



Introduction
Worldwide, gastric cancer (GC) is one of the most common malignant tumors [1–2]. Despite advances in the development of surgery and chemotherapy in recent years, the prognosis of GC patients remains dissatisfactory as a result of recurrence and metastasis rates [3]. Therefore, identifying novel predictive tumor biomarkers as well as understanding the mechanisms responsible for GC development may provide targets useful for improving GC outcomes.
Recently, growing evidence has revealed that epithelial-mesenchymal transition (EMT) is a fundamental process of tumor initiation and progression. During EMT, epithelial cells lose their intercellular junction, reprogram gene expression and undergo a substantial change in signalling programme; this switch modifies the cell shape to promote motility and invasion [4]. Molecularly, a hallmark of EMT is E-cadherin level downregulation and N-cadherin level upregulation. These changes in gene expression are regulated by transcription factors as well as an intricate network of signalling pathways, including TGF-β, JAK-STAT, PI3K/Akt, MAPK, and NF-κB [5].
Human ERCC6L is recognized as an embryonic development-related protein [6]. Recently, ERCC6L also appears to play an important role in cancer progression. As reported, ERCC6L upregulation was demonstrated to be correlated with tumor progression and poor prognosis in hepatocellular carcinoma (HCC) [7]. Additionally, ERCC6L was found to promote colorectal cancer (CRC) cell growth and invasion [8]. However, the pathophysiologic role and the mechanism of ERCC6L-induced GC cell growth and metastasis are still unclear.
In this study, we investigated the role and the potential molecular of ERCC6L in GC progression.
Results
ERCC6L expression is elevated in GC patients and is closely correlated with poor clinical outcomes
We found that ERCC6L mRNA and protein expressions were evidently higher in cancer tissues compared with those in adjacent normal tissues in 45 pairs of cases (Figure 1A and 1B). Likewise, The IHC analysis indicated the similar result (Figure 1C and 1D).
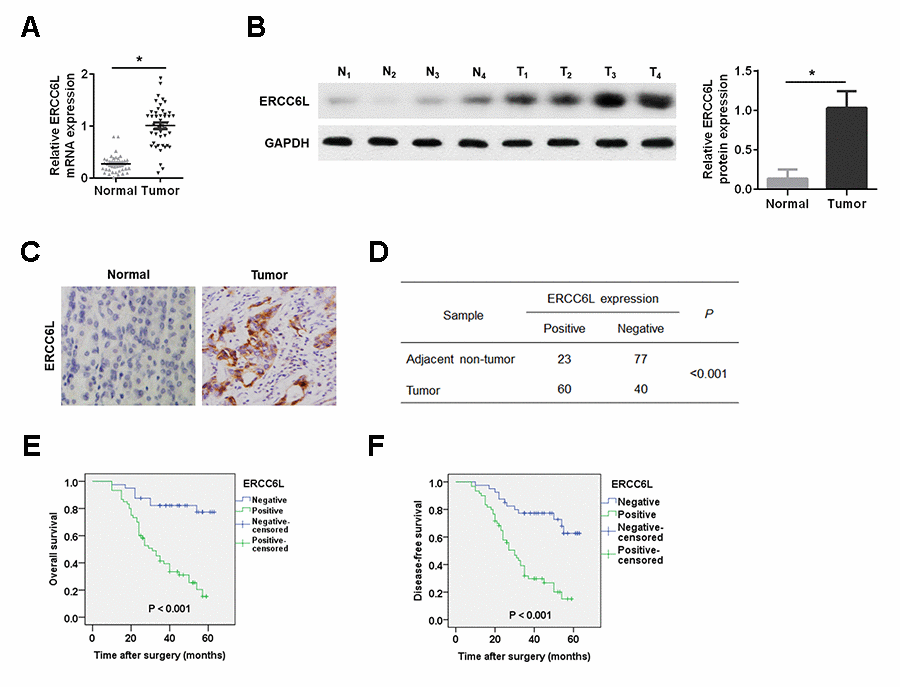
Figure 1. ERCC6L expression in GC tissues. (A) qRT-PCR detection of ERCC6L mRNA expression in GC tissues and adjacent normal tissues. (B) Western blot analysis of ERCC6L protein expression in tumor tissues (T) and matched normal tissues (N). (C) IHC analysis of ERCC6L expression in GC tissues and matched normal tissues. (D) Chi-square analysis of ERCC6L expression in GC tissues and adjacent non-tumor tissues based on IHC staining scores. (E) Kaplan-Meier analysis of overall survival for GC patients with different levels of ERCC6L expression. (F) Kaplan-Meier analysis of disease-free survival for GC patients with different levels of ERCC6L expression. *P < 0.05.
Next, we found that high ERCC6L expression indicated poor clinical outcomes in GC patients (Table 1). Kaplan-Meier analysis turned out that high ERCC6L expression in GC patients led to poor overall survival (Figure 1E) and/or disease-free survival (Figure 1F).
Table 1. Relationship between ERCC6L expression and clinicopathological parameters in gastric cancer.
| Clinicopathological Parameters | n | ERCC6L | P-value | |
| + | – | |||
| Age (years) | ||||
| ≥60 | 69 | 41 | 28 | 0.860 |
| <60 | 31 | 19 | 12 | |
| Gender | ||||
| Male | 72 | 41 | 31 | 0.317 |
| Female | 28 | 19 | 9 | |
| Tumor size (cm) | ||||
| ≥5 | 64 | 44 | 20 | 0.017 |
| <5 | 36 | 16 | 20 | |
| Lauren’s classification | ||||
| Diffuse | 29 | 19 | 10 | 0.472 |
| Intestinal | 71 | 41 | 30 | |
| Lymphatic vessel invasion | ||||
| With | 38 | 24 | 14 | 0.614 |
| Without | 52 | 36 | 26 | |
| T stage | ||||
| T1 + T2 | 45 | 19 | 26 | 0.001 |
| T3 + T4 | 55 | 41 | 14 | |
| pTNM stage | ||||
| I + II | 39 | 16 | 23 | 0.002 |
| III + IV | 61 | 44 | 17 | |
| Lymph node metastasis | ||||
| With (N1 + N2 + N3) | 59 | 46 | 13 | <0.001 |
| Without (N0) | 41 | 14 | 27 | |
ERCC6L induces EMT in GC cells
Compared with GES-1, the mRNA and protein expressions of ERCC6L were significantly elevated in GC cells (AGS, MKN28, SGC7901, BGC823, KATO-III, MGC803 and MKN45) (Figure 2A and 2B). Subsequently, we found that ERCC6L overexpression increased p-NF-κB p65 and N-cadherin expression levels, and decreased E-cadherin expression in AGS cells and MKN28 cells. On the contrary, ERCC6L downregulation in MKN45 and SGC7901 cells led to the opposite outcomes (Figure 2C).
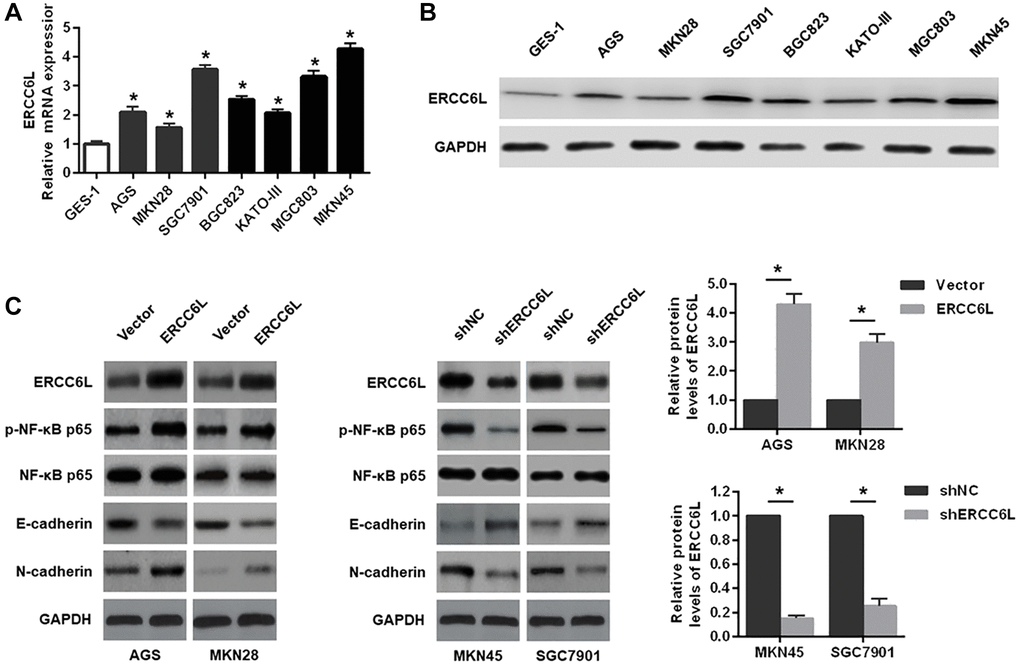
Figure 2. The effects of ERCC6L overexpression or knockdown on the EMT and NF-κB signaling-related markers in GC cells. (A) qRT-PCR detection of relative ERCC6L mRNA expression in human GC cell lines and a normal gastric epithelial cell line. (B) Western blot analysis of ERCC6L protein expression in human GC cell lines and a normal gastric epithelial cell line. (C) Western blot analysis of the effects of ERCC6L overexpression or knockdown on the EMT and NF-κB signaling-related markers in GC cells. *P < 0.05.
ERCC6L promotes proliferation, migration and invasion of GC cells in vitro
We found that ERCC6L overexpression markedly promoted cell growth in AGS cells, while ERCC6L knockdown significantly suppressed cell growth in MKN45 cells (Figure 3A–3C). Additionally, we found that ERCC6L overexpression markedly promoted cell migration and invasion in AGS cells, whereas ERCC6L knockdown in MKN45 cells observably led to the opposite outcomes (Figure 3D and 3E). In summary, these data suggested that ERCC6L promoted proliferation, migration and invasion of GC cells in vitro.
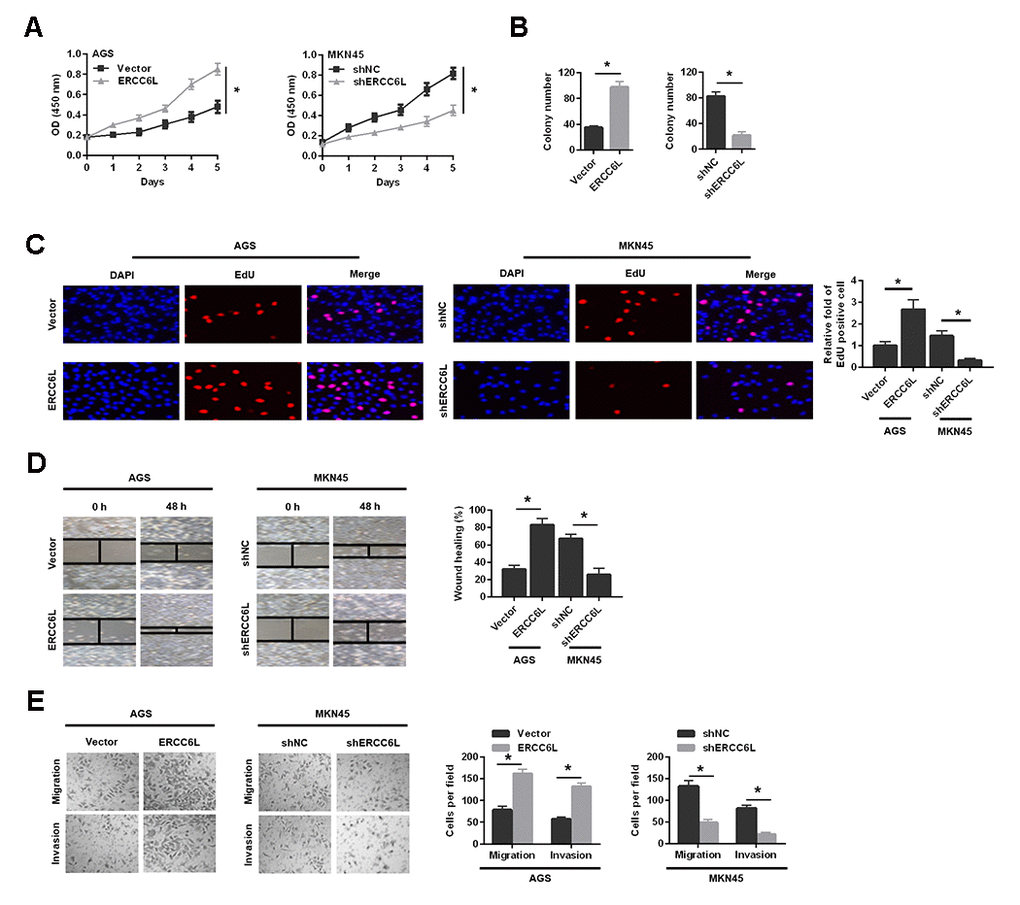
Figure 3. The effects of ERCC6L overexpression or knockdown on GC cells growth, migration and invasion in vitro. Following ERCC6L overexpression or knockdown treatment, CCK-8 assay (A), colony formation assay (B) and EdU assay (C) were used to evaluate the effects of ERCC6L expression on the proliferation ability of GC cells. The wound-healing assay (D), transwell migration and invasion assays (E) were employed to assess the effects of ERCC6L overexpression or knockdown on the migration and invasion ability of GC cells. *P < 0.05.
ERCC6L promotes tumor formation and metastatic potential in vivo
These results demonstrated ERCC6L overexpression in AGS cells markedly promoted cell proliferation on the basis of the volume and weight of the transplanted tumor (Figure 4A). The Western blot analysis of the xenograft tissues revealed that ERCC6L overexpression in AGS cells resulted in higher levels of Ki-67, p-NF-κB p65 and N-cadherin, and lower E-cadherin level (Figure 4B). Conversely, ERCC6L knockdown in MKN45 cells accomplished exactly the opposite (Figure 4A–4B).
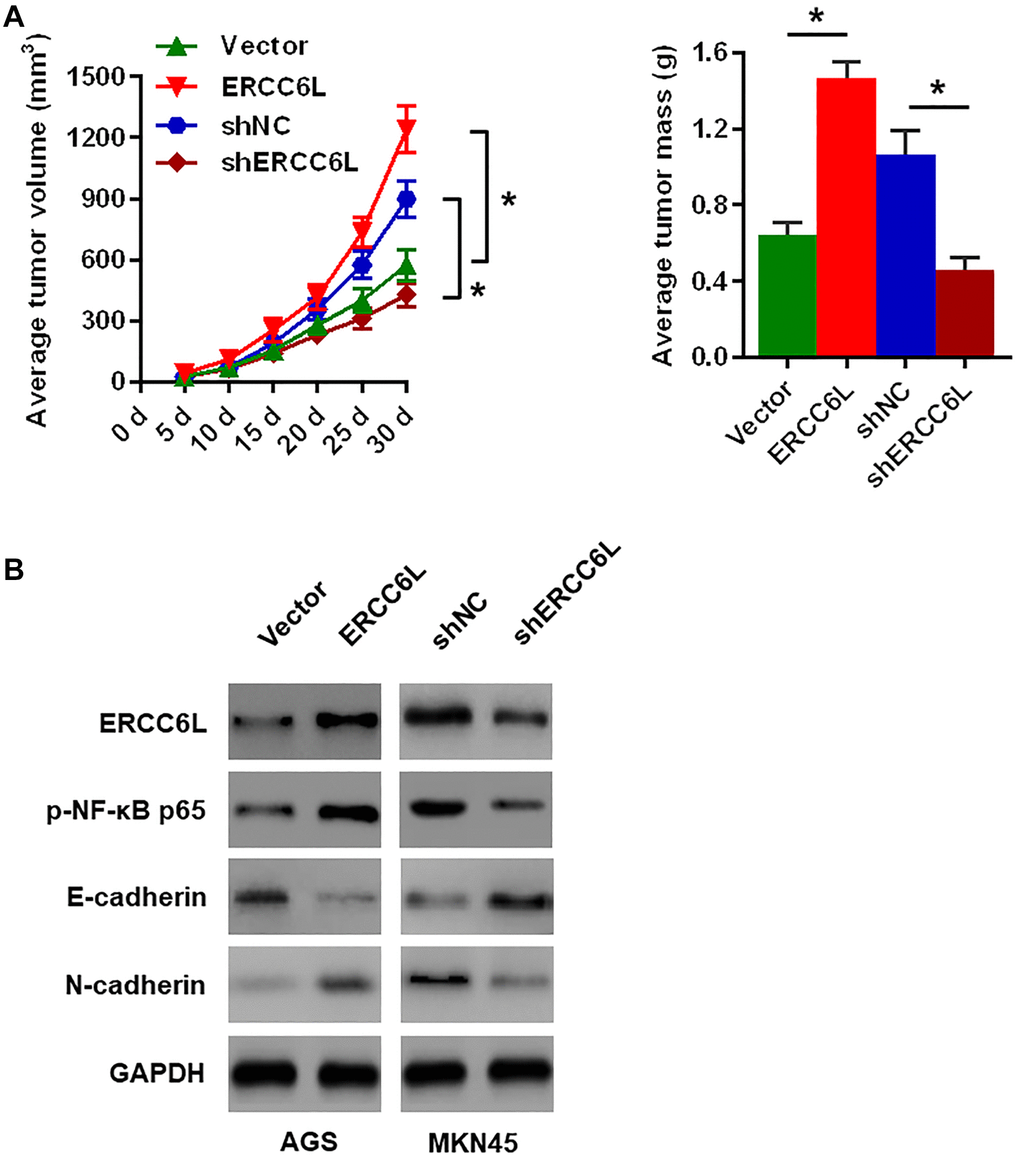
Figure 4. The effects of ERCC6L overexpression or knockdown on GC cells growth in vivo. (A) Representative images of the dissected xenograft tumors. Tumor volume was measured every 5 days. Tumors were weighed at the endpoint (30 days). (B) Western blot analysis of the levels of ERCC6L, p-NF-κB p65, E-cadherin and N-cadherin in the dissected xenograft tumors. (n = 6, each group). *P < 0.05.
In addition, the result revealed that ERCC6L overexpression in AGS cell promoted lung metastasis, as evidenced by the more pulmonary metastases (Figure 5A) and EMT reversal (Figure 5B). As expected, ERCC6L downregulation in MKN45 cells resulted in a significantly opposite effect on cell proliferation and metastasis in vivo (Figure 5A–5B).
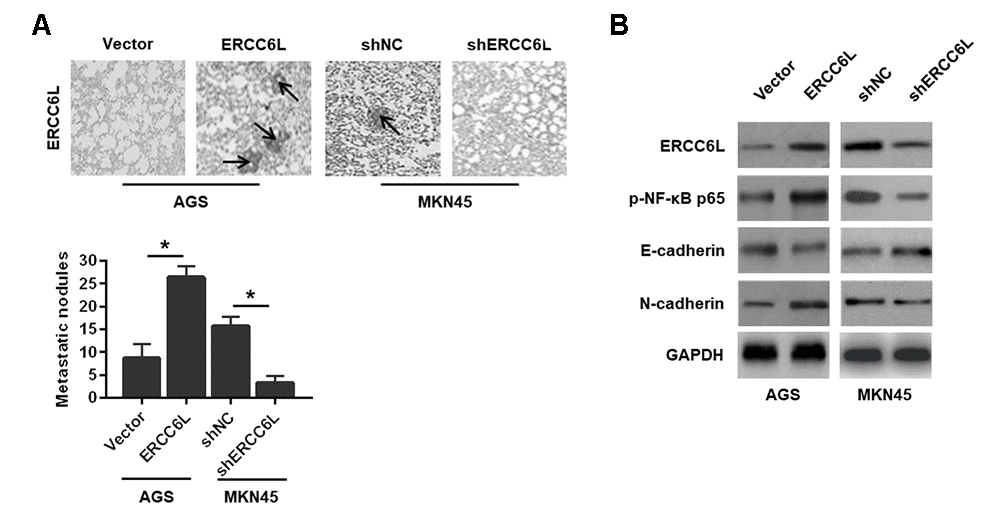
Figure 5. The effects of ERCC6L overexpression or knockdown on GC cells metastasis in vivo. (A) Representative images of the numbers of metastatic foci in lung of individual mouse. (B) Western blot analysis of the levels of ERCC6L, p-NF-κB p65, E-cadherin and N-cadherin in lung metastatic nodules. (n = 6, each group). *P < 0.05.
Taken together, these findings verified the promotive role of ERCC6L in GC cell growth and metastasis.
ERCC6L facilitates GC cell growth, migration and invasion via the NF-κB signaling pathway-induced EMT
The NF-κB signaling pathway is responsible for in tumor progression [9]. We reasoned that ERCC6L might promote EMT by activating NF-κB pathway in GC. We revealed that ERCC6L overexpression increased the levels of p-NF-κB p65 and N-cadherin; whereas, ERCC6L knockdown decreased their levels. To explore whether NF-κB signaling pathway involved in ERCC6L-induced cellular changes, helenalin, the inhibitor of NF-κB, was used. The results showed that helenalin could reverse EMT, decrease p-NF-κB p65 (Figure 6A) and suppress cell growth (Figure 6B), migration and invasion (Figure 6C) induced by ERCC6L. Altogether, these data demonstrated that NF-κB pathway-mediated EMT was involved in the ERCC6L-induced oncogenic function.
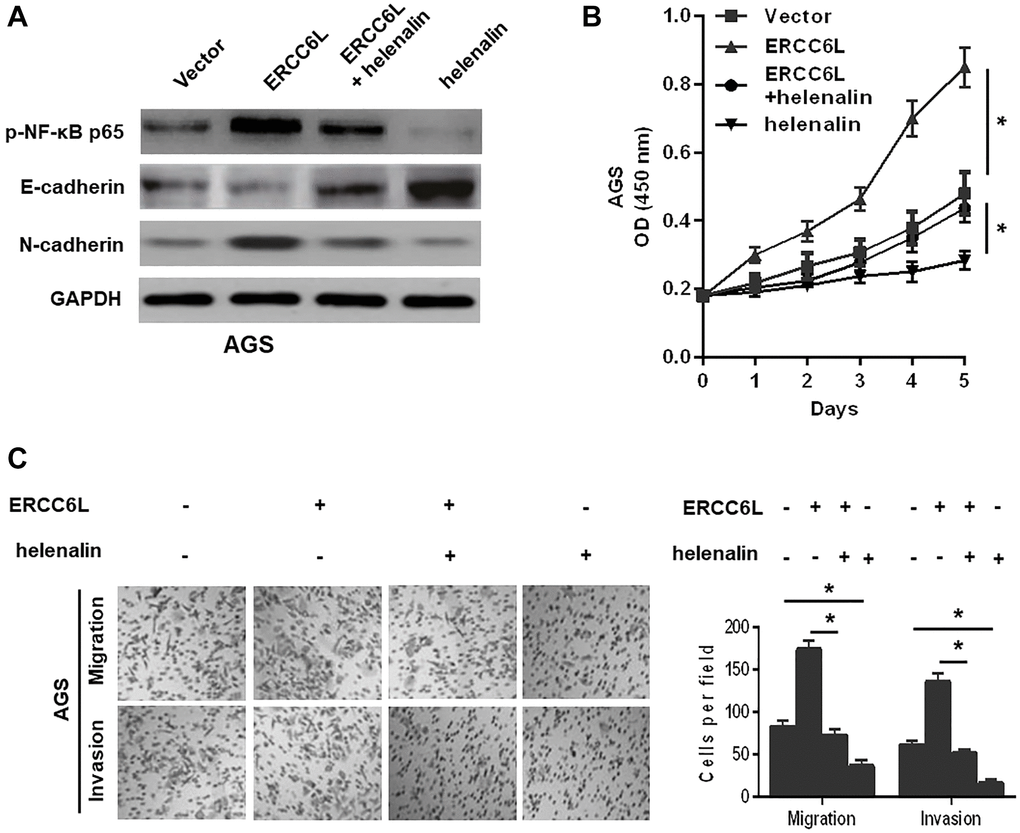
Figure 6. NF-κB signal involved in ERCC6L-facilitated GC cell EMT, growth, migration, and invasion. (A) Western blot analysis of the levels of p-NF-κB p65, E-cadherin and N-cadherin in modified AGS cells treated with helenalin (15 μM, NF-κB signaling inhibitor). (B) Determination of cell proliferation ability followed by the treatment with helenalin by CCK-8 assay. (C) Determination of cell migration and invasion abilities followed by the treatment with helenalin by transwell assay. *P < 0.05.
Discussion
GC remains a common and aggressive malignancy worldwide. Given that tumorigenesis and metastasis of GC is a complicated process and is affected by numerous genetic changes [10–11], it is urgently required to determine novel molecular targets and the potential mechanisms responsible for this process. Remarkably, recent reports have identified ERCC6L as a critical mediator in the malignant biological behavior of various cancers [7–8, 12]. For example, ERCC6L expression is increased in HCC tissue, which significantly correlates with poor clinical outcomes [7]. Furthermore, ERCC6L promotes tumor progression via the PI3K/AKT and NF-κB pathway in HCC [12]. In CRC, ERCC6L is significantly elevated in tumor tissues, and functions importantly in CRC cell growth, describing ERCC6L as a target for CRC [8]. Nevertheless, little is known about its function, especially in GC, and the molecular mechanisms of how ERCC6L influences GC progression and development.
Herein, we evaluated ERCC6L levels in GC tissues and GC cell. Clinical data showed that ERCC6L expression was positively associated with poor clinical outcomes. Survival analysis showed that GC patients with high ERCC6L expression had poor prognosis, suggesting that ERCC6L may be a valuable biomarker for GC. Given that ERCC6L correlates with unfavorable clinicopathological parameters in GC patients, we subsequently assessed the effect of ERCC6L on the malignant process of GC both. The result revealed that ERCC6L facilitated the growth, migration, invasion, and metastasis of GC cells. Mechanistically, we found that ERCC6L overexpression increased the expression of N-cadherin and decreased the expression of E-cadherin; while ERCC6L knockdown significantly reversed these events, suggesting that ERCC6L affected the malignant biological behavior of GC cells by modulating EMT.
As mentioned in the introduction, it is believed that EMT is a hallmark of cancer and a key part in tumor progression and metastasis in multiple types of malignancies, [5]. In recent years, more and more attention has been paid to EMT pathway in tumor therapy, and it is expected to become a target for the prevention of tumor development to some extent [5]. Herein, we identified important roles for the NF-κB pathway in the ERCC6L-induced EMT and aggressive behavior of GC cells. As mentioned earlier, NF-κB pathway has been considered as not only a critical element in cancer progression and metastasis but also an important modulator of EMT-related transcription factors [13–14]. Mechanistically, there was a dramatic increase in the phosphorylation of NF-κB p65 in GC ERCC6L-transfected cells, implying the activation of NF-κB pathway. Conversely, ERCC6L knockdown markedly inactivated the NF-κB pathway. Furthermore, to further ascertain the involvement of NF-κB signaling in ERCC6L-induced EMT, pretreatment of GC cells with helenalin (an NF-κB inhibitor) [15] resulted in blockade of the increase in ERCC6L-induced EMT, and cell growth, migration and invasion. Taking these factors together, we could assume that ERCC6L contributed to GC cell growth and metastasis through activation of NF-κB-induced EMT.
Nevertheless, it is worth further investigating the indirect or direct relationship between ERCC6L and NF-κB signal. As reported, ERCC6L could be involved in centromeric chromatin remodeling as a binding partner and substrate of PLK1, which is a key kinase in cell cycle progression [16]. Thus, it is of great importance to evaluate whether ERCC6L-induced EMT involves other genes or some signaling pathways in subsequent studies.
In conclusion, this study demonstrates that high ERCC6L expression significantly correlates with clinical proliferation and metastasis, and unfavourable prognosis in GC patients. Besides, our work identifies a critical role of ERCC6L in promoting the EMT process through NF-κB signaling pathway, which further facilitates GC cell growth, invasion, and metastasis, possibly providing a promising target to suppress GC progression and development.
Materials and Methods
Tissue samples
100 pairs of tumor tissues were obtained from patients with GC undergoing radical resection at the Department of General Surgery of Taizhou People’s Hospital. The study was approved by the Ethics Committee of our Hospital. According to the Helsinki declaration, the written informed consent of all participants was obtained.
Cell lines
GC cell lines (AGS, MKN28, SGC7901, BGC823, KATO-III, MGC803, and MKN45) andGES-1 were purchased from American Type Culture Collection (ATCC, USA). All cell lines were maintained in DMEM containing 10% serum at 37°C with 5% CO2.
qRT-PCR
Total RNA was extracted from cells or GC tissues, and was reversely transcribed into cDNA using PrimeScript RT Reagent Kit (TaKaRa, Japan) according to the manufacturer’s instructions. All reactions were conducted using the SYBR Green assay kit (Takara, Japan). The mRNA level was evaluated according to the 2-ΔΔCt method. Primers are summarized in Supplementary Table 1 [12].
Western blot assay
Briefly, whole-cell or tissue lysates were prepared and equivalent amounts of proteins were transferred to PVDF membranes. The immune blots were blocked in 5% skimmed milk and then incubated with primary antibody and appropriate secondary antibody. The blots were visualized in accordance with instructions of an enhanced chemiluminescence kit. The primary antibody against ERCC6L was obtained from Abcam (Cambridge, UK). Primary antibodies against p-NF-κB p65, NF-κB p65, E-cadherin, and N-cadherin were purchased from CST (Danvers, MA, USA). [17].
IHC and evaluation
In short, paraffin embedded sections are cut, deparaffined, rehydrated, and then heat treated for antigen recovery. Next, the slices were incubated with 3% hydrogen peroxide to inactivate endogenous peroxidase activity, and then incubated with 5% normal blocking serum. After that, the sections were treated with specific primary antibody overnight, then biotinylated secondary antibody (boster, Wuhan, China) and SABC solution. Finally, the sections were observed and imaged by light microscope.
IHC staining intensity (0, negative; 1, Weak; 2, Medium; 3, strong) and immunostaining area (0, 0–5%; 1, 6–25%; 2, 26–50%). If the total score is less than 3, it is negative. If the total score is more than 3, it is positive.
Lentivirus infection
The commercial ERCC6L specific short hairpin RNA (shRNA) construction and overexpression construction were from genechem (Shanghai, China). Cells transfected with control shRNA or vector were used as control. According to the manufacturer’s instructions, Lipofectamine 2000 (Invitrogen) was used for cell transfection. The transfection efficiency was detected by Western blot.
Cell proliferation assay
The cells were seeded in 96 well plates. Cell proliferation was detected by cell counting kit-8 (CCK-8) on day 0, 1, 2, 3, 4 and 5 after culture followed the manufacturer's instructions. The absorbance at 450 nm was detected and the cell growth rate was calculated.
Colony formation assay
In short, the cells were cultured in 6-well plates for 14 days, then fixed in paraformaldehyde. Then, the cells were stained with crystal violet and counted under light microscope. The number of colonies (> 50 cells/colony) was calculated.
EdU assay
In short, the cells were cultured with Edu for 2 h, fixed in paraformaldehyde, infiltrated into Triton X-100, and then stained with Apollo solution for 0.5 h. The cells were then stained with DAPI solution and observed under fluorescence microscope.
Wound healing assay
In short, the cells were cultured into a monolayer. After that, the tip of the micropipette was used to form the wound space, and then the cell debris is removed. At the designated time points (0 h and 48 h), the wound images were taken to evaluate the remaining distance. The wound coverage rate was calculated as follows: wound closure rate = (0 h width–48 h width)/0 h width × 100%.
Cell migration and invasion assays
Transwell chambers (Corning, New York, USA) and BD matrixtm basement membrane (BD Biosciences, San Jose, California, USA) were used to measure cell migration and invasion. These cells were maintained in serum-free medium and seeded in the upper cavity with or without matrix gel. The medium containing 20% fetal bovine serum was added to the bottom chamber. After incubation for 24 h, the cells were fixed in paraformaldehyde, stained with crystal violet solution, and counted in 5 random areas under the microscope [17].
Animal studies
The cells were subcutaneously injected into nude mice (6-week-old, male) and the tumor volume was measured using the following formula: volume = (short diameter)2 × (Long diameter)/2 [17]. After 30 days, all mice were euthanized, the xenograft tissues were dissected, weighed and analyzed by Western blot. In addition, the cells were injected into nude mice via tail vein. Similarly, the lung of nude mice was excised 30 days after injection to count metastatic nodules, and then prepared for Western blot analysis [17]. The living animal experiment was approved by the ethics committee of the Fifth Affiliated Hospital of Nantong University.
Statistical analysis
Data were presented as the mean ± standard deviation (SD), and were analyzed by Student’s t-test. All statistical tests were conducted using SPSS version 21.0 software. P < 0.05 was considered significant.
Supplementary Materials
Author Contributions
Dehu Chen and Gan Cao wrote the paper and conceived and designed the experiments; Dehu Chen and Qinghong Liu analyzed the data; Dehu Chen and Gan Cao collected and provided the sample for this study. All authors have read and approved the final submitted manuscript.
Acknowledgments
We sincerely thank all the involvers for their supports.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Funding
This work was supported in part by funding from Taizhou Science and Technology Support Plan (Social Development) Project (TS201732).
References
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018; 68:394–424. https://doi.org/10.3322/caac.21492 [PubMed]
- 2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018; 68:7–30. https://doi.org/10.3322/caac.21442 [PubMed]
- 3. Catalano V, Labianca R, Beretta GD, Gatta G, de Braud F, Van Cutsem E. Gastric cancer. Crit Rev Oncol Hematol. 2009; 71:127–64. https://doi.org/10.1016/j.critrevonc.2009.01.004 [PubMed]
- 4. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014; 15:178–96. https://doi.org/10.1038/nrm3758 [PubMed]
- 5. Singh M, Yelle N, Venugopal C, Singh SK. EMT: Mechanisms and therapeutic implications. Pharmacol Ther. 2018; 182:80–94. https://doi.org/10.1016/j.pharmthera.2017.08.009 [PubMed]
- 6. Albers E, Sbroggiò M, Pladevall-Morera D, Bizard AH, Avram A, Gonzalez P, Martin-Gonzalez J, Hickson ID, Lopez-Contreras AJ. Loss of PICH Results in Chromosomal Instability, p53 Activation, and Embryonic Lethality. Cell Rep. 2018; 24:3274–84. https://doi.org/10.1016/j.celrep.2018.08.071 [PubMed]
- 7. Yu B, Liang H, Ye Q, Wang Y. Upregulation of ERCC6L is associated with tumor progression and unfavorable prognosis in hepatocellular carcinoma. J Gastrointest Oncol. 2020; 11:1009–23. https://doi.org/10.21037/jgo-20-192 [PubMed]
- 8. Xie Y, Yu J, Wang F, Li M, Qiu X, Liu Y, Qi J. ERCC6L promotes cell growth and invasion in human colorectal cancer. Oncol Lett. 2019; 18:237–46. https://doi.org/10.3892/ol.2019.10297 [PubMed]
- 9. Taniguchi K, Karin M. NF-κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018; 18:309–24. https://doi.org/10.1038/nri.2017.142 [PubMed]
- 10. Thrift AP, El-Serag HB. Burden of Gastric Cancer. Clin Gastroenterol Hepatol. 2020; 18:534–42. https://doi.org/10.1016/j.cgh.2019.07.045 [PubMed]
- 11. Xia Y, Wei Z, Huang W, Wei X, He Y. Trim47 overexpression correlates with poor prognosis in gastric cancer. Neoplasma. 2021; 68:307–16. https://doi.org/10.4149/neo_2020_200708N706 [PubMed]
- 12. Chen H, Wang H, Yu X, Zhou S, Zhang Y, Wang Z, Huang S, Wang Z. ERCC6L promotes the progression of hepatocellular carcinoma through activating PI3K/AKT and NF-κB signaling pathway. BMC Cancer. 2020; 20:853. https://doi.org/10.1186/s12885-020-07367-2 [PubMed]
- 13. Pal S, Bhattacharjee A, Ali A, Mandal NC, Mandal SC, Pal M. Chronic inflammation and cancer: potential chemoprevention through nuclear factor kappa B and p53 mutual antagonism. J Inflamm (Lond). 2014; 11:23. https://doi.org/10.1186/1476-9255-11-23 [PubMed]
- 14. Sánchez-Tilló E, Liu Y, de Barrios O, Siles L, Fanlo L, Cuatrecasas M, Darling DS, Dean DC, Castells A, Postigo A. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci. 2012; 69:3429–56. https://doi.org/10.1007/s00018-012-1122-2 [PubMed]
- 15. He Z, Dong W, Hu J, Ren X. AQP5 promotes hepatocellular carcinoma metastasis via NF-κB-regulated epithelial-mesenchymal transition. Biochem Biophys Res Commun. 2017; 490:343–48. https://doi.org/10.1016/j.bbrc.2017.06.046 [PubMed]
- 16. Baumann C, Körner R, Hofmann K, Nigg EA. PICH, a centromere-associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell. 2007; 128:101–14. https://doi.org/10.1016/j.cell.2006.11.041 [PubMed]
- 17. Chen D, Liu Q, Cao G, Zhang W. TYRO3 facilitates cell growth and metastasis via activation of the Wnt/β-catenin signaling pathway in human gastric cancer cells. Aging (Albany NY). 2020; 12:2261–74. https://doi.org/10.18632/aging.102744 [PubMed]