



Introduction
Hepatocellular carcinoma (HCC) is a common digestive cancer and the second leading cause of cancer-related death around the world. The number of deaths of HCC patients gradually increases every year, indicating its high lethality [1, 2]. This tumor always emerges in the background of chronic liver disease (e.g., cirrhosis) and is correlated with several well-known factors, such as hepatitis B virus (HBV), hepatitis C virus (HCV), alcohol consumption, diabetes mellitus, and metabolic syndrome [2]. In recent years, novel and fast-growing medical technologies have illuminated the molecular mechanisms underlying the occurrence and development of HCC; however, the current clinical therapeutic methods are limited [2, 3]. Only a subset of HCC patients diagnosed at an early stage obtain favorable effects when receiving conventional therapies, such as surgical resection, liver transplantation, or local ablation [2]. While for patients at advanced or metastatic stages, the effective treatment that prolongs HCC survival is limited to multiple tyrosine kinase inhibitors, including first- (e.g., sorafenib) [4] and second-line agents (e.g., regorafenib) [5]. Although clinical benefits have been reported using these drugs, the median survival interval is still less than 2 years. Therefore, more effective therapeutic approaches are urgently needed for advanced HCC patients.
In the past few years, immune checkpoint inhibitors (ICI) therapies, which reactivate the related regulatory signaling of T cells and revive the immune system of tumor patients to kill tumor cells, have remarkably extended the life expectancy in patients with distinct solid tumors [6, 7]. Owing to the favorable clinical efficacy, the Food and Drug Administration (FDA) has approved 4 immune checkpoint inhibition-based agents (i.e., ipilimumab, nivolumab, pembrolizumab and atezolimumab) for treatment of advanced stage cancers or metastases, such as melanoma and non-small cell lung cancer (NSCLC) [8]. The immune checkpoints directed against monoclonal antibodies from the above agents include cytotoxic T-lymphocyte protein 4 (CTLA-4), the programmed cell death protein 1 (PD-1), and its ligand, PD-L1 [9]. Nevertheless, only a minority of patients could obtain a durable treatment response to these regimens [10]. High PD-L1 expression is a frequently-used indicator to predict the efficacy of anti-PD-1 therapy [11–13]. Previous experimental evidence revealed that the presence of a high T cell infiltration, an interferon-gamma (IFN-γ) signature, checkpoint gene (e.g., PD-1 and PD-L1) expression, or a high tumor mutational load (TML) could favor a treatment response [14–16]. Conversely, several immune-suppressive factors, such as stromal cells and M2 macrophages infiltration, may lead to a reduction in the anti-tumor immune response, and resistance to ICI therapy [17]. In a phase I/II HCC clinical trial, remarkable responses were reported when patients were treated with nivolumab, a monoclonal antibody targeting PD-1 [18]. Unfortunately, there is less evidence relevant to the immunologic subtypes of HCC and how to make use of this information to achieve the best efficacy from immune checkpoint-based treatment.
The HCC microenvironment is a mixture of distinct cell types, including malignant hepatocytes, immune cells, endothelial cells, and stromal cells. A variety of analytic methods have been established to virtually extract molecular features from the tumor-immune microenvironment [19, 20]. By applying a non-negative matrix factorization (NMF) algorithm [21], we deconvoluted the gene expression profile of 719 HCC patients and dissected the signals related to the immune microenvironment, which allowed us to determine a potential immune subtype of HCC with specific immunologic features. The key traits of this subtype include infiltration of immune cells, increased enrichment of the IFN-γ signature, a T cell-inflamed signature and cytolytic activity, elevated expression of immune checkpoints, and most importantly, a favorable prognosis. The HCC immune subtype in our study may provide a novel strategy for evaluating survival and immunotherapy implications. Further in-depth investigations are warranted based on the HCC patients who received immunotherapy.
Results
Identification of an immune class for HCC
Coefficient of variation (CV) analysis showed that 8163 genes had CV values less than 0.1 in the TCGA cohort. Based on the gene expression profile of these genes, 9 classes were identified using NMF clustering analysis (Figure 1A). We found that one class harbored the highest immune enrichment score than others (Figure 1A), thus designated as the ‘immune class’. To verify the functionality of this immune class, we obtained the top 100 genes (Supplementary Table 1) that had the greatest contribution to this class to perform pathway annotation. Based on the results of pathway analysis, we observed that antigen processing and presentation, and signaling mediated by immune cells (e.g., T cells, B cells and NK cells) were significantly enriched (all P < 0.05; Figure 1B). Biology processes, such as innate and adaptive immune responses, T cell and B cell receptor signaling, T cell activation, and cytolysis were also observed (all P < 0.05; Figure 1B). Together, these findings further confirm that this immune class is immunogenic.

Figure 1. Identification of HCC immune class and its pathway analysis in the TCGA cohort. (A) The association of identified 9 HCC classes with immune enrichment score. (B) Pathway analysis of the top 100 genes contributed to immune class.
Identification and validation of an immune HCC subtype
To obtain more accurate HCC subtypes, we performed NMF clustering based on the gene expression profile of the aforementioned top 100 genes of the immune class in the TCGA cohort. We separately evaluated the model parameters with clustering numbers set as 2-6. Cophenetic, dispersion, residuals, and RSS coefficients could obtain the maximum values when the cluster number was selected as 2 (Figure 2A) Consistently, heatmap analysis also exhibited the best clustering effect when the number was 2 (Supplementary Figure 1). We consider that two subtypes potentially exist in HCC patients.
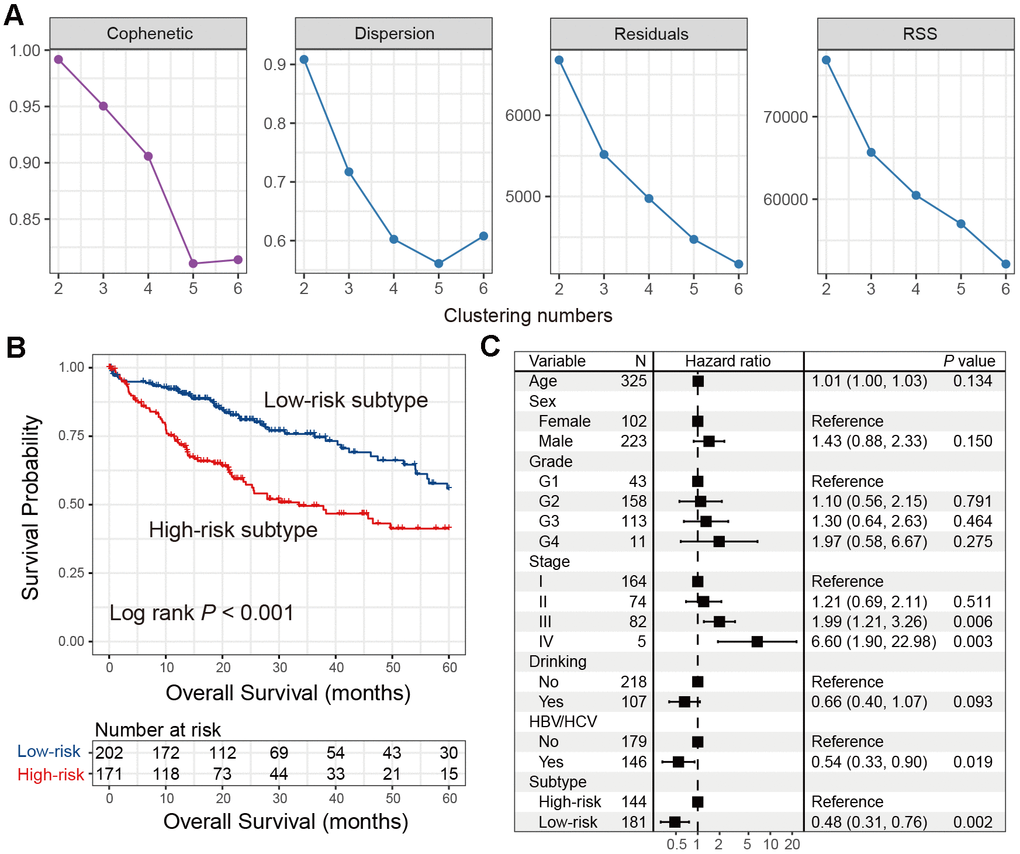
Figure 2. Identification of the immune low-risk subtype of HCC in TCGA. (A) Associations between NMF coefficients and clustering numbers. (B) Kaplan-Meier survival analysis of identified low-risk and high-risk subtypes. (C) Forest plot of multivariate Cox regression model with HCC clinical factors taken into account.
Kaplan-Meier survival analysis showed that these two subtypes were statistically prognostically different (Log-rank test P < 0.001; Figure 2B). The subtype with better prognosis was designated as the ‘low-risk’ subtype (n = 202), and the subtype with poor survival was designated as the ‘high-risk’ subtype (n = 171). Multivariate Cox regression model with clinical characteristics and confounding factors (i.e., age, sex, grade, stage, drinking status, and HBV/HCV status) taken into account was still statistically significant (HR: 0.48, 95% CI: 0.31-0.76, P = 0.002; Figure 2C). Differential analyses results of the top 100 immune genes between low- and high-risk HCC subtypes in the TCGA cohort were shown in Supplementary Table 2.
Two independent HCC cohorts were utilized to validate the prognostic ability of the two subtypes identified from TCGA. Low- and high-risk subtypes were also observed via univariate analysis and the multivariate Cox regression model in ICGC cohort (Log-rank test P = 0.031; HR: 0.64, 95% CI: 0.34-1.01, P = 0.048; Figure 3A, 3B), as well as the GEO cohort (Log-rank test P = 0.005; HR: 0.46, 95% CI: 0.22-0.99, P = 0.042; Figure 3C, 3D).
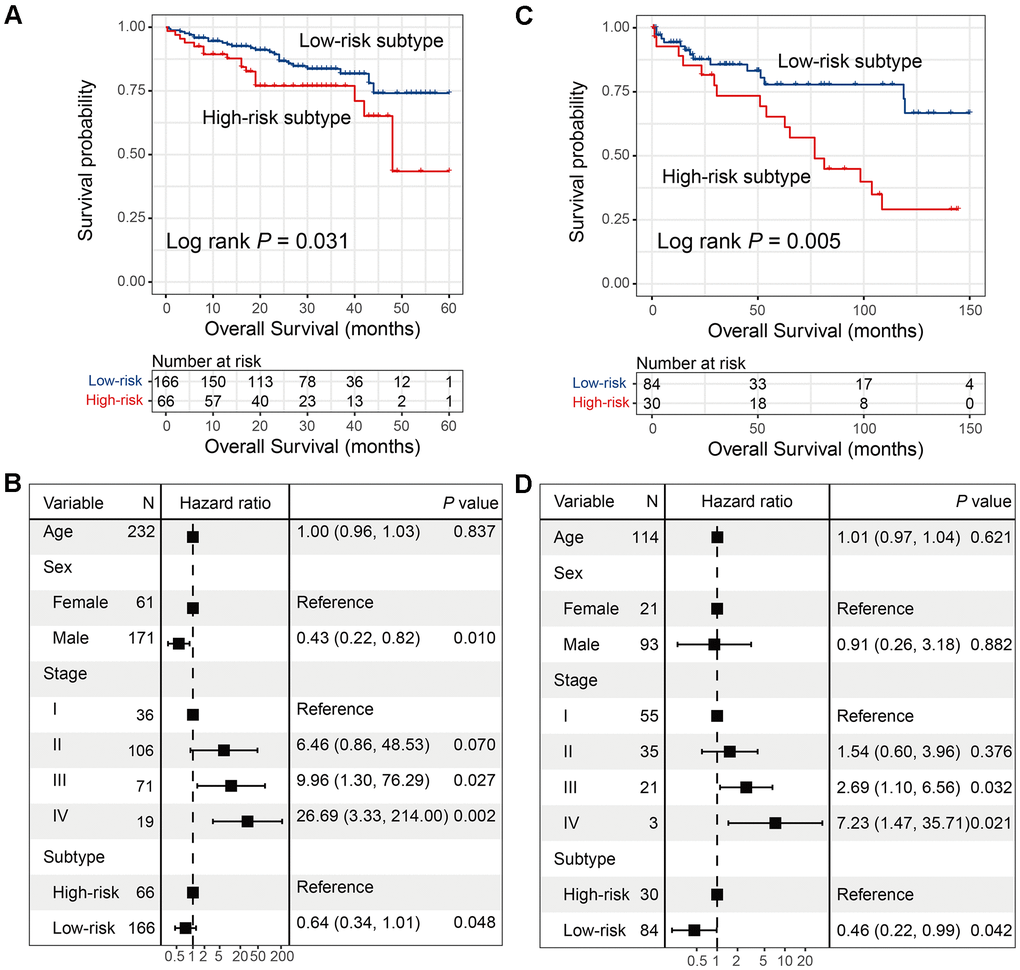
Figure 3. Validation for the 2 HCC subtypes with additional 2 independent datasets. (A, B) Univariate and multivariate survival analysis of 2 HCC subtypes in the ICGC cohort. (C, D) Univariate and multivariate survival analysis of 2 HCC subtypes in GSE76427.
Patients from the low-risk HCC subtype harbored immune-activated microenvironment
To elucidate the association of low-risk subtype with better prognosis, we explored the vital factors in the microenvironment in relation to the low-risk subtype.
For the infiltration of immune cells, we found that the low-risk subtype had significantly higher CD8 T cell infiltration than the high-risk subtype (P < 0.01; Figure 4A). In addition, resting CD4 memory T cells, monocytes, and resting mast cells were also significantly enriched in the low-risk subtype (all P < 0.05; Figure 4A). Enrichment of the regulatory T cells, which exhibit the immune suppression, was markedly decreased in the low-risk subtype (P < 0.05; Figure 4A). Interestingly, the low-risk subtype had significantly elevated infiltration of M1 macrophages (P < 0.05; Figure 4A), which is one subtype of macrophages that promotes the inflammation. M2 macrophages, which are associated with tumor growth and immune inhibition, were significantly enriched in the high-risk subtype, compared to the low-risk subtype (P < 0.001; Figure 4A).
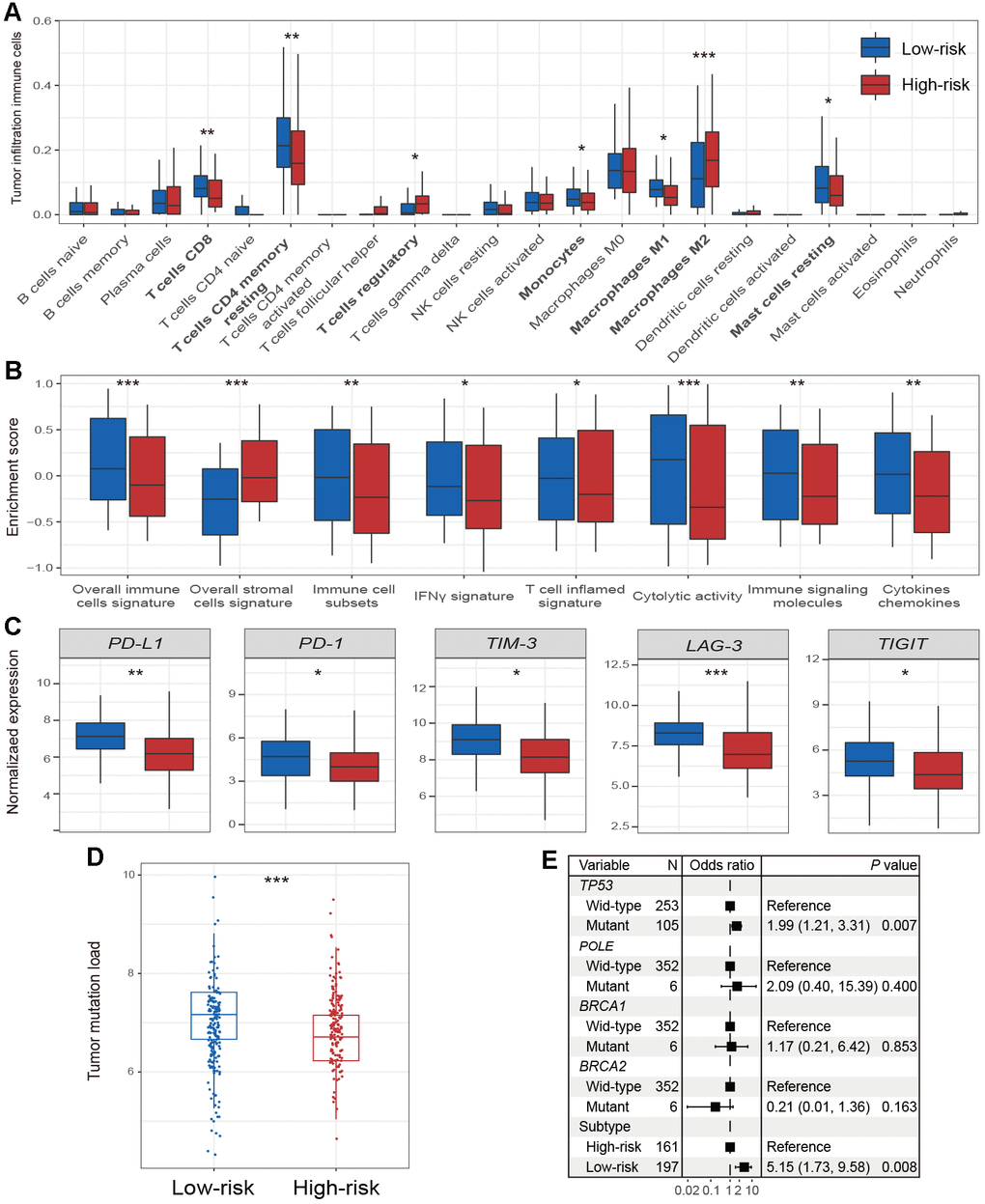
Figure 4. Immune microenvironment and genomic features in relation to 2 HCC subtypes. Distinct enrichment of (A) infiltration immune cells, (B) immune-related signatures, and (C) immune checkpoints in the 2 HCC subtypes. (D) The association of 2 identified subtypes with TML. (E) Forest plot representation of the association between the 2 identified subtypes and TML. * P < 0.05, ** P < 0.01, *** P < 0.001.
Patients of low-risk subtype had significantly higher enrichment of total immune cells, and immune cell subsets (i.e., T cells, B cells, and NK cells) (all P < 0.01; Figure 4B). The enrichment of immune-suppressive stromal cells was significantly decreased in the low-risk subtype (P < 0.001; Figure 4B). In the low-risk subtype, we also found markedly increased enrichment of the IFN-γ signature and the T cell-inflamed gene signature (both P < 0.05; Figure 4B), which were recently reported to be correlated with ICI efficacy [22]. In addition, enhanced cytolytic activity, and elevated enrichment of cytokines and chemokines were all observed in patients from the low-risk subtype (all P < 0.01; Figure 4B).
We found that expression of PD-L1 and PD-1 was significantly upregulated in low-risk subtype patients (both P < 0.05; Figure 4C). Other checkpoints, including TIM-3, LAG-3, and TIGIT also obtained similar results (all P < 0.05; Figure 4C); however, no differences were detected in CTLA-4 and IDO1 expression between low- and high-risk subtypes (both P > 0.05; Supplementary Figure 2).
We also observed that the low-risk HCC subtype harbored a significantly higher TML than high-risk subtype (P < 0.001; Figure 4D). Multivariate Logistic regression model with mutations of DNA repair genes (i.e., TP53, POLE, BRCA1, and BRCA2) taken into consideration was still significant (OR: 5.15, 95% CI: 1.73-9.58, P = 0.008; Figure 4E).
Pathways significantly enriched in the low-risk HCC subtype
GSEA analysis between the two subtypes showed that immune cell-related pathways, such as NK cell-mediated cytotoxicity, and T and B cell receptor signaling pathways were markedly enriched in the low-risk subtype (all FDR < 0.05; Figure 5). Immune response pathways including antigen processing and presentation, and the inflammatory response were also enriched (all FDR < 0.05; Figure 5). Patients of low-risk subtype harbored the enrichment of IFN-γ-related pathways, which are associated with anti-tumor immunity and immunotherapy efficacy (all FDR < 0.05; Figure 5).
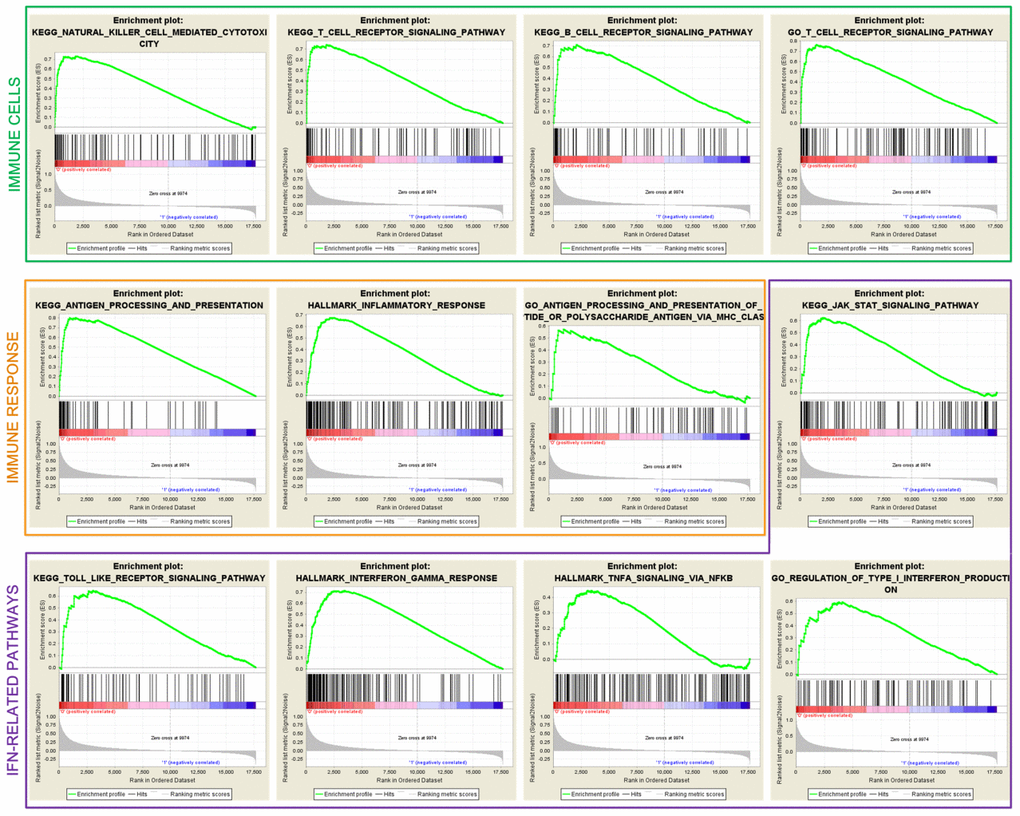
Figure 5. Significantly enriched immune cells, immune response, and IFN-related pathways in the low-risk subtype.
SMGs of the low-risk HCC subtype
A total of 33 significantly mutated genes (SMGs) were identified using the MutSigCV algorithm. Differential analyses of the SMGs mutation rates between the two subtypes showed that TP53, MUC16, RB1, NBEA, SPEG, and DNAH10 mutations were significantly enriched in the patients of low-risk subtype (Fisher exact test, all P < 0.05; Figure 6). Among them, TP53 mutations were previously reported to be associated with the ICI response in lung adenocarcinoma (LUAD), and MUC16 mutations harbored potential immunotherapy implications for gastric cancer (GC) patients.
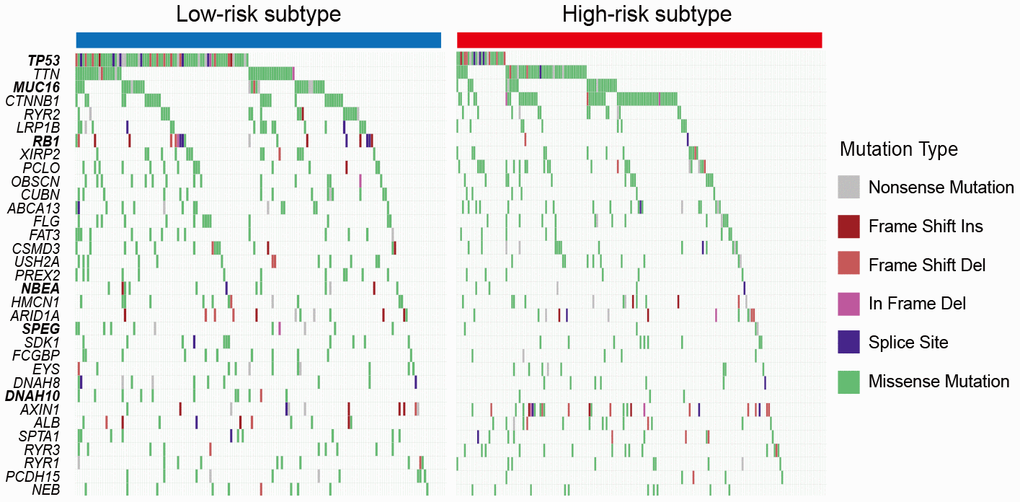
Figure 6. Mutation rates of SMGs stratified with the 2 HCC subclasses. Genes with bold and italic font were observed to be significantly differentially mutated in the 2 HCC subtypes. * P < 0.05, ** P < 0.01, *** P < 0.001.
Discussion
By using virtual dissection analytic methods, we deconvoluted the gene expression profile from mixed HCC tissues, and thus determined an undiscovered immune pattern and a moderate immunology subtype of HCC, herein designated the low-risk subtype. The low-risk subtype harbored favorable survival outcomes, a better immune microenvironment, and genomic features compared to the high-risk subtype. The identified low-risk subtype may be a promising indicator for prognosis prediction and clinical immunotherapy of HCC patients.
The prognoses of patients with melanoma and NSCLC have dramatically changed owing to the approval of ICI agents by the FDA. Long-range clinical benefits and durable remissions induced by these agents have been observed in a subset of patients with metastatic or advanced stage cancer [10, 23]. Taking into consideration that the directed targets of these drugs are immune cells instead of tumor cells, the effective responses could be detected in multiple cancers, such as colorectal [13, 16] and bladder cancer [24]. In the phase II clinical trial that involved 214 HCC patients who received nivolumab, the objective response rate and median survival interval were 16% and 14 months, respectively [18]. In this trial, patients with clinical responses were not shown to be associated with high PD-L1 expression [18]. Therefore, the determination of more reliable indicators to select the appropriate sub-population for receiving ICI therapy is urgently needed for HCC. Patients negative for PD-L1 sometimes exhibit durable benefits. This observation further indicates the instability of PD-L1 expression as a biomarker; novel moderate biomarkers are needed to be investigated.
In this study, a novel immune subtype, herein designated the low-risk subtype of HCC, was identified and crucial insights into the immunologic features of this subtype were provided. Patients with the low-risk subtype, whose molecular traits, including an abundance of infiltration immune cells, enhanced enrichment of immune-related signatures, high expression of immune checkpoints, and an elevated TML, highly resemble the tumors that are responsive to ICI agents [14–16]. IFN-γ and the T cell-inflamed gene signature, which were previously reported to predict the efficacy of pembrolizumab [25], were demonstrated in HCC patients with the immune subtype. This finding reinforces the inference that ICI responses may appear in tumors with a pre-existing IFN-mediated immune signaling. Interestingly, the presence of high TML was observed in the low-risk immune subtype, indicating that, unlike melanoma [16, 26] and NSCLC [26], distinct potential mechanisms may actuate HCC immune responses. Prostate, ovarian, and pancreatic tumors with modest TML also exhibit a similar lack of association [27]. In these situations, the immunogenicity of tumors may be influenced by neoantigen quality, rather than quantity [26]. In addition, several mutation-independent signals, for example, HCC-related antigens expression, may induce a vital effect on the anti-tumor immune response [8].
Differential analysis of SMG mutations showed that six genes exhibited higher mutation rates in the low-risk subtype than high-risk subtype. Among these six SMGs, TP53 mutations were reported to be associated with high expression of immune checkpoints, an active IFNγ signature, and effector T cell signature, and favorable anti-PD-1 efficacy in LUAD [28]. A recent study revealed that MUC16 mutations are significantly correlated with a higher TML and better survival outcomes in patients with GC [29]. We demonstrated that patients with the low-risk subtype harbored higher mutation rates of the 2 SMGs further verify the predictive roles of this subtype in immune checkpoint-based therapy.
By using the Nearest Template Prediction (NTP) algorithm with 1950 representative meta-genes, Hoshida et al. identified three HCC subtypes (i.e., S1, S2, and S3) that were correlated with distinct biological processes [30]. We also employed the NTP method with the same meta-genes to determine Hoshida et al. subtypes in the three HCC datasets included in our study, and compared them with the low- and high-risk subtypes we identified. In the TCGA and ICGC cohorts, we showed that the low-risk subtype harbored a significantly decreased proportion of S1subclass (Supplementary Figure 2A, 2B), which was characterized by WNT-TGFβ pathway activation. Recent studies have demonstrated the WNT-TGFβ signal functions in immune suppression [31, 32]. The lower proportion of S1 further verified the immunotherapy implications of patients from the low- risk subtype; however, no significant distribution differences were observed between the low/high-risk subtypes and the S2/S3 subclasses. The distribution differences of the 3 subclasses in low- vs. high-risk subtypes were not statistically significant in GSE76427 (Supplementary Figure 2C); this may be owing to the smaller sample size of this cohort.
Taking into consideration the important effects of ethnicity on precision medicine, we therefore compared the race distribution between HCC low- and high-risk subtypes. We observed that there is no significantly distinct race distribution in low- vs. high-risk subtypes (Fisher exact test P = 0.271; Supplementary Figure 3).
Meanwhile, several inadequacies existed in our study. First, the gene expression data were acquired from distinct sequencing or microarray platforms, which may introduce result biases in the analysis. Second, somatic mutation data used in this work were only from the TCGA project; no additional HCC datasets contained mutation profiles were available.
Our study discovered a novel immune subtype of HCC patients that was associated with favorable survival outcomes and a better immune microenvironment, who may represent the appropriate sub-populations to receive ICI agents. In-depth explorations of this immune subtype in larger immunotherapy cohorts are needed to validate its potential utility as a predictive indicator of response to ICI therapies.
Materials and Methods
Gene expression profile, somatic mutation data, and clinical information of used HCC patients
A total of 373 HCC patients with mRNA expression profile and follow-up information in the Cancer Genome Atlas (TCGA) were obtained from Genomic Data Commons (https://gdc.cancer.gov/). Among them, 325 patients had complete clinical characteristics (i.e., age, sex, grade, stage, drinking status, and HBV/HCV status). From International Cancer Genome Consortium (ICGC) [LIRI-JP cohort] and Gene Expression Omnibus (GEO) [accession number: GSE76427], we respectively acquired 232 and 114 patients with gene expression and clinical data for further validation (Supplementary Table 3). All gene expression data were normalized for subsequent analyses. For genes with multiple probe sets, the mean gene expression was utilized as the expression level. Somatic mutation data of 364 patients with mRNA expression were obtained from the TCGA cohort. In this study, non-synonymous mutational types, including missense mutation, nonsense mutation, frame shift del/ins, in frame del/ins, and splice site mutation were included to perform related analyses.
NMF clustering analysis
Clustering analyses based on mRNA expression profile were conducted with non-negative matrix factorization (NMF) method embedded in the R NMF package [21]. A binary matrix A representing gene expression levels (rows) across HCC patients (columns) was generated. Then, expression matrix A was divided into two non-negative matrices W and H (i.e., A≈WH). Distinct classes or subtypes were identified with a clustering approach based on Matrix H. Optimal clustering number was determined according to the values of cophenetic, dispersion, residuals and RSS coefficient.
Identification of immune expression patterns and potential immune subtypes
Tumor, stromal and immune cell gene expression data from the TCGA cohort were virtually microdissected using abovementioned NMF algorithm. In this study, genes with a CV less than 0.1 were selected to reduce the biases of results [33]. We selected the number of clustering factor as 9, as it could effectively divide the expression data in the TCGA cohort, and thus exhibit a high cophenetic coefficient [34].
Then we took the following steps to identify an immune class as previously reported by Sia et al. [8]. Firstly, an immune enrichment score gene signature [20], which represents the proportion of infiltration immune cells in tumor tissue, was utilized to determine the potential immune relevant class (or expression pattern). By integrating all 9 NMF-identified clusters with the immune enrichment score gene signature, we observed the NMF cluster with the highest immune enrichment score, and named this as the immune class. Then, we curated the top 100 genes based on their contributions to the immune class, and these 100 genes were annotated with the DAVID tool (https://david.ncifcrf.gov/) to further verify their immune functionalities. Finally, the top 100 genes were utilized to perform unsupervised NMF clustering analysis to divide the TCGA HCC patients into distinct subtypes.
Gene set enrichment analysis
Single sample gene set enrichment analysis (ssGSEA) method embedded in R package GSVA (version 1.32.0) was utilized to calculate the enrichment scores of above immune signatures for each sample [44]. We used gene set enrichment analysis (GSEA) from fgsea package (version 1.10.0, https://bioconductor.org/packages/fgsea/) to explore the dysregulated pathways in distinct subgroups. Annotated pathways in Molecular Signature Database (MSigDB, version 3.0) [45] were utilized as the background signals.
Significantly mutated genes
Significantly mutated genes (SMGs) were determined by applying MutSigCV method [46]. The significant enrichment of non-silent somatic variants of a specific gene was calculated by MutSigCV via addressing mutational context specific background mutation rates. The following criteria were needed to authenticate SMGs: statistically significant (i.e., q value less than 0.1), expressed in TCGA HCC data [47] and encyclopedia of cell lines [48], and mutation rate greater than 5%.
Statistical analyses
R software (version 4.0.3) and its packages were utilized to perform statistical analyses and produce relevant figures. Kaplan-Meier survival curves were generated with R survival (version 2.44-1.1) and Survminer (version 0.4.5) packages, and the Log-rank test was used to compare the difference between survival curves. In addition to univariate analyses, multivariate Logistic and Cox regression models with confounding factors taken into consideration were performed using Forestmodel package (version 0.5.0) to control the false positive. Associations of distinct subtypes with continuous and categorical variables were calculated with Wilcoxon rank-sum test and Fisher exact test, respectively. In this study, P values less than 0.05 were considered to be statistically significant, unless special instructions.
Supplementary Materials
Author Contributions
LX and YZ designed this study; LX, YZ and JS developed the methodology and acquired the related data; JS, NX and YZ performed data analysis and interpretation; LX, YZ and JS drafted and revised the manuscript; LX and YZ supervised this study.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1. Murray CJ, Vos T, Lozano R, Naghavi M, Flaxman AD, Michaud C, Ezzati M, Shibuya K, Salomon JA, Abdalla S, Aboyans V, Abraham J, Ackerman I, et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012; 380:2197–223. https://doi.org/10.1016/S0140-6736(12)61689-4 [PubMed]
- 2. Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, Gores G. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016; 2:16018. https://doi.org/10.1038/nrdp.2016.18 [PubMed]
- 3. Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology. 2015; 149:1226–39.e4. https://doi.org/10.1053/j.gastro.2015.05.061 [PubMed]
- 4. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, et al, and SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008; 359:378–90. https://doi.org/10.1056/NEJMoa0708857 [PubMed]
- 5. Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, Pracht M, Yokosuka O, Rosmorduc O, Breder V, Gerolami R, Masi G, Ross PJ, et al, and RESORCE Investigators. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017; 389:56–66. https://doi.org/10.1016/S0140-6736(16)32453-9 [PubMed]
- 6. Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. 2018; 50:1–11. https://doi.org/10.1038/s12276-018-0191-1 [PubMed]
- 7. Abril-Rodriguez G, Ribas A. SnapShot: Immune Checkpoint Inhibitors. Cancer Cell. 2017; 31:848–48.e1. https://doi.org/10.1016/j.ccell.2017.05.010 [PubMed]
- 8. Sia D, Jiao Y, Martinez-Quetglas I, Kuchuk O, Villacorta-Martin C, Castro de Moura M, Putra J, Camprecios G, Bassaganyas L, Akers N, Losic B, Waxman S, Thung SN, et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology. 2017; 153:812–26. https://doi.org/10.1053/j.gastro.2017.06.007 [PubMed]
- 9. Llovet JM, Villanueva A, Lachenmayer A, Finn RS. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol. 2015; 12:408–24. https://doi.org/10.1038/nrclinonc.2015.103 [PubMed]
- 10. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016; 8:328rv4. https://doi.org/10.1126/scitranslmed.aad7118 [PubMed]
- 11. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn MJ, Felip E, et al, and KEYNOTE-001 Investigators. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015; 372:2018–28. https://doi.org/10.1056/NEJMoa1501824 [PubMed]
- 12. Herbst RS, Baas P, Kim DW, Felip E, Pérez-Gracia JL, Han JY, Molina J, Kim JH, Arvis CD, Ahn MJ, Majem M, Fidler MJ, de Castro G
Jr , et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016; 387:1540–50. https://doi.org/10.1016/S0140-6736(15)01281-7 [PubMed] - 13. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012; 366:2443–54. https://doi.org/10.1056/NEJMoa1200690 [PubMed]
- 14. Bald T, Landsberg J, Lopez-Ramos D, Renn M, Glodde N, Jansen P, Gaffal E, Steitz J, Tolba R, Kalinke U, Limmer A, Jönsson G, Hölzel M, Tüting T. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov. 2014; 4:674–87. https://doi.org/10.1158/2159-8290.CD-13-0458 [PubMed]
- 15. Ji RR, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, Alaparthy S, Berman D, Jure-Kunkel M, Siemers NO, Jackson JR, Shahabi V. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. 2012; 61:1019–31. https://doi.org/10.1007/s00262-011-1172-6 [PubMed]
- 16. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015; 372:2509–20. https://doi.org/10.1056/NEJMoa1500596 [PubMed]
- 17. Li W, Zhang X, Wu F, Zhou Y, Bao Z, Li H, Zheng P, Zhao S. Gastric cancer-derived mesenchymal stromal cells trigger M2 macrophage polarization that promotes metastasis and EMT in gastric cancer. Cell Death Dis. 2019; 10:918. https://doi.org/10.1038/s41419-019-2131-y [PubMed]
- 18. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, Kim TY, Choo SP, Trojan J, Welling TH
3rd , Meyer T, Kang YK, Yeo W, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017; 389:2492–502. https://doi.org/10.1016/S0140-6736(17)31046-2 [PubMed] - 19. Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, Rashid NU, Williams LA, Eaton SC, Chung AH, Smyla JK, Anderson JM, Kim HJ, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. 2015; 47:1168–78. https://doi.org/10.1038/ng.3398 [PubMed]
- 20. Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, Carter SL, Getz G, Stemke-Hale K, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013; 4:2612. https://doi.org/10.1038/ncomms3612 [PubMed]
- 21. Gaujoux R, Seoighe C. A flexible R package for nonnegative matrix factorization. BMC Bioinformatics. 2010; 11:367. https://doi.org/10.1186/1471-2105-11-367 [PubMed]
- 22. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019; 19:133–50. https://doi.org/10.1038/s41568-019-0116-x [PubMed]
- 23. Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol. 2016; 13:273–90. https://doi.org/10.1038/nrclinonc.2016.25 [PubMed]
- 24. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, Bellmunt J, Burris HA, Petrylak DP, Teng SL, Shen X, Boyd Z, Hegde PS, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014; 515:558–62. https://doi.org/10.1038/nature13904 [PubMed]
- 25. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, Piha-Paul SA, Yearley J, Seiwert TY, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017; 127:2930–40. https://doi.org/10.1172/JCI91190 [PubMed]
- 26. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, Watkins TB, Shafi S, Murugaesu N, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016; 351:1463–69. https://doi.org/10.1126/science.aaf1490 [PubMed]
- 27. Balli D, Rech AJ, Stanger BZ, Vonderheide RH. Immune Cytolytic Activity Stratifies Molecular Subsets of Human Pancreatic Cancer. Clin Cancer Res. 2017; 23:3129–38. https://doi.org/10.1158/1078-0432.CCR-16-2128 [PubMed]
- 28. Dong ZY, Zhong WZ, Zhang XC, Su J, Xie Z, Liu SY, Tu HY, Chen HJ, Sun YL, Zhou Q, Yang JJ, Yang XN, Lin JX, et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin Cancer Res. 2017; 23:3012–24. https://doi.org/10.1158/1078-0432.CCR-16-2554 [PubMed]
- 29. Li X, Pasche B, Zhang W, Chen K. Association of MUC16 Mutation With Tumor Mutation Load and Outcomes in Patients With Gastric Cancer. JAMA Oncol. 2018; 4:1691–98. https://doi.org/10.1001/jamaoncol.2018.2805 [PubMed]
- 30. Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, Villanueva A, Newell P, Ikeda K, Hashimoto M, Watanabe G, Gabriel S, Friedman SL, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009; 69:7385–92. https://doi.org/10.1158/0008-5472.CAN-09-1089 [PubMed]
- 31. Pai SG, Carneiro BA, Mota JM, Costa R, Leite CA, Barroso-Sousa R, Kaplan JB, Chae YK, Giles FJ. Wnt/beta-catenin pathway: modulating anticancer immune response. J Hematol Oncol. 2017; 10:101. https://doi.org/10.1186/s13045-017-0471-6 [PubMed]
- 32. Galluzzi L, Spranger S, Fuchs E, López-Soto A. WNT Signaling in Cancer Immunosurveillance. Trends Cell Biol. 2019; 29:44–65. https://doi.org/10.1016/j.tcb.2018.08.005 [PubMed]
- 33. Aja-Fernández S, Alberola-López C. On the estimation of the coefficient of variation for anisotropic diffusion speckle filtering. IEEE Trans Image Process. 2006; 15:2694–701. https://doi.org/10.1109/TIP.2006.877360 [PubMed]
- 34. Chen YP, Wang YQ, Lv JW, Li YQ, Chua ML, Le QT, Lee N, Colevas AD, Seiwert T, Hayes DN, Riaz N, Vermorken JB, O’Sullivan B, et al. Identification and validation of novel microenvironment-based immune molecular subgroups of head and neck squamous cell carcinoma: implications for immunotherapy. Ann Oncol. 2019; 30:68–75. https://doi.org/10.1093/annonc/mdy470 [PubMed]
- 35. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015; 12:453–57. https://doi.org/10.1038/nmeth.3337 [PubMed]
- 36. Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell. 2015; 161:1681–96. https://doi.org/10.1016/j.cell.2015.05.044 [PubMed]
- 37. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015; 160:48–61. https://doi.org/10.1016/j.cell.2014.12.033 [PubMed]
- 38. Tsai KK, Zarzoso I, Daud AI. PD-1 and PD-L1 antibodies for melanoma. Hum Vaccin Immunother. 2014; 10:3111–16. https://doi.org/10.4161/21645515.2014.983409 [PubMed]
- 39. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MH, Goldinger SM, Utikal J, Hassel JC, Weide B, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015; 350:207–11. https://doi.org/10.1126/science.aad0095 [PubMed]
- 40. Long L, Zhang X, Chen F, Pan Q, Phiphatwatchara P, Zeng Y, Chen H. The promising immune checkpoint LAG-3: from tumor microenvironment to cancer immunotherapy. Genes Cancer. 2018; 9:176–89. https://doi.org/10.18632/genesandcancer.180 [PubMed]
- 41. Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev. 2017; 276:97–111. https://doi.org/10.1111/imr.12520 [PubMed]
- 42. Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev. 2017; 276:112–20. https://doi.org/10.1111/imr.12518 [PubMed]
- 43. Blair AB, Kleponis J, Thomas DL 2nd, Muth ST, Murphy AG, Kim V, Zheng L. IDO1 inhibition potentiates vaccine-induced immunity against pancreatic adenocarcinoma. J Clin Invest. 2019; 129:1742–55. https://doi.org/10.1172/JCI124077 [PubMed]
- 44. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013; 14:7. https://doi.org/10.1186/1471-2105-14-7 [PubMed]
- 45. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015; 1:417–25. https://doi.org/10.1016/j.cels.2015.12.004 [PubMed]
- 46. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013; 499:214–18. https://doi.org/10.1038/nature12213 [PubMed]
- 47. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MD, Miller CA, Welch JS, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013; 502:333–39. https://doi.org/10.1038/nature12634 [PubMed]
- 48. Klijn C, Durinck S, Stawiski EW, Haverty PM, Jiang Z, Liu H, Degenhardt J, Mayba O, Gnad F, Liu J, Pau G, Reeder J, Cao Y, et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat Biotechnol. 2015; 33:306–12. https://doi.org/10.1038/nbt.3080 [PubMed]