0.001; psham vs. NC-siRNA < 0.001), while no remarkable differences were observed in the expression levels of IL-1β (psham vs. NLRC4-siRNA = 0.085), TNF-α (psham vs. NLRC4-siRNA = 0.382) or IL-6 (psham vs. NLRC4-siRNA = 0.889) in the NLRC4-siRNA group. Reduced levels of IL-1β, TNF-α and IL-6 were detected in the NLRC4-siRNA group, compared with the sham and NC-siRNA groups (all pCLP vs. NLRC4-siRNA < 0.001; pNC-siRNA vs. NLRC4-siRNA < 0.001), which suggested that NLRC4 gene silencing alleviated the inflammatory reaction induced by septic shock. As reflected by Figure 4B, immunohistochemistry results revealed that the CLP and NC-siRNA groups exhibited markedly elevated levels of F4/80 (psham vs. CLP < 0.001; psham vs. NC-siRNA < 0.001) and CD45 (psham vs. CLP < 0.001; psham vs. NC-siRNA < 0.001) compared to the sham group (all psham vs. CLP < 0.001; psham vs. NC-siRNA < 0.001), whereas no marked changes were seen on the levels of F4/80 (psham vs.NLRC4-siRNA = 0.602) and CD45 (psham vs. NLRC4-siRNA = 0.619) in the NLRC4-siRNA group. The levels of F4/80 (pCLP vs. NLRC4-siRNA < 0.001; pNC-siRNA vs. NLRC4-siRNA < 0.001) and CD45 (pCLP vs. NLRC4-siRNA < 0.001; pNC-siRNA vs. NLRC4-siRNA < 0.001) in the NLRC4-siRNA group were markedly reduced in comparison to that of the CLP and NC-siRNA groups. Thus, these results indicated that NLRC4 gene silencing reduced infiltration of inflammatory cells.
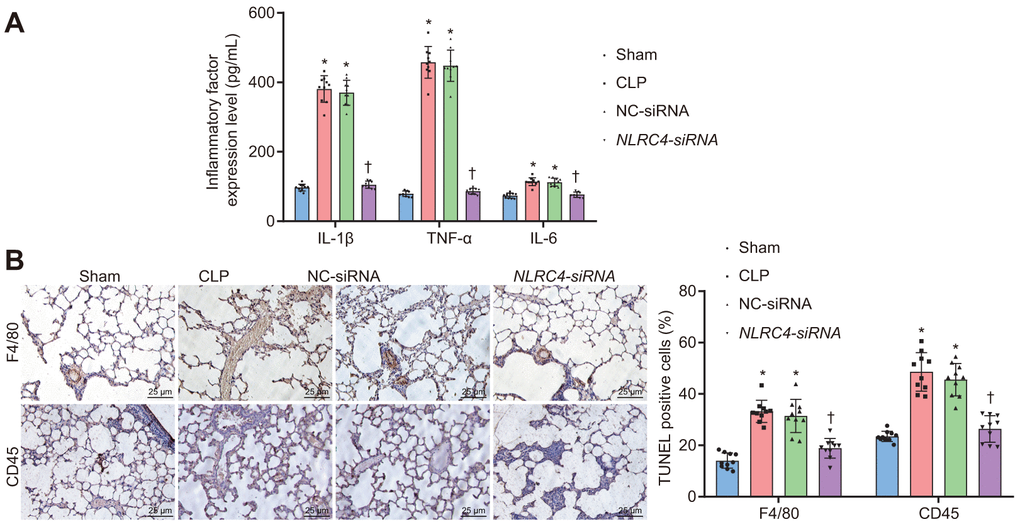
Figure 4. NLRC4 downregulation alleviates inflammatory reaction and reduces inflammatory cell infiltration. (A) the levels of IL-1β, TNF-α and IL-6 in lung tissues of mice with septic shock detected by ELISA. (B) inflammatory cell infiltration evidenced by F4/80 and CD45 positive levels using TUNEL staining (scale bar = 25 μm). n = 10. * p < 0.05 vs. the sham group. † p < 0.05 vs. the CLP and NC-siRNA groups. Data comparison among multiple groups was analyzed by one-way ANOVA, followed by Tukey’s post hoc test. The experiment was repeated 3 times independently.
DCs exhibit positive morphological changes after being cultured and NLRC4 gene silencing inhibits DC maturation and proliferation
Morphological changes in DCs were observed under an inverted microscope. After 3 h of adherence, small round cells and a portion of the closely aggregated cells became apparent. On the 3rd day after culture, a pattern of small densely arranged round cells were observed with a trivial amount of bud-shaped protuberances. On the 5th day, adherent cells in dense arrangements were observed, with an increase in the bud-shaped protuberances. On the 7th day, DCs appeared to be spindle shaped, closely-arranged with a large number of distinct protuberances (Figure 5A). RT-qPCR was employed to detect the NLRC4 expression in DCs in each group (Figure 5B). The results demonstrated that NLRC4 expressions were elevated in the CLP and NC-siRNA groups in contrast to that of the sham group (psham vs. CLP < 0.001; psham vs. NC-siRNA < 0.001), whereas the expressions of NLRC4 did not differ significantly in the NLRC4-siRNA group (psham vs. NLRC4-siRNA = 0.719). Significantly diminished NLRC4 expressions were also detected in the NLRC4-siRNA group compared to that of the CLP and NC-siRNA groups (pCLP vs. NLRC4-siRNA < 0.001; pNC-siRNA vs. NLRC4-siRNA = 0.002). The findings illustrated that morphological changes in DCs induced by septic shock were improved by the silencing of NLRC4.
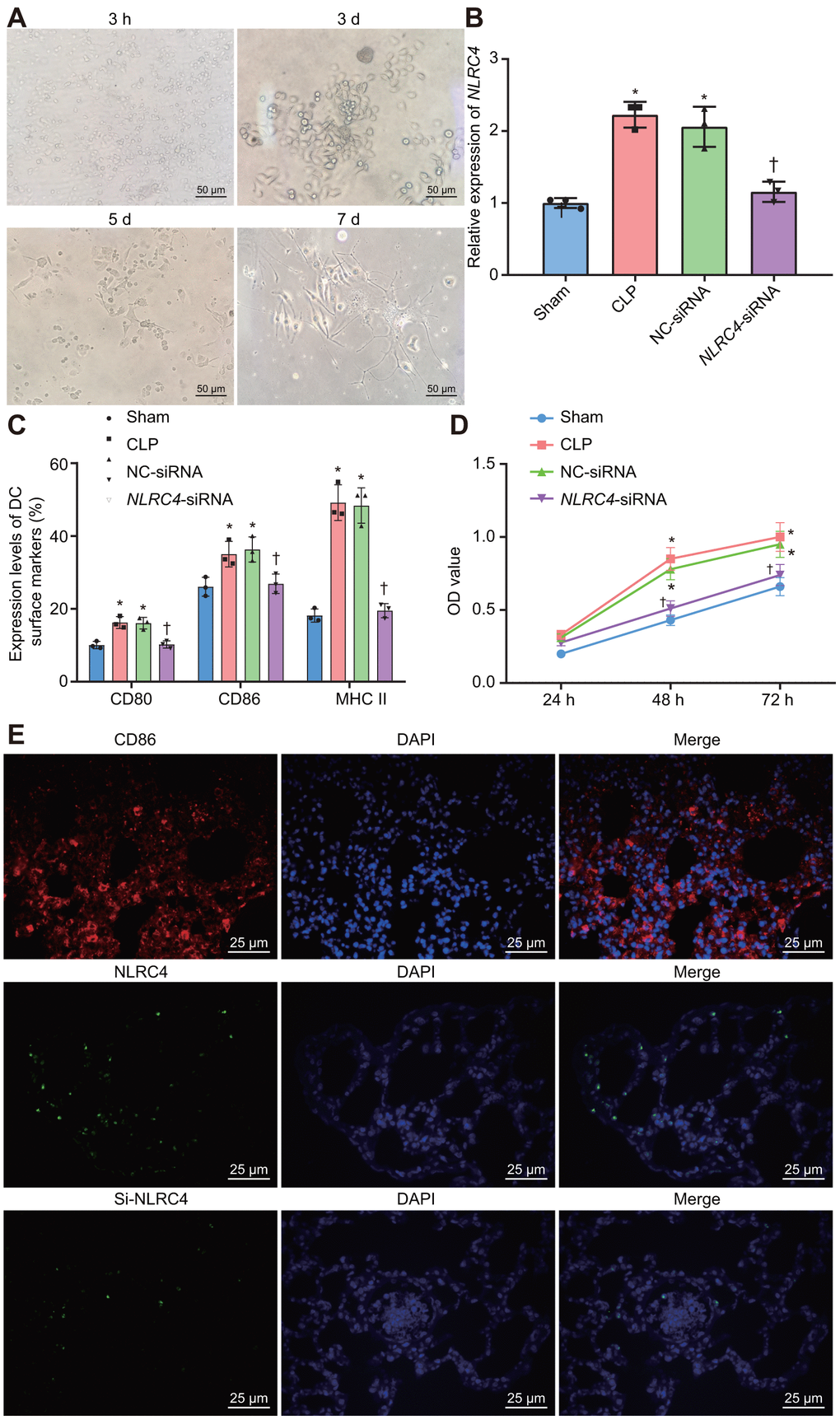
Figure 5. Improved morphological changes in DCs after being cultured for 3 h, 3 d, 5 d, and 7 d and suppressed NLRC4 reduces DC proliferation. (A) Morphological changes of DCs observed under an inverted microscope (scale bar = 50 μm). (B) The expression of NLRC4 in DCs determined by RT-qPCR. (C) The induction of DC surface markers, CD80, CD86, and MHC II, detected by flow cytometry. (D) The DC apoptosis as indicative of OD values detected by flow cytometry. (E) The immunofluorescence labeling of NLRC4 and DC surface marker CD86 (scale bar = 25 μm). The bone marrow-derived DCs from the same group of mice were adopted in the in vivo experiments. * p < 0.05 vs. the sham group. † p < 0.05 vs. the CLP and NC-siRNA groups. Data comparison among multiple groups was analyzed by one-way ANOVA, followed by Tukey’s post-hoc test. Data at different time points were compared by repeated measures ANOVA, followed by Bonferroni test. The experiment was repeated 3 times independently.
Furthermore, flow cytometry was utilized to investigate the influence of NLRC4 gene silencing on the growth and maturation of DCs. The induction of DC surface markers [histocompatibility complex II (MHC II), CD80, and CD86] was detected. The results (Figure 5C) revealed that compared with the sham group, the induction of MHC II (psham vs. CLP < 0.001; psham vs. NC-siRNA < 0.001), CD80 (psham vs. CLP = 0.002; psham vs. NC-siRNA = 0.027) and CD86 (psham vs. CLP = 0.030; psham vs. NC-siRNA = 0.015) were all noticeably elevated in the CLP and NC-siRNA groups. However, no significant differences were detected regarding the induction of MHC II (psham vs. NLRC4-siRNA = 0.968), CD80 (psham vs. NLRC4-siRNA = 0.999) or CD86 (psham vs. NLRC4-siRNA = 0.988) in the NLRC4-siRNA group. In comparison with the CLP and NC-siRNA groups, reduced induction of MHC II (pCLP vs. NLRC4-siRNA < 0.001; pNC-siRNA vs. NLRC4-siRNA < 0.001), CD80 (pCLP vs. NLRC4-siRNA = 0.025; pNC-siRNA vs. NLRC4-siRNA = 0.031) and CD86 (pCLP vs. NLRC4-siRNA = 0.047; pNC-siRNA vs. NLRC4-siRNA = 0.023) were observed in the NLRC4-siRNA group. These results indicated that NLRC4 gene silencing suppressed the maturation of DCs.
Lastly, 3-(4,5-Dimethyl-2-Thiazyl)-2,5-Diphenyl-2H-Tetrazolium Bromide (MTT) assay was performed to detect the effects of NLRC4 gene silencing on DC proliferation. The results (Figure 5D) revealed that the cell proliferation ability was enhanced in the CLP and NC-siRNA groups compared to that of the sham group (48 h: psham vs. CLP < 0.001; psham vs. NC-siRNA < 0.001; 72 h: psham vs. CLP < 0.001; psham vs. NC-siRNA < 0.001), while no notable discrepancies in proliferation ability were detected in the NLRC4-siRNA group (48 h: psham vs. NLRC4-siRNA = 0.394; 72 h: psham vs. NLRC4-siRNA = 0.394). When compared with the CLP and NC-siRNA groups, significantly decreased cell proliferation ability was noted in the NLRC4-siRNA group (48 h: pCLP vs. NLRC4-siRNA < 0.001; pNC-siRNA vs. NLRC4-siRNA < 0.001; 72 h: pCLP vs. NLRC4-siRNA < 0.001; pNC-siRNA vs. NLRC4-siRNA = 0.002). These findings suggested that NLRC4 gene silencing inhibited DC proliferation. Immunofluorescence (Figure 5E) was adopted to label the DC surface marker, CD86, on DCs and the results verified that the isolated cells were DCs. Moreover, the expression of the NLRC4 was effectively knocked down in the NLRC4-siRNA group compared with NC-siRNA group.
Repression of NLRC4 prevents cell cycle entry and increases the DC apoptosis rate in mice with septic shock
PI single staining and AnnexinV-FITC/PI double staining methods were finally adopted to observe changes in the DC cell cycle distribution post-transfection with different vectors. The results of PI single staining revealed that when compared with the sham group, the percentage of cells arrested at the G1 phase was notably reduced (shortened G1 phase), whereas the percentages of cells arrested at the G2 and S phase were significantly elevated (prolonged G2 and S phases) in the CLP and NC-siRNA groups (all psham vs. CLP < 0.001; psham vs. NC-siRNA < 0.001). No significant differences were detected in regards to cell cycle distribution in G1 (psham vs. NLRC4-siRNA = 0.663), G2 (psham vs. NLRC4-siRNA = 0.640) or S phase (psham vs. NLRC4-siRNA = 0.999) in the NLRC4-siRNA group. In comparison with the CLP and NC-siRNA groups, whereas the percentage of cells arrested at the G1 phase was elevated (prolonged G1 phase) and the percentages of cells arrested at the G2 and S phases were reduced (shortened G2 and S phases) in the NLRC4-siRNA group (all pCLP vs. NLRC4-siRNA < 0.001; pNC-siRNA vs. NLRC4-siRNA < 0.001).
In addition, AnnexinV-FITC/PI double staining results indicated that the DC apoptotic rate was lower in the CLP (14.14 ± 1.41%) and NC-siRNA (18.74 ± 1.87%) groups when compared with the sham group (34.87 ± 3.48) (all psham vs. CLP < 0.001; psham vs. NC-siRNA < 0.001), with no significant differences observed in the NLRC4- siRNA group (28.63 ± 2.86%) (psham vs. NLRC4-siRNA = 0.066). DC apoptotic rate was elevated in the NLRC4-siRNA group (pCLP vs. NLRC4-siRNA < 0.001; pNC-siRNA vs. NLRC4-siRNA = 0.006) when compared with the CLP and NC-siRNA groups. These results suggested that NLRC4 gene silencing increased the DC apoptotic rate among mice with septic shock (Figure 6).
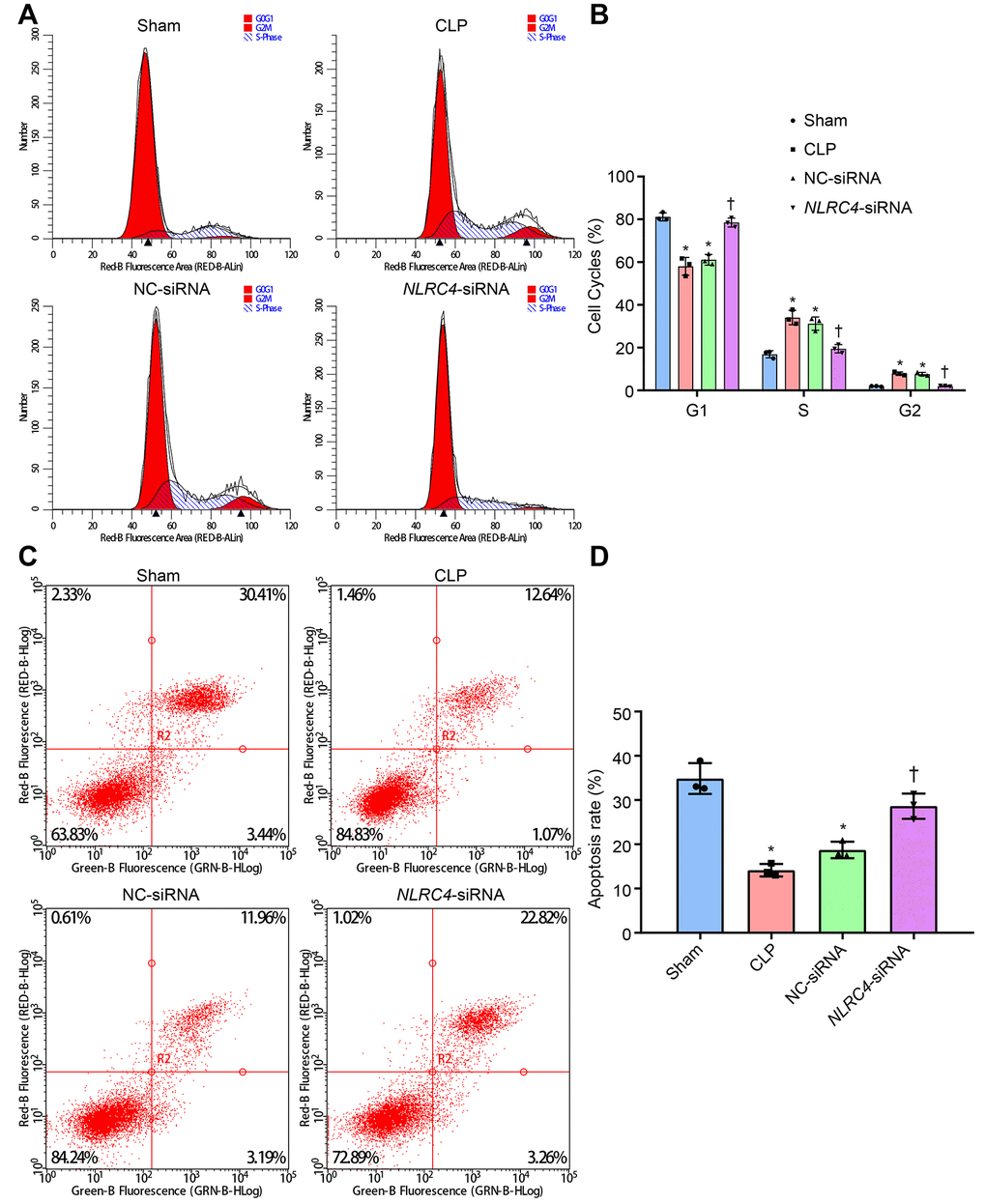
Figure 6. siRNA-mediated silencing of NLRC4 elevates DC cell apoptosis rate in mice with septic shock as evidenced by PI single staining and AnnexinV-FITC/PI double staining. (A, B), DC cycle distribution in the sham, CLP, NC-siRNA, and NLRC4-siRNA groups detected by PI single staining. (C, D) DC apoptosis in the sham, CLP, NC-siRNA, NLRC4-siRNA groups revealed by AnnexinV-FITC/PI double staining. The bone marrow-derived DCs from the same group of mice were adopted in the in vivo experiments. * p < 0.05 vs. the sham group. † p < 0.05 vs. the CLP and NC-siRNA groups. Data comparison among multiple groups was analyzed by one-way ANOVA, followed by Tukey’s post hoc test. The experiment was repeated 3 times independently.
Discussion
Septic shock is a common complication among patients in the ICU, which can lead to various organ failures and deaths [16]. A large array of cytokines are released as a result of systemic sepsis, with TNF-α being a crucial cytokine implicated in the induction of septic shock [17]. Studies have identified that NLRP3, a member of the NLR family is induced by TNF-α based on its own induction of Caspase-1 activation [18]. With the aim of exploring the relationship between the NLR family and septic shock, we investigated the effects of the NLRC4 gene in mice models of septic shock. Our findings suggested that the silencing of NLRC4 could alleviate septic shock-induced inflammatory reactions and lung tissue injury, as well as DC-mediated immune response by negatively mediating the NLR pathway.
Our results initially demonstrated that NLRC4 silencing significantly reduces the degree of lung tissue injury induced by septic shock in mice models. The elevated expression of NLRC4 was observed in septic patients compared to that of healthy individuals [19]. Consistently, CA has been previously reported to improve the survival rate of septic shock mouse models, accompanied by inhibited inflammasome activation including NLRC4 [20]. NLRs are intracellular immunosensors associated with pathogen and damage associated molecular patterns that have unique roles in lung antibacterial immune responses during bacterial infection [21, 22]. In addition, NLRC4 has been implicated in the process of injury and inflammation during brain ischemia, thus serving as a potential treatment target for various pathological conditions [23]. The available evidence and our results suggest that NLRC4 silencing could potentially alleviate lung tissue injury induced by septic shock.
Our findings also highlighted that NLRC4 silencing could negatively regulate the NLR pathway. NLRC4 is a member of the NLR family, which are cytosolic receptors that target bacterial molecules [24]. These NLRs participate in a variety of innate immune pathways, including the adjustment of the NF-κB pathway for NOD1 and NOD2, and the assembling of complexes for NLR proteins NLRP1, NLRC4, and NLRP3 [25]. Substantial evidence has been indicated that NLRP3 and NLRC4 inflammasomes possess well-characterized protective functions in alcoholic-induced liver injury [25]. NOD1 and NOD2 are recognition receptors related to cytosolic patterns, which are important for inherent immune signaling [26]. Moreover, NOD1 and NOD2 were discovered to share a relation with RIP2, which is a downstream signaling molecule receptor [27]. The activation of NOD1 results in septic shock and various organ injury/dysfunction in animal models [28]. Our findings illustrated that silencing NLRC4 resulted in significant reductions in the expressions of the NLR pathway key players including NLRC4, NOD1, NOD2, and RIP2. This suggests NLRC4 silencing was able to negatively regulate the NLR pathway.
Furthermore, we identified that NLRC4 silencing could attenuate inflammatory reactions induced by septic shock. TNF-α, IL-1β, and IL-6 are all well-known pro-inflammatory cytokines as previously reported [29]. Studies have also demonstrated that the NLRC4 inflammasome functions in the regulation and release of pro-inflammatory cytokines in response to an array of microbial stimuli [8]. The macrophage NLRC4 inflammasome initiates potent innate immune responses against Salmonella by eliciting caspase-1-dependent pro-inflammatory cytokine production [30]. A previous study elucidated that NLRP3 and NLRP4 work to regulate production and pyroptosis of IL-18 and IL-1β, thus testifying that NLCR4 silencing causes the inhibition of IL-1β, which is in accordance with our findings [31]. A prior study also noted that gentiopicroside conferred protection against shock and decreased inflammatory cytokine production of IL-1β, IL-6 and TNF-α in lung tissues [32]. Given the above key findings, it is reasonable to regard silenced NLRC4 as an important gene when inhibiting inflammatory reactions.
Lastly, NLRC4 gene silencing was observed to suppress the maturation and proliferation of DCs, while promoting DCs apoptosis. DCs are widely recognized as specialized antigen-presenting cells which begin maturing upon coming in contact with antigens via recognized receptors [33]. Elevated DCs have been identified in the colonic mucosa of patients suffering from Crohn’s disease, another chronic inflammatory bowel disease [34]. Previous studies have further confirmed that NLRC4 promotes the cleavage and maturation of pro-inflammatory cytokines [24]. In a previous study, the activation of NLRC4 inflammasome has been documented in splenic DCs [35]. We speculated that NLRC4 silencing could inhibit maturation of DCs as well as inhibit their population. NLRC3 has been proposed to inhibit the processes related to colorectal cancer through the regulation of cellular proliferation and apoptosis levels [21]. A functional study demonstrated that NLRC5 gene silencing inhibits TGF-β1-induced proliferation, while promoting the apoptosis of LX-2 cells [36]. We acquired the basic understanding of the relationship between NLRC4 gene silencing and maturation, proliferation, and apoptosis of DCs, in accordance with the investigated literature as well as our obtained results.
Overall, the current study demonstrated that siRNA-mediated silencing of NLRC4 blocks the NLR signaling pathway to inhibit DC maturation and immune response, which consequently alleviates lung tissue injury induced by septic shock (Figure 7). However, due to limited time and funding, we solely relied on a single technique (siRNA) to inhibit NLRC4 in our study, highlighting the need for alternative methods such as the use of NLRC4 knockout mice and/or pharmacological inhibitors or transient introduction of cas9 to knock down NLRC4 in vitro to validate the current findings, which will be carried out in our future ventures. Moreover, further studies of larger sample sizes would be conducted.
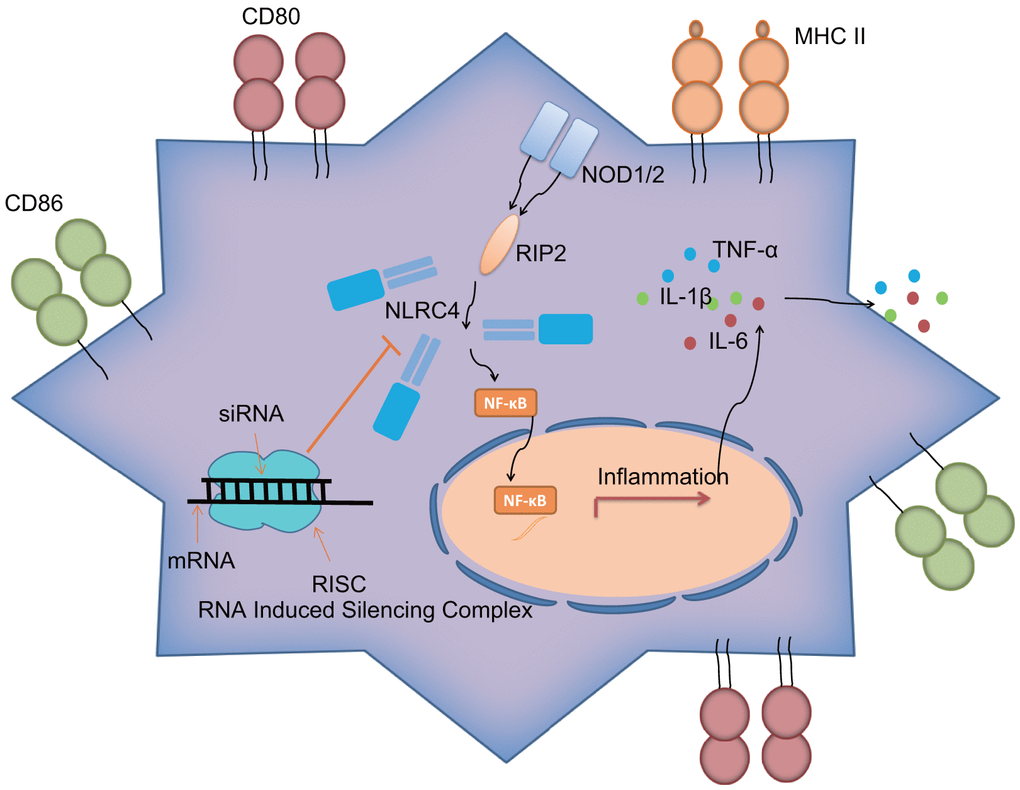
Figure 7. NLRC4 gene regulates inflammatory reaction and immune response of DCs in septic shock by mediating the NLR pathway.NLRC4 is the receptor of the NLR pathway. Activated NF-κB pathway induces inflammatory reaction with secretion of inflammatory factors: IL-1β, TNF-α and IL-6 outside cells, as well as increases in CD80, CD86 and MHC II on the cytomembrane, thus activating DC immune response. Importantly, siRNA-mediated silencing of NLRC4 blocks the NLR signaling pathway to inhibit DC maturation and immune response, which alleviates the lung tissue injury of mice induced by septic shock.
Materials and Methods
Ethics statement
All animal experiment protocols were approved by the Ethics Committee of the Second Affiliated Hospital of Nanchang University and conducted in strict accordance with the recommendations in the Guide for the Care and Use of local Laboratory Animals. All efforts were made to minimize the number and suffering of the included mice in the study.
Experimental animals
Forty healthy C57BL/6 male mice (age: 6 - 8 weeks; weight: 18 - 22 g) were purchased from the Laboratory Animal Center of Jiangxi University of Traditional Chinese Medicine (Nanchang, Jiangxi, China).
All procured mice were raised under specific pathogen-free conditions at a room temperature of 23° C with a relative humidity of 65%, following 12-h light-dark cycles. The mice were not fed but were allowed free access to water for a period over 12 h. Septic models were established in 30 mice by CLP as previously described [37]. Meanwhile, 10 mice were sham-operated with cecum without ligation and puncture. The 30 CLP mice were then assigned into three subgroups: CLP (mice with CLP-induced septic shock), negative control (NC)-siRNA (CLP mice injected with NC-siRNA plasmid), and NLRC4-siRNA (CLP mice injected with NLRC4-siRNA plasmid) groups (n = 10/group). Based on the known sequence of NLRC4 in the National Center for Biotechnology Information, the NC-siRNA plasmid and NLRC4-siRNA plasmid were constructed by Sangon Biotechnology Co., Ltd. (Shanghai, China) using the pCDNA3.1 vector (Shanghai GenePharma, Co., Ltd., Shanghai, China). The sequence of the NC-siRNA was 5’-TACGTCCAAGGTCGGGCAGGAAGA-3’, while the sequence of NLRC4-siRNA was 5’-TACGTCCAAGGTCGGGCAGGAAGA-3’. Then, 0.1 mL recombinant retroviral vector (1.95 × 108 PFU) expressing NC-siRNA (scrambled siRNA) or NLRC4-siRNA was injected into the successfully modeled mice via the tail vein 2 h after CLP surgery [38, 39]. The mice were then euthanatized 24 h after CLP surgery with their lungs promptly collected under sterile conditions. Parts of the lung tissues were fixed in 4% formalin for HE staining, while the remaining were stored in liquid nitrogen for further use in the following procedures: RT-qPCR, Western blot analysis and ELISA. The femur medullary cavity of all mice was collected following the aforementioned aseptic procedures for collection of bone marrow-derived DCs for further use.
HE staining
The formalin-fixed lung tissues were rinsed with tap water 72 h later, routinely dehydrated for 1 min/time, permeabilized with xylene twice (5 min/time), soaked in wax, placed in a paraffin mold, and cooled in a paraffin embedding machine. The embedded tissue pieces were sectioned (5μm) on a microtome before the sections were dried at 70° C for 1 h, and heated at 60° C for 5 h. The paraffin sections were dewaxed to liquid form and subsequently stained with hematoxylin at room temperature for 10 min. The sections were rinsed by running water for 30-60 s, differentiated using 1% hydrochloric acid alcohol for 30 s, and then rinsed under running water for 5 min. The sections were stained with eosin (0001-H, Beijing Xinhualvyuan Science and Technology Ltd., Beijing, China) at room temperature for 1 min [40], dehydrated using varied concentrations of alcohol solutions (concentrations of 70%, 80%, 90%, 95%, and 100%, respectively; 1 min/time), and treated with xylene phthalate before the sections were permeabilized with xylene I and II (GD-RY1215-12, Shanghai, China; 1 min/time) twice and finally mounted using neutral gum. Subsequently, the lung tissues were observed and photographed with a Cai Si fluorescence microscope (PrimoStar iLED; Bioresearch Technology Co., Ltd., Beijing, China). The lung tissue injury scores were evaluated based on the Mikawar method by assessing alveolar congestion, hemorrhage, infiltration or aggregation of neutrophils in airspace or vessel wall, and the thickness of alveolar wall hyaline membrane formation. These items were scored according to the following: 0 minimal damage, 1 mild damage, 2 moderate damage, 3 severe damage, and 4 maximal damage [41].
RT-qPCR
A total of 100 μL prepared frozen lung tissue homogenate was collected and added with l mL TRIzol reagents (15596-018, Beijing Solarbio Science and Technology Co., Ltd., Beijing, China) in order to extract the total RNA content from the frozen tissues. Next, the concentration and quality of the extracted total RNA were determined using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Rockford, IL, USA). When the ratio of 260/280 was about 2.0 and concentration was 1 μg - 5 μg, the RNA was used for the follow-up experiments. The RNA (2 μg) was then reverse transcribed into cDNA using TaqMan Reverse Transcription Reagents (Roche Ltd., Basel, Switzerland) at 42° C for 50 min. Gene fragments of NOD1, NOD2, NLRC4, receptor-interacting protein 2 (RIP2) and Nuclear factor-kappaB (NF-κB) were amplified using PCR. The primer sequences for RT-qPCR were synthesized by Sigma-Aldrich Chemical Company (St Louis, MO, USA) (Table 2). PCR amplification was conducted in real-time quantitative PCR instrument (CFX96, Bio-Rad Laboratories, Hercules, CA, USA). With GAPDH as the internal reference, the relative transcript level of target gene mRNA was calculated using the 2-ΔΔCT method [42].
Table 2. Primer sequences for RT-qPCR.
| Gene | Primer sequence (5' - 3') |
| NOD1 | F: 5'- ACTCAGCG-TCAACCAGATCAC-3' |
| R: 5'-ACGATGGAGGTGCTGTTCTTC-3' |
| NOD2 | F: 5'-CTCAGTCTCGCTTCCTCAGTAC-3' |
| R: 5'-TGCAGA-AGAGTGCTCTTGCC-3' |
| NLRC4 | F: 5'-ATCGTCATCACCGTGTGGAG-3' |
| R: 5'-GCCAGACTCGCCTTCAATCA-3' |
| RIP2 | F: 5'-TCCAGAGTAAGAGGGAAGCC-3' |
| R: 5'-TTGGATGTCAGACGTATCTAGC-3' |
| NF-κB | F: 5'-CCT CTGGCGAATGGCTTTAC-3' |
| R: 5'-GCTATGGAT ACTGCGGTCTGG-3' |
| GAPDH | F: 5'-TTCACCACCATGGAGAAGGC-3' |
| R: 5'-GGCATGGACTGTGGTCATGA-3' |
| Note: RT-qPCR, reverse transcription quantitative polymerase chain reaction; NOD1, nucleotide binding and oligomerization domain-1; NOD2, nucleotide binding and oligomerization domain-2; NLRC4, NLR family CARD domain containing 4; RIP2, receptor-interacting protein 2; NF-κB, Nuclear factor-kappaB; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; F, forward; R, reverse. |
Western blot analysis
First, 100 μL of the prepared frozen lung tissue homogenate was extracted and placed into reaction tubes, which were then added with 1 mL of cell lysis solution and left to react 30 min at 4° C, with a shaking every 10 min. Next, the sample was centrifuged at 12000 r/min and 4° C for 20 min and the lipid layer was discarded. The supernatant was collected as the protein extraction solution. A bicinchoninic acid kit (20201ES76, YEASEN Biotechnology Co., Ltd., Shanghai, China) was used to determine the concentration of the extracted protein. The total protein was subsequently treated with sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane. The membrane was then blocked with 5% skimmed milk powder at room temperature for 1 h and subsequently probed with the following primary antibodies (Abcam Inc., Cambridge, MA, USA): rabbit anti-mouse NLRC4 (ab201792, 1 : 1000), NOD1 (ab22143, 1 : 1000), NOD2 (ab124348, 2 μg/mL, 1 : 1000), anti-RIP2 (ab8428, 1 : 1000), anti-NF-κB (ab28856, 1 : 1000) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (ab181602, 1 : 10000) at 4° C overnight. The membrane was later rinsed with Tris-Buffered Saline Tween-20 (TBST) and re-probed with the diluted horseradish peroxidase-labeled goat anti-rabbit immunoglobulin G (IgG) and left at room temperature for 1 h. After developing, the gray values of each protein band in the membrane were analyzed using the Quantity One v4.6.6 software, followed by the quantitative analysis of proteins (GAPDH as the internal reference) [43].
ELISA
A total of 100 μL of frozen lung tissue homogenate was extracted and transferred to a reaction tube. The expressions of IL-1β, TNF-α and IL-6 in lung tissues of mice were detected in strict accordance with the manual of mouse IL-1β, TNF-α, and IL-6 ELISA kits (Wuhan Zhongzhi Biotechnologies Ltd., Wuhan, Hubei, China), respectively [44].
Isolation and culture of DCs
The separated femur was repeatedly washed and cleaned until the white part of the bone could be seen, followed by the collection of the bone marrow. The bone marrow was centrifuged at 256 × g for 5 min. After the removal of the supernatant, the bone marrow cells were obtained from the precipitate at the bottom of the tube, added with erythrocyte lysate (1 : 10, preheated at 37° C), and mixed thoroughly. The mixture was then left to sit at room temperature for 2 - 4 min, followed by the removal of excess red blood cells. The bone marrow cells were subsequently suspended in Roswell Park Memorial Institute (RPMI) 1640 medium (Wuhan Alpha Biotechnologies Co., Ltd., Wuhan, Hubei, China) containing 10% fetal calf serum (FCS) (Hangzhou Sijiqing Company, Zhejiang, China) and adjusted to a suspension of 2 × 106 cells/mL. The suspension was then placed into a glass cell culture flake and incubated at room temperature for 2 - 3 h. After removing detached cells, the concentration of the bone marrow cell suspension was readjusted to 1 × 108 cells/L using RPMI 1640 complete medium (sodium pyruvate, Glutamine, Hepes, and NaHCO3 at a ratio of 1 : 2 : 10 : 8.9), penicillin, streptomycin (200,000 μ/L each), GM-CSF, IL-4 (50 ng/L each, Yarewell Technology Inc., Shenzhen, China), mercaptoethanol (50 μmol/L) and FCS (20%). Afterwards, the cell suspension (1.5 - 2 mL per well) was added to a 6-well plate for incubation with 5% CO2 at 37° C. Half of the culture medium was renewed every other day, and the non-adherent cells were removed at the 3rd h and on the 3rd d following incubation. The cell incubation condition was observed on the 5th - 7th days, while the morphology and quantity of DCs were observed on the 3rd h, 3rd d, 5th d, and 7th d under an inverted microscope. After 7 d of incubation, DCs were collected for further experimentation [45].
Immunofluorescence
Cells (2 × 104 cells/well) were cultured overnight. When the cells reached 50-70% confluence, they were washed twice with PBS, fixed in a 4% paraformaldehyde and permeabilized in a 0.03% Triton X-100 (Sigma) PBS solution for 20 min. Cells were then washed 3 times with PBS for 5 min each and blocked with 5% bovine serum albumin (BSA) in PBS for 1 h at room temperature. Cells were incubated with their corresponding primary antibodies CD86 (1: 100; Abcam) and NLRC4 (1: 100; Abcam) in a humidified cabinet overnight at 4° C. The cells were later washed 3 times in PBS for 5 min each and incubated with a CY3-conjugated secondary antibody (1: 50; CWBIO, Beijing, China) at room temperature for 1 h in the dark. Finally, the cells were washed three times in PBS and incubated with 1 μg/ml of 4,6-diamidino-2-phenylindole (DAPI, Roche, Basel, Switzerland) for 5 min at room temperature in the dark. The slides were thoroughly washed with PBS, and observed with an inverted fluorescence microscope (400 ×; Nikon, Tokyo, Japan) [46].
Immunohistochemistry
Immunohistochemical staining of F4/80, CD45 were performed adhering to instructions provided by UltraSensitive™ SP Kit (KIT-9710, Maxim Bio, Fuzhou, China). The primary antibodies used were as follows: anti-CD45 (1: 8000, Abcam), and anti-F4/80 (GB11027, 1: 1000, Servicebio, Wuhan, China). Sections were incubated with non-immune serum instead of the primary antibody that was designated as NCs. Images were analyzed with Image-Pro Plus 5.1. software (Media Cybernetics, Inc., Rockville, MD, USA). Immunoactivity of F4/80 and CD45 were quantified with the percent of positive cells [46].
Flow cytometry
After culture for 7 d, DCs were obtained from the mice in each group and detached with 0.25% trypsin to harvest the adherent cells. DCs were then dispersed into a single-cell suspension, re-suspended with 100 μL PBS and incubated with the following Fluorescein isothiocyanate (FITC)-labeled monoclonal antibodies: FITC-labeled anti-mouse CD80 (0.5 μL), and CD86 (0.5 μL) (BioLegend Inc., San Diego, CA, USA) and FITC-labeled anti-mouse I-Aα (0.5 μL) (BD Pharmingen Inc., San Diego, CA, USA) at 4° C for 30 min without any exposure to light. The supernatant was removed after centrifugation and the cells were rinsed with 1 mL PBS and re-suspended with 0.5 mL PBS at 4° C without any exposure to light. Finally, the cells were detected using a flow cytometer (BD Pharmingen Inc., San Diego, CA, USA) and analyzed with the CellQuest 5.1 software [47].
MTT assay
DCs were seeded in 96-well plates (3 × 103 - 6 × 103 cells/well) at a cell volume of 0.2 mL in each well, with 6 duplicate wells set up for each group. At the 24th h, 48th h, and 72th h time periods of incubation, the culture plates were further cultured for another 4 h with 5 g/L 10% MTT solution (GD-Y1317, Shanghai Guduo Bio-technology Co., Ltd., Shanghai, China). Dimethyl sulfoxide (D5879-100ml, 100 μL, Sigma-Aldrich Chemical Company, St Louis, MO, USA) was added to each well and gently shaken for 10 min to dissolve the formazan crystals. The OD value of each well at an excitation wavelength of 570 nm was measured using a microplate reader (BS-1101, Detie Lab, Nanjing, Jiangsu, China). Subsequently, a cell viability curve was plotted with time as the abscissa and OD value as the ordinate [48].
AnnexinV-FITC/PI staining
The cell cycle distribution of DCs was analyzed using PI single staining. After 7-d incubation, the DCs were collected, washed 3 times with cold PBS and centrifuged. After the removal of the supernatant, the DCs were re-suspended with PBS with the cell concentration adjusted to 1 × 105 cells/mL, fixed in pre-cooled 70% ethanol solution and incubated at 4° C overnight. With the supernatant removed through centrifugation at 800 × g at 4° C, the cells were rinsed twice with PBS containing 1% fetal bovine serum, re-suspended in 400 μL binding buffer and incubated with 50 μL RNase A (R4875, Wegene Bio-Technology Co., Ltd., Shanghai, China) at 37° C for 30 min. A total of 50 μL PI (50 mg/L) (GK3601-50T, Beijing Dingguo Changsheng Biotechnology Co., Ltd., Beijing, China) was added for 30-min incubation devoid of light. A flow cytometry was later performed to detect the cell cycle distribution.
AnnexinV-FITC/PI double staining methods were applied to assess DC cell apoptosis. After 7 d of incubation, DCs were obtained, detached with ethylenediaminetetraacetate-free 0.25% trypsin (YB15050057, Yubo Biological Technology Co., Ltd., Shanghai, China), collected into a flow tube, and centrifuged, followed by removal of the supernatant. The cells were then rinsed 3 times with cold PBS, and centrifuged with the supernatant discarded. Annexin-V-FITC/PI dye liquor [Annexin-V-FITC : PI : N-(2-hydroxyethyl) piperazine-N'-2-ethanesulfonic acid (HEPES) = 1 : 2 : 50] was prepared in accordance to the instructions of the Annexin V-FITC apoptosis kit (K201-100, BioVision, CA, USA). Every 100 μL dye liquor was used to resuspend 1 × 106 cells, incubated at room temperature for 15 min and uniformly mixed with 1 mL HEPES buffer solution (PB180325, Procell, Wuhan, China). DC cell apoptosis was then analyzed at wavelengths of 488 nm as well as 525 and 620 nm bandpass filter to detect FITC and PI fluorescence [49].
Statistical analysis
Data analyses were performed using the SPSS 19.0 software (IBM Corp., Armonk, NY, USA). Measurement data were expressed as mean ± standard deviation of at least three independent experiments. Comparisons among multiple groups were analyzed by one-way analysis of variance (ANOVA) with Tukey's tests. The repeated measures ANOVA with Bonferroni corrections was applied for the comparison of data at different time points. A p value of < 0.05 was indicative of statistical significance.
Shi-Sheng Wang and Chun-Song Yan designed the study. Chun-Song Yan and Jun-Ming Luo collated the data, carried out data analyses and produced the initial draft of the manuscript. Jun-Ming Luo revised the figures. Shi-Sheng Wang contributed to drafting the manuscript. All authors have read and approved the final submitted manuscript.
We acknowledge and appreciate our colleagues for their valuable efforts and comments on this paper.
The authors declare that they have no conflicts of interest.