



Introduction
Cellular senescence is a process that imposes permanent proliferative arrest in response to various stresses [1,2]. A wide array of evidence suggests that in vivo cellular senescence causes a loss of tissue stem/progenitor cells, and extracellular environment and cells surrounding senescent cells could be functionally disrupted by inflammatory cytokines, growth factors and proteases, which are secreted by senescent cells. In addition, senescent cells have been found at sites of age-related pathologies e.g. atherosclerosis and osteoarthritis [3]. Recent studies demonstrated that pharmacological or genetic eliminations of senescent cells from aging mice extend median lifespan, delayed tumorigenesis and attenuated progression of already established age-related disorders [4–10]. These findings strongly suggest that senescent cells play a key role in these pathological conditions and hence cellular senescence has been termed as the basic driver of the aging phenotype [1]. Despite these clear negative implications, one system on which the impacts of cellular senescence still remain unexplored is the circadian clock.
The circadian clock is driven by the circadian clock gene expressions with approximately a 24-hr rhythm. In mammals, a master clock resides in the hypothalamic suprachiasmatic nucleus (SCN) [11,12]. In contrast, peripheral clocks are distributed among most of the peripheral tissues and even in cultured cells. Both the master and peripheral clocks are controlled by various stimuli to adapt to environmental rhythms generated by the earth’s rotation [13–15]. Malfunction of circadian clock not only disrupts sleep/wake cycles, but also brings about many physiological abnormalities, leading to a wide variety of age-related diseases and premature aging in mice and humans [16–21]. One example is that Bmal1-/- mice accelerates premature aging accompanied with short lifespan [20]. On the other hand, several studies from model animals and humans have demonstrated that aging can also lead to alteration of the circadian clock [22,23]. Circadian free running periods and days required to re-entrain following new light-dark schedule have been shown to be altered by aging [24–28]. These reports clearly demonstrate that the mechanisms of circadian clock and aging mutually regulate each other at the animal levels.
Although evaluations of impact of aging on the circadian clock in vivo provide valuable information and exceedingly contribute to the progress of circadian biology, it is still unclear how the molecular mechanisms of aging affect the circadian clock and vice versa. In vitro evaluations could have advantages to address that, however, in contrast to various in vivo studies few researches at the cellular level have been performed. We have recently reported that primary fibroblast cells derived from Bmal1-/- mouse embryo, in which circadian clock is completely disrupted, do not display the acceleration of the process of in vitro cellular senescence, suggesting that cell-autonomous circadian clock is not implicated in the process of cellular senescence [29]. On the other hand, Kunieda et al. have demonstrated that circadian gene expressions display low amplitude in senescent primary cultured human aortic vascular smooth muscle cells [30]. However, they did not mention about the period length probably due to the sample collections with low time-resolution (4 hr). In this study, we utilize TIG-3 cells, normal primary human diploid fibroblast cells [31], and the real-time luciferase monitoring system with high time-resolution (10 min) [32,33] to address whether cellular senescence alters circadian clock properties. We show for the first time that senescent TIG-3 cells possess a longer circadian period with delayed peak-time compared to their proliferative counterparts. Moreover, we observe that senescent TIG-3 cells have defective entrainment mechanism in the circadian oscillator, presumably through altered PER1 and PER2 rapid inductions, causing the delayed peak-time. These findings demonstrate that senescent cells possess the altered circadian clock, at least in TIG-3 cells.
Results
Replicative senescence of TIG-3 cells
To evaluate the changes in the circadian clock during senescence, we chose replicative senescence, in which cell cycle would be irreversibly arrested due to the limitation of cell division. TIG-3 cells, which are normal primary diploid human fibroblast cells derived from fetal male lung [31], were serially passaged until they ceased proliferation, that is, reached replicative senescence. When we obtained TIG-3 cells, they had already been passaged 20 times. Therefore, we performed serial passaging starting from Passage 22 (P22). In order to assess at which passage cells begin to cease proliferation, individual and cumulative population doubling level (iPDL and cPDL, respectively) were recorded (Fig.1A and B). Here, we defined a cease of cell proliferation (CCP) as the point at which iPDL value dips below 1.0. Proliferation of TIG-3 cells were fairly constant up to P32, before which iPDLs were above 2. However, iPDLs started to decrease from P33 and TIG-3 cells reached CCP at P37 (Fig.1B).
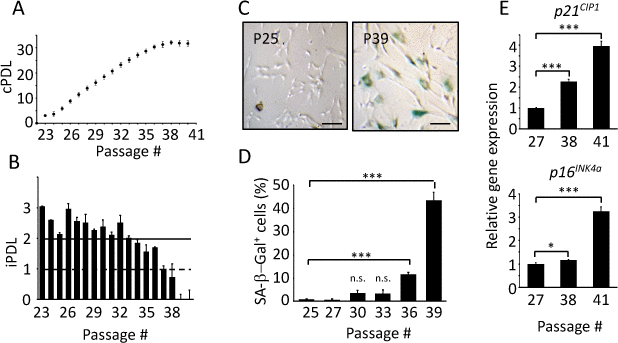
Figure 1. Replicative senescence of TIG-3 cells. Cumulative (A) and individual (B) population doubling level (PDL) of TIG-3 cells were measured. (C) Pictures of SA-β-Gal positive cells (blue) at passage25 (P25) and 39 (P39) were taken. Scale bars represent 100 μm. (D) The percentage of SA-β-Gal positive cells were quantified at different passages. (E) p21CIP1 (upper panel) and p16INK4a (bottom panel) genes expression at different passages were analyzed by qPCR. The lowest expression level was set to 1. Three independent proliferation assays were performed. n.s., not significant, *p<0.05, ***p<0.001, compared to each lowest passage for (D) and (E), by Student’s two-tailed t test.
Senescent cells have unique features, such as enlarged, flattened cell shape, increased senescence-associated β-galactosidase (SA-β-Gal) activity [34,35], and increased expressions of cell cycle inhibitors (p16INK4a, p19ARF and p21CIP1) [35–37]. We therefore investigated the aforementioned features to confirm that serially passaged TIG-3 cells indeed exhibit senescence features, and to use these features as the basis to group the cells into two categories: proliferative and senescent cells. Representative pictures of SA-β-Gal assay at P25 and P39 are shown in Fig.1C. Senescent cells, which were stained in blue, were observed at P39, in which 43.5% of cells were SA-β-Gal positive, but only 0.8% were positive at P25. Fig.1D represents the percentages of SA-β-Gal positive cells at several passages. SA-β-Gal positive cell number was dramatically increased from P36, at which iPDL was already below 2.0 (Fig.1B), suggesting that decrease in proliferation is due to replicative senescence. Moreover, we observed enlarged, flattened cells at P39 compared to the cells at P25 (Fig.1C), supporting that many cells have already turned senescent at P39. We next measured the expression levels of senescence marker genes, p21CIP1 and p16INK4a, which are known to initiate and maintain cellular senescence, respectively [38], at P27, P38 and P41. Expression levels of p21CIP1 increased with passages (Fig.1E upper panel). Expression levels of p16INK4a was high at P41, but not at P27 and P38 (Fig.1E bottom panel). These results indicated that P38 and P41 were at the senescence phase. All the results above indicate that the serially passaged TIG-3 cells indeed reached replicative senescence. Furthermore, we deduced that TIG-3 cells are in the proliferative phase until P32, followed by the transition phase from P33 to P35, and the senescence phase after P36. Hence, in the subsequent parts of this study, TIG-3 cells of P25-29 and of P36-41 were utilized as proliferative and senescent cells, respectively. We performed three independent proliferation assays and could reproduce similar results as shown in Fig.1.
Senescent TIG-3 cells show altered characteristics of circadian clock
To address the changes in the circadian clock during senescence, virus containing bmal1 promoter-driven luciferase gene [39] was infected to the proliferative and senescent TIG-3 cells obtained from three independent proliferation assays. The infected cells were synchronized with dexamethasone (Dex) and subjected to the real-time luciferase assay. Fig.2A shows the superimposition of relative oscillatory patterns of luciferase (See Fig. S1 for raw data of oscillation patterns). In order to exclude the possibility that the phase and/or luciferase intensity of the first peak are modified by acute effect by Dex treatment, we normalized oscillation patterns using the first trough value. The oscillation pattern in the senescent cells did not overlap with that in the proliferative cells and seemed to be delayed (Fig.2A). To investigate the trough-time difference that occurs between the proliferative and senescent cells, trough-times were extracted from each data. The 1st trough-time of the proliferative and senescent cells were 23.7 ± 0.1 hr and 26.1 ± 0.2 hr, respectively (Fig.2B). The 1st trough-time of the senescent cells was 2.4 ± 0.2 hr delayed with significant difference (Fig.2C). Differences of 2nd trough-time between the proliferative and senescent cells were 4.0 ± 0.2 hr (Fig.2C), indicating that trough-time differences have increased. This result suggests that the period length of circadian clock in the senescent cells is longer compared to that in the proliferative cells. To evaluate the period length of the proliferative and senescent cells, we analyzed the duration between two subsequent trough-times. The 1st period, which is the duration between 1st and 2nd trough-times, was 23.1 ± 0.1 hr in the proliferative cells. In contrast, the 1st period in the senescent cells was significantly longer than that in the proliferative cells (24.7 ± 0.1 hr; p-value, 5.35 x 10-14; Fig.2D). We also analyzed the peak-time differences and the period using 2nd and 3rd peak-times (Fig.S2). Consistent with trough-time differences, differences of 2nd and 3rd peak-time between the proliferative and senescent cells were 3.6 ± 0.2 and 4.0 ± 0.3 hr, respectively (Fig.S2). Again, the 2nd period, which is the duration between 2nd and 3rd peak-times, in the senescent cells also demonstrated a longer period compared to the proliferative cells (23.6 ± 0.1 hr vs 24.2 ± 0.2 hr; p-value, 0.03; Fig.S2).
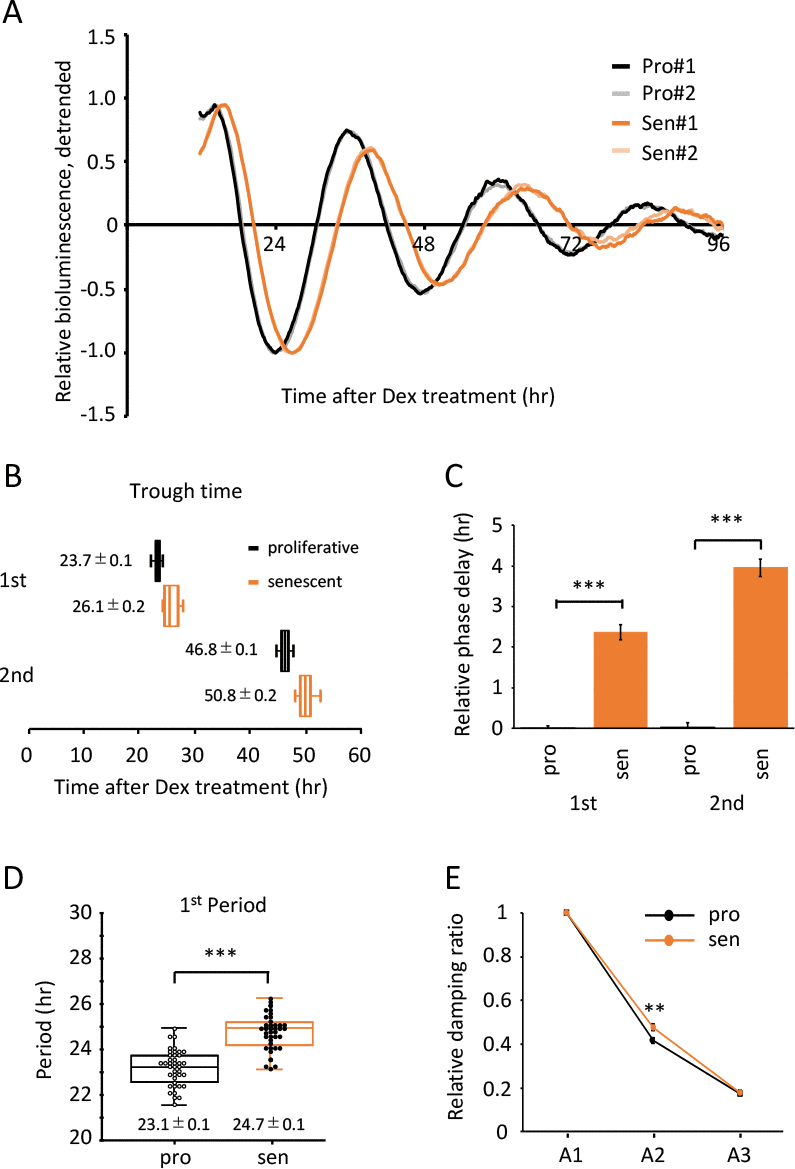
Figure 2. Alteration of circadian clock in the senescent cells was observed by a Dex-induced entrainment. (A) Relative oscillation patterns of luciferase in the proliferative and senescent cells were monitored by using a real-time luciferase monitoring system. Lowest intensity of each sample was set to -1. (B) Box-whisker plots of trough-times are displayed, n=41. Values are mean ± SEM. (C) Relative trough-time differences were measured, n=41. (D) Box-whisker plots of period lengths in the proliferative (pro) and senescent (sen) cells are displayed, n=41. Values are mean ± SEM. (E) Relative damping ratio in the proliferative (pro) and the senescent (sen) cells were analyzed. Data are mean ± SEM. See Fig.S3 for more information. *p<0.05, **p<0.01, ***p<0.001, compared to each of the proliferative cells by Student’s two-tailed t test.
Since the amplitude of circadian clock has been reported to decline with aging at transcriptional [30,40], neural activity [41] and locomotor activity levels [24–26,28], we proceeded to analyze the range of oscillations in TIG-3 cells. To measure the range of oscillation, the detrended luciferase intensity between top to nadir was determined (Fig.S3). Range of the 1st oscillation (A1) was set to 1 and the damping ratios of A2 and A3 were calculated. We found that, unlike previous reports at the tissue and animal levels, the damping ratios between the proliferative and senescent TIG-3 cells were comparable (Fig.2E).
To prove that period lengths and acrophase are different between the proliferative and senescent cells, we mathematically analyzed the data of Fig.2 using the Cosinor software, provided by Dr. R Refinetti [42]. Period lengths were 24.09 ± 0.20 hr and 25.47 ± 0.28 hr for the proliferative and senescent cells, respectively (p-value, 1.56 x 10-4; Table 1). The acrophases were -238.71 ± 1.23 degree and -272.54 ± 1.69 degree for the proliferative and senescent cells, respectively (p-value, 5.79 x 10-27; Table 1), indicating that the phase of senescent cells is significantly delayed compared to that of proliferative cells.
Table 1. Cosinor Analysis of Experiments in Figure 2.
| Period (hr) | acrophase (degree) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| proliferative | 24.09 +/- 0.20 | -238.71 +/- 1.23 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| senescent | 25.47 +/- 0.28 | -272.54 +/- 1.69 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| p value | 1.56 x 10-4 | 5.79 x 10-27 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Student’s two-tailed t test were performed, n=41. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In summary, we revealed that the Dex-synchronized senescent TIG-3 cells display a longer period length and the delay of peak-time compared with the proliferative cells, suggesting that senescent cells possess the altered circadian clock.
Input pathways are comparable between the proliferative and senescent cells
Circadian clock consists of three components; an oscillator with a period of 24 hr, input pathways to the oscillator to allow entrainment of the clock, and output pathways to regulate circadian biochemical, physiological and behavioral rhythms [43,44]. In order to evaluate whether a longer circadian period and delayed peak-time in the senescent TIG-3 cells is the result of the altered oscillator or input pathways, we compared the circadian oscillation patterns of luciferase synchronized by Dex and Forskolin (Fsk). Forskolin is another well-known clock-entrainment factor [14,45,46], activates cAMP-CREB signaling pathway and transcribes per1 gene via the cAMP response elements (CRE) in per1 promoter [47]. In contrast, Dex binds to and activates glucocorticoid receptor and transcribes per1 and per2 genes via the glucocorticoid response element (GRE) in per1 and per2 promoters [48,49]. In the proliferative cells, real-time luciferase monitoring assays demonstrated that circadian oscillation patterns of luciferase activity synchronized by Dex or Fsk show almost similar phase with the same intensity (Fig.3A left panel and Fig.S4). However, consistent with the previous report [46], there was a significant peak-time difference triggered by these two treatments. The second peak of Fsk-treated proliferative cells appeared 2.07 hr earlier than that of Dex-treated proliferative cells (p-value, 0.02; Fig.3B). In the senescent cells, the peak-time of the Fsk-treated cells was also significantly earlier compared to the Dex-treated cells (the difference was 1.67 hr; p-value, 0.03; Fig.3B). Importantly, the peak-time difference between the proliferative and senescent cells were similar (Fig.3B). Although the first peak and the amplitude of Fsk-treated senescent cells was small and weak, respectively, the above result suggests that both input pathways triggered by Dex or Fsk might not be functionally impaired in senescent cells. Rather, the oscillator of circadian clock was altered with senescence.
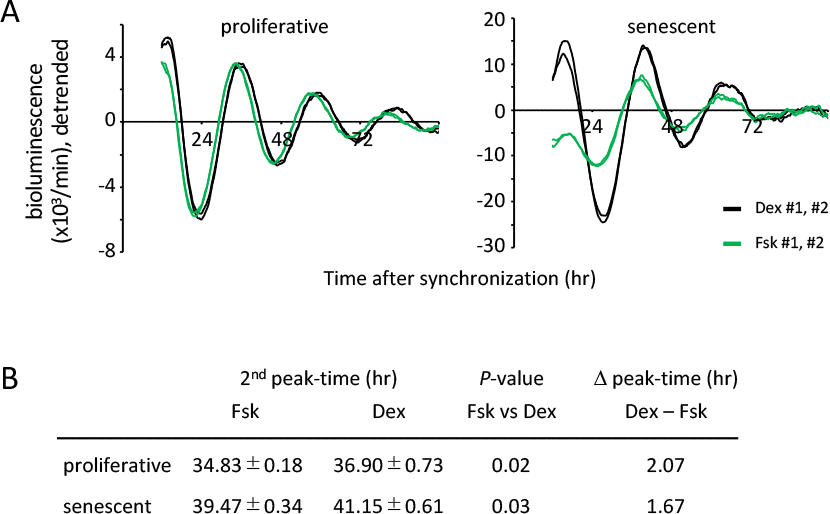
Figure 3. Input pathways to the oscillator of circadian clocks are comparable between the proliferative and senescent cells. (A) Oscillation patterns of luciferase synchronized by dexamethasone (Dex) or forskolin (Fsk) were monitored by using a real-time luciferase monitoring system. Three independent experiments were performed. (B) Summary of peak-times and the difference of peak-time in the proliferative and senescent cells shown in (A). Statistical analyses were performed by Student’s two-tailed t test, n=8.
Infection efficiency does not affect the oscillation patterns of luciferase
It is also interesting to note that the luciferase activity was higher in the senescent cells than in the proliferative cells (Fig.3A, S1 and S4), giving rise to the possibility that BMAL1 gene expression is upregulated in the senescent cells, because the luciferase gene used in this study was driven by the mouse bmal1 promoter [39]. To confirm this possibility, we performed qPCR to compare endogenous BMAL1 transcripts in the non-entrained proliferative and senescent TIG-3 cells. The transcript level of BMAL1 in the senescent cells was, however, similar or slightly downregulated compared to that in the proliferative cells (Fig.4A). Moreover, circadian expression profile of BMAL1 in the senescent cells after Dex-entrainment were also similar or lower than those in the proliferative cells (Fig.7D). These results indicate that the high luciferase activity in the senescent cells might not be due to the upregulation of luciferase transcription, suggesting that infection efficiency is high in the senescent TIG-3 cells. Therefore, to rule out the possibility that the infection efficiency affects luciferase oscillation patterns, cells were then infected with serially diluted viruses. As expected, luciferase intensities were decreased in a virus-dose dependent manner in both the proliferative and senescent cells (Fig.S5). However, the relative oscillation patterns completely overlapped within the same passages (Fig.4B), demonstrating that the infection efficiency has no effect on the oscillation patterns of luciferase.
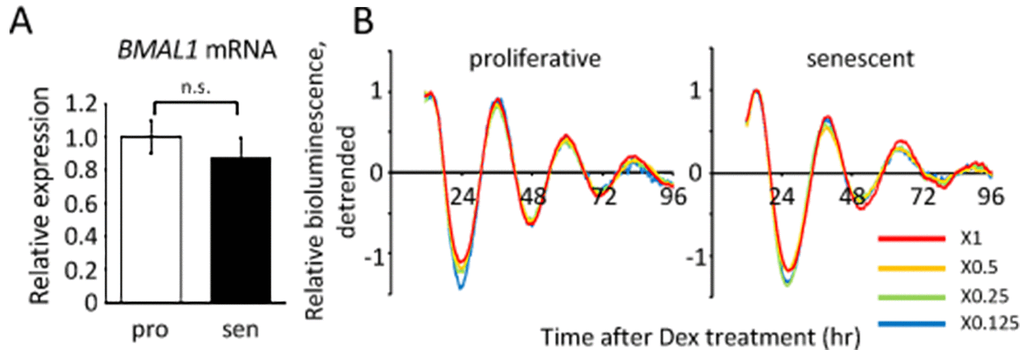
Figure 4. Infection efficiency does not affect the oscillation patterns of luciferase. (A) BMAL1 mRNA levels in the proliferative (pro) and senescent (sen) TIG-3 cells were quantified by qPCR. Each sample was normalized by 18S rRNA. p = 0.47; n.s., not significant; by Student’s two-tailed t test; n=3. Three independent experiments were performed. (B) Relative oscillation patterns of luciferase in serially diluted virus-infected TIG-3 cells are shown. Highest intensity of each sample was set to 1. Two independent experiments were performed.
Half-life of luciferase activity is comparable between the proliferative and senescent cells
Various cellular metabolisms/homeostasis are altered in senescent cells [50], for example, energy production in senescent fibroblast cells shifts to glycolysis with little to no change in oxidative phosphorylation [51]; while protein homeostasis through both ubiquitin proteasome pathway and autophagy is also impaired [2,52,53]. These findings raised the possibility that altered metabolisms/homeostasis in the senescent cells might have effects on luciferase activity, which is independent from the circadian clock. To address this possibility, the half-life of luciferase activity in the proliferative and senescent cells were monitored by performing a real-time luciferase monitoring assay (Fig.5A). Similar to the previous report [54], half-life of luciferase activity in the proliferative cells was 2.30 ± 0.10 hr, which was comparable to that in the senescent cells, 2.54 ± 0.08 hr (Fig.5B). This result showed that the senescent TIG-3 cells have no effect of altered metabolisms/homeostasis on the half-life of luciferase activity, demonstrating that circadian clock-independent, senescence-induced unknown metabolic alterations would not affect the circadian oscillation patterns monitored by luciferase intensity.
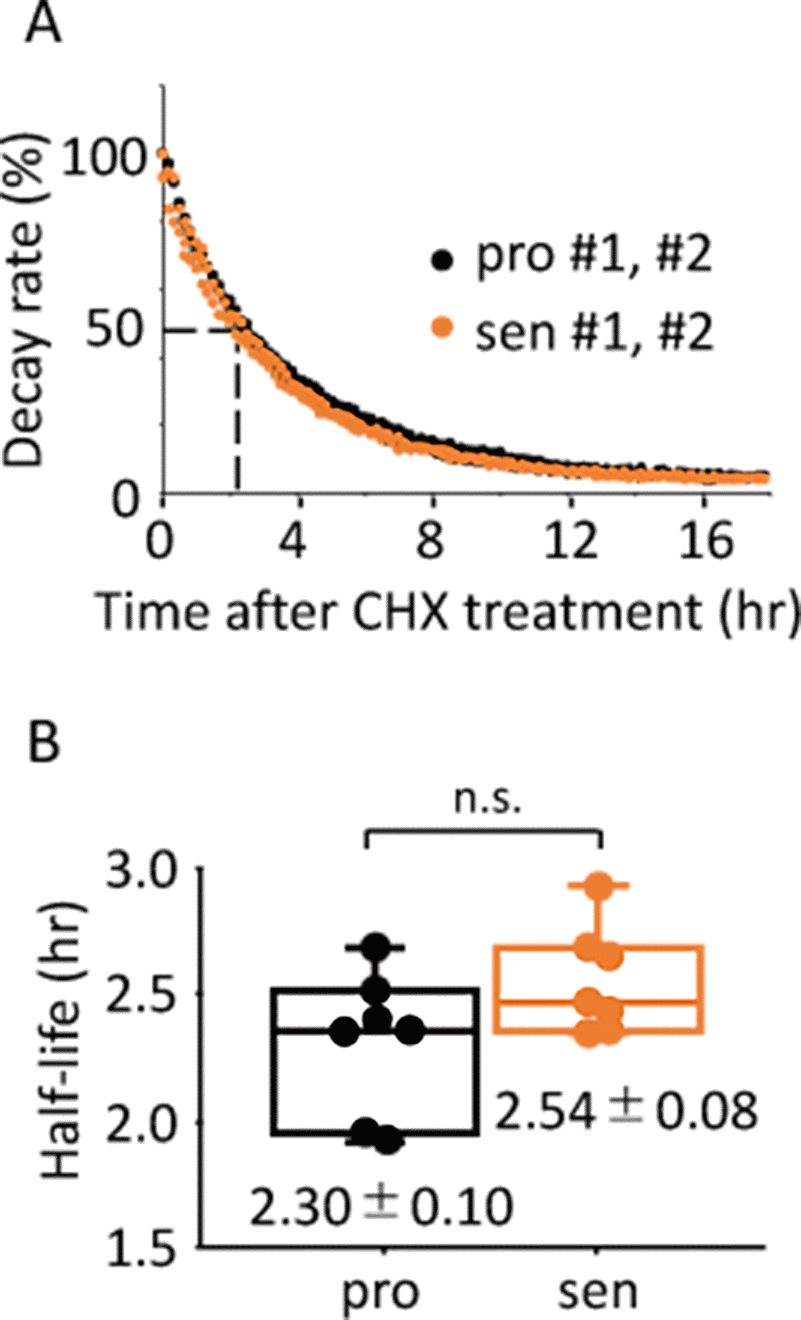
Figure 5. Half-life of luciferase activity is comparable between the proliferative and senescent cells. (A) Representative results of activity decay of luciferase in the proliferative (pro) and senescent (sen) cells are shown. Two independent experiments were performed. (B) Half-life of luciferase activity in the proliferative and senescent cells were quantified. Values are mean ± SEM. p = 0.09; n.s., not significant; n=7, by Student’s two-tailed t test.
Taking all results in Figs.3-5, we concluded that a longer circadian period and delayed peak-time elicited by Dex are due to the altered oscillator of the circadian clock in senescent cells.
Alteration to features of circadian clock in the senescent cells is a general hallmark of senescence of the circadian clock
To elucidate whether the findings shown in Fig.2 are Dex-synchronization specific or can be generalized, we further analyzed the trough-time difference and the period of Fsk-synchronized TIG-3 cells. Superimposed relative oscillation patterns of luciferase demonstrated that the oscillation patterns in the senescent cells seemed to be delayed compared to the proliferative cells (Fig.6A, B and S1), similar to the results synchronized by Dex (Fig.2A and B). Statistical analyses revealed that the 1st and 2nd trough-times of senescent cells were significantly delayed (Fig.6C). In addition, the 1st period length, the duration between 1st and 2nd trough-times, was longer in the senescent cells (23.7 ± 0.3 hr vs 25.8 ± 0.3 hr; p-value, 1.50 x 10-4; Fig.6D). We also analyzed the peak-time differences and the period using 2nd and 3rd peak-times (Fig.S6). Consistent with trough-time differences, differences of 2nd and 3rd peak-time between the proliferative and senescent cells were 4.7 ± 0.3 and 7.1 ± 1.0 hr, respectively (Fig.S6B). Again, the 2nd period, which is the duration between 2nd and 3rd peak-times, in the senescent cells also demonstrated a longer period compared to the proliferative cells (23.5 ± 0.4 hr vs 25.6 ± 0.8 hr; p-value, 0.01; Fig.S6C). Finally, cosiner analyses revealed that the period length in the senescent cells is longer than that in the proliferative cells (23.79 ± 0.05 hr vs 26.91 ± 0.27 hr; p-value, 1.52 x 10-8; Table 2), and the acrophase in the senescent cells is delayed compared to that in the proliferative cells (-227.13 ± 2.37 degree and -257.50 ± 3.12 degree; p-value, 1.99 x 10-6; Table 2). Consistent with the result of Dex-induced entrainment, we again found that the damping ratios between the proliferative and senescent TIG-3 cells were comparable (Fig.6E).
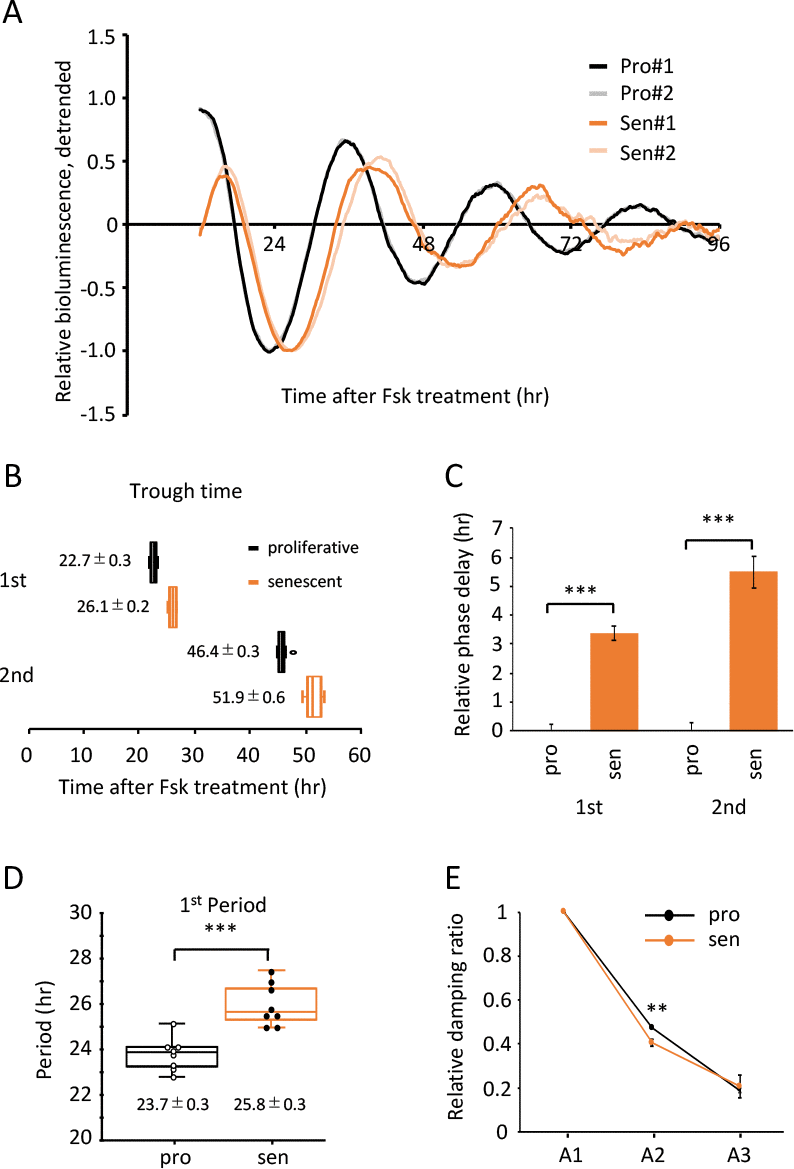
Figure 6. Alteration of circadian clock in the senescent cells was also observed by a Fsk-induced entrainment. (A) Relative oscillation patterns of luciferase in the proliferative (pro) and senescent (sen) cells entrained by Fsk were monitored by using a real-time luciferase monitoring system. Lowest intensity of each sample was set to -1. (B) Box-whisker plots of trough-times are displayed, n=8. Values are mean ± SEM. (C) Relative trough-time differences were measured, n=8. (D) Box-whisker plots of period lengths in the proliferative (pro) and senescent (sen) cells are displayed, n=8. Values are mean ± SEM. (E) Relative damping ratio in the proliferative (pro) and the senescent (sen) cells are analyzed. Data are mean ± SEM. **p<0.01, ***p<0.001, compared to each of the proliferative cells by Student’s two-tailed t test.
Table 2. Cosinor Analysis of Experiments in Figure 6.
| Period (hr) | acrophase (degree) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| proliferative | 23.79 +/- 0.05 | -227.13 +/- 2.37 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| senescent | 26.91 +/- 0.27 | -257.50 +/- 3.12 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| p value | 1.52 x 10-8 | 1.99 x 10-6 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Student’s two-tailed t test were performed, n=8. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Taking all results together, we propose that the alteration to features of circadian clock in the senescent cells is not a Dex-synchronization specific phenomenon, but rather the general hallmark of senescence of the oscillator of circadian clock.
Acute response and circadian expression profiles of endogenous clock genes are attenuated in the senescent cells
Finally, we addressed expression profiles of endogenous circadian clock genes in TIG-3 cells. We first investigated the acute response of clock genes by Dex treatment. Although PER1 and PER2 are known to possess glucocorticoid response elements (GRE) in their promoters [48,49], their acute responses to Dex treatment in the senescent cells were different from those in the proliferative cells. Acute response of PER1 was delayed, the peak-time was 3 hr after Dex treatment in the senescent cells, although the peak-time was approximately 2.5 hr in the proliferative cells (Fig. 7A). In contrast, acute response of PER2 was severely weakened in the senescent cells (Fig.7B). Distinct responses of PER1 and PER2 were also observed by Fsk treatment (Fig.S7). Interestingly, acute responses of other clock genes, which have not been reported to possess GRE on their promoters, were grouped into two categories; one was BMAL1 and CRY2, which showed similar expression levels and responses between the proliferative and the senescent cells (Fig.S8), the other was CRY1 and REV-ERBA, which showed similar responses with different expression levels (Fig.S8). We further monitored circadian expression profiles of the endogenous clock genes in the proliferative and senescent cells. Expression levels of PER2, CRY1 and REV-ERBA mRNAs in the senescent cells decreased to less than half compared to the proliferative cells throughout the time course, and amplitudes of these genes were attenuated (Fig.7C and Fig.S9). On the other hand, expression levels and oscillation patterns of BMAL1, PER1 and CRY2 were similar or moderately weakened in the senescent cells (Fig.7D and Fig.S9). In summary, we revealed that in the senescent cells the acute response of PER1 and PER2 are altered, which may correspond to the following delayed-phase, and PER2, CRY1 and REV-ERBA demonstrate attenuated expressions and oscillation, which may give rise to the extension of circadian period in the senescent cells.
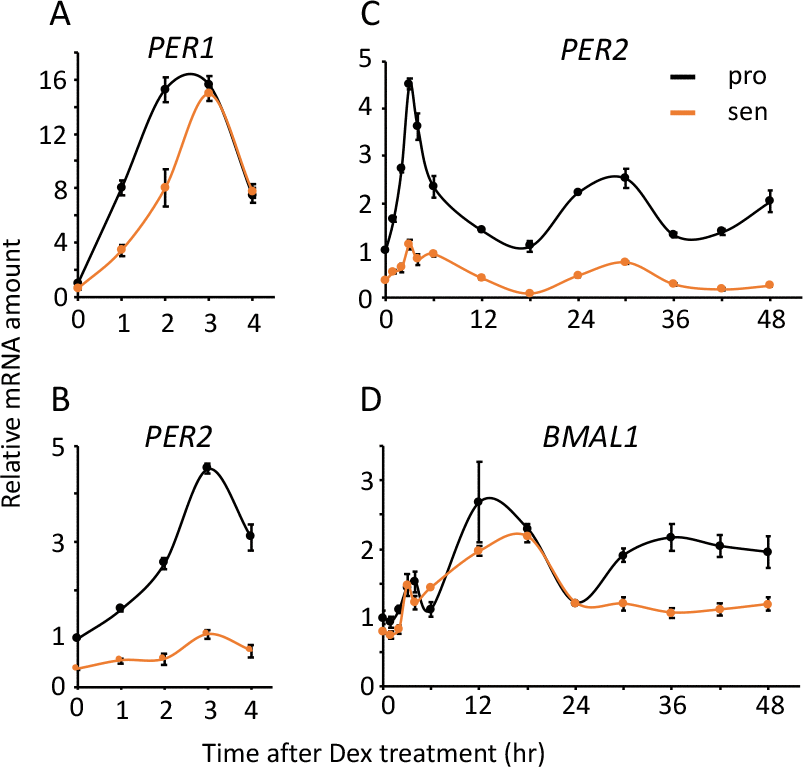
Figure 7. Acute and circadian profiles of endogenous circadian genes were altered in the senescent cells. Acute responses of PER1 (A) and PER2 (B), and circadian profiles of PER2 (C) and BMAL1 (D) induced by Dex were quantified. Each sample was normalized by 18S rRNA. Time 0 of proliferative cells were set to 1 for each gene.
Discussion
Aging has been identified as one of the risk factors linked to the alteration of circadian physiology and behavior [24–28]. On the other hand, disruption of circadian clock has been recognized to bring about many physiological abnormalities, leading to a wide variety of age-related diseases and premature aging [16,17]. These pieces of information demonstrate that the mechanisms of aging and circadian clock are intimately regulated by each other at the animal level. However, little is known about their mutual regulations at the cellular level. Intriguingly, we have recently reported that the primary fibroblast cells derived from Bmal1-/- mouse embryo, in which circadian clock is completely disrupted, do not demonstrate the acceleration of replicative and stress-induced senescence [29], suggesting that circadian clock does not control cell-autonomous cellular senescence. Therefore, the current study addressed the reverse question, i.e. whether cellular senescence could affect the circadian clock, and provided evidence for the first time that senescent cells demonstrate a prolonged period and delayed peak-time (Figs. 2 and 6), which was successfully achieved due to the performance of high time-resolution (10 min) real-time luciferase monitoring system. Importantly, these findings were observed by two different entrainment factors, Dex and Fsk, and senescence had no effect on input pathways to the oscillator (Fig.3). Our current study, therefore, indicate that the altered circadian clock with a prolonged period and delayed peak-time is the general hallmark of senescence of the “oscillator” of circadian clock. Furthermore, we uncovered at the cellular level that aging could affect the circadian clock, but not vice versa.
In the present study, luciferase intensity in the senescent cells was much higher than that in the proliferative cells (Fig.3A, S1 and S4). Since endogenous BMAL1 mRNA levels, promoter of which drives luciferase in this study, were comparable between the proliferative and senescent cells (Fig.4A and 7D), the high luciferase intensity could be due to high infection efficiency in the senescent cells. We used VSV-G pseudotype lentivirus in this study, and cellular receptor for the envelope protein VSV-G has been known to be membrane lipids [55]. Some literature has reported that certain lipid families accumulate during replicative senescence [56,57], suggesting that the accumulation of lipids may contribute to the high susceptibility in the senescent TIG-3 cells. Importantly, we showed that infection efficiency has no effect on the oscillation pattern of luciferase (Fig.4B).
Nakamura et al. performed well-designed ex vivo SCN tissue study [40]. In that paper, they demonstrated that i) peak phase was delayed in the aged SCN, ii) phase differences gradually increased, and iii) period length in the aged SCN was longer than that in young SCN; all of which are consistent with our current study. In addition, it has been reported that aged animals display more variability in the time of activity onset, and require more days to be re-entrained following new light-dark schedule than young animals [26–28], which are similar to the current results in that the variability in trough-time was wider and the trough-time was delayed in the synchronized senescent cells (Fig.2B-C and 6B-C). Our novel findings at the cellular level are consistent with the phenotypes at the tissue and animal levels, indicating that the alternation of circadian clock at tissue and animal levels is caused by the alterations of the oscillator of circadian clock in the cells, although we cannot deny the possibility that aged animals may have altered cell to cell and/or organ to organ communications for the circadian clock.
It is well known that photic stimulation induces rapid expressions of per1 and per2 in the SCN to give rise to the light-induced entrainment [58,59]. Light-induced rapid inductions of per1 and per2 in the SCN are also known to be attenuated with aging [60,61]. Consistent with these reports, we demonstrated in the current study that PER1 and PER2 rapid induction by Dex-induced entrainment is attenuated in the senescent TIG-3 cells. However, their profiles are not same; PER1 induction is delayed, but its expression level is comparable to that in the proliferative cells, on the other hand, PER2 induction is almost completely repressed. This difference suggests that gene regulations of PER1 and PER2 are distinct. Epigenetic changes such as DNA methylation, which has been proposed to play a central role in senescence and aging [2], is one candidate regulation. Furthermore, our finding suggests that not only acute response of PER2, but circadian expression profiles of PER2, CRY1 and REV-ERBA are also epigenetically modified. Further investigations will reveal molecular mechanisms of how the circadian gene expressions are regulated in the senescent cells.
Understanding circadian biology of aging in humans is a pressing challenge for the global aging society. Two groups have developed virus-based circadian reporter and succeeded to characterize circadian rhythms in human skin biopsy or human hair follicle tissue [62,63] for investigating the impact of aging on circadian clock, instead of using cultured primary cells such as the current study. Both reports demonstrated that the circadian period lengths have no difference between young and elder subjects. This discrepancy in their and our results is probably due to reasons as follows; samples from human subjects have the inter-individual differences of circadian period length, which were much more variable than different samples of the same individual. Moreover, samples from human elder subjects may have more inter-individual variations of the ratio of senescent and non-senescent cells than those from our current study. Although their assay systems reflect more physiological circadian characteristics of aging, our assay system can control the genetic/environmental backgrounds and the ratio of senescent cells. We believe that our current study demonstrated the possibility of primary cells for studying in vitro circadian biology of aging. Further investigations using both primary cells and biopsy samples will be needed to assess in-depth mechanisms of aging in circadian clock.
In conclusion, in our current study at the cellular level, we revealed for the first time that replicative senescent cells demonstrate the delayed peak-time and prolonged period compared to the proliferative cells, uncovering that the circadian clock is altered with the establishment of cellular senescence. These findings are consistent with the prior studies at tissue and animal levels. Although, in this study, we have not demonstrated any molecular mechanisms of how aging alters the circadian clock, which is still largely unknown, we propose that investigation at the cellular level is a powerful and useful approach to dissect the molecular mechanisms of how aging alters the circadian clock. Further investigations using primary cells could shed light on the molecular mechanisms of aging in circadian clock, and perhaps help to promote healthy aging and longevity in the elderly.
Materials and Methods
Cell culture
TIG-3 cells, which were kindly provided by Drs. T. Takumi and T. Akagi, were grown in Dulbecco's Modified Eagle Medium (4.5 g/L glucose) (Nacalai Tesque, Kyoto, Japan) supplemented with 10% fetal bovine serum (biowest, Nuaillé, France) and 10 U/ml penicillin/10 µg/ml streptomycin (Nacalai Tesque, Kyoto, Japan) at 37 °C and 5% CO2.
Cell growth assay
TIG-3 cells were cultured in triplicates in 10 cm dishes at seeding density = 5x105 cells/10 cm dish. Upon reaching semi-confluence; every 3 days till passage 34 and 4 days or more from passage 35, the cells were trypsinized, counted and seeded again at 5x105 cells/10cm dish. This process was repeated until the cells stopped proliferation and three independent assays were performed. To calculate the Population Doubling Level (PDL), the following formula was used: PDL=3.32log(Nharvest/Nseeding), where Nseeding is the seeding cell number and Nharvest is the cell number at harvest when cells reach semi-confluence. Cumulative PDL (cPDL) of a particular passage number is the accumulation of all the previous PDL values, including that of the passage being measured. In this study cells are defined as senescent when the PDL value of the cells is below 1.
Senescence associated β-galactosidase (SA-β-Gal) activity
Cells were fixed with 2% formaldehyde and 0.2% glutaraldehyde in H2O for 5 min and incubated at 37 °C without CO2 for 16 hr in X-Gal staining solution (40 mM sodium citrate (pH 6.0), 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 1 mg/ml 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal), 150 mM sodium chloride, and 2 mM magnesium chloride). 200 cells were counted under a bright-field microscope for each sample.
RNA extraction and qPCR
Total RNA from the TIG-3 cells were extracted using Sepasol RNA-I Super G (Nacalai Tesque, Kyoto, Japan). Total RNA was reverse-transcribed by using SuperScriptII Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) with random primers in accordance with the manufacturer’s instructions. Quantitative PCR was performed in the presence of KAPA SYBR FAST Universal 2X qPCR Master Mix (Nippon Genetics, Tokyo, Japan) on the Light Cycler 480 (Roche, Basel, Switzerland) under the following conditions: 95 °C for 3 min, and 40 cycles at 95 °C for 10 sec, 60 °C for 20 sec and 72 °C for 1 sec. The sequences of the forward and reverse primers are as follows. p16INK4a FW; ACC AGA GGC AGT AAC CAT GC, p16INK4a RV; GGA CCT TCG GTG ACT GAT GA. p21CIP FW; AGC ATG ACA GAT TTC TAC CAC TCC, p21CIP RV; GCA GAA GAT GTA GAG CGG GC. PER1 FW; GAG AGC AGC AAG AGC ACA AA, PER1 RV; AGC TCT CGA AGT GCT GTC AT. PER2 FW;TCC AGT GGA CAT GAG ACC AA, PER2 RV; CGC TAC TGC AGC CAC TTG TA. CRY1 FW; CAG CAG TGG AAG TTG CTC TC, CRY1 RV; CTA GGA CGT TTC CCA CCA CT. CRY2 FW; GGG AGG AGA GAC AGA AGC TC, CRY2 RV; GAG GGA GTT GGC GTT CAT TC. REV-ERBA FW; GAC ATG ACG ACC CTG GAC TC, REV-ERBA RV; GCT GCC ATT GGA GTT GTC AC. BMAL1 FW; CAT GCA ACG CAA TGT CCA G, BMAL1 RV; GTG TAT GGA TTG GTG GCA CCT. The primer set of 18S rRNA was described preciously [35].
Lentivirus production and infection to TIG-3 cells
HEK293T cells were seeded at 4x106 cells in 10 cm dish. After 24 hr of seeding, when the cells reached 80-90% confluency, transfection was performed as follows: two 1.5 mL tubes were taken and 200 μL of Opti-MEM (WAKO, Osaka, Japan) was put into each tube. Into one tube, 9 μg of psPAX2, 6 μg of pMD2.G and 30 μL of 1 mg/ml polyethylenimine (PEI) (Polysciences, Warrington, PA, USA) were added. Into the other tube, 14 μg of pLV6-Bmal1-luc and 28 μL of PEI were added. Then the components of each tube were mixed by repeated inversion and then incubated at room temperature for 15 minutes. The contents of each tube were then added to the HEK293T cell dish. After 24 hr, the medium was removed from the cells, washed with PBS once and 5 mL of fresh medium was added. 24 hr later, the viral supernatant was collected and stored in 4°C, and 5 mL of fresh medium was added to the dish again. After 24 hr the viral supernatant was collected and pooled with the viral supernatant of the previous day. The total volume of viral supernatant was centrifuged at 167.7 x g at room temperature for 5 min to get rid of any cellular debris. The supernatant was further filtered through a 0.2 μm filter to remove any remaining debris of HEK293T cells. The filtered viral suspension was then centrifuged at 8,000 x g at 4°C for 4 hr to concentrate the lentivirus particles. After 4 hr, the supernatant was removed, and the lentivirus pellet was resuspended in 2 mL of medium to obtain the viral suspension.
For infection of the target TIG-3 cells, which had been seeded on the previous day at 1.5x105 cells/35 mm dish, the culture medium was replaced with the viral suspension supplemented with protamine sulfate. 24 hr later the cells were washed with PBS once and 2 mL of fresh medium was added. The cells were then cultured for an additional 48 hr.
Synchronization of circadian clock in TIG-3 cells for Real-time luciferase monitoring assay and qPCR
48 hr after medium change of the infected TIG-3 cells, the medium was replaced with fresh 2 mL medium containing 0.1 μM dexamethasone (Dex; Nacalai Tesque, Japan) or 20 μM forskolin (Fsk; Nacalai Tesque, Japan), and incubated for 1 hr at 37°C 5% CO2 for the entrainment of the circadian clock. Afterwards, the medium was removed, cells washed with PBS twice and medium containing 100 μM D-luciferin (Nacalai Tesque, Kyoto, Japan) was added. The cells were placed in the KronosDio luminometer (ATTO, Tokyo, Japan) and bioluminescence of the cells were measured for 5 days continuously. After 5 days, raw data were collected and in order to observe the circadian gene expression pattern more precisely, detrended data were also retrieved from the KronosDio software.
For the evaluation of endogenous circadian expression profiles, TIG-3 cells at confluent conditions were treated with 0.1 μM dexamethasone for 1 hr. Then the medium was replaced with normal culture medium and cells were cultured until indicated time points. RNA extraction, cDNA synthesis and qPCR were described above.
Cosinor analysis
To perform Cosinor analyses, data of three-day detrended samples from real-time luciferase monitoring assay were used. Cosinor software was kindly provided by Dr. R Refinetti [42].
Half-life of luciferase activity
Bmal1-luc-infected TIG-3 cells were treated with 50 μg/ml cycloheximide, then cells were set to the real-time luciferase monitoring system. Half-life of luciferase activity was evaluated at the time when decay rate became 50%.
Statistics
Values are reported as mean ± SEM. Statistical differences were determined by a Student's two-tailed t test. Statistical significance is displayed as * (p < 0.05), ** (p < 0.01), or *** (p < 0.001).
Supplementary Materials
Acknowledgments
We thank Drs. T. Takumi and T. Akagi for providing TIG-3 cells and Dr. SA Brown for providing pLV6-Bmal-luc, pMD2.G and psPAX2 vectors. We also thank Dr. FD Khaidizar for his critical comments on the manuscript.
Conflicts of Interest
The authors declare that they have no conflicts of interest with the contents of this article.
Funding
This work was supported by JSPS KAKENHI Grant Numbers JP26830074 (Y.N.) and JP17K08569 (Y.N.). This work was also supported, in part, by Takeda Science Foundation, Mochida Memorial Foundation for Medical and Pharmaceutical Research and Suzuken Memorial Foundation.
References
- 1. Bhatia-Dey N, Kanherkar RR, Stair SE, Makarev EO, Csoka AB. Cellular senescence as the causal nexus of aging. Front Genet. 2016; 7:13. https://doi.org/10.3389/fgene.2016.00013 [PubMed]
- 2. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013; 153:1194–217. https://doi.org/10.1016/j.cell.2013.05.039 [PubMed]
- 3. Childs BG, Gluscevic M, Baker DJ, Laberge RM, Marquess D, Dananberg J, van Deursen JM. Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov. 2017; 16:718–35. https://doi.org/10.1038/nrd.2017.116 [PubMed]
- 4. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, Khazaie K, Miller JD, van Deursen JM. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016; 530:184–89. https://doi.org/10.1038/nature16932 [PubMed]
- 5. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011; 479:232–36. https://doi.org/10.1038/nature10600 [PubMed]
- 6. Hashimoto M, Asai A, Kawagishi H, Mikawa R, Iwashita Y, Kanayama K, Sugimoto K, Sato T, Maruyama M, Sugimoto M. Elimination of p19ARF-expressing cells enhances pulmonary function in mice. JCI Insight. 2016; 1:e87732. https://doi.org/10.1172/jci.insight.87732 [PubMed]
- 7. Baar MP, Brandt RM, Putavet DA, Klein JD, Derks KW, Bourgeois BR, Stryeck S, Rijksen Y, van Willigenburg H, Feijtel DA, van der Pluijm I, Essers J, van Cappellen WA, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell. 2017; 169:132–147.e16. https://doi.org/10.1016/j.cell.2017.02.031 [PubMed]
- 8. Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, Luo Y, Wang X, Aykin-Burns N, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016; 22:78–83. https://doi.org/10.1038/nm.4010 [PubMed]
- 9. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 2017; 8:14532. https://doi.org/10.1038/ncomms14532 [PubMed]
- 10. Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, Vadai E, Dassa L, Shahar E, Condiotti R, Ben-Porath I, Krizhanovsky V. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun. 2016; 7:11190. https://doi.org/10.1038/ncomms11190 [PubMed]
- 11. Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science. 2010; 330:1349–54. https://doi.org/10.1126/science.1195027 [PubMed]
- 12. Patton AP, Hastings MH. The suprachiasmatic nucleus. Curr Biol. 2018; 28:R816–22. https://doi.org/10.1016/j.cub.2018.06.052 [PubMed]
- 13. Balsalobre A, Damiola F, Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell. 1998; 93:929–37. https://doi.org/10.1016/S0092-8674(00)81199-X [PubMed]
- 14. Balsalobre A, Brown SA, Marcacci L, Tronche F, Kellendonk C, Reichardt HM, Schütz G, Schibler U. Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science. 2000; 289:2344–47. https://doi.org/10.1126/science.289.5488.2344 [PubMed]
- 15. Asher G, Sassone-Corsi P. Time for food: the intimate interplay between nutrition, metabolism, and the circadian clock. Cell. 2015; 161:84–92. https://doi.org/10.1016/j.cell.2015.03.015 [PubMed]
- 16. Nakahata Y, Bessho Y. The circadian NAD+ metabolism: impact on chromatin remodeling and aging. BioMed Res Int. 2016; 2016:3208429. https://doi.org/10.1155/2016/3208429 [PubMed]
- 17. Sahar S, Sassone-Corsi P. Regulation of metabolism: the circadian clock dictates the time. Trends Endocrinol Metab. 2012; 23:1–8. https://doi.org/10.1016/j.tem.2011.10.005 [PubMed]
- 18. Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, Laposky A, Losee-Olson S, Easton A, Jensen DR, Eckel RH, Takahashi JS, Bass J. Obesity and metabolic syndrome in circadian Clock mutant mice. Science. 2005; 308:1043–45. https://doi.org/10.1126/science.1108750 [PubMed]
- 19. Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, Ivanova G, Omura C, Mo S, Vitaterna MH, Lopez JP, Philipson LH, Bradfield CA, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 2010; 466:627–31. https://doi.org/10.1038/nature09253 [PubMed]
- 20. Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 2006; 20:1868–73. https://doi.org/10.1101/gad.1432206 [PubMed]
- 21. Roenneberg T, Merrow M. The circadian clock and human health. Curr Biol. 2016; 26:R432–43. https://doi.org/10.1016/j.cub.2016.04.011 [PubMed]
- 22. Mattis J, Sehgal A. Circadian rhythms, sleep, and disorders of aging. Trends Endocrinol Metab. 2016; 27:192–203. https://doi.org/10.1016/j.tem.2016.02.003 [PubMed]
- 23. Van Someren EJ. Circadian rhythms and sleep in human aging. Chronobiol Int. 2000; 17:233–43. https://doi.org/10.1081/CBI-100101046 [PubMed]
- 24. Pittendrigh CS, Daan S. Circadian oscillations in rodents: a systematic increase of their frequency with age. Science. 1974; 186:548–50. https://doi.org/10.1126/science.186.4163.548 [PubMed]
- 25. Witting W, Mirmiran M, Bos NP, Swaab DF. The effect of old age on the free-running period of circadian rhythms in rat. Chronobiol Int. 1994; 11:103–12. https://doi.org/10.3109/07420529409055896 [PubMed]
- 26. Valentinuzzi VS, Scarbrough K, Takahashi JS, Turek FW. Effects of aging on the circadian rhythm of wheel-running activity in C57BL/6 mice. Am J Physiol. 1997; 273:R1957–64. [PubMed]
- 27. Davidson AJ, Yamazaki S, Arble DM, Menaker M, Block GD. Resetting of central and peripheral circadian oscillators in aged rats. Neurobiol Aging. 2008; 29:471–77. https://doi.org/10.1016/j.neurobiolaging.2006.10.018 [PubMed]
- 28. Sellix MT, Evans JA, Leise TL, Castanon-Cervantes O, Hill DD, DeLisser P, Block GD, Menaker M, Davidson AJ. Aging differentially affects the re-entrainment response of central and peripheral circadian oscillators. J Neurosci. 2012; 32:16193–202. https://doi.org/10.1523/JNEUROSCI.3559-12.2012 [PubMed]
- 29. Nakahata Y, Yasukawa S, Khaidizar FD, Shimba S, Matsui T, Bessho Y. Bmal1-deficient mouse fibroblast cells do not provide premature cellular senescence in vitro. Chronobiol Int. 2018; 35:730–38. https://doi.org/10.1080/07420528.2018.1430038 [PubMed]
- 30. Kunieda T, Minamino T, Katsuno T, Tateno K, Nishi J, Miyauchi H, Orimo M, Okada S, Komuro I. Cellular senescence impairs circadian expression of clock genes in vitro and in vivo. Circ Res. 2006; 98:532–39. https://doi.org/10.1161/01.RES.0000204504.25798.a8 [PubMed]
- 31. Matsuo M, Kaji K, Utakoji T, Hosoda K. Ploidy of human embryonic fibroblasts during in vitro aging. J Gerontol. 1982; 37:33–37. https://doi.org/10.1093/geronj/37.1.33 [PubMed]
- 32. Yamazaki S, Straume M, Tei H, Sakaki Y, Menaker M, Block GD. Effects of aging on central and peripheral mammalian clocks. Proc Natl Acad Sci USA. 2002; 99:10801–06. https://doi.org/10.1073/pnas.152318499 [PubMed]
- 33. Nakahata Y, Akashi M, Trcka D, Yasuda A, Takumi T. The in vitro real-time oscillation monitoring system identifies potential entrainment factors for circadian clocks. BMC Mol Biol. 2006; 7:5. https://doi.org/10.1186/1471-2199-7-5 [PubMed]
- 34. Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009; 4:1798–806. https://doi.org/10.1038/nprot.2009.191 [PubMed]
- 35. Khaidizar FD, Nakahata Y, Kume A, Sumizawa K, Kohno K, Matsui T, Bessho Y. Nicotinamide phosphoribosyltransferase delays cellular senescence by upregulating SIRT1 activity and antioxidant gene expression in mouse cells. Genes to cells. 2017; 22:982–992. https://doi.org/10.1111/gtc.12542 [PubMed]
- 36. Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004; 114:1299–307. https://doi.org/10.1172/JCI22475 [PubMed]
- 37. Stein GH, Drullinger LF, Soulard A, Dulić V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol. 1999; 19:2109–17. https://doi.org/10.1128/MCB.19.3.2109 [PubMed]
- 38. van Deursen JM. The role of senescent cells in ageing. Nature. 2014; 509:439–46. https://doi.org/10.1038/nature13193 [PubMed]
- 39. Brown SA, Fleury-Olela F, Nagoshi E, Hauser C, Juge C, Meier CA, Chicheportiche R, Dayer JM, Albrecht U, Schibler U. The period length of fibroblast circadian gene expression varies widely among human individuals. PLoS Biol. 2005; 3:e338. https://doi.org/10.1371/journal.pbio.0030338 [PubMed]
- 40. Nakamura TJ, Nakamura W, Tokuda IT, Ishikawa T, Kudo T, Colwell CS, Block GD. Age-related changes in the circadian system unmasked by constant conditions. eNeuro. 2015; 2. https://doi.org/10.1523/ENEURO.0064-15.2015 [PubMed]
- 41. Nakamura TJ, Nakamura W, Yamazaki S, Kudo T, Cutler T, Colwell CS, Block GD. Age-related decline in circadian output. J Neurosci. 2011; 31:10201–05. https://doi.org/10.1523/JNEUROSCI.0451-11.2011 [PubMed]
- 42. https://www.circadian.org/softwar.html.
- 43. Bellet MM, Sassone-Corsi P. Mammalian circadian clock and metabolism - the epigenetic link. J Cell Sci. 2010; 123:3837–48. https://doi.org/10.1242/jcs.051649 [PubMed]
- 44. Buhr ED, Takahashi JS. Molecular components of the Mammalian circadian clock. Handb Exp Pharmacol. 2013; 217:3–27. https://doi.org/10.1007/978-3-642-25950-0_1 [PubMed]
- 45. Yagita K, Okamura H. Forskolin induces circadian gene expression of rPer1, rPer2 and dbp in mammalian rat-1 fibroblasts. FEBS Lett. 2000; 465:79–82. https://doi.org/10.1016/S0014-5793(99)01724-X [PubMed]
- 46. Izumo M, Sato TR, Straume M, Johnson CH. Quantitative analyses of circadian gene expression in mammalian cell cultures. PLOS Comput Biol. 2006; 2:e136. https://doi.org/10.1371/journal.pcbi.0020136 [PubMed]
- 47. Travnickova-Bendova Z, Cermakian N, Reppert SM, Sassone-Corsi P. Bimodal regulation of mPeriod promoters by CREB-dependent signaling and CLOCK/BMAL1 activity. Proc Natl Acad Sci USA. 2002; 99:7728–33. https://doi.org/10.1073/pnas.102075599 [PubMed]
- 48. Yamamoto T, Nakahata Y, Tanaka M, Yoshida M, Soma H, Shinohara K, Yasuda A, Mamine T, Takumi T. Acute physical stress elevates mouse period1 mRNA expression in mouse peripheral tissues via a glucocorticoid-responsive element. J Biol Chem. 2005; 280:42036–43. https://doi.org/10.1074/jbc.M509600200 [PubMed]
- 49. Cheon S, Park N, Cho S, Kim K. Glucocorticoid-mediated Period2 induction delays the phase of circadian rhythm. Nucleic Acids Res. 2013; 41:6161–74. https://doi.org/10.1093/nar/gkt307 [PubMed]
- 50. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018; 28:436–53. https://doi.org/10.1016/j.tcb.2018.02.001 [PubMed]
- 51. James EL, Michalek RD, Pitiyage GN, de Castro AM, Vignola KS, Jones J, Mohney RP, Karoly ED, Prime SS, Parkinson EK. Senescent human fibroblasts show increased glycolysis and redox homeostasis with extracellular metabolomes that overlap with those of irreparable DNA damage, aging, and disease. J Proteome Res. 2015; 14:1854–71. https://doi.org/10.1021/pr501221g [PubMed]
- 52. Chondrogianni N, Stratford FL, Trougakos IP, Friguet B, Rivett AJ, Gonos ES. Central role of the proteasome in senescence and survival of human fibroblasts: induction of a senescence-like phenotype upon its inhibition and resistance to stress upon its activation. J Biol Chem. 2003; 278:28026–37. https://doi.org/10.1074/jbc.M301048200 [PubMed]
- 53. Chondrogianni N, Trougakos IP, Kletsas D, Chen QM, Gonos ES. Partial proteasome inhibition in human fibroblasts triggers accelerated M1 senescence or M2 crisis depending on p53 and Rb status. Aging Cell. 2008; 7:717–32. https://doi.org/10.1111/j.1474-9726.2008.00425.x [PubMed]
- 54. Ignowski JM, Schaffer DV. Kinetic analysis and modeling of firefly luciferase as a quantitative reporter gene in live mammalian cells. Biotechnol Bioeng. 2004; 86:827–34. https://doi.org/10.1002/bit.20059 [PubMed]
- 55. Schlegel R, Tralka TS, Willingham MC, Pastan I. Inhibition of VSV binding and infectivity by phosphatidylserine: is phosphatidylserine a VSV-binding site? Cell. 1983; 32:639–46. https://doi.org/10.1016/0092-8674(83)90483-X [PubMed]
- 56. Venable ME, Lee JY, Smyth MJ, Bielawska A, Obeid LM. Role of ceramide in cellular senescence. J Biol Chem. 1995; 270:30701–08. https://doi.org/10.1074/jbc.270.51.30701 [PubMed]
- 57. Lizardo DY, Lin YL, Gokcumen O, Atilla-Gokcumen GE. Regulation of lipids is central to replicative senescence. Mol Biosyst. 2017; 13:498–509. https://doi.org/10.1039/C6MB00842A [PubMed]
- 58. Akiyama M, Kouzu Y, Takahashi S, Wakamatsu H, Moriya T, Maetani M, Watanabe S, Tei H, Sakaki Y, Shibata S. Inhibition of light- or glutamate-induced mPer1 expression represses the phase shifts into the mouse circadian locomotor and suprachiasmatic firing rhythms. J Neurosci. 1999; 19:1115–21. https://doi.org/10.1523/JNEUROSCI.19-03-01115.1999 [PubMed]
- 59. Shigeyoshi Y, Taguchi K, Yamamoto S, Takekida S, Yan L, Tei H, Moriya T, Shibata S, Loros JJ, Dunlap JC, Okamura H. Light-induced resetting of a mammalian circadian clock is associated with rapid induction of the mPer1 transcript. Cell. 1997; 91:1043–53. https://doi.org/10.1016/S0092-8674(00)80494-8 [PubMed]
- 60. Kolker DE, Vitaterna MH, Fruechte EM, Takahashi JS, Turek FW. Effects of age on circadian rhythms are similar in wild-type and heterozygous Clock mutant mice. Neurobiol Aging. 2004; 25:517–23. https://doi.org/10.1016/j.neurobiolaging.2003.06.007 [PubMed]
- 61. Asai M, Yoshinobu Y, Kaneko S, Mori A, Nikaido T, Moriya T, Akiyama M, Shibata S. Circadian profile of Per gene mRNA expression in the suprachiasmatic nucleus, paraventricular nucleus, and pineal body of aged rats. J Neurosci Res. 2001; 66:1133–39. https://doi.org/10.1002/jnr.10010 [PubMed]
- 62. Yamaguchi A, Matsumura R, Matsuzaki T, Nakamura W, Node K, Akashi M. A simple method using ex vivo culture of hair follicle tissue to investigate intrinsic circadian characteristics in humans. Sci Rep. 2017; 7:6824. https://doi.org/10.1038/s41598-017-07268-8 [PubMed]
- 63. Pagani L, Schmitt K, Meier F, Izakovic J, Roemer K, Viola A, Cajochen C, Wirz-Justice A, Brown SA, Eckert A. Serum factors in older individuals change cellular clock properties. Proc Natl Acad Sci USA. 2011; 108:7218–23. https://doi.org/10.1073/pnas.1008882108 [PubMed]