



Introduction
The function of N-methyl-D-aspartate receptors (NMDARs) have a profound influence on synaptic plasticity, cognition, psychiatric diseases, and the connectivity of neural networks [1,2]. For example, redox regulation of NMDAR function during development influences the formation of synaptic connections and neuronal circuits involved in schizophrenia [3,4]. In adults, a redox-mediated NMDAR hypofunction results in depressive-like behavior [5]. With advancing age, metabolic redox stress induces NMDAR hypofunction, weakening synaptic plasticity, and impairing cognition [6–10].
Redox regulation of NMDAR function can be studied by examining the effects of oxidizing or reducing agents on the NMDAR component of synaptic transmission. In aged animals, the reducing agent, dithiothreitol (DTT), increases the NMDAR synaptic response and rescues synaptic plasticity [6–9,11–13]. In contrast, application of oxidizing agents decreases NMDAR responses and impairs the induction of synaptic plasticity, specifically in young animals [6,14]. The results point to a redox sensitive mechanism in mediating the well-characterized decrease in the CA3-CA1 NMDAR synaptic response of older-memory impaired animals, and suggests that redox regulation of NMDARs influences synaptic plasticity during aging [15–18].
The exact mechanism for redox regulation of NMDARs during aging is unclear, but likely involves thiol S-nitrosylation of cysteine residues or formation of disulfide bonds between cysteine residues of NMDAR subunits or proteins involved in NMDAR regulatory processes [19,20]. NMDARs are heterotetramers and previous research has focused on diheteromeric NMDARs with two GluN1 subunits and two identical GluN2 subunits, either GluN2A or GluN2B. The diheteromeric GluN2 subunits have different kinetics and are differentially sensitive to Zn2+ and redox reagents. For example, the GluN1 and GluN2A subunits have extracellular cysteine residues, and under oxidizing conditions, S-nitrosylation or disulfide bond formation of cysteine residues decreases receptor function [21–24]. Over the course of development, many brain regions exhibit an increase in the decay rate of NMDAR synaptic responses resulting from an increased contribution of GluN2A to NMDAR responses [25,26]. Due to the redox sensitive cysteine residues of GluN2A, a shift in the ratio of GluN2A/GluN2B could render older synapses more susceptible to redox regulation. In this case, DTT should increase the GluN2A contribution to the synaptic response.
In addition, an intracellular oxidized redox state is predicted to impair signaling involved in regulating NMDAR function and receptor trafficking. The DTT-mediated increase in the NMDAR response is blocked by inhibition of Ca2+/calmodulin-dependent protein kinase II (CaMKII) [6]. In turn, CaMKII regulates NMDAR trafficking to the synapse [27,28] and CaMKII increases the contribution of GluN2B to the synaptic response [28–32]. If redox regulation is acting through NMDAR plasticity involving GluN2B, DTT application should increase the GluN2B contribution to the synaptic response.
The current study, recorded synaptically evoked excitatory postsynaptic currents (EPSCs) from CA1 hippocampal pyramidal neurons and field excitatory postsynaptic potentials (fEPSPs) from CA3-CA1 synapses, and examined the contribution of GluN2A and GluN2B subunits to the decline in NMDAR synaptic function during aging, and the DTT-induced enhancement of NMDAR-mediated synaptic transmission. The results indicate that the age-related decrease in the NMDAR response is not due to a shift in the ratio of diheteromeric GluN2A/GluN2B subunits at the synapse. Furthermore, the DTT-mediated increase in the synaptically evoked NMDAR current involves an increase contribution of GluN2B. The redox regulation of the NMDAR response was dependent on the level of NMDAR activity and kinase activation. Together, the results suggest that increased oxidative stress during aging suppresses NMDAR activity-dependent plasticity.
Results
Decreased NMDAR synaptic currents during aging
Whole-cell patch-clamp recordings of synaptically evoked NMDAR-mediated EPSCs were obtained from CA1 pyramidal cells of hippocampal slices obtained from young (11/4 cells/animals) and aged (9/4 cells/animals) animals. For a subset of cells, isolation of NMDA currents was confirmed by bath application of AP-5 (100 µM) (Suppl. Fig. 1). The EPSCs were recorded at holding voltages between -60 and +60 mV in 20 mV steps. No age-related difference was observed for intrinsic properties, including access resistance, membrane resistance, and capacitance (Table 1). For negative voltages, currents were inward with reduced amplitudes, consistent with Mg2+ blockade of the NMDAR channel. The reversal potential was calculated from a regression of responses through -20, 0, and +20 mV. The reversal potential of the synaptically evoked current was near 0 mV, consistent with NMDAR permeability to multiple cations, and no age-related difference was observed for the reversal potential (young: -6.9 ± 3.1 mV; aged: -3.4 ± 3.2 mV). A repeated measures Analysis of variance (ANOVA) on EPSC amplitudes across the voltage steps indicated a tendency (p = 0.051) for an interaction of age and membrane potential due to an increase in outward currents for young animals at positive potentials (Fig. 1 A, Suppl. Fig. 2 A & B).
Table 1. Intrinsic properties of CA1 hippocampal pyramidal neurons.
| Cell Properties | Young Control | Aged Control | Young DTT | Aged DTT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Access resistance (MΩ) | 33.94 ± 2.3 | 32.82 ± 2.5 | 27.51 ± 3.1 | 33.41 ± 7.9 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Membrane resistance (MΩ) | 159.29 ± 7.5 | 147.77 ± 6.9 | 152.28 ± 14.7 | 138.37 ± 7.0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Capacitance (pF) | 141.58 ± 5.7 | 140.14 ± 8.7 | 141.58 ± 5.7 | 158.54 ± 16.6 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Intrinsic properties of CA1 hippocampal pyramidal neurons recorded by whole-cell patch clamp from young control (n = 25/11 cells/animals), young DTT (n = 8/2 cells/animals), aged (n = 24/14, cells/animals), and aged DTT (n = 7/3 cells/animals) animals. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
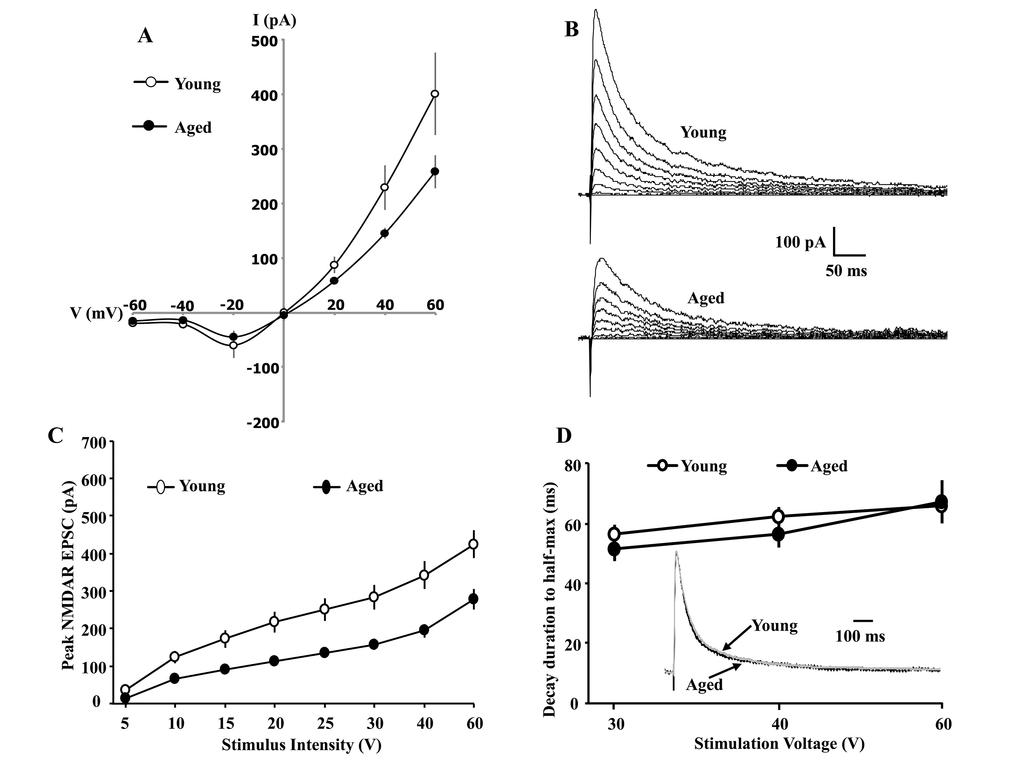
Figure 1. Whole-cell patch clamp recording from CA1 hippocampal pyramidal neurons of aged and young animals demonstrating the current-voltage relationship and synaptic decay duration. (A) The current-voltage relationship was recorded from CA1 pyramidal neurons from young (11/4 cells/animals) and aged (9/4 cells/animals) animals. When cells are clamped at positive voltages, the currents are outward and larger currents are observed for young animals. The reversal potential is near 0 mV for both age groups. When cells are clamped at negative voltages, currents are inward and reduced, consistent with Mg2+ blockade of the NMDAR channel. Examination of peak amplitude and time to half-decay of the NMDAR EPSC during aging. The cells were voltage clamped at +40 mV. (B) Representative traces evoked by the eight different stimulation intensities and recorded from young (top) and aged animals (bottom). (C) A decrease in the peak NMDAR EPSC was observed across the range of stimulation intensities for CA1 pyramidal cells recorded from aged animals (filled circle, n = 26/14 cells/animals), relative to cells from young animals (open circle, n = 20/9 cells/animals). (D) The mean (±SEM) time for the EPSC to decay to 50% of the peak for the three highest stimulation intensities. The inset shows the time course of the EPSC, evoked by 40 V stimulation, across all CA1 pyramidal cells recorded from young (gray trace, n = 20/9 cells/animals) and aged (dark trace, n = 26/14 cells/animals) animals. For each cell, the response amplitude evoked by 40 V stimulation was normalized to the peak of the response.
To examine synaptic input-output relationships, cells were held at +40 mV and NMDAR EPSCs were recorded across a range of stimulation intensities (5, 10, 15, 20, 25, 30, 40, and 60 V). For each stimulation intensity, the peak amplitude of the EPSC was obtained from CA1 cells of aged (n = 24/14 cells/animals) and young (n = 25/11 cells/animals) animals. A repeated measures ANOVA across stimulation intensities indicated an interaction of age and stimulation intensity [F(7,329) = 6.58, p< 0.0001] and the age difference was due to larger peak EPSC in young animals (Fig. 1 B, C). The results confirm that NMDAR synaptic responses are reduced during aging [15–18].
The GluN2A/GluN2B subunit composition is not altered with advanced age
The subunit composition can be studied by examining the time course of NMDAR synaptic responses. An increase in the ratio of GluN2A/GluN2B subunits results in a more rapid decay, producing a shortened synaptic response [25,33]. To examine age differences in the decay of the synaptic response, the EPSC responses for 30, 40, and 60 V stimulation were normalized to the peak of the response, and the time to decay to 50% was calculated. A small, nonsignificant, decrease in the decay rate was observed for aged animals, suggesting that aging is not associated with a large shift in the GluN2A/ GluN2B ratio (Fig. 1 D).
Next, we employed GluN2 selective antagonists to examine age differences in the contribution of GluN2 subtypes to the EPSC peak synaptic response. Again, cells were held at +40 mV. Following at least five-minutes of stable baseline recording of the isolated NMDAR EPSC, the GluN2B selective antagonist, ifenprodil (5 µM), was bath applied. Ifenprodil decreased the peak EPSC by ~30%, and an ANOVA on percent of baseline for the peak EPSC, measured 15 min following application of ifenprodil, indicated no age difference (young: 70.59 ± 8.14 mean ± SEM% of baseline, n = 4/4 cells/animals; aged: 71.58 ± 10.39%, n = 6/5 cells/animals) (Fig. 2 A & B). The decrease was specific to ifenprodil, as no effect was observed following application of vehicle alone.
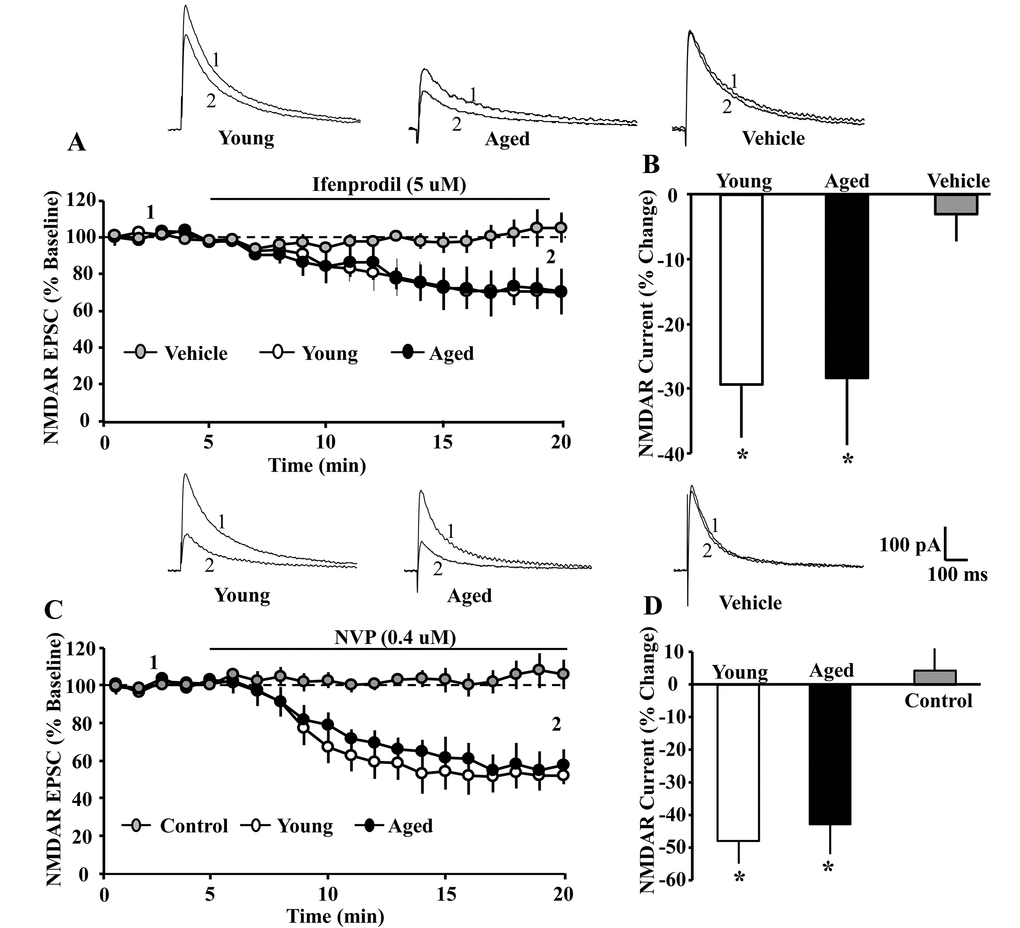
Figure 2. The GluN2A and GluN2B selective antagonists attenuated the NMDAR EPSC amplitude to a similar extent in young and aged CA1 pyramidal neurons. For each cell, the peak response was normalized to the 5 min pre-drug baseline. (A) Time course of the decrease in the NMDAR EPSCs recorded from CA1 hippocampal pyramidal neurons 5 min before and 15 min after bath application of ifenprodil (5 µM, solid line) in young (open circle, n = 4/4 cells/animals) and aged (filled circle, n = 6/5 cells/animals) animals. For the control condition (gray circle, n = 7/6 cells/animals, young-aged combined) recordings were obtained before and after application of ethanol vehicle. (B) Bar graph demonstrates percentage decrease in NMDAR EPSCs for young and aged animals following application of ifenprodil or vehicle. Asterisks indicate a significant difference from baseline. The top panel provides representative traces illustrating the NMDAR EPSC at baseline (1) and at the end of a 15 min of ifenprodil application (2) recorded from a young (left) or aged (middle) cell, and for a cell recorded in the vehicle control condition (right). The GluN2A selective antagonist, NVP, attenuated the NMDAR EPSC to a similar extent in young and aged CA1 pyramidal neurons. For each cell, the peak response was normalized to the 5 min pre-drug baseline. (C) Time course of the decrease in the NMDAR EPSCs recorded from CA1 hippocampal pyramidal neurons 5 min before and 15 min after bath application of NVP (0.4 µM, solid line) in young (open circle, n = 4/4 cells/animals) and aged (filled circle, n = 5/5 cells/animals) animals. For the control condition (gray circle, n = 6/6 cells/animals, young-aged combined) recordings were maintained for the same duration in the absence of NVP application. (D) Bar graph demonstrates percentage decrease in NMDA EPSCs during the last 5 min of recording. Asterisks indicate a significant difference from baseline. Representative traces on the top illustrating the NMDAR EPSC at baseline (1) and at the end of a 15 min NVP application (2) recorded from a young (left) or aged (middle) cell, and for a cell in the control condition (right).
Similarly, bath application of the GluN2A selective antagonist, NVP (0.4 µM), reduced the peak NMDAR EPSC by ~45% over the next 15 min. No change in the peak EPSC was observed in the absence of NVP, recorded over the same duration. An ANOVA on percent of baseline for the peak EPSC, measured 15 min following application of NVP, indicated no age difference in the reduction of the EPSC (young: 52.10 ± 7.28% of baseline, n = 4/2 cells/animals; aged: 57.14 ± 9.2%, n = 6/3 cells/animals) (Fig. 2 C & D).
The DTT-mediated potentiation in NMDAR currents involves an increased contribution of GluN2B
Following bath application of the reducing agent DTT (0.5 mM), input-output curves of the peak NMDAR EPSC were again obtained (aged: n = 7/3 cells/animals, young: n = 8/2 cells/animals) and compared to input-output curves for the respective control condition for young and aged animals (Fig. 1C). A repeated measures ANOVA across stimulation intensities for the control condition and DTT conditions, within each age group, indicated an effect of treatment on the peak EPSC for aged animals [F(1,203) = 53.24, p < 0.0001], but not for young animals [F(1,217) = 0.49, p > 0.05] (Fig. 3A & B), confirming an age-related difference in the DTT-induced potentiation of the NMDAR response.
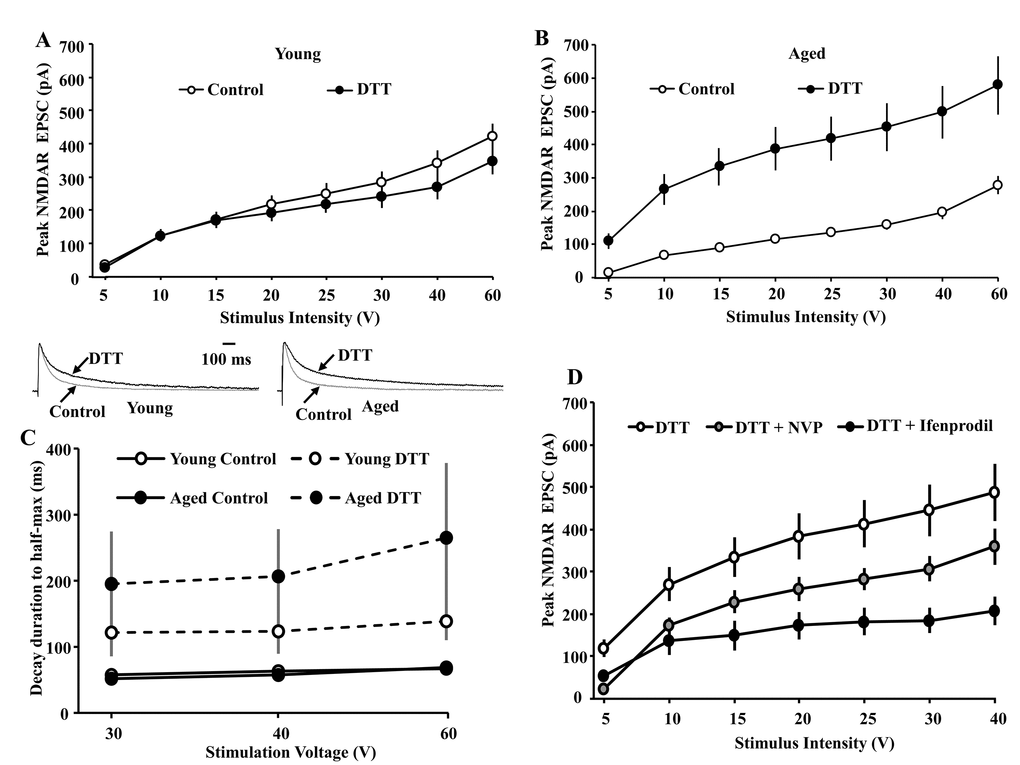
Figure 3. Input-output curves examining age-related differences in the peak NMDAR EPSCs under control conditions and in the presence of DTT (0.5 mM). The cells were voltage clamped at +40 mV. (A) Bath application of DTT (filled circle, n = 8/2 cells/animals) failed to increase NMDAR EPSCs for CA1 pyramidal cells recorded from young animals relative to the control condition (open circle, n = 25/11 cells/animals). (B) Across the range of stimulation intensities, DTT (filled circle, n = 7/3 cells/animals) significantly augmented NMDAR EPSCs in CA1 cells recorded from aged animals relative to the control condition (open circle, n = 26/14 cells/animals). (C) DTT increases the time to half-decay of the NMDAR synaptic response. The symbols represent the mean (±SEM) time of NMDAR-mediated EPSC to decay to 50% of the peak under control conditions and in the presence of DTT for the three highest stimulation intensities. The inset (left) shows time course of the EPSC, evoked by 40 V stimulation, across all CA1 pyramidal cells recorded from young animals under the control condition (gray trace, n = 20/9 cells/animals) and in the presence of DTT (black trace, n = 8/2 cells/animals). The inset (right) time course of the EPSC, evoked by 40 V stimulation, across all CA1 pyramidal cells recorded from aged animals under the control condition (gray trace, n = 26/14 cells/animals) and in the presence of DTT (black trace, n = 7/3 cells/animals). For each cell, the response amplitude evoked by 40 V stimulation was normalized to the peak of the response. (D) Increased contribution of the GluN2B subunit to the peak NMDAR EPSC following DTT-induced potentiation of NMDAR EPSCs in slices obtained from aged animals. The cells were voltage clamped at +40 mV and input-output curves of the peak NMDAR EPSCs were generated in presence of DTT (open circle, n = 7/3 cells/animals), DTT+NVP (gray circle, n = 5/2 cells/animals), and DTT+ifenprodil (filled circle, n = 5/3 cells/animals).
To determine if DTT influenced the decay rate of the NMDAR response, the NMDAR EPSC in response to 30, 40, and 60 V stimulation was normalized to the peak of the response at each stimulation intensity, and the time to decay to 50% of the peak was calculated in the presence of DTT and compared to the control condition. Bath application of DTT increased the time to half-decay ~4 fold in aged animals and ~2 fold in young animals (Fig. 3 C). An ANOVA repeated across stimulation intensities indicated a treatment effect [F(1,106) = 22.76, p < 0.0001] in the absence of an age difference or an interaction of age and treatment. ANOVAs within each age group confirmed that DTT increased the time to half-decay of the EPSC for aged [F(1,52) = 12.61, p < 0.005] and young [F(1,54) = 11.55, p < 0.005] animals.
To examine the contribution of GluN2A/GluN2B subunit to DTT-induced potentiation in NMDAR function, subunit selective antagonists were applied to slices obtained from aged animals in the presence of DTT. Figure 3D shows input-output curves collected from cells of aged animals exposed to DTT alone (n = 7/3 cells/animals), or in the presence of DTT followed by application of NVP (n = 5/2 cells/animals) or ifenprodil (n = 5/3 cells/animals). A repeated measures ANOVA across stimulation intensities indicated an interaction of stimulation intensities and treatment [F(12,84) = 5.01, p < 0.0001]. Post hoc comparisons for treatment effects indicated NVP tended (p = 0.055) to decrease the peak response to ~65%, relative to the DTT alone condition. The effect of ifenprodil was more robust, decreasing the peak response (p < 0.005) to ~45% of the DTT alone condition (Fig. 3D). Accordingly, in contrast to the greater effect of NVP under controls conditions, ifenprodil reduced the peak response to a greater extent in the presence of DTT. Thus, DTT application increased decay time and increased sensitivity to ifenprodil in aged animals, suggest an increase contribution of GluN2B to the NMDAR synaptic response.
Redox regulation of NMDAR function depends on NMDAR activity
Next, we sought to determine if the DTT-mediated growth of the NMDAR response in aged animals represents an activity-dependent NMDAR plasticity. An influx of Ca2+ following NMDAR activation induces long-term modifications in NMDAR function in young animals [34,35]. Due to the inhibition of Ca2+ entry for cells held at +40 mV, we employed extracellular fEPSP recordings of isolated NMDAR synaptic responses to examine the role of NMDAR activity and Ca2+ on the DTT-mediated potentiation of the NMDAR synaptic response in slices from aged animals. Control recordings, examining the effects of DTT alone, were interleaved between recordings in which the GluN2A or GluN2B selective antagonist were applied, prior to DTT application. For control slices (n = 31slices/26 aged animals), DTT (0.5 mM) induced an increase (161.2 ± 6.2% mean ± SEM of baseline) in the NMDAR-mediated synaptic response, similar to previous reports [6,8,9,13] (Fig. 4).
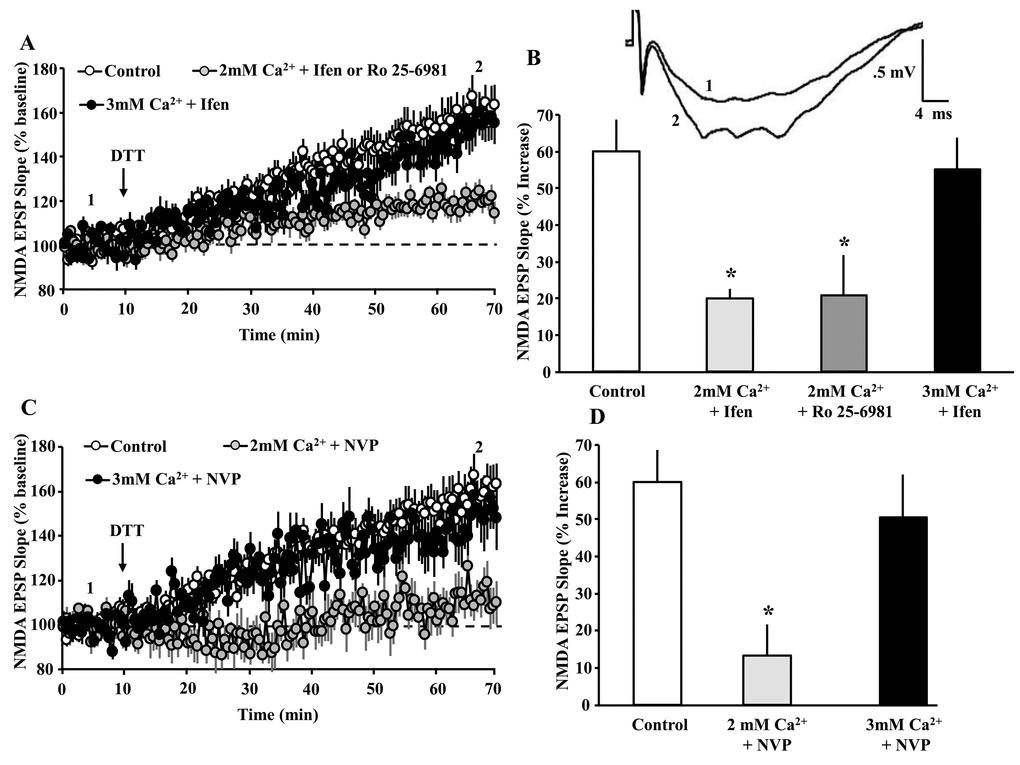
Figure 4. NMDAR activity and Ca2+ are required for the DTT-induced potentiation of NMDAR synaptic function. (A) Time course of mean (±SEM) NMDAR-fEPSP slope normalized to the baseline (dashed line) for the control condition (open circles), in the presence of ifenprodil or Ro 25-6981 in 2 mM Ca2+ recording medium (gray circles), and ifenprodil in 3 mM Ca2+ recording medium (filled circles). For clarity, the responses for the GluN2B antagonists (ifenprodil and Ro 25-6981) in 2 mM Ca2+ recording medium were combined. The arrow indicates the time of DTT (0.5 mM) application. The insert provides an example of the growth of the NMDAR-mediated fEPSP during baseline (1) and 60 min following application of the DTT (2) under the control condition. (B) Bar graph demonstrates the percent change in NMDAR-mediated fEPSP response during the last 5 min of recording, due to DTT application under the control condition (open bar, n = 31/26 slices/ animals), 2 mM Ca2+ + ifenprodil (light gray bar, n = 8/8 slices/animals), 2 mM Ca2+ + Ro 25-6981 (gray bar, n = 5/5 slices/animals), and 3 mM Ca2+ + ifenprodil (black bar, n = 8/4 slices/animals). (C) Time course of mean (±SEM) NMDAR-fEPSP slope normalized to the baseline (dashed line) for the control condition (open circles), in the presence of NVP in 2 mM Ca2+ recording medium (gray circles), and NVP in 3 mM Ca2+ recording medium (filled circles). The arrow indicates the time of DTT (0.5 mM) application. (D) Bar graph demonstrates the percent change in NMDAR-mediated fEPSP response during the last 5 min of recording, due to DTT application under the various conditions including control (open bar, n = 31/26 slices/animals), NVP (light gray bar, n = 7/6 slices/animals), and 3 mM Ca2+ + NVP (black bar, n= 8/4 slices/animals). For B & D, the asterisks indicate a significant difference relative to control.
The GluN2B antagonists, ifenprodil (5 µM, n = 8/8 slices/animals) or Ro 25-6981 (5 µM, n = 5/5 slices/animals) were added to the bath at least 45 min prior to application of DTT. In another set of slices (8 slices from 4 animals), the concentration of Ca2+ in the bath was increased from 2 mM to 3 mM, and ifenprodil was added to the bath at least 45 min prior to application of DTT. An ANOVA on the percent increase in the synaptic response, 60 min following application of DTT indicated a treatment effect [F(3,48) = 5.7, p = 0.02]. Post hoc tests indicated that, in 2 mM Ca2+ recording medium, ifenprodil (120.0 ± 2.5%) and Ro 25-6981 (120.8 ± 10.0%) inhibited the DTT-induced growth of the NMDAR-mediated synaptic responses, relative to the control condition (Fig. 4 A & B). In contrast, when the level of Ca2+ was increased to 3 mM, ifenprodil failed to attenuate the DTT-induced potentiation of NMDAR-mediated synaptic responses (155.2 ± 8.7%) (Fig. 4 A & B).
To determine the contribution of GluN2A to the DTT-induced potentiation of the NMDAR response, NVP (0.4 µM, n = 7/6 slices/animals) was added to the bath 45 min prior to application of DTT. For some slices (n = 8/4 slices/animals), the concentration of Ca2+ in the bath was increased from 2 mM to 3 mM, and NVP was added to the bath 45 min prior to application of DTT. An ANOVA on the percent change in the response, 60 min after DTT application, indicated a difference across treatment groups [F(3,45) = 4.48, p = 0.0078]. Post hoc tests indicated that NVP in 2 mM Ca2+ significantly attenuated the DTT-mediated growth (113.3 ± 6.4%) relative to controls (Fig. 4 C & D). Again, raising the level of Ca2+ was able to overcome the NMDAR antagonist inhibition of the DTT-mediated growth in the synaptic response. Together, the results indicate that the DTT-mediated growth of the NMDAR response depends on the level of NMDAR activation and Ca2+ entry.
One possibility is that DTT increased Ca2+ entry through GluN2A containing receptors by chelating Zn2+ [24,36–38]. However, addition of excess Zn2+ (ZnCl2 1 µM, n = 3/3 slices/animals) failed to block the effects of DTT, which increased the response (160.3 ± 8.4%) (Fig. 5A). In addition, bath application of the Zn2+ chelating agent ZX1 (100 µM, n = 11 slices/6 animals) failed to enhance the NMDAR synaptic response (103.5 ± 3.99%) (Fig. 5B). In contrast, when DTT was subsequently applied 60 min following application of ZX1, a significant (p < 0.0001) increase (143.6 ± 8.79%, n = 11 slices/6 animals) in NMDAR response was observed, suggesting that DTT effects are not due to Zn2+ chelation (Fig. 5C).
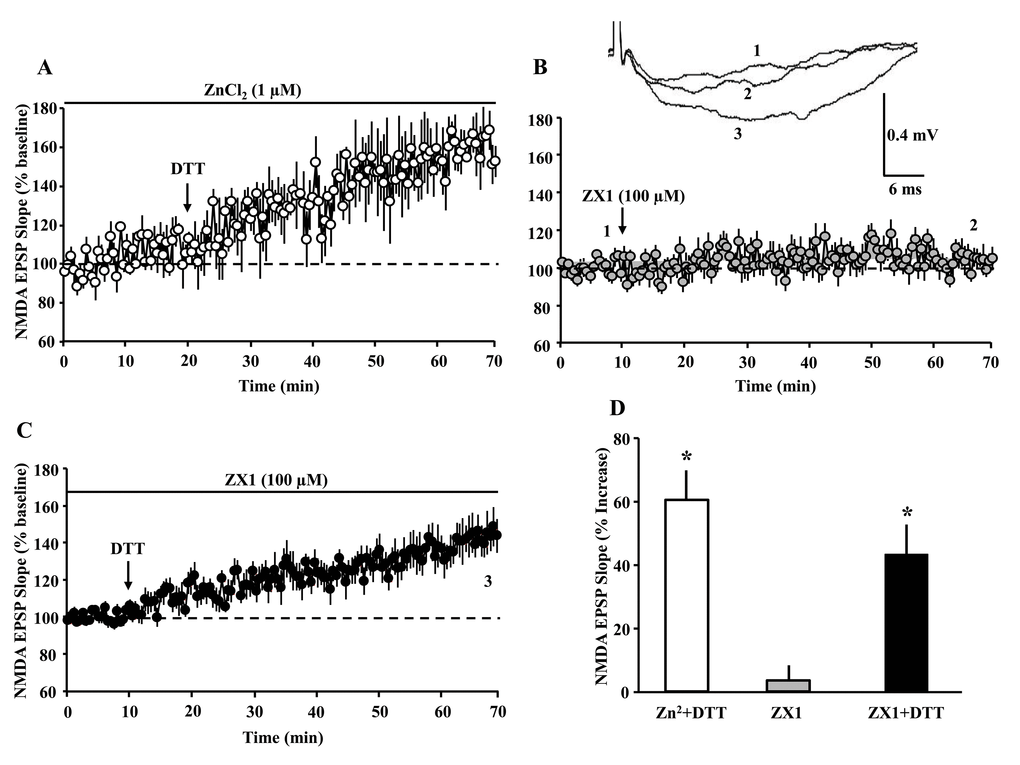
Figure 5. The DTT-induced potentiation of the NMDAR-synaptic response is not due to zinc chelation. The panels A-C illustrate the time course for the NMDAR-fEPSP slope; each point represents the mean (±SEM), normalized to the baseline (dashed line). (A) The arrow indicates the time of bath application of DTT (0.5 mM) in presence of ZnCl2 (1 µM). (B) The arrow indicates the time of bath application of ZX1 (100 µM). (C) The last ten min of NMDAR-fEPSP slope recording in presence of ZX1 was renormalized and DTT was added (arrow). (D) Bar graph represents the mean (+SEM) percent change in NMDAR-mediated fEPSP during the last 5 min of recording, in response to Zn2+ plus DTT (open bar, n = 3/3 slices/animals), ZX1 alone (gray bar, n = 11/6 slices/animals,) and ZX1 plus DTT (filled bar, n = 11/6 slices/animals). Asterisks indicate significant potentiation relative to baseline.
In young animals, influx of Ca+2 through NMDARs results in CaMKII-mediated trafficking and insertion of NMDAR subunits into the postsynaptic membrane [27,28], and the DTT-mediated increase in the NMDAR response in older animals is blocked by inhibition of CaMKII [6]. In addition, CaMKII activity determines the long-term maintenance of synaptic strength [32,39]. Oxidizing agents decrease the NMDAR synaptic response, specifically in young animals [6,14,40]. To determine whether ongoing CaMKII activity is involved in the decrease of NMDAR responses under oxidizing conditions, slices from young animals were exposed to the CaMKII inhibitor KN-62 (10 µM) or dimethyl sulfoxide (DMSO) vehicle for at least 60 min before application of the oxidizing agent, 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) (0.5 mM). Bath application of DTNB in the presence of vehicle decreased the NMDAR-fEPSP (66.3 ± 4.9%, 7/7 slices/animals) 60 min following drug application. The DTNB-induced decrease in the NMDAR-fEPSP was blocked by the CaMKII selective antagonist, KN-62 (101.6 ± 2.9%, 7/7 slices/animals) [F(1,12) = 37.97, p < 0.0001], suggesting that the basal level of kinase activity determines the effectiveness of redox modulators on the NMDAR response (Fig. 6 A & B).
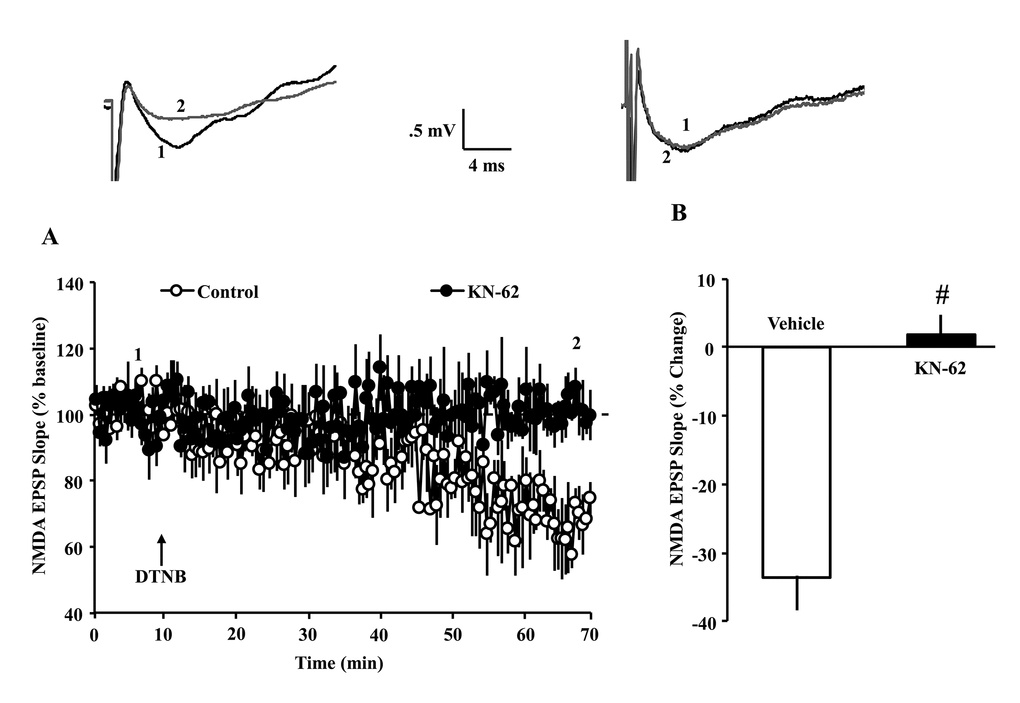
Figure 6. Decrease in NMDAR synaptic responses in young animals, under oxidizing conditions, depends on CaMKII activity. (A) Time course of normalized NMDAR-fEPSP slope following application of DTNB (0.5 mM, arrow) in the young animals. Each point represents the mean (±SEM), normalized to the baseline (dashed line), for slices in the control condition (open circles) or following pre-incubation with the CaMKII inhibitor, KN-62 (10 µM, filled circles). DTNB reduced NMDAR synaptic response. Pre-incubation with KN-62 blocked the decrease in the NMDAR response associated with DTNB application. (B) Quantification of the mean percent change in the NMDAR-fEPSP slope during the last 5 min of recording in the presence of vehicle control (open, n = 7/7 slices/animals) and KN-62 (, n = 7/7 slices/animals). Pound sign indicates a significant difference between the two groups. The waveforms represent examples of NMDAR-fEPSPs recorded during baseline (1) and 60 min following application of DTNB (2) in the control condition (left) and following pre-incubation in KN-62 (right).
Discussion
An understanding of the mechanisms for regulation of NMDAR function is important, due to the critical role of NMDARs in synaptic plasticity and cognition. Redox-mediated NMDAR hypofunction is linked to cognitive deficits for a range of illnesses including Alzheimer’s disease, depression, and schizophrenia [4,41,42]. However, the molecular mechanism for redox regulation of NMDARs in aging and disease is unknown. The age-related decrease in the NMDAR synaptic response is well characterized and contributes to altered synaptic plasticity and impaired cognition [15–18]. Recent studies point to redox regulation as a mechanism contributing to the decline in NMDAR function with age [6,8,11,12,43].
On possible mechanism involves a shift in the subunit composition during aging, which would determine the ability of redox reagents to modify NMDAR function [24,44]. NMDARs are heterotetramers and much of the previous research has focused on diheteromeric NMDARs with two GluN1 subunits and two identical GluN2 subunits, either GluN2A or GluN2B. The diheteromeric GluN2 subunits have different kinetics and are differentially sensitive to Zn2+ and redox reagents. The GluN2A subunit is more sensitive to Zn2+ inhibition and has unique extracellular cysteine residues. Under oxidizing conditions, S-nitrosylation of cysteine residues or disulfide bonds between pairs of cysteine residues decreases receptor function [21,23,24]. During the first two weeks of postnatal development, the synaptic GluN2B subunits may be switched for GluN2A subunits [45]. If this switch continues with advancing age, metabolic oxidative stress during aging could act on extracellular cysteine residues of GluN2A to decrease NMDAR function and increase responsiveness to reducing agents. In contrast, the results of the current study suggest that the ratio of diheteromeric GluN2A/GluN2B receptors remain stable with advancing age. An increase in the proportion of GluN2A subunits would decrease the decay time, shortening the synaptic response [25,33]. Conversely, we found that the decay of the synaptic response was not different between young and aged animals. In addition, the proportional decrease in the NMDAR EPSCs, in response to selective GluN2 antagonists, was similar for young and aged animals, indicating that diheteromeric GluN1-GluN2A receptors did not increase with age.
Previous work suggests that the majority of NMDARs in the hippocampus are triheteromeric, having two different GluN2 subunits (GluN1/GluN2A/GluN2B) [37,46,47]. Indeed, under control conditions, ifenprodil and NVP decreased the synaptic response by ~30% and ~45%, respectively, which is similar to previous reports examining cells that express a combination of diheteromeric and triheteromeric NMDARs [37,47–49]. The absence of an age difference for the synaptic decay and the similarity in the response inhibition by GluN2 selective antagonists suggest that the proportion of GluN2A/GluN2B subunits is not altered with advancing age. This conclusion is consistent with studies that examine expression of both GluN2A and GluN2B subunits in the same animal. When both subunits are examined, GluN2A and GluN2B are observed to decline equally during aging [50,51]. Together, the results suggest that the age difference in NMDAR synaptic function and redox sensitivity does not involve a large shift in the expression ratio of GluN2A/GluN2B at the synapse. Nevertheless, it is still possible that the decrease in the NMDAR responses and increased sensitivity to DTT involves redox regulation of extracellular cysteine residues on GluN1 or GluN2A [21]. However, extracellular application of glutathione, which can potentiate GluN1-GluN2A channels, does not increase the NMDAR response in older animals [6,21]. Finally, if the age difference was due to redox of GluN2A subunits, then we would expect that DTT would specifically increase the contribution of GluN2A subunits to the synaptic response. In contrast, the results of the current study indicate that GluN2B is a major contributor to the DTT-induced potentiation of the NMDAR response.
DTT markedly increased the decay time of NMDAR EPSCs in cells from young and aged animals. The increased in the decay time is consistent with an increased contribution of GluN2B [25,33,46,52,53]. An increased contribution of GluN2B was also evident in the effectiveness of selective GluN2 antagonists in reducing the peak response. The ifenprodil-induced decrease in the peak response of aged animals was greater in the presence of DTT, reducing the EPSC to ~45% of baseline, relative to the control condition, which was ~70% of baseline. Similarly, the contribution of GluN2A to the synaptic currents diminished following application of DTT. NVP deceased the EPSC peak response to ~55% of baseline in the control condition and to ~65% of baseline in the presence of DTT. The results are consistent with a DTT-mediated increase in the contribution of GluN2B, particularly for aged animals.
In younger animals, an NMDAR activity-dependent long-term potentiation (LTP) of NMDAR synaptic transmission involves trafficking of NMDARs to the synapse [54]. The mechanism for expression of LTP of NMDAR function and redox regulation of NMDAR function across the lifespan may involve similar mechanisms. In both cases, the increase in the synaptic response is not associated with a change in paired-pulse facilitation, and does not require an increase in the α-amino-3-hydroxy-5-mehtyl-4-isoxazolepropionic (AMPA) receptor component of synaptic transmission [6,34,35]. The results suggest that expression of LTP of NMDAR and DTT-mediated potentiation are not due to presynaptic changes.
Postsynaptic Ca+2 influx through NMDARs, results in CaMKII-mediated trafficking and insertion of NMDAR subunits into the postsynaptic membrane [27,28], modifying the NMDAR subunit composition at the synapse [55,56]. Furthermore, activated CaMKII associates with GluN2B subunit, increasing the contribution of GluN2B to NMDAR function [28–32]. Similar to activity-induced potentiation of NMDAR synaptic transmission, the DTT-induced growth of the NMDAR synaptic response is also CaMKII-dependent [6] and our current results point to an increased contribution of GluN2B to the NMDAR response.
The induction of LTP of NMDARs requires NMDAR activity and Ca2+ influx [34,35,57–61]. Using selective antagonists and different levels of extracellular Ca2+, we observed that the DTT-mediated increase in the NMDAR response was also dependent on NMDAR activity and level of Ca2+. The results suggest that the DTT-mediated increase in NMDAR function in aged animals involves a redox-mediated increase in the activity of CaMKII to activate mechanisms similar to LTP of NMDARs. As such, redox regulation of CaMKII activity may contribute to inhibition of LTP of NMDAR function under oxidizing conditions [14] and impaired LTP of NMDAR function and NMDAR trafficking in advanced age [62].
In the current study, we observed that prior inhibition of CaMKII in young animals blocked a decrease in the NMDAR synaptic response under oxidizing conditions, suggesting that the redox-CaMKII pathway is also important for the redox-mediated decrease in NMDAR function. As such, the redox-CaMKII pathway may contribute to the age-related difference in basal NMDAR function in a manner similar to CaMKII regulation of the maintenance of synaptic strength [32,39]. However, more research is required to determine if the decrease response, due to oxidizing agents, involves a differential influence on GluN2A and GluN2B. CaMKII activity is involved in regulated trafficking and synaptic insertion of GluN2A subunits [27,28,63].
Redox mediated NMDAR hypofunction can act as metaplasticity mechanism, regulating synaptic modifiability required for synaptic networks that underlie cognition [2,3,64]. Furthermore, redox-mediated NMDAR hypofunction, and the interaction of CaMKII and GluN2B are thought to provide a link between altered synaptic plasticity and cognitive deficits for a range of illnesses including Alzheimer’s disease, depression, schizophrenia, and impaired episodic memory during aging [4,8,10,41,42,65,66]. Thus, therapeutic interventions to alleviate the redox-mediated NMDAR hypofunction may rescue synaptic plasticity and improve cognitive function during aging and neurological or psychiatric diseases.
Materials and Methods
Animals
Procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of University of Florida and were in accordance with guidelines established by the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals. Male Fischer 344 rats, young (4–6 months, n = 30) and aged (24-26 months, n = 95), were obtained from the National Institute on Aging colony at Harlan (Indianapolis, IN, USA).
Whole Cell Patch Clamp Recordings
Rats were anaesthetized with intraperitoneal injection of xylazine (10 mg/kg) and ketamine (100 mg/kg). Isoflurane (5%) was administered for 5 min, and following no indication of a withdrawal reflex, animals were perfused transcardially with an ice-cold sucrose-cutting solution (~50 ml) containing (in mM): 206 sucrose, 2 KCl, 25 NaHCO3, 1.2 NaH2PO4, 1 CaCl2, 1 MgSO4, 0.01 glycine, and 10 D-glucose saturated with 95% O2/5% CO2. Following decapitation, brains were removed and placed in ice-cold sucrose-cutting solution for 2 minutes. Horizontal hippocampal sections of 350 µm were generated using a Lecia vibratome 3000 (Buffalo Grove, IL, USA) and sections were immediately transferred to a holding chamber with artificial cerebrospinal fluid, at 32-36ºC, containing (in mM): 124 NaCl, 2.5 KCl, 25 NaHCO3, 1.23 NaH2PO4, 1 CaCl, 3 MgSO4, and 10 D-glucose saturated with 95% O2/5% CO2. Sections were allowed to equilibrate for at least 30 min prior to being transferred into the recording chamber. The recording chamber was perfused (2ml/min) with recording medium (in mM): 126 NaCl, 3 KCl, 25 NaHCO3, 1.2 NaH2PO4, 2.4 CaCl2, 1.5 MgSO4, and 11 D-glucose, saturated with 95% O2/5% CO2, warmed to 30ºC. Prior to patch-clamp recording, sections were allowed to equilibrate for at least 20-minute in the recording chamber.
The recording chamber was mounted on a Burleigh-Gibarltar stage (Thor labs, Newton, NJ, USA) attached to an Olympus BX51WI microscope with infrared differential interference contrast optics (Shinjuku, Tokyo, Japan). Sections were visualized at 10x using a Hamamatsu C4742-95 digital camera (Hamamatsu City, Shizuoka Pref., Japan) attached to a Dell Optiplex 7010 running HCImage Live software (Hamamatsu) on a Windows 7 OS. Glass micropipettes (Sutter, Novato, CA, USA) were filled with a CsMeS03- based solution containing (in mM): 140 CsMeS03, 8 NaCl, 1 MgCl2, 0.2 EGTA, 10 HEPES, 2 Mg-ATP, 0.3 Na-GTP, 5 QX-314, pH 7.3 (CsOH). NMDAR EPSCs were isolated by addition of 6,7-dinitroquinoxaline-2,3-dione (DNQX, 30 μM, Cayman Chemical) and picrotoxin (PTX, 20 μM, Tocris) to the recording medium. Open-tip glass pipette resistances ranged from 3-6 MΩ. Pipettes were placed directly over CA1 pyramidal cell bodies at 10x (~0.2mm from the stimulator electrode) and then a 40x water-emersion objective was used to visualize contact with cell membranes. Whole-cell voltage clamp recordings were obtained with an Axon Multiclamp700B (Molecular Devices, Sunnyvale, CA, USA). Signals were sampled at 10kHz, filtered at 1kHz, and digitized with a Digidata 1440A (Molecular Devices) using Clampex (v.10.2, Molecular Devices). Cells were held at -60mV while access resistance, membrane resistance, and whole-cell capacitance were measured using a -10mV step protocol. Average whole-cell parameters are presented in Table 1. Cells were not included in the final analyses if access resistance changed more than ~35% during the experiment. A concentric bipolar stainless steel electrode was positioned in stratum radiatum and 50 µsec bipolar pulses (0.033 Hz) from a Grass stimulator (SD9) were used to evoke EPSCs and generate stimulation voltage-peak EPSC curves. For pharmacological studies, stimulation intensity was adjusted in order to obtain ~50% maximum EPSC and a baseline response was recorded prior to drug application.
Extracellular field potential recordings
Methods for hippocampal slice preparation and electrophysiological recording of total and NMDAR-mediated synaptic responses have been published previously [6,8]. Briefly, hippocampi were harvested and slices (~400 µm) cut parallel to the alvear fibers. Slices were placed in a recording chamber and bathed in 30 ± 0.5oC oxygenated recording medium (in mM): NaCl 124, KCl 2, KH2PO4 1.25, MgSO4 2, CaCl2 2, NaHCO3 26, and glucose 10. Extracellular fEPSP from stratum radiatum of CA1 were recorded with glass micropipettes (4-6 MΩ) filled with recording medium. A concentric bipolar stainless steel electrode was positioned ~1 mm away in the middle of the stratum radiatum. Field potentials (0.033 Hz) were evoked by biphasic stimulus pulses (100 μsec). Signals were amplified, filtered (1 Hz and 1 kHz), and stored on computer for off-line analysis. For analysis, two cursors were placed around the initial descending phase of the waveform and the maximum slope (mV/ms) of the fEPSP was determined by a computer algorithm that found the maximum change across all sets of consecutively recorded points (20 kHz sampling rate) between the 2 cursors.
The NMDAR-mediated component of synaptic transmission (NMDAR-fEPSP) was isolated by incubating slices in recording medium containing low Mg2+ (0.5 mM), DNQX (30 μM), and PTX (10 μM) for at least 60 minutes. For pharmacological studies, the stimulation intensity was adjusted to evoke a response ~50% of maximum and baseline NMDAR-mediated synaptic responses were recorded for at least 10 min prior to and for 60 min after drug application.
Pharmacological agents
Other pharmacological agents were bath applied at final concentrations of: DTT (0.5 mM, Sigma), DTNB (0.5 mM, Sigma), 2-amino-5-phosphonopentanoic acid (AP5, Sigma, 100 µM), NVP-AAM077 (NVP, 0.4 µM, Sigma), ifenprodil (5 µM, Sigma), Ro 25-6981 (5 µM, Tocris), ZnCl2 (1 µM, Sigma), ZX1 (100 µM, Strem Chemicals), and 4-[(2S)-2-[(5-isoquinolinylsulfonyl)-methylamino]-3-oxo-3-(4-phenyl-1-piperazinyl)propyl] phenyl isoquinolinesulfonic acid ester (KN-62, 10 µM, Tocris). DTT, Ro 25-6981, NVP, ZnCl2, and ZX1 were directly dissolved in the recording medium. DNQX was initially dissolved in DMSO (Sigma) and diluted in recording medium to a final DMSO concentration of <0.01%. PTX, DTNB, and ifenprodil were dissolved in ethanol and diluted in recording medium to a final ethanol concentration of 0.0001%.
Statistical analysis
ANOVAs were carried out using StatView 5.0 (SAS Institute) in order to determine significant main effects and interactions. Post hoc ANOVAs and Fisher’s protected least significant difference comparisons, with the p-value set at 0.05, were used to further localize significant differences. In cases of multiple comparisons (e.g. baseline responses relative to DTT or NMDAR antagonists), Bonferroni corrections were applied.
Supplementary Materials
Author Contributions
AK, designed and performed experiments, analyze data, constructed illustrations, wrote manuscript, TCF, designed the experiments, analyzed data, and wrote the manuscript. JST, performed the experiments.
Acknowledgments
The authors are extremely grateful to Dr. Habibeh Khoshbouei for comments on the manuscript.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
Financial support by National Institutes of Aging Grant R01AG049711, R01AG052258, R01AG037984, and R37AG036800, and the Evelyn F. McKnight Brain Research Foundation is highly appreciated. This work was partially supported by the University of Florida Claude D. Pepper Older Americans Independence Center (P30-AG028740).
References
- 1. Dore K, Stein IS, Brock JA, Castillo PE, Zito K, Sjöström PJ. Unconventional NMDA Receptor Signaling. J Neurosci. 2017; 37:10800–07. https://doi.org/10.1523/JNEUROSCI.1825-17.2017 [PubMed]
- 2. Zorumski CF, Izumi Y. NMDA receptors and metaplasticity: mechanisms and possible roles in neuropsychiatric disorders. Neurosci Biobehav Rev. 2012; 36:989–1000. https://doi.org/10.1016/j.neubiorev.2011.12.011 [PubMed]
- 3. Francis-Oliveira J, Vilar Higa GS, Mendonça Munhoz Dati L, Carvalho Shieh I, De Pasquale R. Metaplasticity in the Visual Cortex: Crosstalk Between Visual Experience and Reactive Oxygen Species. J Neurosci. 2018; 38:5649–65. https://doi.org/10.1523/JNEUROSCI.2617-17.2018 [PubMed]
- 4. Steullet P, Cabungcal JH, Monin A, Dwir D, O’Donnell P, Cuenod M, Do KQ. Redox dysregulation, neuroinflammation, and NMDA receptor hypofunction: A “central hub” in schizophrenia pathophysiology? Schizophr Res. 2016; 176:41–51. https://doi.org/10.1016/j.schres.2014.06.021 [PubMed]
- 5. Ibi M, Liu J, Arakawa N, Kitaoka S, Kawaji A, Matsuda KI, Iwata K, Matsumoto M, Katsuyama M, Zhu K, Teramukai S, Furuyashiki T, Yabe-Nishimura C. Depressive-Like Behaviors Are Regulated by NOX1/NADPH Oxidase by Redox Modification of NMDA Receptor 1. J Neurosci. 2017; 37:4200–12. https://doi.org/10.1523/JNEUROSCI.2988-16.2017 [PubMed]
- 6. Bodhinathan K, Kumar A, Foster TC. Intracellular redox state alters NMDA receptor response during aging through Ca2+/calmodulin-dependent protein kinase II. J Neurosci. 2010; 30:1914–24. https://doi.org/10.1523/JNEUROSCI.5485-09.2010 [PubMed]
- 7. Guidi M, Kumar A, Foster TC. Impaired attention and synaptic senescence of the prefrontal cortex involves redox regulation of NMDA receptors. J Neurosci. 2015; 35:3966–77. https://doi.org/10.1523/JNEUROSCI.3523-14.2015 [PubMed]
- 8. Kumar A, Foster TC. Linking redox regulation of NMDAR synaptic function to cognitive decline during aging. J Neurosci. 2013; 33:15710–15. https://doi.org/10.1523/JNEUROSCI.2176-13.2013 [PubMed]
- 9. Lee WH, Kumar A, Rani A, Foster TC. Role of antioxidant enzymes in redox regulation of N-methyl-D-aspartate receptor function and memory in middle-aged rats. Neurobiol Aging. 2014; 35:1459–68. https://doi.org/10.1016/j.neurobiolaging.2013.12.002 [PubMed]
- 10. Lee WH, Kumar A, Rani A, Herrera J, Xu J, Someya S, Foster TC. Influence of viral vector-mediated delivery of superoxide dismutase and catalase to the hippocampus on spatial learning and memory during aging. Antioxid Redox Signal. 2012; 16:339–50. https://doi.org/10.1089/ars.2011.4054 [PubMed]
- 11. Haxaire C, Turpin FR, Potier B, Kervern M, Sinet PM, Barbanel G, Mothet JP, Dutar P, Billard JM. Reversal of age-related oxidative stress prevents hippocampal synaptic plasticity deficits by protecting D-serine-dependent NMDA receptor activation. Aging Cell. 2012; 11:336–44. https://doi.org/10.1111/j.1474-9726.2012.00792.x [PubMed]
- 12. Yang YJ, Wu PF, Long LH, Yu DF, Wu WN, Hu ZL, Fu H, Xie N, Jin Y, Ni L, Wang JZ, Wang F, Chen JG. Reversal of aging-associated hippocampal synaptic plasticity deficits by reductants via regulation of thiol redox and NMDA receptor function. Aging Cell. 2010; 9:709–21. https://doi.org/10.1111/j.1474-9726.2010.00595.x [PubMed]
- 13. Kumar A, Rani A, Scheinert RB, Ormerod BK, Foster TC. Nonsteroidal anti-inflammatory drug, indomethacin improves spatial memory and NMDA receptor function in aged animals. Neurobiol Aging. 2018; 70:184–93. https://doi.org/10.1016/j.neurobiolaging.2018.06.026 [PubMed]
- 14. Bernard CL, Hirsch JC, Khazipov R, Ben-Ari Y, Gozlan H. Redox modulation of synaptic responses and plasticity in rat CA1 hippocampal neurons. Exp Brain Res. 1997; 113:343–52. https://doi.org/10.1007/BF02450332 [PubMed]
- 15. Foster TC. Dissecting the age-related decline on spatial learning and memory tasks in rodent models: N-methyl-D-aspartate receptors and voltage-dependent Ca2+ channels in senescent synaptic plasticity. Prog Neurobiol. 2012; 96:283–303. https://doi.org/10.1016/j.pneurobio.2012.01.007 [PubMed]
- 16. Kumar A. NMDA Receptor Function During Senescence: Implication on Cognitive Performance. Front Neurosci. 2015; 9:473. https://doi.org/10.3389/fnins.2015.00473 [PubMed]
- 17. Magnusson KR, Brim BL, Das SR. Selective Vulnerabilities of N-methyl-D-aspartate (NMDA) Receptors During Brain Aging. Front Aging Neurosci. 2010; 2:11. [PubMed]
- 18. Rosenzweig ES, Barnes CA. Impact of aging on hippocampal function: plasticity, network dynamics, and cognition. Prog Neurobiol. 2003; 69:143–79. https://doi.org/10.1016/S0301-0082(02)00126-0 [PubMed]
- 19. Kumar A, Yegla B, Foster TC. Redox Signaling in Neurotransmission and Cognition During Aging. Antioxid Redox Signal. 2018; 28:1724–45. https://doi.org/10.1089/ars.2017.7111 [PubMed]
- 20. Morris G, Walder K, Carvalho AF, Tye SJ, Lucas K, Berk M, Maes M. The role of hypernitrosylation in the pathogenesis and pathophysiology of neuroprogressive diseases. Neurosci Biobehav Rev. 2018; 84:453–69. https://doi.org/10.1016/j.neubiorev.2017.07.017 [PubMed]
- 21. Köhr G, Eckardt S, Lüddens H, Monyer H, Seeburg PH. NMDA receptor channels: subunit-specific potentiation by reducing agents. Neuron. 1994; 12:1031–40. https://doi.org/10.1016/0896-6273(94)90311-5 [PubMed]
- 22. Lipton SA, Choi YB, Takahashi H, Zhang D, Li W, Godzik A, Bankston LA. Cysteine regulation of protein function--as exemplified by NMDA-receptor modulation. Trends Neurosci. 2002; 25:474–80. https://doi.org/10.1016/S0166-2236(02)02245-2 [PubMed]
- 23. Sullivan JM, Traynelis SF, Chen HS, Escobar W, Heinemann SF, Lipton SA. Identification of two cysteine residues that are required for redox modulation of the NMDA subtype of glutamate receptor. Neuron. 1994; 13:929–36. https://doi.org/10.1016/0896-6273(94)90258-5 [PubMed]
- 24. Choi Y, Chen HV, Lipton SA. Three pairs of cysteine residues mediate both redox and zn2+ modulation of the nmda receptor. J Neurosci. 2001; 21:392–400. https://doi.org/10.1523/JNEUROSCI.21-02-00392.2001 [PubMed]
- 25. Flint AC, Maisch US, Weishaupt JH, Kriegstein AR, Monyer H. NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. J Neurosci. 1997; 17:2469–76. https://doi.org/10.1523/JNEUROSCI.17-07-02469.1997 [PubMed]
- 26. Stocca G, Vicini S. Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J Physiol. 1998; 507:13–24. https://doi.org/10.1111/j.1469-7793.1998.013bu.x [PubMed]
- 27. Mauceri D, Gardoni F, Marcello E, Di Luca M. Dual role of CaMKII-dependent SAP97 phosphorylation in mediating trafficking and insertion of NMDA receptor subunit NR2A. J Neurochem. 2007; 100:1032–46. https://doi.org/10.1111/j.1471-4159.2006.04267.x [PubMed]
- 28. Yan JZ, Xu Z, Ren SQ, Hu B, Yao W, Wang SH, Liu SY, Lu W. Protein kinase C promotes N-methyl-D-aspartate (NMDA) receptor trafficking by indirectly triggering calcium/calmodulin-dependent protein kinase II (CaMKII) autophosphorylation. J Biol Chem. 2011; 286:25187–200. https://doi.org/10.1074/jbc.M110.192708 [PubMed]
- 29. Sobczyk A, Svoboda K. Activity-dependent plasticity of the NMDA-receptor fractional Ca2+ current. Neuron. 2007; 53:17–24. https://doi.org/10.1016/j.neuron.2006.11.016 [PubMed]
- 30. Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005; 48:289–301. https://doi.org/10.1016/j.neuron.2005.08.034 [PubMed]
- 31. Wang N, Chen L, Cheng N, Zhang J, Tian T, Lu W. Active calcium/calmodulin-dependent protein kinase II (CaMKII) regulates NMDA receptor mediated postischemic long-term potentiation (i-LTP) by promoting the interaction between CaMKII and NMDA receptors in ischemia. Neural Plast. 2014; 2014:827161. https://doi.org/10.1155/2014/827161 [PubMed]
- 32. Barcomb K, Hell JW, Benke TA, Bayer KU. The CaMKII/GluN2B Protein Interaction Maintains Synaptic Strength. J Biol Chem. 2016; 291:16082–89. https://doi.org/10.1074/jbc.M116.734822 [PubMed]
- 33. Cathala L, Misra C, Cull-Candy S. Developmental profile of the changing properties of NMDA receptors at cerebellar mossy fiber-granule cell synapses. J Neurosci. 2000; 20:5899–905. https://doi.org/10.1523/JNEUROSCI.20-16-05899.2000 [PubMed]
- 34. Aniksztejn L, Ben-Ari Y. Expression of LTP by AMPA and/or NMDA receptors is determined by the extent of NMDA receptors activation during the tetanus. J Neurophysiol. 1995; 74:2349–57. https://doi.org/10.1152/jn.1995.74.6.2349 [PubMed]
- 35. Li HB, Jackson MF, Yang K, Trepanier C, Salter MW, Orser BA, Macdonald JF. Plasticity of synaptic GluN receptors is required for the Src-dependent induction of long-term potentiation at CA3-CA1 synapses. Hippocampus. 2011; 21:1053–61. https://doi.org/10.1002/hipo.20818 [PubMed]
- 36. Anderson CT, Radford RJ, Zastrow ML, Zhang DY, Apfel UP, Lippard SJ, Tzounopoulos T. Modulation of extrasynaptic NMDA receptors by synaptic and tonic zinc. Proc Natl Acad Sci USA. 2015; 112:E2705–14. https://doi.org/10.1073/pnas.1503348112 [PubMed]
- 37. Hatton CJ, Paoletti P. Modulation of triheteromeric NMDA receptors by N-terminal domain ligands. Neuron. 2005; 46:261–74. https://doi.org/10.1016/j.neuron.2005.03.005 [PubMed]
- 38. Sullivan JA, Zhang XL, Sullivan AP, Vose LR, Moghadam AA, Fried VA, Stanton PK. Zinc enhances hippocampal long-term potentiation at CA1 synapses through NR2B containing NMDA receptors. PLoS One. 2018; 13:e0205907. https://doi.org/10.1371/journal.pone.0205907 [PubMed]
- 39. Sanhueza M, Fernandez-Villalobos G, Stein IS, Kasumova G, Zhang P, Bayer KU, Otmakhov N, Hell JW, Lisman J. Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength. J Neurosci. 2011; 31:9170–78. https://doi.org/10.1523/JNEUROSCI.1250-11.2011 [PubMed]
- 40. Aizenman E, Hartnett KA, Reynolds IJ. Oxygen free radicals regulate NMDA receptor function via a redox modulatory site. Neuron. 1990; 5:841–46. https://doi.org/10.1016/0896-6273(90)90343-E [PubMed]
- 41. Foster TC, Kyritsopoulos C, Kumar A. Central role for NMDA receptors in redox mediated impairment of synaptic function during aging and Alzheimer’s disease. Behav Brain Res. 2017; 322:223–32. https://doi.org/10.1016/j.bbr.2016.05.012 [PubMed]
- 42. Hardingham GE, Do KQ. Linking early-life NMDAR hypofunction and oxidative stress in schizophrenia pathogenesis. Nat Rev Neurosci. 2016; 17:125–34. https://doi.org/10.1038/nrn.2015.19 [PubMed]
- 43. Robillard JM, Gordon GR, Choi HB, Christie BR, MacVicar BA. Glutathione restores the mechanism of synaptic plasticity in aged mice to that of the adult. PLoS One. 2011; 6:e20676. https://doi.org/10.1371/journal.pone.0020676 [PubMed]
- 44. Williams K. Extracellular Modulation of NMDA Receptors. In: Van Dongen AM, editor. Biology of the NMDA Receptor. Boca Raton (FL)2009.
- 45. Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008; 55:1081–94. https://doi.org/10.1016/j.neuropharm.2008.07.046 [PubMed]
- 46. Rauner C, Köhr G. Triheteromeric NR1/NR2A/NR2B receptors constitute the major N-methyl-D-aspartate receptor population in adult hippocampal synapses. J Biol Chem. 2011; 286:7558–66. https://doi.org/10.1074/jbc.M110.182600 [PubMed]
- 47. Tovar KR, McGinley MJ, Westbrook GL. Triheteromeric NMDA receptors at hippocampal synapses. J Neurosci. 2013; 33:9150–60. https://doi.org/10.1523/JNEUROSCI.0829-13.2013 [PubMed]
- 48. Hansen KB, Ogden KK, Yuan H, Traynelis SF. Distinct functional and pharmacological properties of Triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron. 2014; 81:1084–96. https://doi.org/10.1016/j.neuron.2014.01.035 [PubMed]
- 49. Lind GE, Mou TC, Tamborini L, Pomper MG, De Micheli C, Conti P, Pinto A, Hansen KB. Structural basis of subunit selectivity for competitive NMDA receptor antagonists with preference for GluN2A over GluN2B subunits. Proc Natl Acad Sci USA. 2017; 114:E6942–51. https://doi.org/10.1073/pnas.1707752114 [PubMed]
- 50. Magnusson KR, Nelson SE, Young AB. Age-related changes in the protein expression of subunits of the NMDA receptor. Brain Res Mol Brain Res. 2002; 99:40–45. https://doi.org/10.1016/S0169-328X(01)00344-8 [PubMed]
- 51. Sonntag WE, Bennett SA, Khan AS, Thornton PL, Xu X, Ingram RL, Brunso-Bechtold JK. Age and insulin-like growth factor-1 modulate N-methyl-D-aspartate receptor subtype expression in rats. Brain Res Bull. 2000; 51:331–38. https://doi.org/10.1016/S0361-9230(99)00259-2 [PubMed]
- 52. Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001; 11:327–35. https://doi.org/10.1016/S0959-4388(00)00215-4 [PubMed]
- 53. Roberts EB, Ramoa AS. Enhanced NR2A subunit expression and decreased NMDA receptor decay time at the onset of ocular dominance plasticity in the ferret. J Neurophysiol. 1999; 81:2587–91. https://doi.org/10.1152/jn.1999.81.5.2587 [PubMed]
- 54. Grosshans DR, Clayton DA, Coultrap SJ, Browning MD. LTP leads to rapid surface expression of NMDA but not AMPA receptors in adult rat CA1. Nat Neurosci. 2002; 5:27–33. https://doi.org/10.1038/nn779 [PubMed]
- 55. Bellone C, Nicoll RA. Rapid bidirectional switching of synaptic NMDA receptors. Neuron. 2007; 55:779–85. https://doi.org/10.1016/j.neuron.2007.07.035 [PubMed]
- 56. Xu Z, Chen RQ, Gu QH, Yan JZ, Wang SH, Liu SY, Lu W. Metaplastic regulation of long-term potentiation/long-term depression threshold by activity-dependent changes of NR2A/NR2B ratio. J Neurosci. 2009; 29:8764–73. https://doi.org/10.1523/JNEUROSCI.1014-09.2009 [PubMed]
- 57. Bashir ZI, Alford S, Davies SN, Randall AD, Collingridge GL. Long-term potentiation of NMDA receptor-mediated synaptic transmission in the hippocampus. Nature. 1991; 349:156–58. https://doi.org/10.1038/349156a0 [PubMed]
- 58. Berretta N, Berton F, Bianchi R, Brunelli M, Capogna M, Francesconi W. Long-term Potentiation of NMDA Receptor-mediated EPSP in Guinea-pig Hippocampal Slices. Eur J Neurosci. 1991; 3:850–54. https://doi.org/10.1111/j.1460-9568.1991.tb00096.x [PubMed]
- 59. Muller D, Lynch G. Long-term potentiation differentially affects two components of synaptic responses in hippocampus. Proc Natl Acad Sci USA. 1988; 85:9346–50. https://doi.org/10.1073/pnas.85.23.9346 [PubMed]
- 60. Xie X, Berger TW, Barrionuevo G. Isolated NMDA receptor-mediated synaptic responses express both LTP and LTD. J Neurophysiol. 1992; 67:1009–13. https://doi.org/10.1152/jn.1992.67.4.1009 [PubMed]
- 61. Kotecha SA, Jackson MF, Al-Mahrouki A, Roder JC, Orser BA, MacDonald JF. Co-stimulation of mGluR5 and N-methyl-D-aspartate receptors is required for potentiation of excitatory synaptic transmission in hippocampal neurons. J Biol Chem. 2003; 278:27742–49. https://doi.org/10.1074/jbc.M301946200 [PubMed]
- 62. Clayton DA, Grosshans DR, Browning MD. Aging and surface expression of hippocampal NMDA receptors. J Biol Chem. 2002; 277:14367–69. https://doi.org/10.1074/jbc.C200074200 [PubMed]
- 63. Gardoni F, Schrama LH, van Dalen JJ, Gispen WH, Cattabeni F, Di Luca M. AlphaCaMKII binding to the C-terminal tail of NMDA receptor subunit NR2A and its modulation by autophosphorylation. FEBS Lett. 1999; 456:394–98. https://doi.org/10.1016/S0014-5793(99)00985-0 [PubMed]
- 64. Abraham WC. Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci. 2008; 9:387. https://doi.org/10.1038/nrn2356 [PubMed]
- 65. Opazo P, Viana da Silva S, Carta M, Breillat C, Coultrap SJ, Grillo-Bosch D, Sainlos M, Coussen F, Bayer KU, Mulle C, Choquet D. CaMKII Metaplasticity Drives Aβ Oligomer-Mediated Synaptotoxicity. Cell Reports. 2018; 23:3137–45. https://doi.org/10.1016/j.celrep.2018.05.036 [PubMed]
- 66. Cook SG, Goodell DJ, Restrepo S, Arnold DB, Bayer KU. Simultaneous Live Imaging of Multiple Endogenous Proteins Reveals a Mechanism for Alzheimer’s-Related Plasticity Impairment. Cell Reports. 2019; 27:658–665.e4. https://doi.org/10.1016/j.celrep.2019.03.041 [PubMed]