




Introduction
Ischemic stroke is a major cause of human morbidities and mortalities around the world [1, 2]. The prevalence of this disease is rising in recent years [1, 2]. It is therefore important to further understand the pathological mechanisms of neuronal cell injury in ischemic stroke [3, 4], and to develop novel therapy strategies [2, 5, 6].
In cultured neurons, oxygen and glucose deprivation (OGD) re-oxygenation (OGDR) procedure is applied to mimic ischemia-reperfusion injury [7–10]. Sustained OGD (over 1h) will disrupt mitochondrial functions, and when coupled with re-oxygenation, significant reactive oxygen species (ROS) would be produced to cause severe oxidative injury [9, 11]. These events would lead to protein damage, lipid peroxidation, DNA breaks, and eventually neuronal cell necrosis and apoptosis [9, 11].
CPI-1189 (4-acetamido-N-(tert-butyl)-benzamide) is a tumor necrosis factor alpha (TNFα)-inhibiting compound. It has displayed cytoprotective and anti-inflammatory actions in different cell culture and animal models [12–17]. In animal models of Parkinson's disease (PD) and AIDS dementia, CPI-1189 treatment attenuated the deterioration in cognitive and/or motor function with no relevant side effects [16, 17].
Studies have also implied that CPI-1189 represents a promising neuroprotective compound. As it can protect neuronal cells/primary neurons from various stimuli [12–17]. CPI-1189 was able to mitigate TNFα-induced cell apoptosis and quinolinic acid-induced cell necrosis [14, 16]. In addition, CPI-1189 alleviated cell death caused by supernatants from macrophages of patients with AIDS dementia [14, 16]. Furthermore, CPI-1189 inhibited p38 phosphorylation and suppressed interleukin 1β (IL1β)-induced neuronal cell death [15]. Whether CPI-1189 can protect neuronal cells from OGDR-induced oxidative injury remains unknown.
Results
CPI-1189 protects neuronal cells from OGDR-induced cell death
SH-SY5Y neuronal cells were treated with CPI-1189 at gradually-increased concentrations from 10 to 300 nM. Cells were further cultured for 48h. Using CCK-8 cell viability and Trypan blue staining assays, we showed that CPI-1189, at tested concentrations, failed to significantly inhibit cell viability (Figure 1A) and induce cell death (Figure 1B). Next, OGDR was applied. SH-SY5Ycells were subjected to OGD for 4h, followed by re-oxygenation for another 48h. OGDR procedure led to over 70% viability (CCK-8 OD) reduction (Figure 1A) and significant cell death (increased Trypan blue ratio, Figure 1B). CPI-1189 pretreatment (for 1h) largely alleviated OGDR-induced cytotoxicity (Figure 1A, 1B). CPI-1189 displayed a concentration-dependent manner in protecting SH-SY5Y cells from OGDR (Figure 1A, 1B), especially at 30-300 nM (Figure 1A, 1B). It was however ineffective at 10 nM, the lowest concentration tested (Figure 1A, 1B). Since 100 nM of CPI-1189 displayed a significant effect against OGDR (Figure 1A, 1B), this concentration was selected for further studies.
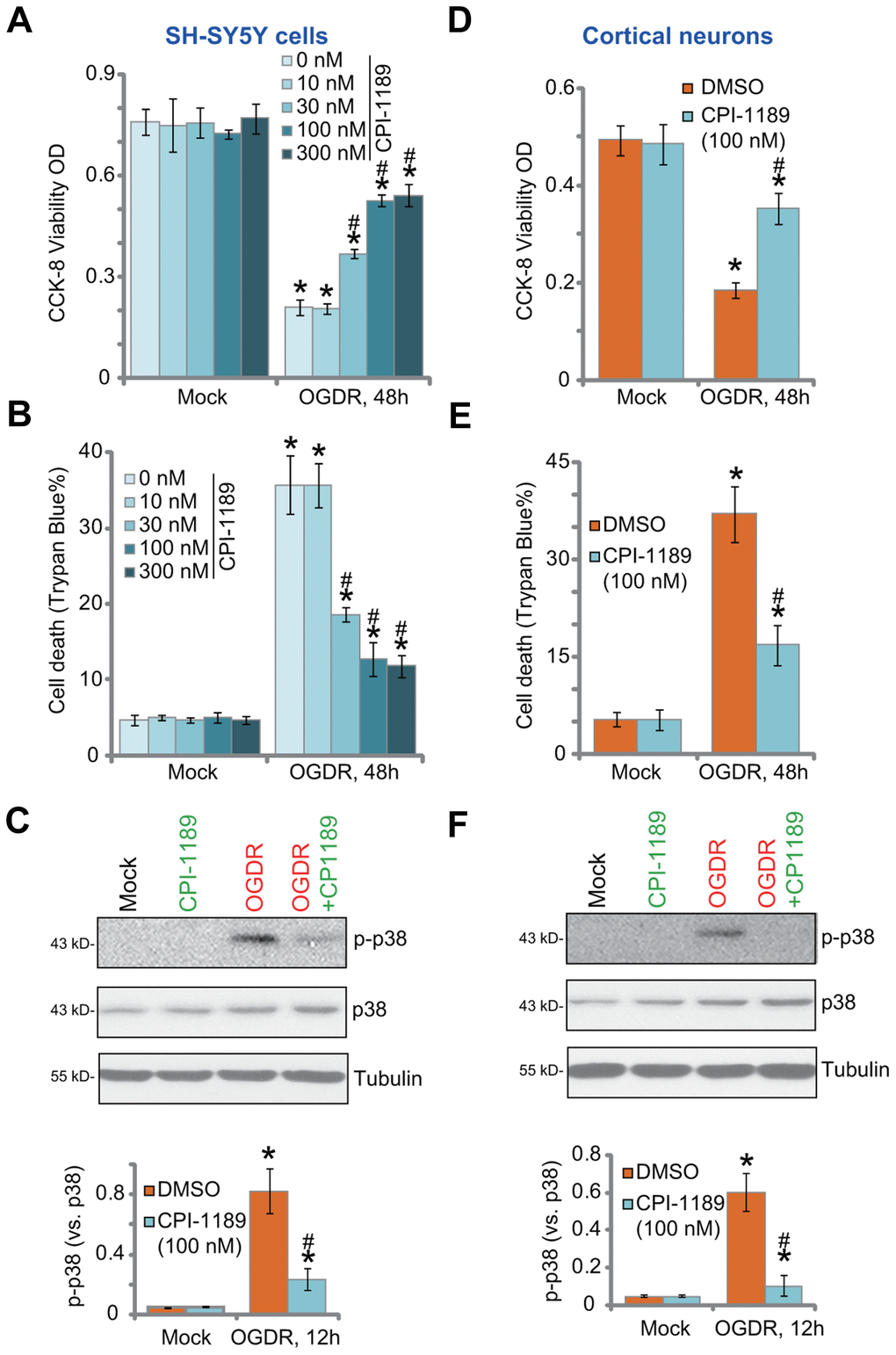
Figure 1. CPI-1189 protects neuronal cells from OGDR-induced cell death. SH-SY5Y neuronal cells (A–C) or primary murine cortical neurons (D–F) were pretreated for 1h with CPI-1189 (at applied concentrations) and subjected to OGDR procedure, cells were cultured for applied time periods, cell viability, cell death and p38 activation were tested by CCK-8 (A–D), Trypan blue staining (B–E) and Western blotting (C–F) assays, respectively. “Mock” stands for neuronal cells placed in norm-oxygenated regular medium containing glucose (same for all Figures). Quantified values were mean ± standard deviation (SD, n=5). * P < 0.05 vs. “Mock” cells. #P < 0.05 vs. cells with OGDR stimulation but “DMSO (0.1%)” pretreatment. Experiments were repeated three times, with similar results obtained.
OGDR-induced neuronal cell death is associated with p38 activation [18–20]. Inhibition of p38 can protect neuronal cells from OGDR-induced oxidative injury and cell death [18–20]. Studies have shown thatCPI-1189 was able to inhibit p38 activation to exert neuroprotective activity [12, 15]. Here we found that OGDR stimulation induced robust p38 activation (p38α Thr180/Tyr182 phosphorylation) in SH-SY5Y cells (Figure 1C). It was largely inhibited by CPI-1189 (100 nM) pretreatment (Figure 1C).
In primary murine cortical neurons, OGDR procedure induced potent viability (CCK-8 OD) reduction (Figure 1D) and cell death (Figure 1E). Both were attenuated by CPI-1189 (100 nM) pretreatment. OGDR-induced p38 activation was almost blocked by CPI-1189 (Figure 1F). CPI-1189 single treatment did not alter cell viability (Figure 1A–1D), cell death (Figure 1B–1E), or p38 activation (Figure 1C–1F) in SH-SY5Y cells and cortical neurons. These results showed that CPI-1189 protected neuronal cells from OGDR-induced cell death.
CPI-1189 inhibits OGDR-induced oxidative injury in neuronal cells
To test whether p38 inhibition is the primary mechanism of CPI-1189-induced neuroprotection against OGDR, we utilized the CRISPR/Cas9 strategy to knockout p38α. As described, a CRISPR/Cas9-p38α-KO-GFP construct was transduced to SH-SY5Y cells. Single stable cells were established following GFP sorting and puromycin selection. These cells were namely as ko-p38α cells. As shown, p38α protein expression was depleted in ko-p38α cells (Figure 2A). OGDR-induced p38 activation, or p38α Thr180/Tyr182 phosphorylation, was blocked (Figure 2A). In ko-p38α SH-SY5Y cells, OGDR-induced viability reduction (Figure 2B) and cell death (Figure 2C) were alleviated. Significantly, CPI-1189 could still protect ko-p38α SH-SY5Y cells from OGDR (Figure 2B, 2C), indicating that p38-independent mechanisms should participate in CPI-1189-induced neuroprotection against OGDR.
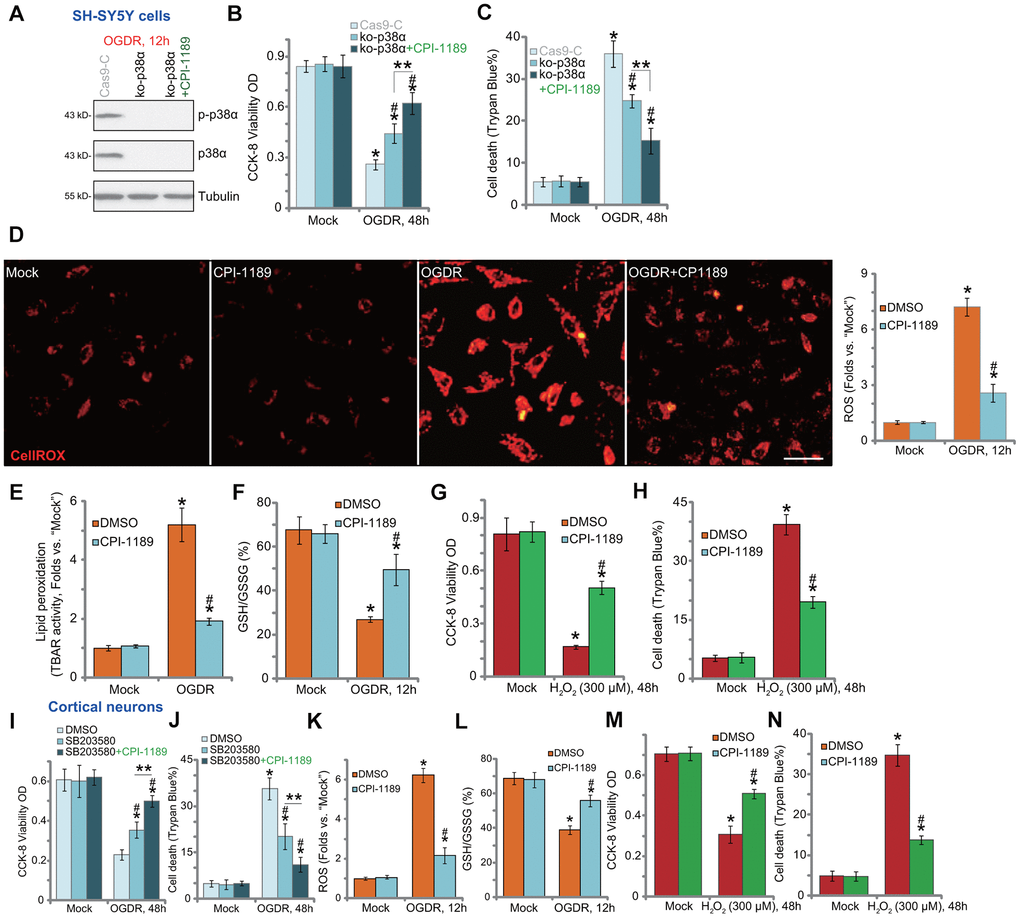
Figure 2. CPI-1189 inhibits OGDR-induced oxidative injury in neuronal cells. Stable SH-SY5Y cells with CRISPR/Cas9-p38α-KO-GFP (ko-p38α cells) were pretreated with or without CPI-1189 (100 nM, 1h pretreatment), control cells were transduced with the empty vector (“Cas9-C”), cells were subjected to OGDR procedure and cultured for applied time periods; Expression of listed proteins was shown (A); Cell viability and death were tested by CCK-8 (B) and Trypan blue staining (C) assays, respectively. SH-SY5Y cells (D–H) or primary murine cortical neurons (K–N) were pretreated for 1h with CPI-1189 (100 nM), followed by OGDR or hydrogen peroxide (H2O2, 300 μM) stimulation, cells were then cultured for applied time periods, cellular ROS contents (CellROX dye intensity, D, K), lipid peroxidation (by recording TBAR activity, E), and GSH/GSSG ratio (F–L) were tested. For cells with H2O2 stimulation, cell viability and death were tested by CCK-8 (G–M) and Trypan blue staining (H–N) assays, respectively. The primary murine cortical neurons were pretreated for 1h with SB203580 (5 μM) or plus CPI-1189 (100 nM), followed by OGDR stimulation and cells were then cultured for 48h; Cell viability and death were tested by CCK-8 (I) and Trypan blue staining (J) assays, respectively. * P < 0.05 vs. “Mock” cells. #P < 0.05 vs. cells with OGDR stimulation/H2O2 treatment but “DMSO (0.1%)” pretreatment. ** P < 0.05 (B, C, I, J). Quantified values were mean ± standard deviation (SD, n=5). Experiments were repeated three times, with similar results obtained. Scale bar= 100 μm (D).
OGDR is able to induce mitochondrial dysfunction and ROS production to mediate neuronal cell death [7–10, 20–22]. Conversely, antioxidants or other ROS scavenging strategies can protect neuronal cells from OGDR [7–10, 20–22]. By applying CellROX dye assay [23, 24], we found that OGDR stimulation in SH-SY5Y cells significantly increased cellular ROS contents (Figure 2D). It was largely inhibited by CPI-1189 (100 nM) pretreatment (Figure 2D). In addition, OGDR-induced lipid peroxidation (TBAR activity increase, Figure 2E) and GSH consumption (reflected by decreased GSH/GSSG ratio, Figure 2F) were inhibited by CPI-1189. These results implied that CPI-1189 inhibited OGDR-induced oxidative injury in SH-SY5Y cells. To mimic oxidative stress, hydrogen peroxide (H2O2) was added to cultured SH-SY5Y cells, resulting in significant viability reduction (Figure 2G) and cell death (Figure 2H). These were also inhibited by CPI-1189 pretreatment (Figure 2G, 2H).
In the primary murine cortical neurons, the p38 inhibitor SB203580 alleviated OGDR-induced cytotoxicity by restoring cell viability (Figure 2I) and inhibiting cell death (Figure 2J). Significantly, CPI-1189 offered additional neuroprotection against OGDR in cortical neurons (Figure 2I, 2J), indicating the existence of p38-independent mechanisms. Indeed, OGDR stimulation induced oxidative injury. It caused increase in CellROX intensity (Figure 2K) and GSH/GSSG ratio reduction (Figure 2L). OGDR-induced oxidative stress was potently inhibited by CPI-1189 (100 nM, 1h pretreatment) (Figure 2K, 2L). Furthermore, H2O2 induced significant viability (CCK-8 OD) reduction (Figure 2M) and cell death (Figure 2N) in cortical neurons, which were also attenuated by CPI-1189 pretreatment (Figure 2M, 2N). Collectively, CPI-1189 inhibited OGDR-induced oxidative injury in neuronal cells.
CPI-1189 inhibits OGDR-induced apoptosis activation in neuronal cells
OGDR-induced oxidative injury would lead to neuronal cell apoptosis [21, 22, 25, 26]. We thus tested the potential effect of CPI-1189 on cell apoptosis. Following OGDR stimulation, caspase-3 activity (Figure 3A) and caspase-9 activity (Figure 3B) were significantly increased in SH-SY5Y cells. In addition, cleavages of caspase-3, PARP (caspase-3’s substrate), and caspase-9 were detected in OGDR-stimulated cells (Figure 3C). Single strand DNA (ssDNA) accumulation was investigated (indicating DNA breaks, Figure 3D). These results implied activation of mitochondrial apoptosis cascade in OGDR-stimulated SH-SY5Y cells. Importantly, CPI-1189 (100 nM, 1h pretreatment) potently inhibited OGDR-induced caspase-3/-9 activation (Figure 3A–3C) and ssDNA accumulation (Figure 3D) in SH-SY5Y cells.
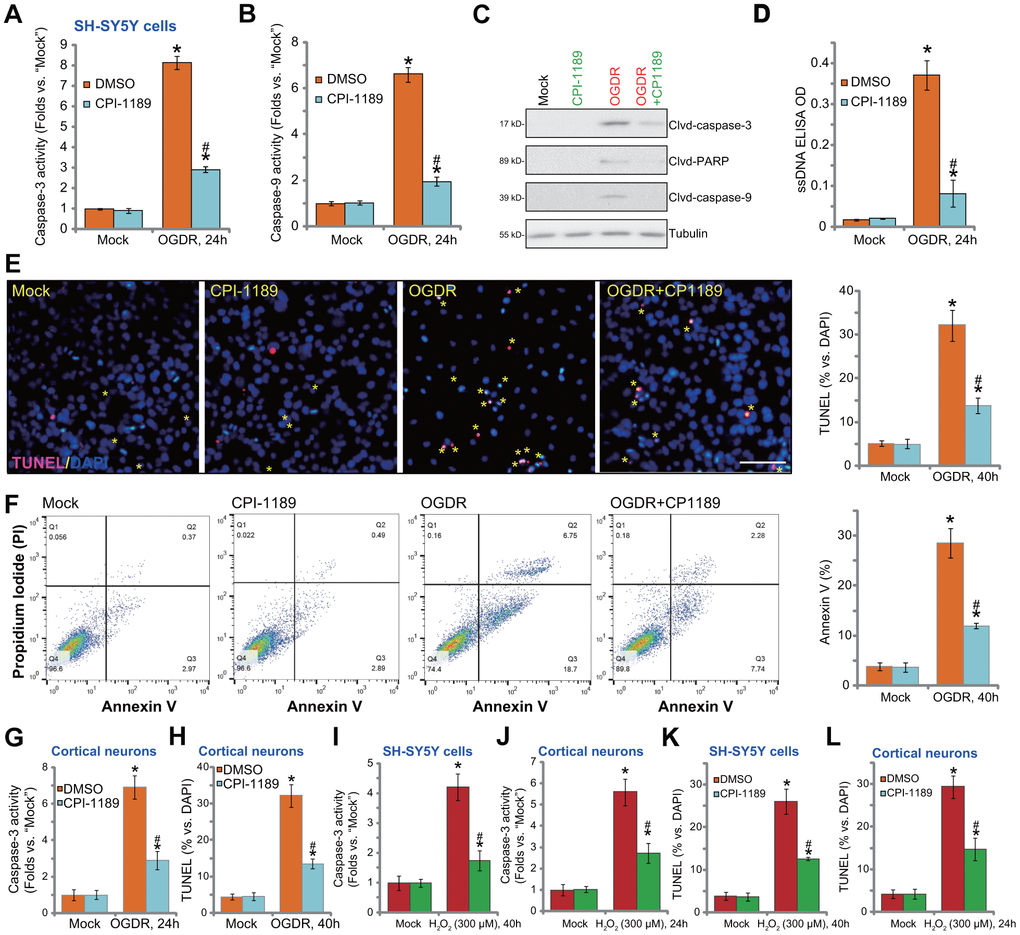
Figure 3. CPI-1189 inhibits OGDR-induced apoptosis activation in neuronal cells. SH-SY5Y neuronal cells (A–F, I–K) or primary murine cortical neurons (G, H, J–L) were pretreated for 1h with CPI-1189 (100 nM) and stimulated with OGDR or hydrogen peroxide (H2O2, 300 μM), cells were cultured for applied time periods, caspase activation and cell apoptosis were tested by the assays mentioned in the text. Quantified values were mean ± standard deviation (SD, n=5). * P < 0.05 vs. “Mock” cells. #P < 0.05 vs. cells with OGDR stimulation/H2O2 treatment but “DMSO (0.1%)” pretreatment. Experiments were repeated three times, with similar results obtained. Scale bar= 100 μm (E).
Further confirming apoptosis activation in SH-SY5Y cells, TUNEL-positive nuclei (labeled with yellow stars) ratio was significantly increased following OGDR stimulation (Figure 3E). Also, cells with positive Annexin V staining were increased after OGDR (Figure 3F). CPI-1189 pretreatment largely attenuated OGDR-induced apoptosis activation in SH-SY5Y cells (Figure 3E, 3F). CPI-1189 single treatment failed to induce caspase (Figure 3A–3D) and apoptosis (Figure 3E, 3F) activation in SH-SY5Y cells.
In primary murine cortical neurons, OGDR stimulation increased caspase-3 activity (Figure 3G) and TUNEL- positive nuclei ratio (Figure 3H), which were largely attenuated byCPI-1189 (100 nM, 1h pretreatment, Figure 3G, 3H). As the positive control, H2O2 was utilized to increase caspase-3 activation (Figure 3I, 3J) and TUNEL-positive nuclei ratio (Figure 3K, 3L) in SH-SY5Y neuronal cells and primary neurons. Importantly, CPI-1189 pretreatment largely inhibited H2O2-induced apoptosis activation in neuronal cells (Figure 3I–3L). Thus, CPI-1189 inhibited OGDR-induced apoptosis activation in neuronal cells.
CPI-1189 inhibits OGDR-induced programmed necrosis in neuronal cells
In SH-SY5Y cells, the caspase-3 inhibitor z-DEVD-fmk and the pan caspase inhibitor z-VAD-fmk were only alleviated and not abolished OGDR-induced viability reduction (Figure 4A) and cell death (Figure 4B). Besides apoptosis studies have shown that OGDR can simultaneously induce programmed necrosis in neuronal cells [22, 25]. In OGDR-treated SH-SY5Y cells, p53 translocated to mitochondria (Figure 4C, “Mito Inputs”) and immunoprecipitated with CyPD and ANT1 (Figure 4C, “Mito-IP”), two key components of mPTP [27, 28]. Furthermore, mitochondrial depolarization, evidenced by mitochondrial JC-1 green monomers accumulation (Figure 4D), was detected in OGDR-treated SH-SY5Y cells. It was followed by medium LDH release (Figure 4E). These results confirmed the activation of mitochondrial programmed necrosis cascade in OGDR-treated SH-SY5Y cells (see other studies reporting the same cascade [28, 29]). Significantly, CPI-1189 pretreatment largely attenuated OGDR-induced programmed necrosis activation in SH-SY5Y cells, inhibiting p53-CyPD-ANT1 association (Figure 4C), mitochondrial depolarization (Figure 4D), and LDH release to the medium (Figure 4E).
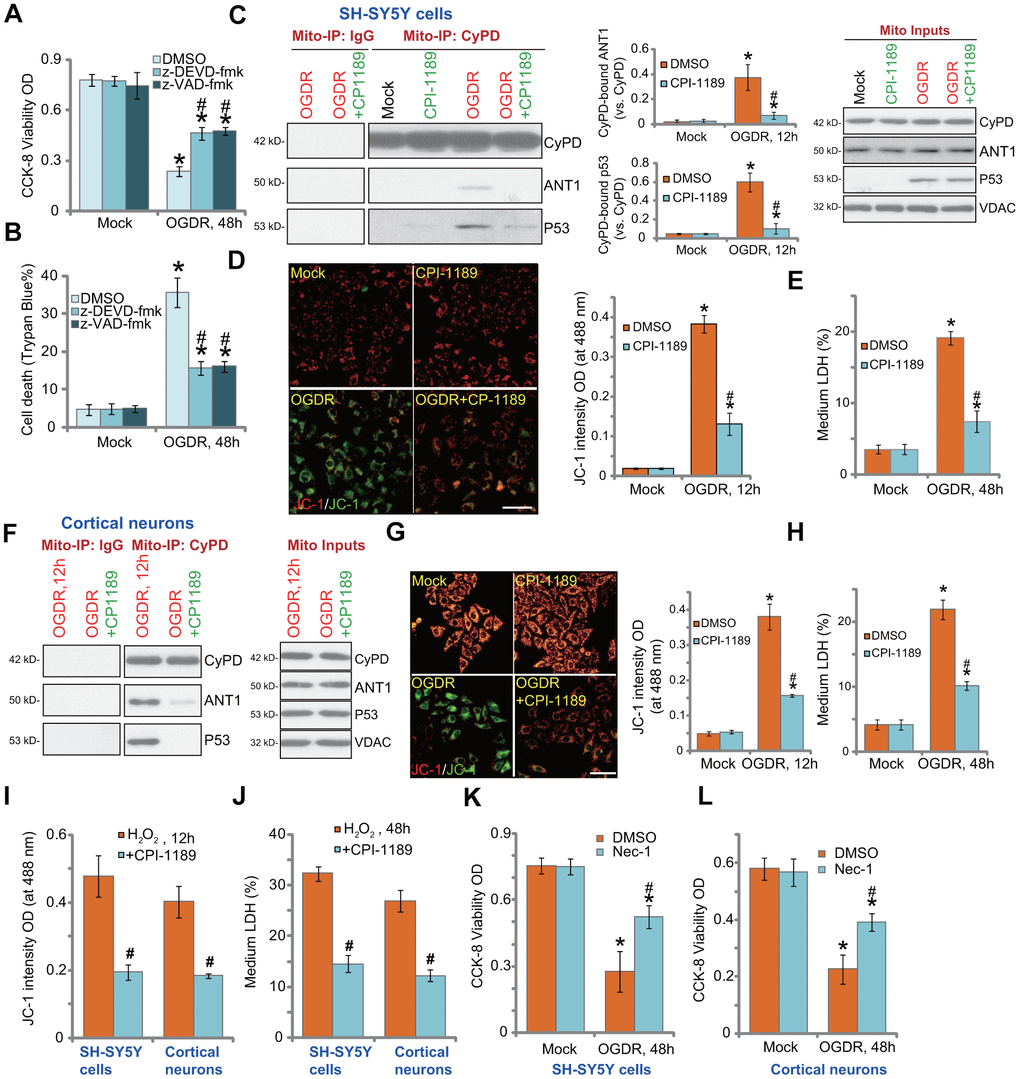
Figure 4. CPI-1189 inhibits OGDR-induced programmed necrosis in neuronal cells. SH-SY5Y cells were pretreated for 1h with z-DEVD-fmk or z-VAD-fmk (each at 50 μM), followed by OGDR stimulation; Cells were cultured for another 48h, cell viability and death were tested by CCK-8 (A) and Trypan blue staining (B) assays, respectively. SH-SY5Y neuronal cells (C–E) or primary murine cortical neurons (F–H) were pretreated for 1h with CPI-1189 (100 nM) and treated with OGDR, cells were cultured for applied time periods, mitochondrial p53-CyPD-ANT1 association (“Mito-IP: CyPD”) and their expression (“Mito Inputs”) were tested (C–F); Mitochondrial depolarization and cell necrosis were tested by JC-1 dye assay (D–G) and medium LDH release (E–H), respectively. SH-SY5Y neuronal cells or primary murine cortical neurons were pretreated for 1h with CPI-1189 (100 nM) and stimulated with hydrogen peroxide (H2O2, 300 μM); Cells were cultured for applied time periods, mitochondrial depolarization (I) and cell necrosis (J) were tested similarly. SH-SY5Y cells or primary cortical neurons were pre-treated for 1 hour with 25 μM of necrostatin-1 (“Nec-1”), followed by OGDR stimulation and cells were then cultured for 48h; Cell viability was tested by CCK-8 assays (K, L). Quantified values were mean ± standard deviation (SD, n=5). * P < 0.05 vs. “Mock” cells. #P < 0.05 vs. cells with OGDR stimulation/H2O2 treatment but “DMSO (0.1%)” pretreatment. Experiments were repeated three times, with similar results obtained. Scale bar= 100 μm (D–G).
Similar results were obtained in primary murine cortical neurons. CPI-1189 pretreatment inhibited OGDR-induced mitochondrial p53-CyPD-ANT1 association (Figure 4F), JC-1 green monomers accumulation (Figure 4G), and cell necrosis (Figure 4H). CPI-1189 by itself, as expected, did not induce programmed necrosis cascade in SH-SY5Y cells and cortical neurons (Figure 4C-H). Following H2O2 stimulation, mitochondrial depolarization (JC-1 green monomers accumulation) was detected in SH-SY5Y cells and primary cortical neurons (Figure 4I). Cell necrosis, evidenced by medium LDH release, was detected as well (Figure 4J). CPI-1189 pretreatment inhibited H2O2-induced actions above in SH-SY5Y cells and cortical neurons (Figure 4I, 4J). Therefore CPI-1189 inhibited OGDR-induced programmed necrosis in neuronal cells. Necrostatin-1 (Nec-1) is a specific necrosis inhibitor and it directly blocks receptor-interacting serine/threonine-protein kinase 1/3 (RIPK1/3) [30]. Further supporting our hypothesis, we found that Nec-1 reduced OGDR-induced viability reduction in SH-SY5Y cells (Figure 4K) and primary cortical neurons (Figure 4L).
Discussion
CPI-1189 is a compound clinically evaluated as a potential therapy for AIDS patients with dementia [13, 17]. Recent in vitro and in vivo studies have proposed the potential neuroprotective property of this compound [14–16]. Supporting its potential function in protecting neurons, we found that CPI-1189 pretreatment, at only nM concentrations, potently inhibited OGDR-induced viability reduction and death in SH-SY5Y cells and murine cortical neurons. CPI-1189 blocked OGDR-induced p38 activation. This compound was also neuroprotective in p38α-KO SH-SY5Y cells and p38-inhibited cortical neurons, indicating the possibility of p38-independent mechanisms.
OGDR-induced neuronal cell injury is associated with oxidative stress [9, 11, 21, 22]. OGD can severely impair mitochondrial functions, and ROS produced by re-oxygenation would then cause significant oxidative stress, DNA breaks, protein damage, inflammation, lipid peroxidation, and eventually neuronal cell death. Conversely, ROS scavenging is able to protect neuronal cells from OGDR [21, 22, 25].
Di et al., showed that in neuronal cells, microRNA-613 silencing upregulated its target SphK2 and inhibited OGDR-induced oxidative stress [21]. The same group reported that the SphK1 activator K6PC-5 provoked SphK1-Nrf2 signaling to inhibit OGDR-induced oxidative injury in neuronal cells [22]. Zhang et al., showed that plumbagin improved OGDR-induced SH-SY5Y cell injury by inhibiting ROS [31].
In the present study, we show that CPI-1189-induced anti-OGDR activity was associated with ROS scavenging. CPI-1189 largely inhibited OGDR-induced ROS production, lipid peroxidation, and GSH consumption in SH-SY5Y cells and cortical neurons. Importantly, OGDR-induced neuronal cell apoptosis, the consequence of oxidative injury [21, 22, 25], was inhibited by CPI-1189 as well. Therefore, ROS scavenging should be one important mechanism of CPI-1189 protecting against OGDR.
Besides apoptosis OGDR can also provoke programmed necrosis in neuronal cells. Wang et al., found that OGDR induced NKILA (NF-κB Interacting LncRNA) upregulation to promote neuronal cell necrosis [25]. SphK1 activation by K6PC-5 inhibited OGDR-induced programmed necrosis in neuronal cells [22]. Here in SH-SY5Y cells and murine cortical neurons, CPI-1189 suppressed OGDR-induced programmed necrosis by inhibiting mitochondrial p53-CyPD-ANT1 association, mitochondrial depolarization, and LDH release to the medium. These results suggested a novel mechanism (inhibition of programmed necrosis) of anti-OGDR by CPI-1189. Future studies with concurrent inhibition of OGDR-induced cell necrosis and apoptosis should explain the superior neuroprotective activity by CPI-1189.
Materials and Methods
Chemicals and reagents
CPI-1189 was provided by Selleck (Shanghai, China). Antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). DMSO, caspase-3 inhibitor z-DEVD-fmk, hydrogen peroxide (H2O2), necrostatin-1 (“Nec-1”), SB203580, the pan caspase inhibitor z-VAD-fmk, and puromycin were provided by Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
SH-SY5Y neuronal cells were provided by Dr. Di [32] and were cultured as described [32]. SH-SY5Y cells were differentiated by the incubation in BDNF plus glutamine medium (serum free) [32]. The primary murine cortical neurons were also provided by Dr. Di, and were cultured using previously described protocols [32]. At day-10 (DIV-10), over 95% of cells were cortical neurons. The protocols of using primary murine cells were approved by the Ethics Committee and IACUC of authors’ institution.
Cell viability
Cell Counting Kit-8 (CCK-8, Dojindo Laboratories, Kumamoto, Japan) was utilized to test cell viability. Neuronal cells were seeded into 96-well plates at 4, 000 cells per well. After treatment, neuronal cells were incubated with CCK-8 reagent for 3h. In each well, CCK-8 optical density (OD) was tested at 450 nm.
Cell death
Neuronal cells were seeded into 96-well plates at 4, 000 cells per well. Following treatment, dead cells were positively stained with Trypan blue. The ratio was recorded by an automatic cell counter (Roche, Shanghai, China).
Lactate dehydrogenase (LDH) assay
Neuronal cells were seeded into six-well plates at 1×105 cells per well. With the applied treatment, LDH contents in culture medium were analyzed through a two-step enzymatic reaction LDH assay kit (Takara, Tokyo, Japan), which were then normalized to total LDH contents.
OGD/re-oxygenation
As reported [7], neuronal cells were placed in an airtight chamber with a continuous flux of gas (95% N2/5% CO2). The chamber was sealed and the cells were incubated under OGD for 4h. Cells were then re-oxygenated (OGDR) and cultured in regular medium for applied time period. “Mock” neuronal cells were placed in norm-oxygenated with regular medium containing glucose.
Lipid peroxidation assay
Following treatment, thiobarbituric acid reactive substances (TBAR) assay was carried out to examine the cellular lipid peroxidation contents. The detailed protocol was reported before [21, 33].
Western blotting
Protocols for Western blotting were described previously [34]. In brief, 30 μg protein lysates per treatment were loaded to 10-12% SDS-PAGE gels and transfected to PVDF blots. The blots were then blocked and incubated with the applied primary and secondary antibodies. ECL reagents were utilized to examine the targeted protein band. The ImageJ software (NIH) was utilized to quantify the protein band.
Caspase-3/-9 activity
Previously describe protocol was used [7, 32]. In brief 20 μg of cytosolic protein lysates from neuronal cells with applied treatment were incubated with caspase-3/-9 substrate in the assay buffer [7]. Substrates were conjugated with 7-amido-4-(trifluoromethyl)-coumarin (AFC) (Calbiochem-EMD Millipore). An Fluoroskan Ascent FL machine was utilized to quantify the intensity of released AFC under 355 nm excitation and 525 nm emission.
TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay
Neuronal cells were seeded into12-well plates (at 5 × 104 cells per well). Following treatment, a TUNEL In Situ Cell Death Detection Kit (Roche) was applied to measure apoptotic nuclei. Nuclei were co-stained with TUNEL and DAPI. Cells were then visualized under a confocal microscope (Leica). For each treatment, 1000 cells in five random views (1×100 magnification) were counted to calculate the average TUNEL ratio (% vs. DAPI).
ROS assay
Neuronal cells were seeded into six-well plates. Following treatment, cells were stained with fluorescent dye CellROX (Sigma, 7.5 μM for 1h). CellROX intensity was tested by a fluorescence spectrofluorometer (Molecular Devices, San Jose, CA, USA). Representative CellROX fluorescence images were presented.
Glutathione (GSH) contents
Following treatment, a GSH/GSSG assay kit (Beyotime, Wuxi, China) was utilized to calculate the ratio of reduced glutathione to oxidized GSSG (GSH/GSSG×100%).
DNA breaks
Neuronal cells were seeded into six-well plates. Following treatment, single strand DNA (ssDNA) contents, indicating DNA breaks, were measured through ssDNA ApoStrandTM ELISA kit (BIOMOL International, Plymouth Meeting, PA, USA). ELISA OD was examined at 450 nm.
CRISPR-Cas9-induced p38α knockout (KO)
A CRISPR-Cas9-p38α-KO-GFP-puromycin construct was designed by Genechem (Shanghai, China) and transfected into cultured SH-SY5Y cells (in polybrene medium). GFP-positive SH-SY5Y cells were sorted by FACS and distributed to 96-well plates to achieve single cells. Stable cells were further selected by puromycin. In the stable cells, p38α KO was verified by Western blotting. The target DNA sequence of p38α sgRNA is TGGACGTTTTTACACCTGCA (PAM: AGG).
Mitochondrial
immunoprecipitation (Mito-IP). As described [25], following the applied treatment, a “Mitochondria Isolation Kit for Cultured Cells” (Pierce, Rockford, IL) was utilized to isolate mitochondria of neuronal cells (via high-speed centrifugation). The resulting mitochondrial fraction lysates (300 μg) were pre-cleared and incubated with anti-Cyclophilin D (CyPD) antibody [35]. Afterwards, protein IgG beads (30 μL per treatment, Sigma) were added to obtain CyPD-immunoprecipitated proteins, which were then tested by Western blotting. The mitochondrial CyPD-ANT1 (adenine nucleotide translocase 1)-p53 association was analyzed.
Mitochondrial depolarization
JC-1 fluorescence dye aggregates into mitochondria to form green monomers in cells with mitochondrial depolarization [36]. Neuronal cells were seeded into 12-well plates. Following treatment, cells were stained with JC-1 (15 μg/mL, Sigma), and then washed and examined under a fluorescence spectrofluorometer (F-7000, Hitachi, Japan) at 488 nm (green). The representative JC-1 fluorescence images integrating both green (at 488 nm) and red (at 625 nm) fluorescence channels were presented as well.
Annexin V-FACS
Neuronal cells were seeded into six-well plates at 1×105 cells per well. With the applied treatment, cells were co-stained with Annexin V (15 μg/mL) and Propidium Iodide (PI, 15 μg/mL), and measured under a FACS machine (BD, Shanghai, China). Annexin V-positive cells (apoptotic cells) were gated and its ratio was recorded.
Statistics
Data were expressed as means ± standard deviation (SD). Statistical analyses among different groups were tested by one-way analysis of variance (ANOVA) and Tukey’s post hoc multiple comparison tests (SPSS 23.0, SPSS, Chicago, IL, USA). The Student t test (Excel2007) was applied to compare statistical difference between two groups. P< 0.05 was considered as statistically significant.
Author Contributions
YL, YZ, CL performed SH-SY5Y cell culture and signaling studies. YL, YZ, CL, JS, CY performed oxidative injury studies and all cell death studies. YL, YZ, CL, JS, CY: Study conception and design, and data analysis, Figure organization, involved in drafting the article and revising it critically for important intellectual content, and with final approval of the version submitted to the journal.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This work was generously supported by grants from Affiliated Hospital of Yangzhou University.
References
- 1. Fuentes B, Tejedor ED. Stroke: the worldwide burden of stroke—a blurred photograph. Nat Rev Neurol. 2014; 10:127–28. https://doi.org/10.1038/nrneurol.2014.17 [PubMed]
- 2. Tymianski M. Stroke in 2013: disappointments and advances in acute stroke intervention. Nat Rev Neurol. 2014; 10:66–68. https://doi.org/10.1038/nrneurol.2013.271 [PubMed]
- 3. Verklan MT. The chilling details: hypoxic-ischemic encephalopathy. J Perinat Neonatal Nurs. 2009; 23:59–68. https://doi.org/10.1097/01.JPN.0000346221.48202.7e [PubMed]
- 4. Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. 2009; 4:461–70. https://doi.org/10.1111/j.1747-4949.2009.00387.x [PubMed]
- 5. Clark WM, Clark TD. Stroke: treatment for acute stroke—the end of the citicoline saga. Nat Rev Neurol. 2012; 8:484–85. https://doi.org/10.1038/nrneurol.2012.166 [PubMed]
- 6. Planas AM. Advances in stroke: translational medicine 2012. Stroke. 2013; 44:318–19. https://doi.org/10.1161/STROKEAHA.111.000495 [PubMed]
- 7. Zhao LP, Ji C, Lu PH, Li C, Xu B, Gao H. Oxygen glucose deprivation (OGD)/re-oxygenation-induced in vitro neuronal cell death involves mitochondrial cyclophilin-D/P53 signaling axis. Neurochem Res. 2013; 38:705–13. https://doi.org/10.1007/s11064-013-0968-5 [PubMed]
- 8. Gu DM, Lu PH, Zhang K, Wang X, Sun M, Chen GQ, Wang Q. EGFR mediates astragaloside IV-induced Nrf2 activation to protect cortical neurons against in vitro ischemia/reperfusion damages. Biochem Biophys Res Commun. 2015; 457:391–97. https://doi.org/10.1016/j.bbrc.2015.01.002 [PubMed]
- 9. Almeida A, Delgado-Esteban M, Bolaños JP, Medina JM. Oxygen and glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones but not in astrocytes in primary culture. J Neurochem. 2002; 81:207–17. https://doi.org/10.1046/j.1471-4159.2002.00827.x [PubMed]
- 10. Zhao H, Mitchell S, Ciechanowicz S, Savage S, Wang T, Ji X, Ma D. Argon protects against hypoxic-ischemic brain injury in neonatal rats through activation of nuclear factor (erythroid-derived 2)-like 2. Oncotarget. 2016; 7:25640–51. https://doi.org/10.18632/oncotarget.8241 [PubMed]
- 11. Blokhina O, Virolainen E, Fagerstedt KV. Antioxidants, oxidative damage and oxygen deprivation stress: a review. Ann Bot. 2003; 91:179–94. https://doi.org/10.1093/aob/mcf118 [PubMed]
- 12. Müller T. CPI-1189. Centaur. Curr Opin Investig Drugs. 2002; 3:1763–67. [PubMed]
- 13. Clifford DB, McArthur JC, Schifitto G, Kieburtz K, McDermott MP, Letendre S, Cohen BA, Marder K, Ellis RJ, Marra CM, and Neurologic AIDS Research Consortium. A randomized clinical trial of CPI-1189 for HIV-associated cognitive-motor impairment. Neurology. 2002; 59:1568–73. https://doi.org/10.1212/01.wnl.0000034177.47015.da [PubMed]
- 14. Pulliam L, Irwin I, Kusdra L, Rempel H, Flitter WD, Garland WA. CPI-1189 attenuates effects of suspected neurotoxins associated with AIDS dementia: a possible role for ERK activation. Brain Res. 2001; 893:95–103. https://doi.org/10.1016/s0006-8993(00)03293-5 [PubMed]
- 15. Hensley K, Robinson KA, Pye QN, Floyd RA, Cheng I, Garland WA, Irwin I. CPI-1189 inhibits interleukin 1beta-induced p38-mitogen-activated protein kinase phosphorylation: an explanation for its neuroprotective properties? Neurosci Lett. 2000; 281:179–82. https://doi.org/10.1016/s0304-3940(00)00861-2 [PubMed]
- 16. Bjugstad KB, Flitter WD, Garland WA, Philpot RM, Kirstein CL, Arendash GW. CPI-1189 prevents apoptosis and reduces glial fibrillary acidic protein immunostaining in a TNF-alpha infusion model for AIDS dementia complex. J Neurovirol. 2000; 6:478–91. https://doi.org/10.3109/13550280009091948 [PubMed]
- 17. Bjugstad KB, Flitter WD, Garland WA, Su GC, Arendash GW. Preventive actions of a synthetic antioxidant in a novel animal model of AIDS dementia. Brain Res. 1998; 795:349–57. https://doi.org/10.1016/s0006-8993(98)00351-5 [PubMed]
- 18. Li CT, Zhang WP, Lu YB, Fang SH, Yuan YM, Qi LL, Zhang LH, Huang XJ, Zhang L, Chen Z, Wei EQ. Oxygen-glucose deprivation activates 5-lipoxygenase mediated by oxidative stress through the p38 mitogen-activated protein kinase pathway in PC12 cells. J Neurosci Res. 2009; 87:991–1001. https://doi.org/10.1002/jnr.21913 [PubMed]
- 19. Shinozaki Y, Sato Y, Koizumi S, Ohno Y, Nagao T, Inoue K. Retinoic acids acting through retinoid receptors protect hippocampal neurons from oxygen-glucose deprivation-mediated cell death by inhibition of c-jun-N-terminal kinase and p38 mitogen-activated protein kinase. Neuroscience. 2007; 147:153–63. https://doi.org/10.1016/j.neuroscience.2007.04.032 [PubMed]
- 20. Lu Q, Rau TF, Harris V, Johnson M, Poulsen DJ, Black SM. Increased p38 mitogen-activated protein kinase signaling is involved in the oxidative stress associated with oxygen and glucose deprivation in neonatal hippocampal slice cultures. Eur J Neurosci. 2011; 34:1093–101. https://doi.org/10.1111/j.1460-9568.2011.07786.x [PubMed]
- 21. Di G, Wang Z, Wang W, Cheng F, Liu H. AntagomiR-613 protects neuronal cells from oxygen glucose deprivation/re-oxygenation via increasing SphK2 expression. Biochem Biophys Res Commun. 2017; 493:188–94. https://doi.org/10.1016/j.bbrc.2017.09.049 [PubMed]
- 22. Liu H, Zhang Z, Xu M, Xu R, Wang Z, Di G. K6PC-5 activates SphK1-Nrf2 signaling to protect neuronal cells from oxygen glucose deprivation/re-oxygenation. Cell Physiol Biochem. 2018; 51:1908–20. https://doi.org/10.1159/000495716 [PubMed]
- 23. Zheng Z, Wang Y, Yu H, Li W, Wu J, Cai C, He Y. Salvianolic acid B inhibits ototoxic drug-induced ototoxicity by suppression of the mitochondrial apoptosis pathway. J Cell Mol Med. 2020; 24:6883–97. https://doi.org/10.1111/jcmm.15345 [PubMed]
- 24. Escada-Rebelo S, Mora FG, Sousa AP, Almeida-Santos T, Paiva A, Ramalho-Santos J. Fluorescent probes for the detection of reactive oxygen species in human spermatozoa. Asian J Androl. 2020; 22:465–71. https://doi.org/10.4103/aja.aja_132_19 [PubMed]
- 25. Wang M, Jiang YM, Xia LY, Wang Y, Li WY, Jin T. LncRNA NKILA upregulation mediates oxygen glucose deprivation/re-oxygenation-induced neuronal cell death by inhibiting NF-κB signaling. Biochem Biophys Res Commun. 2018; 503:2524–30. https://doi.org/10.1016/j.bbrc.2018.07.010 [PubMed]
- 26. Weng Y, Lin J, Liu H, Wu H, Yan Z, Zhao J. AMPK activation by tanshinone IIA protects neuronal cells from oxygen-glucose deprivation. Oncotarget. 2017; 9:4511–21. https://doi.org/10.18632/oncotarget.23391 [PubMed]
- 27. Tang XF, Liu HY, Wu L, Li MH, Li SP, Xu HB. Ginseng Rh2 protects endometrial cells from oxygen glucose deprivation/re-oxygenation. Oncotarget. 2017; 8:105703–13. https://doi.org/10.18632/oncotarget.22390 [PubMed]
- 28. Xu HB, Zheng YF, Wu D, Li Y, Zhou LN, Chen YG. microRNA-1203 targets and silences cyclophilin D to protect human endometrial cells from oxygen and glucose deprivation-re-oxygenation. Aging (Albany NY). 2020; 12:3010–24. https://doi.org/10.18632/aging.102795 [PubMed]
- 29. Sun Q, Shen X, Wang P, Ma J, Sha W. Targeting cyclophilin-D by miR-1281 protects human macrophages from Mycobacterium tuberculosis-induced programmed necrosis and apoptosis. Aging (Albany NY). 2019; 11:12661–73. https://doi.org/10.18632/aging.102593 [PubMed]
- 30. Ito K, Eguchi Y, Imagawa Y, Akai S, Mochizuki H, Tsujimoto Y. MPP+ induces necrostatin-1- and ferrostatin-1-sensitive necrotic death of neuronal SH-SY5Y cells. Cell Death Discov. 2017; 3:17013. https://doi.org/10.1038/cddiscovery.2017.13 [PubMed]
- 31. Zhang Q, Zhao S, Zheng W, Fu H, Wu T, Hu F. Plumbagin attenuated oxygen-glucose deprivation/reoxygenation-induced injury in human SH-SY5Y cells by inhibiting NOX4-derived ROS-activated NLRP3 inflammasome. Biosci Biotechnol Biochem. 2020; 84:134–42. https://doi.org/10.1080/09168451.2019.1664893 [PubMed]
- 32. Liu H, Feng Y, Xu M, Yang J, Wang Z, Di G. Four-octyl itaconate activates Keap1-Nrf2 signaling to protect neuronal cells from hydrogen peroxide. Cell Commun Signal. 2018; 16:81. https://doi.org/10.1186/s12964-018-0294-2 [PubMed]
- 33. Li C, Yan K, Wang W, Bai Q, Dai C, Li X, Huang D. MIND4-17 protects retinal pigment epithelium cells and retinal ganglion cells from UV. Oncotarget. 2017; 8:89793–801. https://doi.org/10.18632/oncotarget.21131 [PubMed]
- 34. Cao C, Huang X, Han Y, Wan Y, Birnbaumer L, Feng GS, Marshall J, Jiang M, Chu WM. Galpha(i1) and Galpha(i3) are required for epidermal growth factor-mediated activation of the Akt-mTORC1 pathway. Sci Signal. 2009; 2:ra17. https://doi.org/10.1126/scisignal.2000118 [PubMed]
- 35. Li ST, Chen NN, Qiao YB, Zhu WL, Ruan JW, Zhou XZ. SC79 rescues osteoblasts from dexamethasone though activating Akt-Nrf2 signaling. Biochem Biophys Res Commun. 2016; 479:54–60. https://doi.org/10.1016/j.bbrc.2016.09.027 [PubMed]
- 36. Brooks MM, Neelam S, Fudala R, Gryczynski I, Cammarata PR. Lenticular mitoprotection. Part A: Monitoring mitochondrial depolarization with JC-1 and artifactual fluorescence by the glycogen synthase kinase-3β inhibitor, SB216763. Mol Vis. 2013; 19:1406–12. [PubMed]