



Introduction
Genome-wide screens in simple model organisms have identified a number of longevity genes with potentially conserved roles in aging in mammals. The validity of this approach is supported by work in the last two decades showing that the principal lifespan-regulating genes and pathways are conserved in species ranging from yeast to mice [1].
Several novel lifespan determinants have previously been identified by screening for all viable yeast deletion mutants [2]. Deletion of one of the genes identified, ACB1, not only doubles the mean lifespan of yeast but also markedly enhances heat resistance, a phenotype often associated with extended longevity in yeast and worms [2]. ACB1 encodes the highly conserved acyl-CoA–binding protein (ACBP), which is found in all eukaryotes and some prokaryotes tested to date [3]. ACBP binds with high affinity and specificity to long- and medium-chain acyl-CoA esters and is thought to protect them from hydrolysis during transport to acyl-CoA consuming processes such as lipid biosynthesis and remodeling, β-oxidation, and protein acylation [4]. Acb1, the only ACBP in S. cerevisiae, has been studied extensively. Its depletion has been shown to retard growth and reduce sphingolipid biosynthesis [5], and the observations that Acb1 deficiency perturbs plasma membrane structure, disrupts vacuole assembly, and causes vesicle accumulation suggest a key role for Acb1 in vesicular trafficking and membrane assembly [5,6].
Although mammals express several ACBP paralogs with varying numbers of functional domains, the majority of studies have focused on the ubiquitously expressed ACBD1 [7]. In both human and bovine epithelial cells, ACBD1 is predominantly localized in the cytosol but is also present in the endoplasmic reticulum (ER), Golgi, and nucleus [8]. ACBD1 knockout mice are fertile and develop normally, but show a delay in the induction of liver lipogenesis at weaning [9]. This is consistent with studies of cultured cells pointing to a role for ACBP in the promotion of adipocyte differentiation, metabolism of triacylglycerides (TAGs), and activation of lipid biosynthetic genes [10–12].
The C. elegans genome encodes seven ACBP paralogs. Four of these, ACBP-1, -3, -4, and -6, contain only the ACBP domain, while ACBP-2, -5, and MAA-1 carry additional domains [13]. Ectopic expression of each C. elegans paralog can complement the slow growth of yeast acb1 deletion mutants, suggesting that the C. elegans ACBPs are functional acyl-CoA–binding proteins [13,14]. However, the paralogs have different expression patterns depending on the developmental stage and/or the tissue examined, suggesting they may each have distinct or only partially overlapping functions [13]. ACBP-2, which contains an enoyl-CoA hydratase/isomerase (ECH) domain, plays an important role in promoting β-oxidation of unsaturated fatty acids [13]. Compared with wild-type worms, ACBP-1–deficient animals contain lower TAG levels and fewer but larger lipid droplets in the intestine [13]. MAA-1 (membrane-associated ACBP 1) is a transmembrane protein detected exclusively in the intestine and hypodermal cells, where it localizes to the Golgi and to the endocytic recycling compartment (ERC) [14]. Loss of maa-1 reduces endocytic recycling rates and alters the morphology of the ERC membrane, consistent with a role for MAA-1 in recruiting long-chain acyl-CoA esters to the endosomal and Golgi systems to promote vesicle fission or fusion [14]. Notably, long-chain acyl-CoA esters are required for Golgi vesicle fission/fusion in vitro, although their function has yet to be elucidated [15,16]. A role for MAA-1 in vesicular trafficking is consistent with one of the proposed roles for yeast Acb1 [5,6] suggesting that MAA-1 in the C. elegans intestine and hypodermis may have evolved to control the formation of an intramembrane acyl-CoA ester pool required for vesicle formation [14]. Taken together, studies of ACBP paralogs in different species have revealed a complex functional repertoire for ACBPs in processes ranging from lipogenesis to vesicle trafficking.
The high interspecies conservation of ACBP and the discovery that yeast Acb1 controls longevity prompted us to investigate the role(s) of ACBP in lifespan regulation in a multicellular organism. We found that of the seven C. elegans paralogs, maa-1 alone played a significant role in regulating lifespan. MAA-1 deficiency prolonged lifespan through a mechanism involving the hypoxia inducible factor 1 (HIF-1), which has recently been shown to play dual roles in limiting and extending the C. elegans lifespan through distinct, but as yet not fully characterized, mechanisms [17–20]. In this study, we identified an ACBP- and HIF-1–dependent pathway for longevity regulation in C. elegans that involves small heat-shock proteins and functions under hypoxia-independent conditions.
Results
maa-1 inactivation promotes lifespan extension and stress resistance in C. elegans
An earlier study failed to show any extension of lifespan in a few ACBP null mutants of C. elegans [13]. Since complete loss of ACBP causes growth defects in yeast, we reasoned that a reduction, rather than a total loss, of gene function might reveal a contribution of ACBP to lifespan in C. elegans. To address this, we used dsRNA-mediated RNAi to individually down-regulate each of the seven ACBP genes. Indeed, the lifespan of wild-type worms was increased markedly by knockdown of maa-1 (Figure 1A; Table S1 and S2) and more modestly by knockdown of either acpb-1 or acbp-3 (Figure 1B; Table S1). We confirmed the effect of maa-1 deficiency on longevity using two maa-1 deletion mutants (ok2033 and sv38) (Figure 1C,D and Figure S1). Only the sv38 allele had previously been established to be null [14]; however, both mutants showed similar survival curves, suggesting that maa-1(ok2033) may also be a null allele (Figure 1C,D; Table S1 and S2). Given that manipulation of maa-1 expression has the most marked effects on C. elegans lifespan, we focused our further analyses on this ACBP paralog.
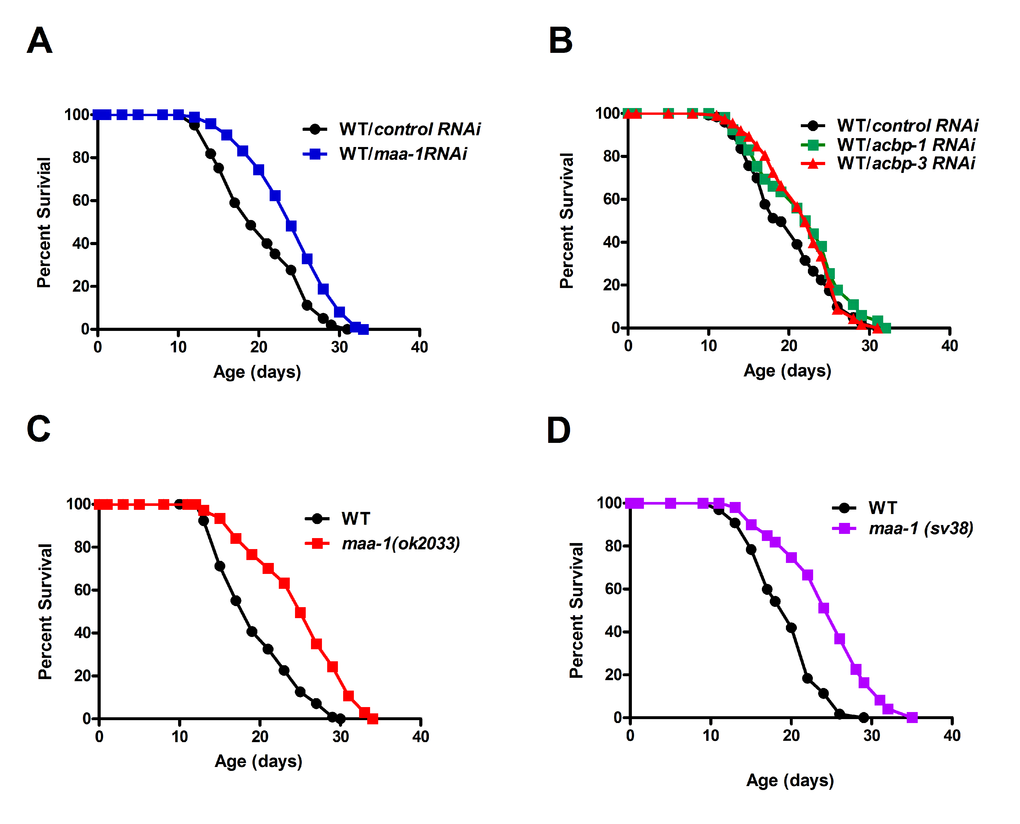
Figure 1. ACBPs regulate C. elegans lifespan. (A) Downregulation of maa-1 promotes longevity. Lifespans of wildtype worms (N2) subjected to control RNAi vs maa-1 dsRNA, P<0.0001. (B) Downregulation of acbp-1 or acbp-3 modestly increases longevity. Lifespans of wild-type worms subjected to control RNAi vs acbp-1 RNAi, P<0.01; and control RNAi vs acbp-3 RNAi, P=0.056. (C-D) Loss-of-function mutations maa-1(ok2033) (C) and maa-1(sv38) (D) prolong lifespan (both P<0.0001). P values were calculated using the log-rank (Mantel-Cox) method. Replicate experiments and details of statistical analyses are shown in Table S1 and S2.
Several lifespan-extending mutations, including deletion of ACB1 in yeast, concomitantly increase resistance to various forms of stress [1]. Consistent with this, we found that maa-1 (ok2033) mutants were more resistant than wild-type animals to both thermal stress induced by incubation at 35°C and oxidative stress induced by the superoxide generator paraquat (Figure 2A,B). Stress induced by protein misfolding and aggregation is thought to contribute to the aging process in many species, and elevated resistance to such proteotoxicity is another feature common to many long-lived C. elegans mutants [21]. Several transgenic models of proteotoxicity have been developed in C. elegans, two of which are expression of polyglutamine repeats fused to YFP (Q35YFP) or human β-amyloid peptide (Aß1-42) in the body wall muscle. In these animals, the effect of proteotoxic stress is conveniently measured as age-dependent paralysis [22,23]. We found that in both of these models, loss of motility was significantly delayed by maa-1 RNAi (Figures 2C,D and Figure S2A,B), suggesting that loss of MAA-1 activity counteracts the age-associated disruption of proteostasis. Collectively, these data indicate a novel role for MAA-1 in stress resistance and longevity in C. elegans.
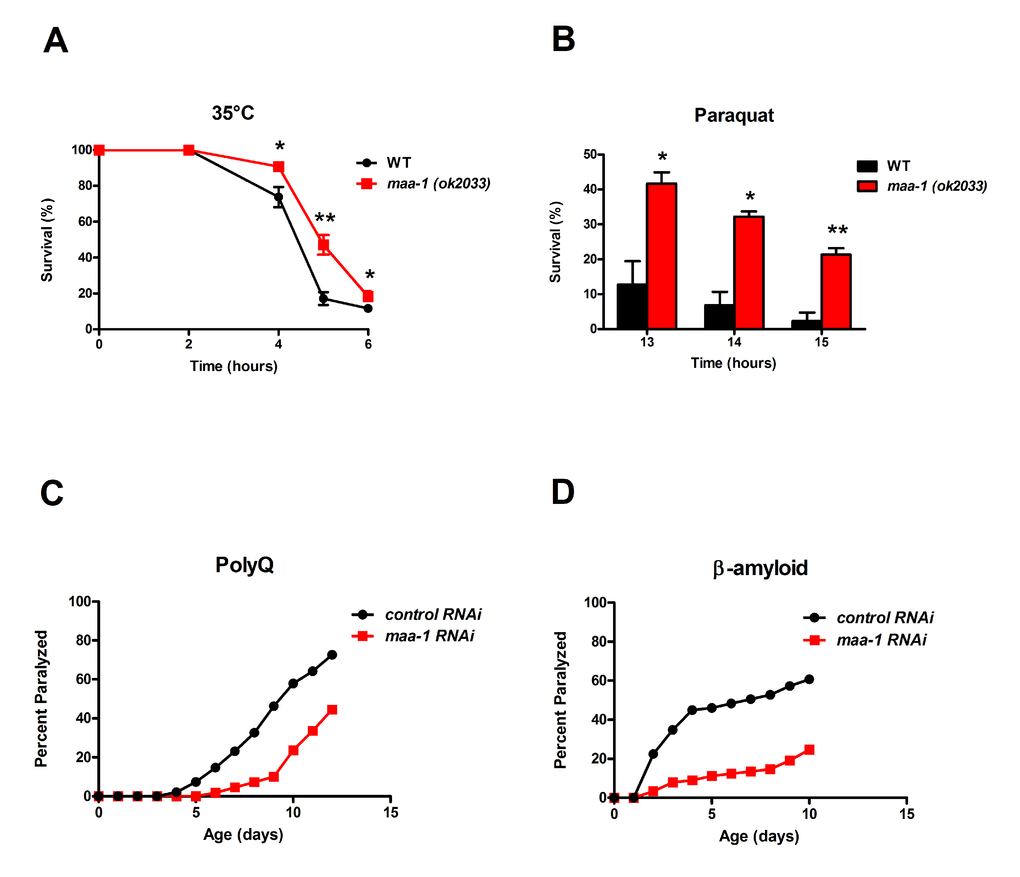
Figure 2. Loss of maa-1 promotes heat, oxidative, and proteotoxic stress resistance. (A-B) maa-1(ok2033) mutants showed enhanced resistance to incubation at 35°C (A) and exposure to 150 mM paraquat (B). Error bars represent SEM from three independent experiments (*P˂0.05, **P˂0.01 compared to wildtype worms by Student’s t-test). (C-D) maa-1 RNAi increases resistance to paralysis induced by aggregation of a 35-residue polyglutamine repeat (C; P<0.0001) or human β-amyloid (D; P<0.0001). P values were calculated using the log-rank (Mantel-Cox) method. Replicate experiment is shown in Figure S2.
MAA-1 functions in the intestine to control longevity
Previous studies of C. elegans lines expressing a MAA-1::GFP fusion protein have shown that MAA-1 is highly expressed in intestinal and hypodermal cells, but is essentially undetectable elsewhere [14]. To identify the tissue(s) in which MAA-1 functions to regulate lifespan, we performed tissue-specific RNAi. RDE-1 is an essential component of the RNAi machinery and inactivation abolishes the RNAi response in all tissues. Re-expressing rde-1(+) under the control of a tissue-specific promoter therefore allows the effects of RNAi to be examined specifically in that tissue. To reduce maa-1 expression exclusively in the intestine or hypodermis, we used rde-1(ne129) mutants in which rde-1(+) had been restored using the elt-2 (intestine) and lin-26 (hypodermis) promoters (R. Roy, unpublished results and [24]). rde-1(ne129) mutants, in which RNAi is inactivated ubiquitously, were used as control animals. Down-regulation of maa-1 in the intestine resulted in a highly significant longevity extension (Figure 3A; Table S1 and S2). Conversely, the hypodermal down-regulation of maa-1 had only a minor effect on longevity (Figure 3B) and, as expected, maa-1 RNAi had no effect on the lifespan of the unrescued rde-1(ne129) mutants (Figure 3C; Table S1). To further substantiate the modest response to hypodermal down-regulation of maa-1, we used an alternative transgenic animal in which the wrt-2 promoter was used to rescue the rde-1(+) function exclusively in the hypodermis [25]. The results were consistent with those obtained with the lin-26p::rde-1 transgenic line (Figure S3, Table S1 and S2). Together, these results suggest that MAA-1 functions to modulate aging predominantly in the intestine and to a much lesser extent in the hypodermis.
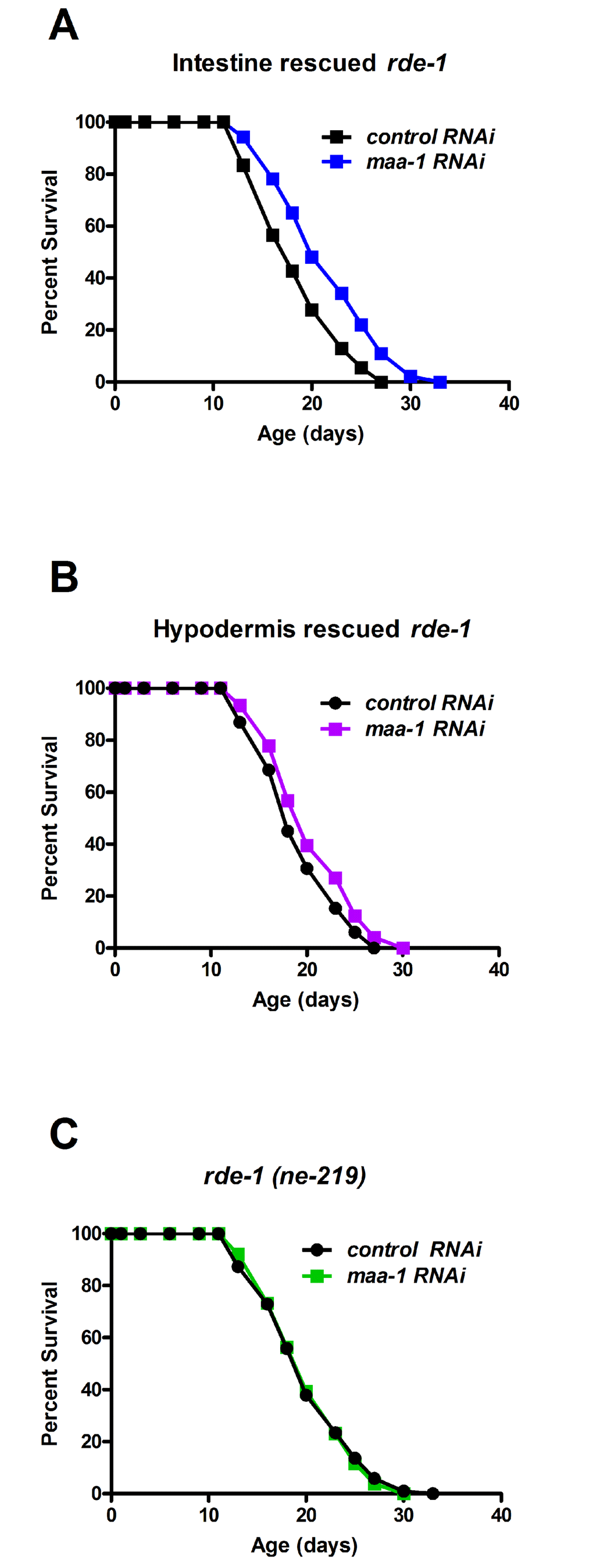
Figure 3. MAA-1 functions predominantly in the intestine to regulate longevity. Intestinal-specific RNAi is sufficient to extend longevity. Lifespans of rde-1(ne219) mutants in which rde-1 expression is restored in the intestine (A) or the hypodermis (B); animals were subjected to control or maa-1 RNAi (P<0.0001 and P<0.05 for A and B, respectively). (C) Lifespans of the control strain rde-1(ne219) subjected to control or maa-1 RNAi (P=0.8513). P values were calculated using the log-rank (Mantel-Cox) method. Replicate experiments are shown and additional statistical analysis are shown in Table S1 and S2.
HIF-1 mediates lifespan extension and proteotoxic stress resistance induced by maa-1 inactivation
As shown above (Figure 2C,D), maa-1 RNAi protects worms against age-dependent proteotoxicity . Since a prominent role in proteome maintenance and lifespan regulation has recently emerged for HIF-1 [26], a highly conserved transcriptional regulator of the metazoan response to hypoxia [27], we investigated a possible role for hif-1 in extending the lifespan of maa-1–deficient animals.
Whereas hif-1 RNAi had no effect on the lifespan of wildtype animals, it partially suppressed the lifespan extension conferred by the maa-1(ok2033) allele (Figure 4A; Table S1). We then generated double mutants combining the maa-1(ok2033) and loss-of-function hif-1(ia4) alleles, and observed complete reversion of the longevity phenotype of the maa-1 (ok2033) single mutants (Figure 4B; Table S1 and S2). Together, these results suggest a role for hif-1 in prolonging the lifespan of maa-1–deficient animals.
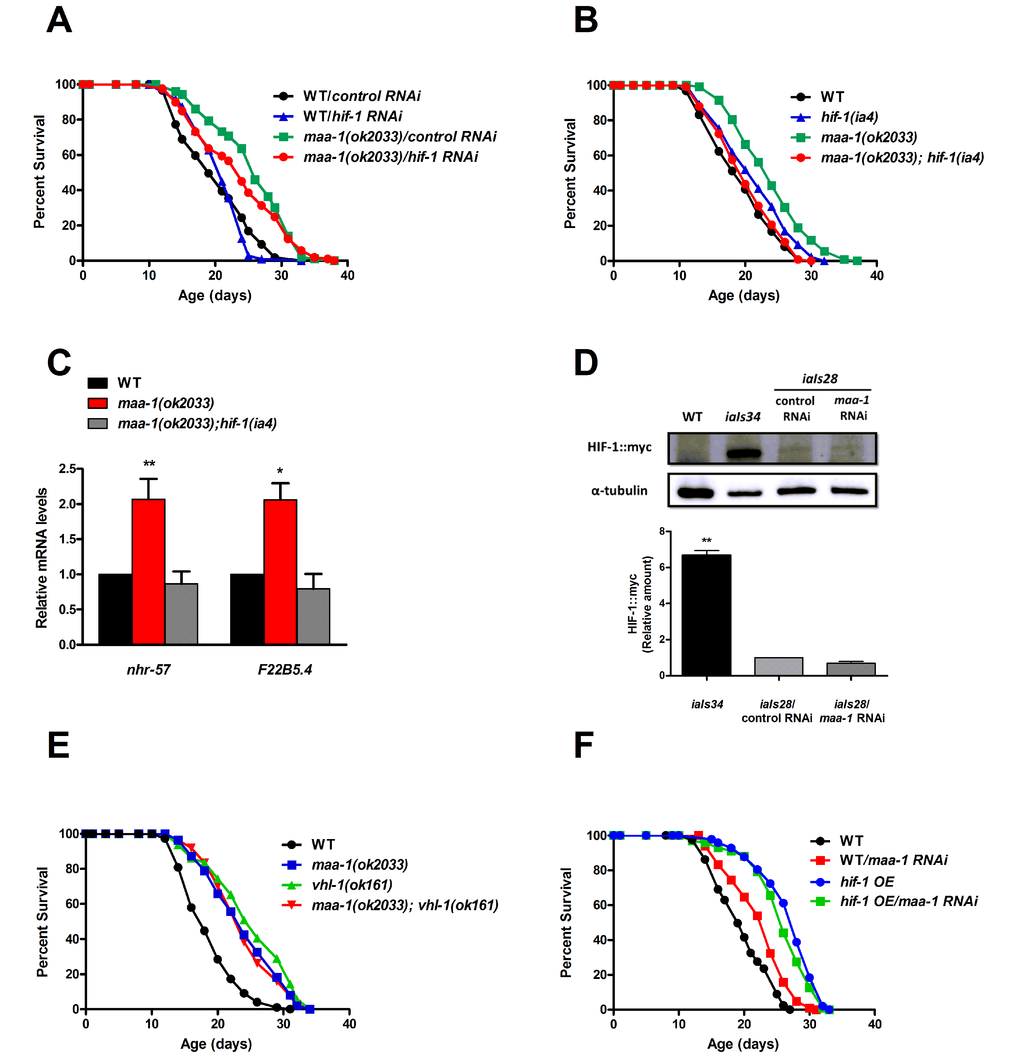
Figure 4. HIF-1 mediates lifespan extension of maa-1–deficient animals. (A) Downregulation of hif-1 reduces the longevity of maa-1(ok2033) mutants. Lifespans of wild-type and maa-1(ok2033) mutants subjected to hif-1 or control RNAi (P=0.1091 for control RNAi vs hif-1 RNAi of maa-1(ok2033) mutants). (B) Deletion of hif-1 reverses the lifespan extension conferred by the maa-1 mutation. (P=0.2661 for wild-type vs maa-1(ok2033); hif-1(ia4) mutants; P<0.0001 for maa-1(ok2033) vs maa-1(ok2033); hif-1(ia4) mutants). (C) qPCR analysis of the HIF-1 targets nhr-57 and F22B5.4 in maa-1(ok2033) and maa-1(ok2033); hif-1(ia4) mutants. Results are relative to levels in wildtype animals. Error bars represent SEM (t-test: *P<0.05, **P<0.001 for maa-1(ok2033) vs wildtype animals). (D) HIF-1 stability is not affected by downregulation of maa-1. Western blot of protein extracts from wildtype, transgenic iaIs34 animals carrying HIF-1 (P621G)::myc, and transgenic iaIs28 animals carrying HIF-1::myc, subjected to control or maa-1 RNAi. Blots were probed with anti-myc and anti-α-tubulin antibodies (upper panel). Quantification of band intensity is shown in lower panel (N=3, **P˂0.001). (E) A maa-1 loss-of-function mutation does not further increase the lifespan of long-lived vhl-1(ok161) mutants (P=0.0692 for vhl-1(ok161) vs maa-1(ok2033); vhl-1(ok161), P=0.8449 for maa-1(ok2033) vs maa-1(ok2033); vhl-1(ok161)). (F) Downregulation of maa-1 does not affect the lifespan of long-lived transgenic animals overexpressing HIF-1(P621G)::myc (hif-1 OE). (P=0.0678 for hif-1 OE on control vs maa-1 RNAi). P values were calculated using the log-rank (Mantel-Cox) method. Replicate experiments are shown in Table S1. Additional statistical analysis is shown in Table S2.
Interestingly, the mean lifespan of the hif-1(ia4) single mutant was significantly extended in each of three independent experiments (Figure 4B and Table S1 and S2) consistently with previous work showing a negative effect of hif-1 on longevity, which is observed most frequently at 25°C but also at 20°C [19,20,28]. In our studies (all performed at 20°C) the pro-longevity effect of the hif-1(ia4) allele is lost in the maa-1(ok2033); hif-1(ia4) double mutant (Figure 4B and Table S1 and S2) suggesting that lack of maa-1 inhibits the anti-aging mechanisms triggered by inactivation of hif-1.
To determine whether HIF-1 transcriptional activity was necessary for its function in maa-1 mutant animals, we analyzed the expression of three known HIF-1-dependent target genes induced by hypoxia; nhr-57, F22B5.4, and fmo-2. The latter, fmo-2, codes for the xenobiotic detoxification enzyme flavin containing monoxygenase-2, which plays a key role in promoting longevity in response to HIF-1 stabilization [29]. Notably, two of the genes tested, nhr-57 and F22B5.4, were significantly up-regulated in maa-1(ok2033) mutants compared to wild-type animals, and the induction was hif-1 dependent (Figure 4C and data not shown). These data are in agreement with an inhibitory role for MAA-1 in the regulation of HIF-1–driven transcription. Because HIF-1 transcriptional activity is tightly regulated by changes in its stability, we asked whether maa-1 RNAi might increase HIF-1 protein levels. To test this, we analyzed a transgenic line carrying integrated copies of myc-tagged hif-1 (iaIs28) [19]. However, we observed no differences between HIF-1∷myc proteins levels in animals subjected to control and maa-1 RNAi, ruling out a major role for MAA-1 in controlling HIF-1 protein stability (Figure 4D).
Previous work has described a role for hif-1 in the long lifespan of animals carrying a mutation in vhl-1, the C. elegans homolog of the mammalian von Hippel-Lindau VHL tumor suppressor gene. Mutants carrying the loss-of-function vhl-1(ok161) allele live longer than wild-type worms (Figure 4E and [17,18]), and the effect is fully dependent on HIF-1 activity [17]. To further investigate the role of HIF-1 in the longevity of maa-1 mutants, we analyzed the genetic interaction between maa-1 and vhl-1. However, we found that each single mutant and the maa-1(ok2033); vhl-1(ok161) double mutants showed similarly extended lifespans, suggesting that both maa-1 and vhl-1 function through hif-1 to modulate longevity (Figure 4E; Table S1 and S2). Finally, we performed maa-1 RNAi on a long-lived transgenic line overexpressing HIF-1 (P621G), a stabilized version of HIF-1 [19]. Consistent with the results of experiments with the genetic mutants, this manipulation had no significant effect on the lifespan of HIF-1–overexpressing animals, confirming that HIF-1 is a key mediator of the effect of maa-1 on longevity (Figure 4F; Table S1 and S2).
Overall, these results suggest a requirement for HIF-1 transcriptional activity in extending the lifespan of the maa-1 (ok2033) via mechanisms independent of both HIF-1 stability and fmo-2 expression.
To test whether HIF-1 was responsible for the enhanced resistance to proteotoxicity in response to maa-1 deficiency we generated β-amyloid peptide (Aß1-42) transgenic animals carrying the hif-1 (ia4) null allele. In contrast with the control line, transgenic worms lacking hif-1 did not show any improvement in age-dependent paralysis when subjected to maa-1 RNAi (Figure S4 A,B) pointing to an important role for HIF-1 in mediating the effect of loss of maa-1 on proteotoxic stress.
DAF-16 is required for lifespan extension induced by maa-1 inactivation
To identify additional mediators of longevity extension downstream of maa-1, we examined the contribution of the forkhead transcription factor DAF-16, a master regulator of longevity in nematodes. DAF-16 nuclear translocation is induced in mutants with reduced insulin/IGF-1-like signaling (IIS) and mediates their longevity response [30–32]. Similarly, DAF-16 transcriptional activity contributes to enhanced longevity induced by germline disruption, TOR inactivation, or temperature reduction [32–34]. To investigate whether daf-16 might be involved in the longevity phenotype of maa-1 mutants, we generated maa-1 (ok2033); daf-16 (mu86) double mutants. The lifespan of daf-16(mu86) mutants was shorter than that of wildtype animals, as expected; however, we found that daf-16 deletion completely eliminated the lifespan extension conferred by loss of maa-1 (Figure 5; Table S1 and S2), pointing to a role for daf-16 in mediating the effect of maa-1 on longevity. To test this further, we asked whether DAF-16 nuclear translocation was affected by the loss of maa-1. However, nuclear localization of DAF-16::GFP in maa-1(ok2033) mutants carrying a daf-16::GFP transgene was comparable to that of wildtype daf-16::GFP animals under standard conditions (Figure S5A). As expected, DAF-16::GFP relocation was increased in both wildtype and maa-1(ok2033) worms subjected to heat-shock at 37°C, demonstrating that DAF-16 is functional in the maa-1(ok2033) mutants (Figure S5A).
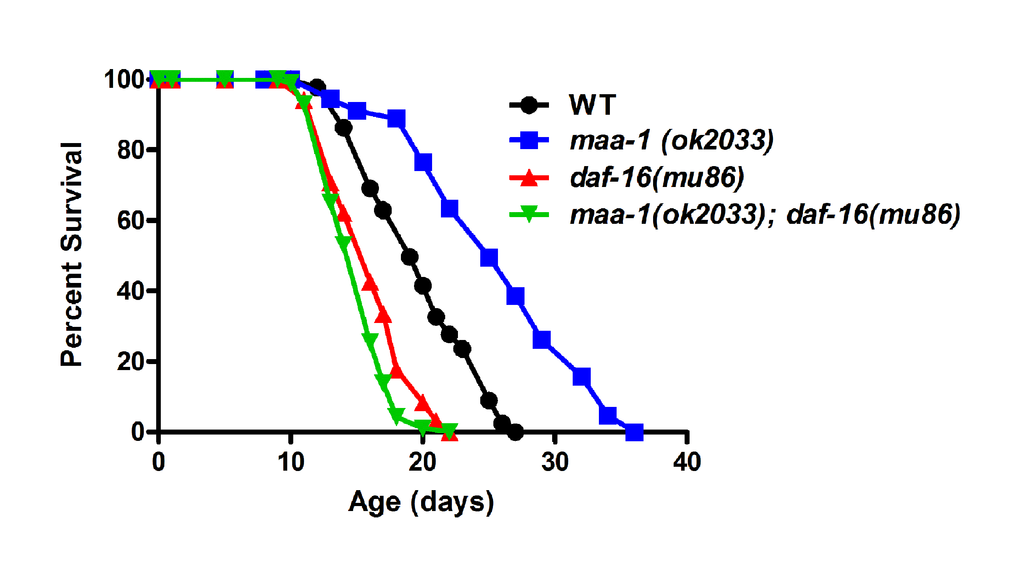
Figure 5. DAF-16 is required for lifespan extension in maa-1–deficient animals. Lifespan of maa-1(ok2033); daf-16(mu86) double mutants is significantly shorter than that of maa-1(ok2033) single mutants (P<0.0001). P values were calculated using the log-rank (Mantel-Cox) method. Replicate experiments and additional statistical analysis are shown in Table S1 and S2.
We next asked whether loss of maa-1 might increase DAF-16 transcriptional activity independently of nuclear translocation [33,35–37]. For this, we examined the expression of sod-3, a well-established transcriptional target of DAF-16 [38]. However, sod-3 mRNA level was not increased in maa-1(ok2033) mutants compared to wild-type worms (Figure S5B). Together, these results suggest that although DAF-16 is required for longevity extension induced by maa-1 deficiency, neither its nuclear translocation nor its transcriptional activity is affected by maa-1 inactivation.
Small heat-shock proteins modulate longevity in maa-1 deficient worms
Although the effects of HIF-1 activity on C. elegans longevity have been reported by several laboratories [26], the HIF-1–dependent transcriptional program responsible for lifespan modulation is not fully understood.
Recent work has shown that the survival of AMPK null mutant dauer larvae is improved by increases in total triglyceride levels and qualitative alterations in fatty acid content triggered by the stabilization of HIF-1 [39]. However, we found no differences in either the total lipid content or the fatty acid content of wild-type and maa-1(ok2033) mutants (Figures S6 and S7), ruling out a role for fatty acid biosynthesis in the extended lifespan of maa-1–deficient worms.
HIF-1 has also been reported to activate the C. elegans ER-associated unfolded protein response (UPRER) [29,40], a compartment-specific stress response implicated in lifespan regulation [41]. We tested the contribution of UPRER activation to the longevity of maa-1 mutants by examining their resistance to the ER stress inducer tunicamycin. For this, worms at the L4 larval stage were grown to adulthood on tunicamycin-containing plates, and the development and survival of their progeny was monitored for 72 h. We observed no difference between maa-1 mutants and wildtype animals in the number of eggs reaching adulthood (Figure S8). Although not conclusive, our results do not support a role for the UPRER in mediating longevity extension in maa-1–deficient worms.
To identify further mediators responsible for extending longevity in response to maa-1 inactivation, we turned to the molecular chaperone family of small heat-shock proteins (sHSPs). sHSPs delay the aggregation of polyglutamine repeat-containing proteins and contribute to the longevity extension of IIS-deficient worms [42]. We thus asked whether shsp genes might be transcriptional targets of HIF-1 and play roles in extending the lifespan of maa-1–deficient worms. We analyzed four shsp genes previously shown to affect lifespan and proteostasis [42]; namely, hsp-12.6, hsp-16.1, hsp-16.49, and sip-1. Expression of hsp-16.1 and hsp-16.49, but not of the other two, was markedly increased in maa-1(ok2033) mutants compared with wild-type worms, and the increases were completely reversed by concomitant deletion of hif-1 (Figure 6A and data not shown) but not of daf-16 (Figure 6A). These findings suggested that HIF-1–dependent transcription of shsp genes may play a direct role in the longevity of maa-1 mutants.
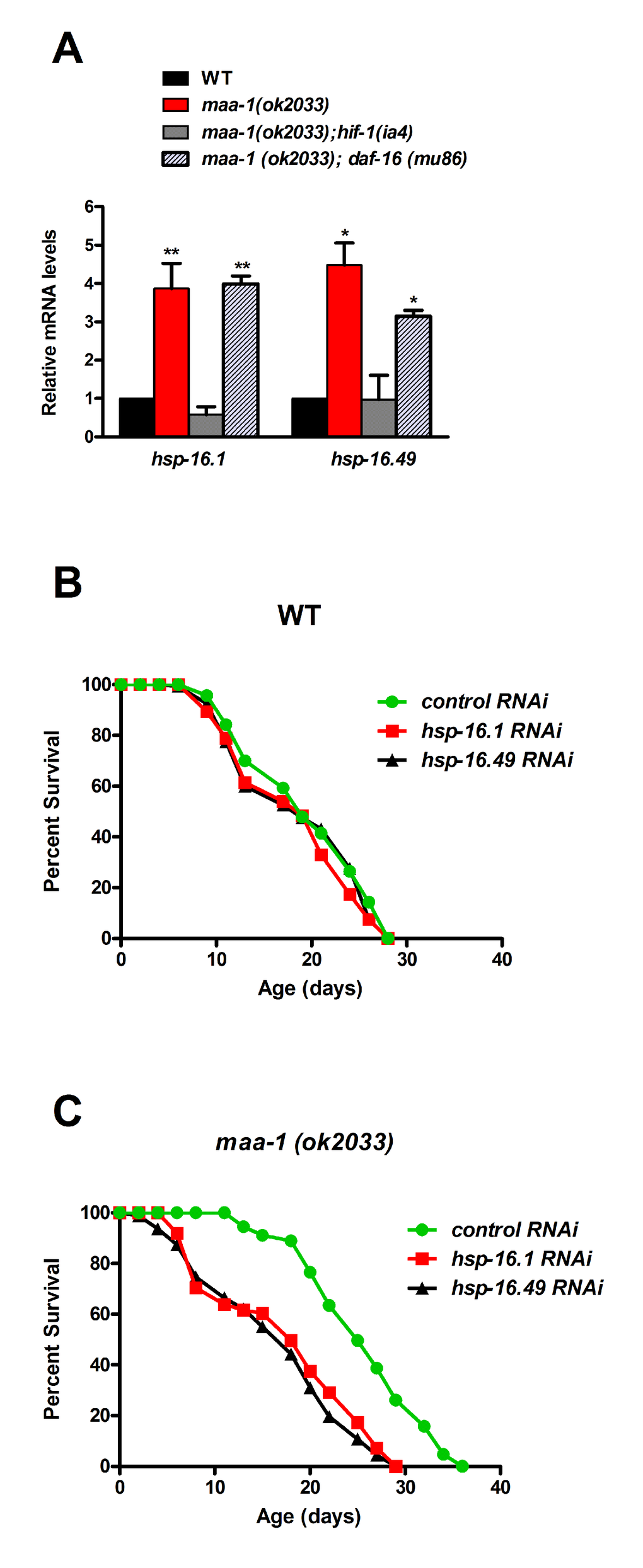
Figure 6. Molecular chaperones promote longevity downstream of HIF-1 in maa-1–deficient mutants. (A) The expression of two shsp genes is induced by maa-1 deficiency. qPCR analysis of hsp-16.1 and hsp-16.49 in wildtype, maa-1(ok2033), maa-1(ok2033); hif-1(ia4), and maa-1(ok2033); daf-16 (mu86) animals. mRNA levels are expressed relative to those in wildtype animals (t-test: *P<0.05 and **P<0.001 for maa-1(ok2033) and maa-1(ok2033); daf-16 (mu86) vs wildtype) respectively. (B) Downregulation of shsp expression has little effect on the lifespan of wildtype animals (P=0.1024 and P=0.2988 for control vs hsp-16.1 and hsp-16.49, respectively). (C) Downregulation of shsp expression markedly shortens the lifespan of maa-1(ok2033) mutants (P<0.0001 for control vs each RNAi). P values for lifespan analyses were calculated using the log-rank (Mantel-Cox) method. Replicate experiments and additional statistical analysis are shown in Table S1 and S2.
To investigate this possibility, we assessed the lifespans of wild-type and maa-1(ok2033) mutants subjected to shsp RNAi. Down-regulation of the individual hsp-16.1 and hsp-16.49 genes only slightly shortened the lifespan of wild-type worms (Figure 6B, Table S1 and S2), but markedly reduced that of the maa-1(ok2033) mutants, strongly suggesting that, while contributing marginally to wild type lifespan, sHSPs may play significant roles downstream of HIF-1 in the control of longevity (Figure 6C, Table S1 and S2).
To further explore the role of proteostasis in extending the lifespan of maa-1 mutants, we examined autophagy, the cellular process for recycling of cytosolic macromolecules and organelles implicated in lifespan extension in a number of species (reviewed in [43]). In C. elegans, autophagy is commonly monitored microscopically by tracking the formation of GFP-positive autophagosomes in transgenic lines expressing GFP-tagged LGG-1, the C. elegans homolog of the mammalian autophagy-related protein LC3. When autophagy is induced, LGG-1 localizes to the autophagosomal membranes, giving rise to a typical punctate staining pattern that is easily detectable in the hypodermal seam cells. We found that LGG-1::GFP transgenic animals subjected to maa-1 RNAi exhibited a modest but significant reduction in GFP-positive autophagosomes (Figure S9), comparable to the numbers in animals subjected to RNAi of lgg-3, an essential autophagy gene. let-363 (CeTOR) RNAi was used as positive control, because down-regulation of the TOR pathway is known to induce autophagy (Figure S9). We conclude that autophagy does not play a role in extending the lifespan of maa-1 mutants. Furthermore, given the anti-aging role of autophagy, a reduction of autophagic activity in maa-1 mutants might limit the beneficial effects associated with HIF-1 activation and responsible for increased longevity.
Collectively, while excluding the activation of autophagy, our results point to the HIF-1–dependent activation of small heat-shock proteins as essential to extend the lifespan of maa-1–deficient animals.
Discussion
In this study, we report that the C. elegans ACBP MAA-1 shares a conserved role in longevity regulation and stress resistance with the yeast ortholog Acb1. While the mechanisms by which ACBP influences yeast lifespan are not yet known, we identified several genes involved in the novel MAA-1 pathway in C. elegans. We found that loss of MAA-1 prolongs lifespan and promotes resistance to heat, oxidative, and proteotoxic stress; identified HIF-1 as the principal transcriptional regulator of longevity; implicated an interaction between maa-1 and daf-16 in lifespan regulation; and showed that HIF-1–dependent activation of shsp gene expression plays a key role in lifespan extension.
Recent work has demonstrated that HIF-1 stabilization in neurons is sufficient to extend worm lifespan via the cell non-autonomous activation of the gene coding the detoxification enzyme FMO-2 in intestinal cells, while stabilization of HIF-1 exclusively in the gut does not affect longevity [29]. The results reported here indicate that down-regulation of maa-1 in the intestine has the major effect on lifespan (Figure 3B). This is in agreement with previous studies demonstrating a central role for intestinal cells in the regulation of longevity [44–46]. Although we can speculate about an intestinal signal leading to the neuronal activation of HIF-1 in maa-1–deficient worms, our results do not show HIF-1 stabilization or induction of fmo-2 mRNA and point instead to shsps as key mediators of the effect of maa-1 deficiency on lifespan. We tend therefore to favor a simpler model in which loss of intestinal maa-1 triggers the activation of HIF-1 directly in the gut through mechanisms independent of protein stability. This activation is likely to induce both cell-autonomous and nonautonomous anti-aging mechanisms including a stress response that counteracts the toxicity of protein aggregation. Although our model may seem inconsistent with the results mentioned above [29], this apparent contradiction can be easily resolved by hypothesizing that the transcriptional response triggered by HIF-1 through protein stabilization differs in part from that induced via yet to be determined alternative mechanisms activated by intestinal loss of maa-1 (see next paragraph).
How are MAA-1 and HIF-1 linked mechanistically? Of the seven ACBP genes in C. elegans, inactivation of maa-1 alone markedly increased lifespan, suggesting a unique role for MAA-1 in the control of aging. Loss of MAA-1 has been proposed to alter the pool of acyl-CoA esters present on the Golgi and endosomal membranes, which are necessary for proper vesicle fusion/fission [14]. One possible direct link between MAA-1 and HIF-1 is that a specific bioactive lipid(s) produced from acyl-CoA esters on vesicular membranes might modulate HIF-1 nuclear localization and/or transcriptional activity. RHY-1 is another negative regulator of HIF-1 that is highly expressed in intestinal cells. RHY-1 is a multipass membrane protein containing an acyltransferase-3 domain, with proposed roles in lipid synthesis, metabolism, and transport [47]. It is tempting to speculate that both MAA-1 and RHY-1 may function in the production of lipid species that regulate HIF-1 activation. Although computational analyses suggest that RHY-1 is present in the ER and plasma membrane [47], it will be of interest to determine whether MAA-1 and RHY-1 colocalize. The fact that neither RHY-1 nor MAA-1 substantially modify HIF-1 stability ([47] and Figure 4D) suggests that they may share a common mechanism for regulating HIF-1 transcriptional activity. Alternatively, MAA-1 and RHY-1 might independently control the metabolism of bioactive lipids that directly or indirectly affect HIF-1–dependent transcription.
The positive effect of maa-1 inactivation on resistance to proteotoxicity prompted us to investigate the contributions of the proteostasis network to the longevity of maa-1–deficient mutants. We focused on the sHSP family of chaperones because of the well-established links between sHSP activation, reduced protein aggregation, and extended lifespan [42]. In C. elegans, the expression of various shsp genes is known to be regulated by the stress response transcription factors DAF-16 and HSF-1 [42]. Our data extend these observations by demonstrating HIF-1–dependent upregulation of hsp-16.1 and hsp-16.49, in maa-1–deficient animals (Figure 6A), consistent with the suggestion that HIF-1, DAF-16, and HSF-1 have common gene targets [48]. RNAi-mediated knockdown of each of the two shsp genes abolished the longevity extension of maa-1 mutants, pointing to the importance of these chaperones as key mediators of longevity downstream of HIF-1. Of note, this is a novel mechanistic explanation for the anti-aging role of HIF-1 in C. elegans. Interestingly, several stress response genes, including HSP104, HSP26, and HSP12, have been reported to be induced by Acb1 depletion in yeast [49]. Hsp104 and Hsp26 function together to counteract protein aggregation, and both Hsp104 and Hsp12 have reported roles in lifespan regulation [50–52]. Moreover, HSP104, HSP26, and HSP12 are targets of transcription factors long known to control yeast lifespan and/or stress resistance; namely, Msn2, Msn4, Gis1, and Hsf1 [1]. It seems reasonable to assume that one or more of these transcription factors replace HIF-1 (not present in yeast) in a conserved ACBP–dependent stress response, which may function to extend longevity – at least in part – by promoting proteostasis.
Our epistasis analyses additionally show that daf-16 is required to extend the lifespan of maa-1–deficient animals (Figure 5, Table S1 and S2), although paradoxically, daf-16 nuclear localization and transcriptional activity (as indicated by expression of its direct transcriptional target sod-3) are unaffected by maa-1 loss (Figure S5A,B). Notably, the expression of hsps triggered by maa-1 deficiency is not reversed by loss of DAF-16 ruling out a direct contribution of DAF-16 to the activation of stress response observed. Together, our results suggest that DAF-16 basal activity is necessary but not sufficient for maa-1(ok2033) animals to reach their full longevity potential. Alternatively, loss of maa-1 may induce the DAF-16–dependent transcription of a set of genes yet to be identified.
In C. elegans the role played by HIF-1 in lifespan regulation is complex. Several articles have demonstrated a positive effect of HIF-1 on longevity [17–19,28]. However, worms carrying hif-1 loss of function alleles live longer than wild type [19,20,28]. These paradoxical findings have not been fully understood [26]. While the results presented here and those of others [29] shed light on the anti-aging role of hif-1, the anti-aging mechanisms triggered by loss of hif-1 are still elusive. Recent work has shown that lack of hif-1 extends longevity significantly only at 25°C whereas a modest effect can be observed at 20°C only if the frequent deaths due to vulval rupture are censored [28]. Intriguingly, in the lifespan studies reported here hif-1(ia4) mutants showed a consistent and significant 10% mean lifespan extension at 20C° (Figure 4B; Table S1 and S2). This effect seems to be independent of the exclusion of animals showing the “exploded through vulva” phenotype, which was negligible in our experiments (data not shown). Although the origin of these discrepancies is unclear, we speculate it may be due the different experimental conditions such as the bacterial strain used or whether it is dead or alive. It has been previously shown that both E. coli HT115 and OP50 differentially modulate the metabolism, behavior, development and aging of worms [53] up to an extent that certain genes may have opposite effects on the lifespan of worms fed on OP50 and HT115 [54–56]. Similarly, the use of UV-killed bacteria may differentially affect the lifespan of certain mutants [57]. Most of the previously published hif-1 (ia4) worm lifespan experiments have been performed on UV-killed OP50 [28]. However, in our case all lifespan experiments have been done on live HT115. Thus, it is not surprising that we obtain slightly different results than previously reported.
Of note, maa-1; hif-1 double mutants live shorter than individual hif-1 mutants (Table S1 and S2). This suggests that loss of maa-1 suppresses the anti-aging response triggered by inactivation of hif-1. A possible explanation for these results is that, in analogy with what observed for autophagy, maa-1 down-regulation has a negative effect on yet to be established anti-aging systems induced by hif-1 deficiency.
In mammals, HIF-1 and HIF-2 are the master regulators of the hypoxia response and have essential roles in embryonic development as well as in the physiopathology of cardiovascular diseases and cancer [27]. Increasing evidence indicates that although HIF-1 activity is protective against ischemia, HIF-1 and/or HIF-2 activation promote metabolic reprogramming, vascularization, and metastasis in the majority of human cancers [27]. HIF stability and transcriptional activity are modulated by several post-translational modifications, and numerous signaling molecules are known to control HIF-dependent transactivation in an oxygen-independent manner [58]. Given this complex scenario, it will be worth exploring the existence of a conserved mammalian stress response pathway involving ACBP and/or acyl-CoA esters, HIF-1, and molecular chaperones such as sHSPs. Mammalian ACBD1 may be a good candidate in this regard, because it is likely to share a role with MAA-1 in vesicle trafficking [8], a function that may be relevant to the regulation of HIF-1. Similarly, it will be important to test tissues/cells isolated from ACBD1 knockout mice for changes in stress resistance, HIF-1 activity, and chaperone gene expression. Notably, studies in mice have shown an age-dependent decline in the inducible HIF-driven transcription in ischemic muscles [27]. Moreover, the age-associated decline in HIF-1 activity observed in rat liver, heart, and skeletal muscle can be reversed by dietary restriction, a well-established life-extending intervention [59]. While it seems clear from studies of cancer that constitutive stabilization of HIF-1 can be detrimental to human health, the preservation of HIF-1 inducibility over time is likely to be beneficial, both to prevent the age-associated decline in tissue function and to protect against ischemia. Thus, a comprehensive understanding of the mechanisms responsible for HIF-1 function may contribute to the development of pharmacotherapeutic strategies aimed at improving human health by modulating HIF function.
Materials and Methods
Nematode maintenance and strains
Strains were cultured under standard laboratory conditions [60]. The wildtype N2 (Bristol) was used as the reference strain. Strains used are listed below (name, genotype, and origin).
RB1644, maa-1(ok2033) III, CGC
VB1051, maa-1(sv38) III, Tuck Lab
AM140, rmIs132 [unc54p::Q-35::YFP], CGC
CL2006 (Aβ1-42), dvIs2 [pCL12 (unc-54p::Aβ1-42) + pRF4], CGC
HGA8024, hif-1 (ia4) V, dvIs2 [pCL12 (unc-54p::Aβ1-42) + pRF4], this work
WM27, rde-1(ne219) V, CGC
MR0931, rde-1(ne129) V, rrIs01 [elt-2::GFP]; Ex236 [end-3p::rde-1; elt-2p::rde-1; inx-6::GFP], Roy Lab
NR222, rde-1(ne129) V, kzIs9 [pKK1260 (lin-26p::nls::GFP); (lin-26p::rde-1) + pRF6], CGC
JM43, rde-1(ne219)V, Is[wrt-2p::rde-1], Melo Lab
AD105, daf-16(mu86) I, CGC
TJ356, zIs356 IV [daf-16::GFP+pRF6], CGC
HGA8020, maa-1(ok2033) III; daf-16(mu86) I, this work
HGA8021, maa-1(ok203 3) III; zIs356 IV [daf-16::GFP + pRF6], this work
ZG31, hif-1(ia4) V, CGC
HGA8022, maa-1(ok2033) III; hif-1(ia4) V, this work
CB5602, vhl-1(ok16) X, CGC
HGA8023, vhl-1(ok16) X; maa-1(ok2033) III, this work
DA2123, adIs2122 [lgg-1::GFP+pRF6], CGC
ZG583, iaIs34 [hif-1p::hif-1a (P621G)::myc + unc-119(+)], Powell-Coffman Lab
ZG580, iaIs28 [hif-1p::hif-1a::myc + unc-119(+)], Powell-Coffman Lab
Lifespan analysis
Lifespan assays were conducted at 20°C according to standard protocols [61]. Worms were synchronized by bleaching and transferred to plates at the L1 stage. Worms were maintained on solid Nematode Growth Medium (NGM) containing 25 µg/ml carbenicillin and 15 µM 5-fluorouracil and seeded with Escherichia coli strain HT115. Animals that failed to display heat-provoked movement were scored as dead. Animals that crawled off the plates were not included in the analysis. P values were calculated using the log-rank (Mantel-Cox) method. Statistics and significance calculations for individual lifespan studies were determined using the Oasis online software [62]. Statistical analysis of experiments shown in the main text and replicate experiments are provided in Table S1. To take into account the variance between replicate experiments we performed pairwise comparisons for both mean and maximum lifespan by two tailed Student’s t-test. Results are shown in Table S2.
RNA-mediated interference
RNAi experiments were carried out by feeding worms with bacteria expressing dsRNA against the gene of interest (or control bacteria carrying the empty L4440 vector). Bacteria transformed with the appropriate vectors were grown at 37°C overnight and then seeded onto NGM plates containing carbenicillin (25 µg/ml). Expression of dsRNA was induced by the addition of 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG; Sigma) before L1 worms were added to the bacterial lawns. RNAi clones were obtained from the Ahringer collection (Source BioScience) and were verified by sequencing prior to use.
Quantification of proteotoxicity-induced paralysis
The paralysis of worms expressing Q35YFP or human Aβ1-42 was assessed visually, as previously described [63]. Briefly, worms were tapped on the head with a platinum wire and were scored as paralyzed if the head moved but the worm failed to make forward progress on the agar surface. The assay was terminated within day 12 to avoid mistakenly scoring old worms as paralyzed [63]. P values were calculated using the log-rank (Mantel-Cox) method.
Resistance to oxidative stress
To measure oxidative stress resistance, fifty day-1 adult worms were transferred to M9 buffer containing 150 mM paraquat (1,1-dimethyl-4,4-bipyridinium dichloride; Sigma-Aldrich) and then incubated at 20°C. Live/dead worms were scored hourly. Every time point shown consists of three independent experiments. Significance was calculated using a two-tailed Student’s t-test.
Resistance to thermal stress
For heat-shock assays, sixty day 1-adult worms on NGM plates seeded with E. coli strain HT115 were exposed to 35°C for 4, 5, and 6 hours. Survival was assayed after 14-16 hours of recovery at 20°C. For each time point an independent set of plates was used. Values shown correspond to the average survival of three independent experiments. P values were calculated using a two-tailed Student’s t-test.
RNA extraction, reverse transcription, and qPCR
Total RNA was isolated from synchronized populations of day-1 adult worms using TRIzol (MRC). Reverse transcription was performed using the iScript cDNA Synthesis kit (Bio-Rad) using 500 ng of RNA per sample. SYBR Green real-time qPCR experiments were performed as described in the StepOnePlus manual using a StepOnePlus Real-Time PCR system (Applied Biosystems). PCR products were amplified using the primers listed below (Table 1). The standard curve method was used to determine the relationship between mRNA abundance and PCR cycle number. Levels of cdc-42 and pmp-3 mRNA were used for normalization of data shown in Figure 4C; cdc-42, act-1, and ama-3 mRNA were used for normalization of data shown in Figure 6A and Figure S5. Each experiment was repeated at least two times using three biological and two technical replicates. ΔCt values were analyzed by unpaired t-test.
Table 1. Sequences of primers used for qPCR analysis
| Gene | Forward primer | Reverse primer | Amplicon length (nt) |
| cdc-42 | CTGCTGGACAGGAAGATTACG | CTCGGACATTCTCGAATGAAG | 111 |
| pmp-3 | GTTCCCGTGTTCATCACTCAT | ACACCGTCGAGAAGCTGTAGA | 115 |
| sod-3 | TCGCACTGCTTCAAAGCTTGTTCAA | CCAAATCTGCATAGTCAGATGGGAGAT | 98 |
| nhr-57 | TTATCGAGTTTCTCGCATTGG | AAGTCTGCAATCACGCTCTGT | 115 |
| hsp-16.49 | GAGAAATGCTGATCACAACTC | GAAACATCGAGTTGAACAGAG | 92 |
| hsp-16.1 | GGCTCAGATGGAACGTCAA | TGGCAAACTTTTGATCATTGTTA | 89 |
| F22B5.4 | CGCCATTCAGAAGGGAGATA | ATGCACTGCAGAAGAGAACG | 119 |
| ama-1 act-1 | CCTACGATGTATCGAGGCAAA GCTGGACGTGATCTTACTGATTACC | CCTCCCTCCGGTGTAATAATG GTAGCAGAGCTTCTCCTTGATGTC | 139 113 |
Western blotting
Approximately 400 L4 worms were collected in M9 buffer and snap frozen in dry ice. Pellets were resuspended in 50 mM Tris, pH 7.5, containing protease inhibitors (Roche). Ceramic beads were added, and worms were lysed using a Precellys 24 homogenizer (Bertin Technologies). Extracts were resolved by SDS-PAGE and transferred to nitrocellulose membrane. Western blot analysis was performed using anti-c-myc (Roche) and anti-α-tubulin (Sigma) primary antibodies. The band intensity was quantified using ImageJ (https://imagej.nih.gov/ij/).
DAF-16 localization assays
On day 1 of adulthood, animals carrying a daf-16::GFP transgene were analyzed for DAF-16 nuclear localization in intestinal cells using a Nikon Eclipse 80i fluorescent microscope at 400x magnification. For the heat-stress challenge, worms were shifted to 37°C for 1 h. Animals were scored as having nuclear DAF-16 if the majority of intestinal cells displayed a distinct concentration of GFP in the nucleus. Approximately 15–30 worms were analyzed for each condition. Each experiment was repeated twice, and a representative experiment is shown in Figure S5.
Analysis of fatty acid composition by GLC
Synchronized L4 stage worms were collected from five to ten 9-cm plates and washed three times in 0.9% NaCl. Worms were left for 20 min to empty their intestines, washed once in sterile water, resuspended in freshly prepared 2.5% (v/v) H2SO4 in water-free methanol (1 ml) supplemented with 10 μg/ml butylated hydroxytoluene, and incubated for 5 h at 80°C. Subsequently, the fatty acid methyl esters (FAMEs) were extracted by the addition of hexane (0.5 ml) and H2O (1.5 ml). The organic phase was transferred to fresh sample vials and dried under a stream of N2. Each sample was dissolved in hexane (40–50 μl), and FAMEs were analyzed by GLC on a Chrompack CP 9002 instrument equipped with a DB-WAX column (Agilent Technologies). FAMEs were identified by comparison with standards (Larodan Fine Chemicals).
Autophagy assay
Autophagy was monitored using an LGG-1::GFP translational reporter [64]. GFP-positive punctae in seam cells were counted in L4 transgenic worms using a Leica DMI 6000 B microscope at 1000× magnification. All animals were kept at 20°C and analyzed by the same experimenter. The number of punctae per seam cell was averaged for each worm and this average was used for calculating the population mean number of LGG-1::GFP-containing punctae per seam cell. Statistical analysis was performed by one-way ANOVA followed by the Dunnett’s multiple comparison test.
ER stress survival assay
L4 worms were placed on plates containing 3 µg/ml tunicamycin (Sigma) and seeded with OP50 bacteria. Eggs laid over 6 h were counted and the number of progeny reaching adulthood was scored after 72 h at 20°C.
Oil Red O staining
Worms were fixed in 2% paraformaldehyde for 1 h at 20˚C, washed twice with PBS, and incubated in 60% isopropanol for 15 min. Worms were stained with a 60% Oil Red O solution overnight, and then observed using an Axioplan microscope (Zeiss) at 100 x magnification.
Supplementary Materials
Author Contributions
Designed research and interpreted results: PF, HA, NJF. Performed experiments: MS, MD, MT, and EBH. Analyzed data: PF and MS. Wrote the paper: PF.
Acknowledgements
Strains were obtained from the Caenorhabditis Genetics Center (CGC), supported by the NIH Office of Infrastructure Programs (P40 OD010440). We would like to thank Michael Hebeisen and Richard Roy for providing strain MR0931, Simon Tuck for providing strain VB1051, and Jo Anne Powell-Coffman for providing strains ZG580 and ZG583. We also thank Francesca Palladino, Anne Laurençon, and Manish Grover for critical reading of the manuscript and Claire-Emmanuelle Indelicato for technical help.
Conflicts of Interest
The authors have no conflict of interests to declare.
Funding
This work was supported by grants from the Fondation ARC pour la Recherche Contre le Cancer and La Ligue Contre le Cancer (Comité du Rhône) to PF, and by a grant from the Fondation pour la Recherche Médicale and an ERC consolidator grant (H2020/2014-2019-N°647003) to HA . MS was supported by a doctoral fellowship from the Ministère de l’Enseignement Supérieure et de la Recherche and the Fondation ARC pour la Recherche Contre le Cancer. MD was supported by a post-doctoral fellowship from the Fondation pour la Recherche Médicale.
References
- 1. Fontana L, Partridge L, Longo VD. Extending healthy life span--from yeast to humans. Science. 2010; 328:321–26. https://doi.org/10.1126/science.1172539 [PubMed]
- 2. Fabrizio P, Hoon S, Shamalnasab M, Galbani A, Wei M, Giaever G, Nislow C, Longo VD. Genome-wide screen in Saccharomyces cerevisiae identifies vacuolar protein sorting, autophagy, biosynthetic, and tRNA methylation genes involved in life span regulation. PLoS Genet. 2010; 6:e1001024. https://doi.org/10.1371/journal.pgen.1001024 [PubMed]
- 3. Burton M, Rose TM, Faergeman NJ, Knudsen J. Evolution of the acyl-CoA binding protein (ACBP). Biochem J. 2005; 392:299–307. https://doi.org/10.1042/BJ20050664 [PubMed]
- 4. Knudsen J, Jensen MV, Hansen JK, Faergeman NJ, Neergaard TB, Gaigg B. Role of acylCoA binding protein in acylCoA transport, metabolism and cell signaling. Mol Cell Biochem. 1999; 192:95–103. https://doi.org/10.1023/A:1006830606060 [PubMed]
- 5. Gaigg B, Neergaard TB, Schneiter R, Hansen JK, Faergeman NJ, Jensen NA, Andersen JR, Friis J, Sandhoff R, Schrøder HD, Knudsen J. Depletion of acyl-coenzyme A-binding protein affects sphingolipid synthesis and causes vesicle accumulation and membrane defects in Saccharomyces cerevisiae.. Mol Biol Cell. 2001; 12:1147–60. https://doi.org/10.1091/mbc.12.4.1147 [PubMed]
- 6. Faergeman NJ, Feddersen S, Christiansen JK, Larsen MK, Schneiter R, Ungermann C, Mutenda K, Roepstorff P, Knudsen J. Acyl-CoA-binding protein, Acb1p, is required for normal vacuole function and ceramide synthesis in Saccharomyces cerevisiae.. Biochem J. 2004; 380:907–18. https://doi.org/10.1042/bj20031949 [PubMed]
- 7. Fan J, Liu J, Culty M, Papadopoulos V. Acyl-coenzyme A binding domain containing 3 (ACBD3; PAP7; GCP60): an emerging signaling molecule. Prog Lipid Res. 2010; 49:218–34. https://doi.org/10.1016/j.plipres.2009.12.003 [PubMed]
- 8. Hansen JS, Faergeman NJ, Kragelund BB, Knudsen J. Acyl-CoA-binding protein (ACBP) localizes to the endoplasmic reticulum and Golgi in a ligand-dependent manner in mammalian cells. Biochem J. 2008; 410:463–72. https://doi.org/10.1042/BJ20070559 [PubMed]
- 9. Neess D, Bloksgaard M, Bek S, Marcher AB, Elle IC, Helledie T, Due M, Pagmantidis V, Finsen B, Wilbertz J, Kruhøffer M, Færgeman N, Mandrup S. Disruption of the acyl-CoA-binding protein gene delays hepatic adaptation to metabolic changes at weaning. J Biol Chem. 2011; 286:3460–72. https://doi.org/10.1074/jbc.M110.161109 [PubMed]
- 10. Mandrup S, Sorensen RV, Helledie T, Nohr J, Baldursson T, Gram C, Knudsen J, Kristiansen K. Inhibition of 3T3-L1 adipocyte differentiation by expression of acyl-CoA-binding protein antisense RNA. J Biol Chem. 1998; 273:23897–903. https://doi.org/10.1074/jbc.273.37.23897 [PubMed]
- 11. Yang Y, Pritchard PH, Bhuiyan J, Seccombe DW, Moghadasian MH. Overexpression of acyl-coA binding protein and its effects on the flux of free fatty acids in McA-RH 7777 cells. Lipids. 2001; 36:595–600. https://doi.org/10.1007/s11745-001-0762-0 [PubMed]
- 12. Vock C, Biedasek K, Boomgaarden I, Heins A, Nitz I, Döring F. ACBP knockdown leads to down-regulation of genes encoding rate-limiting enzymes in cholesterol and fatty acid metabolism. Cell Physiol Biochem. 2010; 25:675–86. https://doi.org/10.1159/000315087 [PubMed]
- 13. Elle IC, Simonsen KT, Olsen LC, Birck PK, Ehmsen S, Tuck S, Le TT, Færgeman NJ. Tissue- and paralogue-specific functions of acyl-CoA-binding proteins in lipid metabolism in Caenorhabditis elegans.. Biochem J. 2011; 437:231–41. https://doi.org/10.1042/BJ20102099 [PubMed]
- 14. Larsen MK, Tuck S, Faergeman NJ, Knudsen J. MAA-1, a novel acyl-CoA-binding protein involved in endosomal vesicle transport in Caenorhabditis elegans.. Mol Biol Cell. 2006; 17:4318–29. https://doi.org/10.1091/mbc.E06-01-0035 [PubMed]
- 15. Pfanner N, Orci L, Glick BS, Amherdt M, Arden SR, Malhotra V, Rothman JE. Fatty acyl-coenzyme A is required for budding of transport vesicles from Golgi cisternae. Cell. 1989; 59:95–102. https://doi.org/10.1016/0092-8674(89)90872-6 [PubMed]
- 16. Pfanner N, Glick BS, Arden SR, Rothman JE. Fatty acylation promotes fusion of transport vesicles with Golgi cisternae. J Cell Biol. 1990; 110:955–61. https://doi.org/10.1083/jcb.110.4.955 [PubMed]
- 17. Mehta R, Steinkraus KA, Sutphin GL, Ramos FJ, Shamieh LS, Huh A, Davis C, Chandler-Brown D, Kaeberlein M. Proteasomal regulation of the hypoxic response modulates aging in C. elegans.. Science. 2009; 324:1196–98. https://doi.org/10.1126/science.1173507 [PubMed]
- 18. Müller RU, Fabretti F, Zank S, Burst V, Benzing T, Schermer B. The von Hippel Lindau tumor suppressor limits longevity. J Am Soc Nephrol. 2009; 20:2513–17. https://doi.org/10.1681/ASN.2009050497 [PubMed]
- 19. Zhang Y, Shao Z, Zhai Z, Shen C, Powell-Coffman JA. The HIF-1 hypoxia-inducible factor modulates lifespan in C. elegans.. PLoS One. 2009; 4:e6348. https://doi.org/10.1371/journal.pone.0006348 [PubMed]
- 20. Chen D, Thomas EL, Kapahi P. HIF-1 modulates dietary restriction-mediated lifespan extension via IRE-1 in Caenorhabditis elegans.. PLoS Genet. 2009; 5:e1000486. https://doi.org/10.1371/journal.pgen.1000486 [PubMed]
- 21. Taylor RC, Dillin A. Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol. 2011; 3:a004440. https://doi.org/10.1101/cshperspect.a004440 [PubMed]
- 22. Morley JF, Brignull HR, Weyers JJ, Morimoto RI. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans.. Proc Natl Acad Sci USA. 2002; 99:10417–22. https://doi.org/10.1073/pnas.152161099 [PubMed]
- 23. Link CD, Johnson CJ, Fonte V, Paupard M, Hall DH, Styren S, Mathis CA, Klunk WE. Visualization of fibrillar amyloid deposits in living, transgenic Caenorhabditis elegans animals using the sensitive amyloid dye, X-34. Neurobiol Aging. 2001; 22:217–26. https://doi.org/10.1016/S0197-4580(00)00237-2 [PubMed]
- 24. Qadota H, Inoue M, Hikita T, Köppen M, Hardin JD, Amano M, Moerman DG, Kaibuchi K. Establishment of a tissue-specific RNAi system in C. elegans.. Gene. 2007; 400:166–73. https://doi.org/10.1016/j.gene.2007.06.020 [PubMed]
- 25. Melo JA, Ruvkun G. Inactivation of conserved C. elegans genes engages pathogen- and xenobiotic-associated defenses. Cell. 2012; 149:452–66. https://doi.org/10.1016/j.cell.2012.02.050 [PubMed]
- 26. Leiser SF, Kaeberlein M. The hypoxia-inducible factor HIF-1 functions as both a positive and negative modulator of aging. Biol Chem. 2010; 391:1131–37. https://doi.org/10.1515/bc.2010.123 [PubMed]
- 27. Semenza GL. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu Rev Pathol. 2014; 9:47–71. https://doi.org/10.1146/annurev-pathol-012513-104720 [PubMed]
- 28. Leiser SF, Begun A, Kaeberlein M. HIF-1 modulates longevity and healthspan in a temperature-dependent manner. Aging Cell. 2011; 10:318–26. https://doi.org/10.1111/j.1474-9726.2011.00672.x [PubMed]
- 29. Leiser SF, Miller H, Rossner R, Fletcher M, Leonard A, Primitivo M, Rintala N, Ramos FJ, Miller DL, Kaeberlein M. Cell nonautonomous activation of flavin-containing monooxygenase promotes longevity and health span. Science. 2015; 350:1375–78. https://doi.org/10.1126/science.aac9257 [PubMed]
- 30. Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: an HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans.. Science. 1997; 278:1319–22. https://doi.org/10.1126/science.278.5341.1319 [PubMed]
- 31. Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans.. Nature. 1997; 389:994–99. https://doi.org/10.1038/40194 [PubMed]
- 32. Lin K, Hsin H, Libina N, Kenyon C. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet. 2001; 28:139–45. https://doi.org/10.1038/88850 [PubMed]
- 33. Xiao R, Zhang B, Dong Y, Gong J, Xu T, Liu J, Xu XZ. A genetic program promotes C. elegans longevity at cold temperatures via a thermosensitive TRP channel. Cell. 2013; 152:806–17. https://doi.org/10.1016/j.cell.2013.01.020 [PubMed]
- 34. Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, Sabatini DM, Blackwell TK. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012; 15:713–24. https://doi.org/10.1016/j.cmet.2012.04.007 [PubMed]
- 35. Alam H, Williams TW, Dumas KJ, Guo C, Yoshina S, Mitani S, Hu PJ. EAK-7 controls development and life span by regulating nuclear DAF-16/FoxO activity. Cell Metab. 2010; 12:30–41. https://doi.org/10.1016/j.cmet.2010.05.004 [PubMed]
- 36. Li J, Ebata A, Dong Y, Rizki G, Iwata T, Lee SS. Caenorhabditis elegans HCF-1 functions in longevity maintenance as a DAF-16 regulator. PLoS Biol. 2008; 6:e233. https://doi.org/10.1371/journal.pbio.0060233 [PubMed]
- 37. Wolff S, Ma H, Burch D, Maciel GA, Hunter T, Dillin A. SMK-1, an essential regulator of DAF-16-mediated longevity. Cell. 2006; 124:1039–53. https://doi.org/10.1016/j.cell.2005.12.042 [PubMed]
- 38. Honda Y, Honda S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans.. FASEB J. 1999; 13:1385–93. [PubMed]
- 39. Xie M, Roy R. Increased levels of hydrogen peroxide induce a HIF-1-dependent modification of lipid metabolism in AMPK compromised C. elegans dauer larvae. Cell Metab. 2012; 16:322–35. https://doi.org/10.1016/j.cmet.2012.07.016 [PubMed]
- 40. Bellier A, Chen CS, Kao CY, Cinar HN, Aroian RV. Hypoxia and the hypoxic response pathway protect against pore-forming toxins in C. elegans.. PLoS Pathog. 2009; 5:e1000689. https://doi.org/10.1371/journal.ppat.1000689 [PubMed]
- 41. Taylor RC, Dillin A. XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell. 2013; 153:1435–47. https://doi.org/10.1016/j.cell.2013.05.042 [PubMed]
- 42. Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003; 300:1142–45. https://doi.org/10.1126/science.1083701 [PubMed]
- 43. Gelino S, Hansen M. Autophagy - An Emerging Anti-Aging Mechanism. J Clin Exp Pathol. 2012 (Suppl 4). [PubMed]
- 44. Libina N, Berman JR, Kenyon C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell. 2003; 115:489–502. https://doi.org/10.1016/S0092-8674(03)00889-4 [PubMed]
- 45. Hsin H, Kenyon C. Signals from the reproductive system regulate the lifespan of C. elegans.. Nature. 1999; 399:362–66. https://doi.org/10.1038/20694 [PubMed]
- 46. Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011; 144:79–91. https://doi.org/10.1016/j.cell.2010.12.016 [PubMed]
- 47. Shen C, Shao Z, Powell-Coffman JA. The Caenorhabditis elegans rhy-1 gene inhibits HIF-1 hypoxia-inducible factor activity in a negative feedback loop that does not include vhl-1. Genetics. 2006; 174:1205–14. https://doi.org/10.1534/genetics.106.063594 [PubMed]
- 48. McElwee JJ, Schuster E, Blanc E, Thomas JH, Gems D. Shared transcriptional signature in Caenorhabditis elegans Dauer larvae and long-lived daf-2 mutants implicates detoxification system in longevity assurance. J Biol Chem. 2004; 279:44533–43. https://doi.org/10.1074/jbc.M406207200 [PubMed]
- 49. Feddersen S, Neergaard TB, Knudsen J, Faergeman NJ. Transcriptional regulation of phospholipid biosynthesis is linked to fatty acid metabolism by an acyl-CoA-binding-protein-dependent mechanism in Saccharomyces cerevisiae.. Biochem J. 2007; 407:219–30. https://doi.org/10.1042/BJ20070315 [PubMed]
- 50. Cashikar AG, Duennwald M, Lindquist SL. A chaperone pathway in protein disaggregation. Hsp26 alters the nature of protein aggregates to facilitate reactivation by Hsp104. J Biol Chem. 2005; 280:23869–75. https://doi.org/10.1074/jbc.M502854200 [PubMed]
- 51. Hill SM, Hao X, Liu B, Nyström T. Life-span extension by a metacaspase in the yeast Saccharomyces cerevisiae.. Science. 2014; 344:1389–92. https://doi.org/10.1126/science.1252634 [PubMed]
- 52. Herbert AP, Riesen M, Bloxam L, Kosmidou E, Wareing BM, Johnson JR, Phelan MM, Pennington SR, Lian LY, Morgan A. NMR structure of Hsp12, a protein induced by and required for dietary restriction-induced lifespan extension in yeast. PLoS One. 2012; 7:e41975. https://doi.org/10.1371/journal.pone.0041975 [PubMed]
- 53. Xiao R, Chun L, Ronan EA, Friedman DI, Liu J, Xu XZ. RNAi Interrogation of Dietary Modulation of Development, Metabolism, Behavior, and Aging in C. elegans. Cell Reports. 2015; 11:1123–33. https://doi.org/10.1016/j.celrep.2015.04.024 [PubMed]
- 54. Maier W, Adilov B, Regenass M, Alcedo J. A neuromedin U receptor acts with the sensory system to modulate food type-dependent effects on C. elegans lifespan. PLoS Biol. 2010; 8:e1000376. https://doi.org/10.1371/journal.pbio.1000376 [PubMed]
- 55. Pang S, Curran SP. Adaptive capacity to bacterial diet modulates aging in C. elegans.. Cell Metab. 2014; 19:221–31. https://doi.org/10.1016/j.cmet.2013.12.005 [PubMed]
- 56. Soukas AA, Kane EA, Carr CE, Melo JA, Ruvkun G. Rictor/TORC2 regulates fat metabolism, feeding, growth, and life span in Caenorhabditis elegans.. Genes Dev. 2009; 23:496–511. https://doi.org/10.1101/gad.1775409 [PubMed]
- 57. Abergel R, Livshits L, Shaked M, Chatterjee AK, Gross E. Synergism between soluble guanylate cyclase signaling and neuropeptides extends lifespan in the nematode Caenorhabditis elegans.. Aging Cell. 2017; 16:401–13. https://doi.org/10.1111/acel.12569 [PubMed]
- 58. Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol. 2006; 70:1469–80. https://doi.org/10.1124/mol.106.027029 [PubMed]
- 59. Rohrbach S, Teichert S, Niemann B, Franke C, Katschinski DM. Caloric restriction counteracts age-dependent changes in prolyl-4-hydroxylase domain (PHD) 3 expression. Biogerontology. 2008; 9:169–76. https://doi.org/10.1007/s10522-008-9126-x [PubMed]
- 60. Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974; 77:71–94. [PubMed]
- 61. Sutphin GL, Kaeberlein M. Measuring Caenorhabditis elegans life span on solid media. J Vis Exp. 20091152. https://doi.org/10.3791/1152 [PubMed]
- 62. Yang JS, Nam HJ, Seo M, Han SK, Choi Y, Nam HG, Lee SJ, Kim S. OASIS: online application for the survival analysis of lifespan assays performed in aging research. PLoS One. 2011; 6:e23525. https://doi.org/10.1371/journal.pone.0023525 [PubMed]
- 63. Cohen E, Du D, Joyce D, Kapernick EA, Volovik Y, Kelly JW, Dillin A. Temporal requirements of insulin/IGF-1 signaling for proteotoxicity protection. Aging Cell. 2010; 9:126–34. https://doi.org/10.1111/j.1474-9726.2009.00541.x [PubMed]
- 64. Meléndez A, Tallóczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans.. Science. 2003; 301:1387–91. https://doi.org/10.1126/science.1087782 [PubMed]