



Introduction
Ischemic brain injury is the second most common cause of mortality and the third cause of disability in humans [1], resulting in neurological and cognitive deficits, and eventually in dementia of Alzheimer’s disease type [2, 3]. The incidence of dementia following the first ischemic stroke is estimated in 10% of survivors and following recurrent stroke in 33-41% [4]. In long-term, 25 years follow-up of stroke-related dementias, the incidence of dementia was estimated at 48% [5]. Neurological deficits following ischemic stroke in survivors tend to progress to a different extent. On the other hand, cognitive functions gradually deteriorate leading to dementia of Alzheimer’s disease type. Presumably, in the global stroke population, the ischemia-reperfusion episodes will soon become the leading cause of death [1, 6] and of the Alzheimer’s disease type dementia [2, 3].
Recent research has shown that the ischemic brain injury could induce the neuropathology of Alzheimer’s disease type, possibly facilitating the development of dementia, due to amyloidogenesis - processing of the amyloid protein precursor into amyloid [7–9], as well as to the changes in the structure of the tau protein [10–13]. It has been documented that in the human brain, following total and focal ischemia-reperfusion episode, an accumulation of amyloid in the intra- and extracellular spaces occurs [14–17]. It has been shown that both diffuse and senile amyloid plaques form mainly in the cortex and hippocampus [14–17].
Following brain ischemia-reperfusion injury in the rat accumulation of amyloid has also been reported in the hippocampus and cortex, as well as in the white matter [18–20]. In the same animal model, amyloid deposition was observed in neuronal as well as in neuroglial cells [18–20]. The accumulation of diffuse amyloid plaques in response to ischemia-reperfusion brain injury in rats was not transient, since it has been documented that these plaques transform into senile amyloid plaques during one year after ischemic episode [21].
Post-ischemic accumulation of tau protein in neuronal and neuroglial cells was found in the hippocampus and cortex [10, 11, 13]. It has been shown that the dysfunctional tau protein may inhibit the transport of amyloid protein precursor into the cells. In animals and humans dysfunctional tau protein could form paired helical filaments leading to the development of neurofibrillary tangle-like or typical tangle structures [22–24].
Elevated amyloid levels in the post-ischemic animal brain increases the inflammatory response, infarct volume and may affect neurological outcomes [25–28]. The elevated level of soluble β-amyloid peptide predisposes neurons to both hyperactivity and excitotoxicity [29], that can be associated with an increased microglial response [30, 31]. Neuroinflammation modulates the processing of the amyloid protein precursor into amyloid by upregulating the amyloid protein precursor and β-secretase, thereby establishing a specific vicious circle [32–34]. Mice overexpressing the extremely aggregation-prone tau protein show activation of microglia in the brain, all leading to extensive neuronal death [35]. Another study shows that the reactive microglia causes tau protein pathology, contributing to the spread of dysfunctional tau protein in the brain, thus creating the self-perpetuating vicious cycle [36]. Several studies suggested that the resident inflammatory cells, microglia, are the first to respond to ischemia-reperfusion injury in the brain [25, 26, 37, 38] and that through cross-talk with astrocytes they expand neuroinflammation. The neuroinflammatory response following stroke in mice is closely related to the progress and prognosis of stroke in patients [39]. However, the exact effect of microglia on the developing neuroinflammation and its involvement in ischemic-reperfusion brain injury in humans and animals has not been investigated for the long run. With the onset of ischemic brain injury astrocytes aggressively participate in the generation of proinflammatory factors [40]. Depending on the phase of post-ischemic brain pathology, astrocytes can also show anti-inflammatory properties such as in the case of glial scar formation [41]. Notably, no studies have examined the mutual response of microglia and astrocytes in different brain regions under post-ischemic conditions, particularly upon survival time of up to 2 years, and of its translational value. Therefore, the purpose of this study was to determine if post-ischemic activity of microglia and astrocytes, 2 years after the insult, shows regional differences and whether these can be associated with previously described neuronal and functional changes.
Results
Neuroinflammatory response in the rat brain two years after ischemia
Two years post-ischemia, in 26 months old rat brain, we found in the hippocampal CA1 (Figure 1) and CA3 (Figure 2) areas, dentate gyrus (DG) (Figure 3), primary motor cortex (pMO) (Figure 4), primary sensory cortex (pSS) (Figure 5), and in striatum-caudoputamen (STR-CP) (Figure 6), as well as in the dorso-lateral nucleus of thalamus (LD) (Figure 7) a significant increase of astrocytes (GFAP-positive cells) activity in post-ischemic animals vs. sham controls. On the other hand, the study also showed significant microglial activation and infiltration in the rat hippocampal CA1 and CA3 regions and motor cortex (Figures 1, 2, 4). Microglia (Iba1-positive cells) of the ramified type was widespread in CA1 and CA3 areas and motor cortex as opposed to its rare appearances in sham controls (Figures 1, 2, 4).
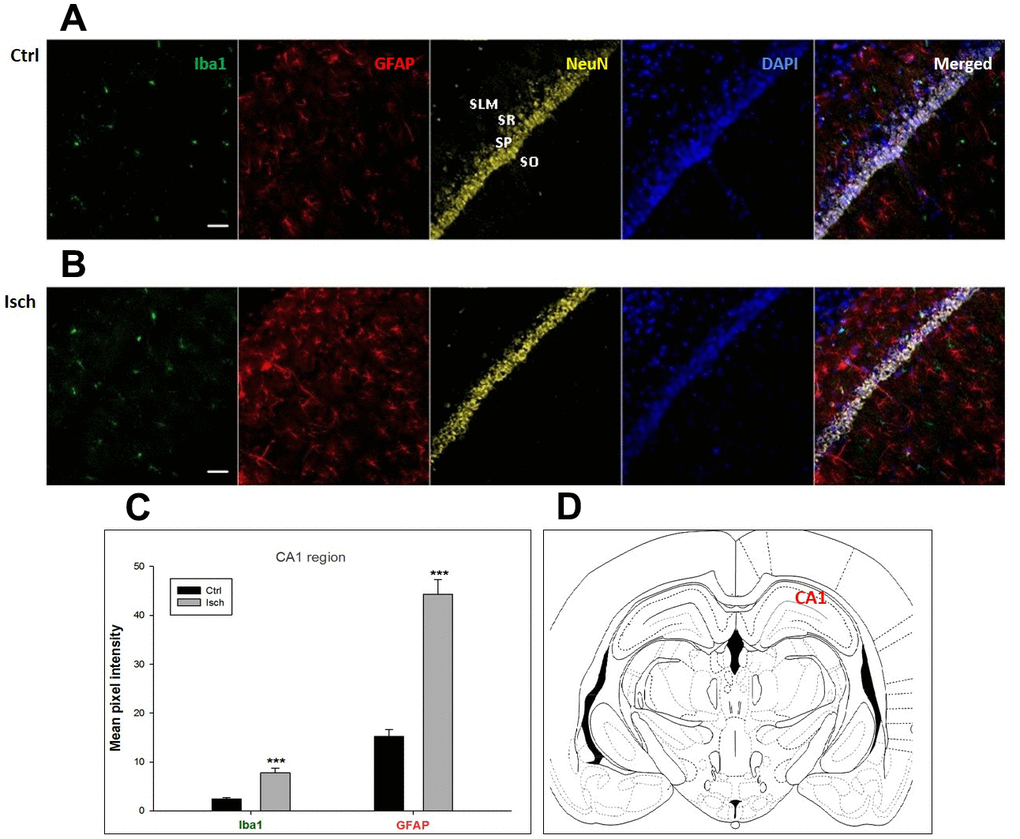
Figure 1. Confocal images of microglia and astrocytes in the post-ischemic CA1 region of the rat brain. Fourfold immunofluorescence labeling microglia with Iba1 (green), astrocytes with GFAP (red), neurons with NeuN (yellow), and nuclei with DAPI (blue). SO - stratum oriens, SP – stratum pyramidale, SR – stratum radiatum, SLM – stratum lacunosum moleculare. The scale bar represents 50 μm. (A) Ctrl – control brain, (B) Isch – post-ischemic brain, (C) Quantification of the mean pixel intensities for Iba1 and GFAP signals of post-ischemic vs. control animals with 2 years survival. Values are presented as mean ± SEM. *** p<0.001. nCtrl = 16, nIsch = 17, n = number of analyzed cross sections. (D) Schematic representation of the rat hippocampus level with CA1 region indicated.
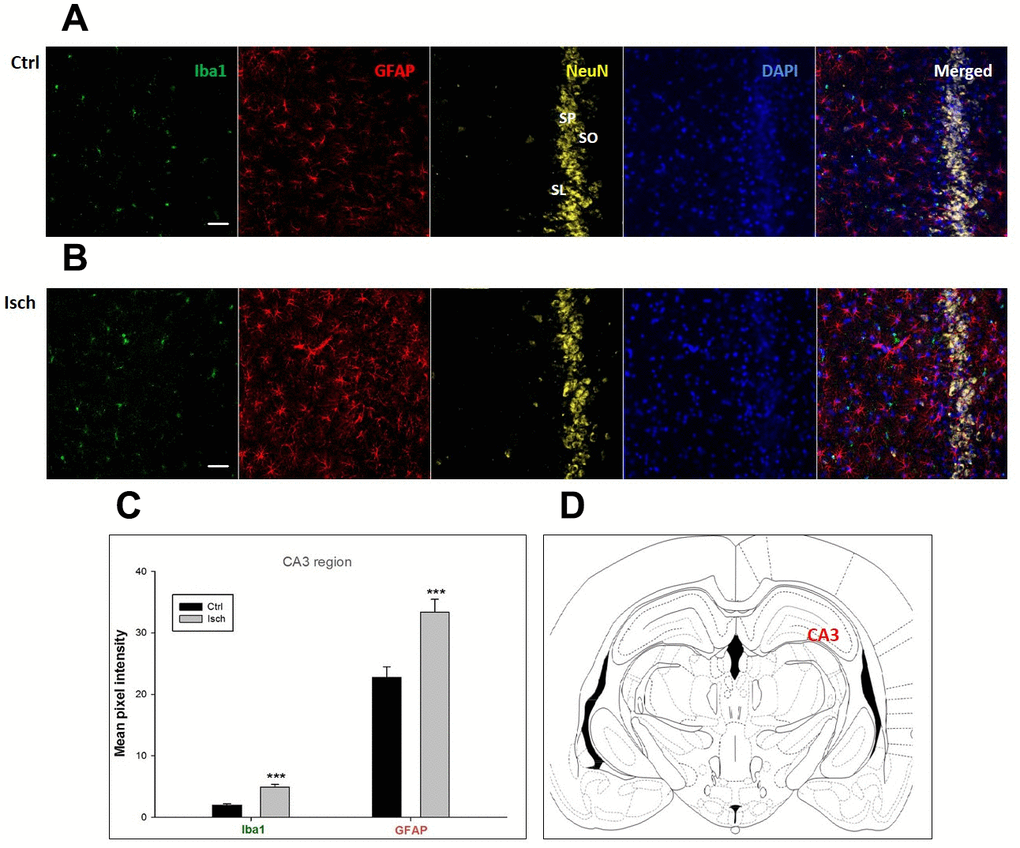
Figure 2. Confocal images of microglia and astrocytes in the post-ischemic CA3 region of the rat brain. Fourfold immunofluorescence labeling microglia with Iba1 (green), astrocytes with GFAP (red), neurons with NeuN (yellow), and nuclei with DAPI (blue). SO – stratum oriens, SP – stratum pyramidale, SL – stratum lacunosum. The scale bar represents 50 μm. (A) Ctrl – control brain, (B) Isch – post-ischemic brain, (C) Quantification of the mean pixel intensities for Iba1 and GFAP signals of post-ischemic vs. control animals with 2 years survival. Values are presented as mean ± SEM. *** p<0.001. nCtrl = 18, nIsch = 18, n = number of analyzed cross sections. (D) Schematic representation of the rat hippocampus level with CA3 region indicated.
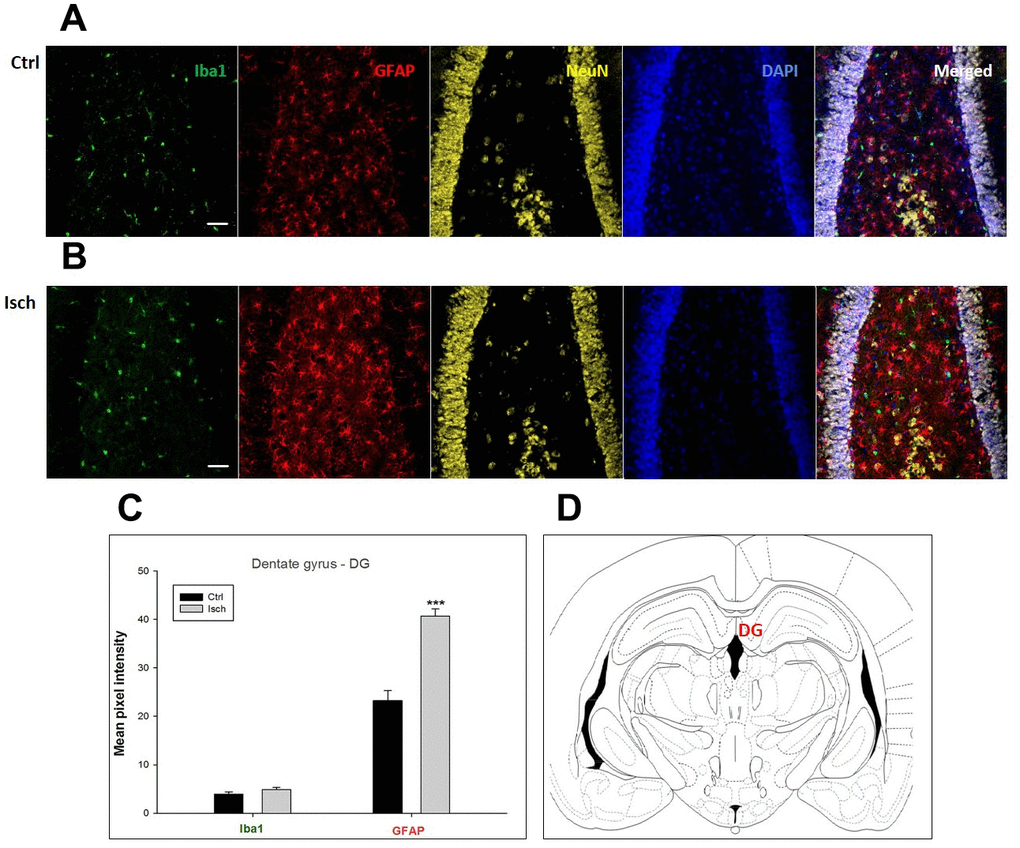
Figure 3. Confocal images of microglia and astrocytes in the post-ischemic dentate gyrus (DG) of the rat brain. Fourfold immunofluorescence labeling microglia with Iba1 (green), astrocytes with GFAP (red), neurons with NeuN (yellow), and nuclei with DAPI (blue). The scale bar represents 50 μm. (A) Ctrl – control brain, (B) Isch – post-ischemic brain, (C) Quantification of the mean pixel intensities for Iba1 and GFAP signals of post-ischemic vs. control animals with 2 years survival. Values are presented as mean ± SEM. *** p<0.001. nCtrl = 20, nIsch = 20, n = number of analyzed cross sections. (D) Schematic representation of the rat hippocampus level with DG region indicated.
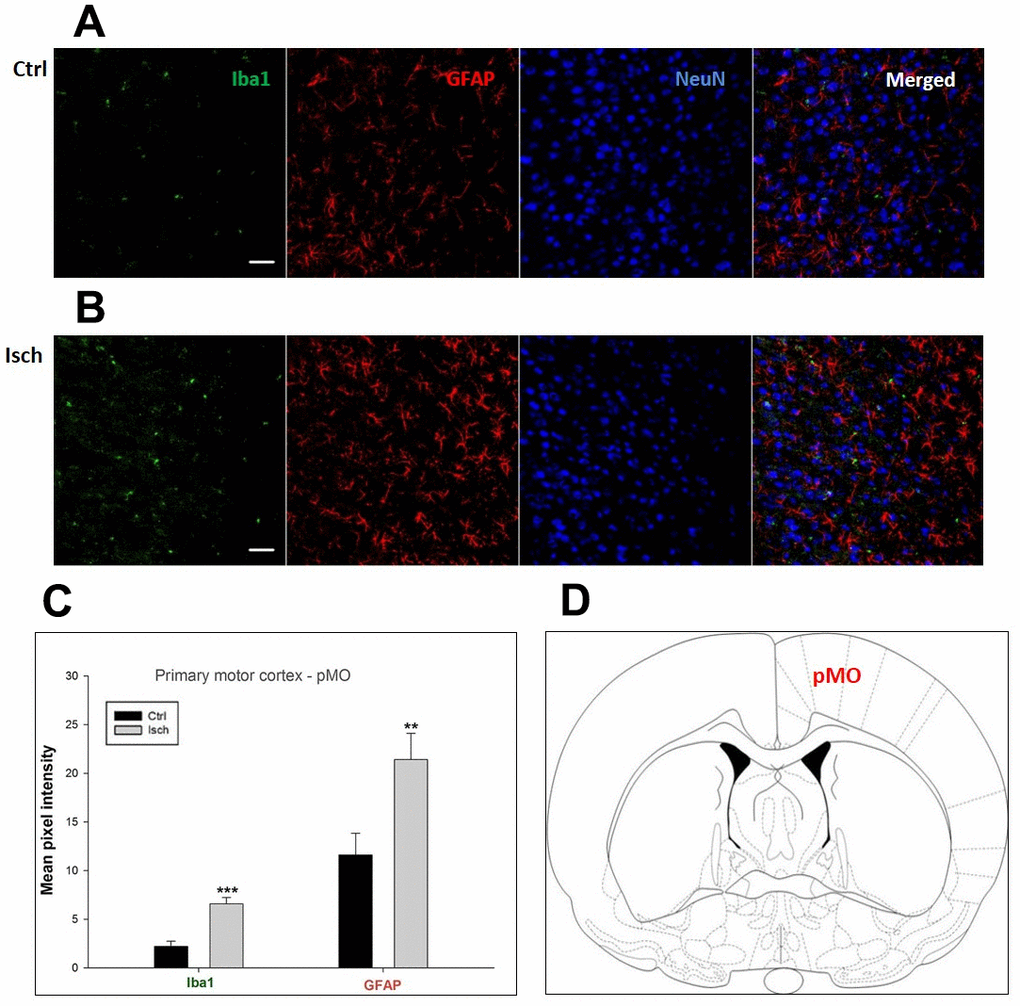
Figure 4. Confocal images of microglia and astrocytes in the post-ischemic primary motor cortex (pMO) of the rat brain. Triple immunofluorescence labeling microglia with Iba1 (green), astrocytes with GFAP (red), and neurons with NeuN (blue). The scale bar represents 50 μm. (A) Ctrl – control brain, (B) Isch – post-ischemic brain, (C) Quantification of the mean pixel intensities for Iba1 and GFAP signals of post-ischemic vs. control animals with 2 years survival. Values are presented as mean ± SEM. ** p<0.01, *** p<0.001. nCtrl = 10, nIsch = 13, n = number of analyzed cross sections. (D) Schematic representation of the rat striatal level with pMO region indicated.
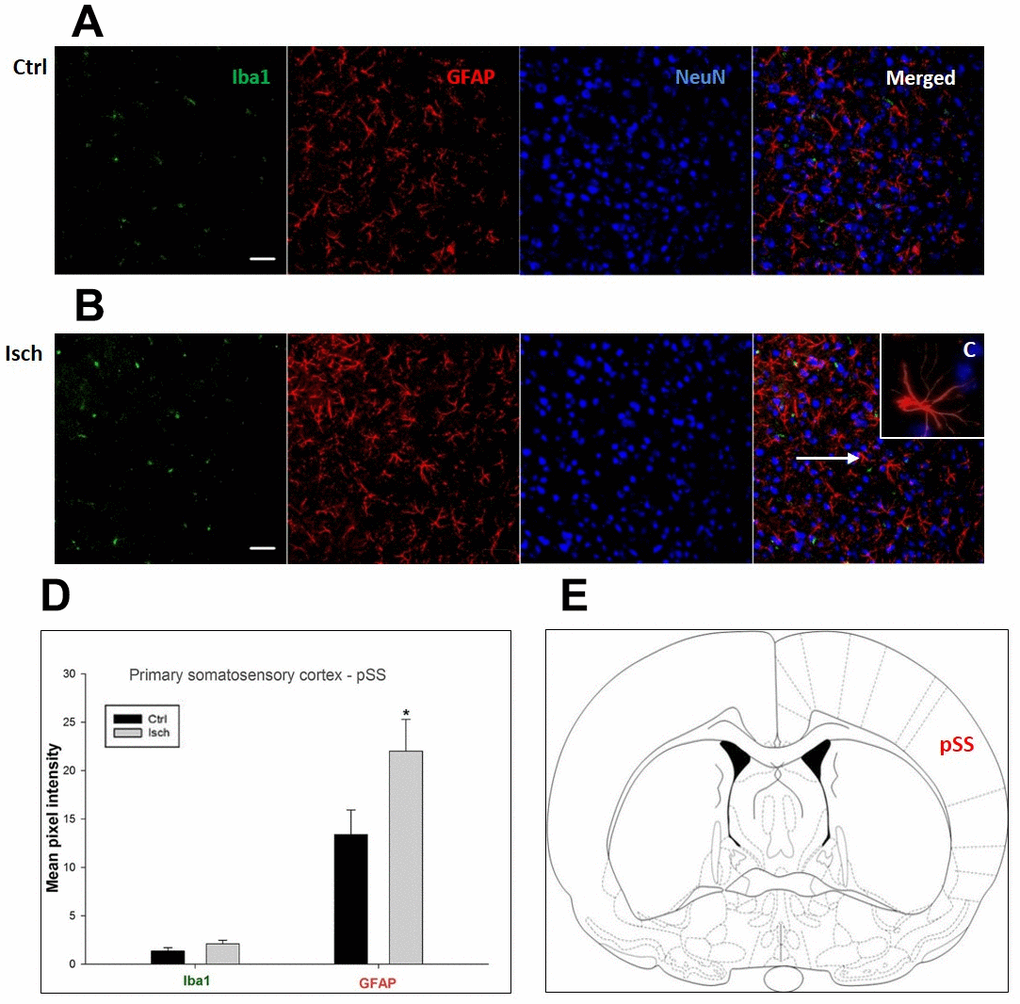
Figure 5. Confocal images of microglia and astrocytes in the post-ischemic primary somatosensory cortex (pSS) of the rat brain. Triple immunofluorescence labeling microglia with Iba1 (green), astrocytes with GFAP (red), and neurons with NeuN (blue). The scale bar represents 50 μm. (A) Ctrl – control brain, (B) Isch – post-ischemic brain, (C) Inset indicating an astrocyte interaction with neurons (8x), (D) Quantification of the mean pixel intensities for Iba1 and GFAP signals of post-ischemic vs. control animals with 2 years survival. Values are presented as mean ± SEM. * p<0.05. nCtrl = 11, nIsch = 11, n = number of analyzed cross sections. (E) Schematic representation of the rat striatal level with pSS region indicated.
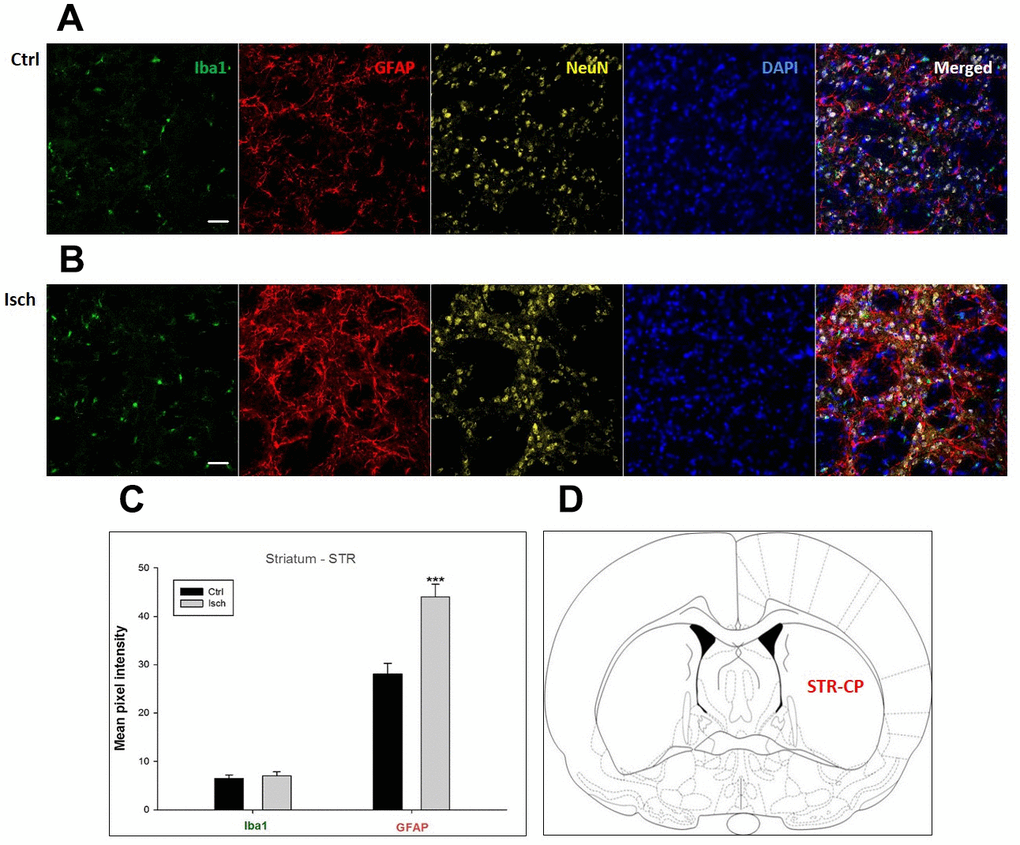
Figure 6. Confocal images of microglia and astrocytes in the post-ischemic striatum-caudoputamen (STR- CP) of the rat brain. Fourfold immunofluorescence labeling microglia with Iba1 (green), astrocytes with GFAP (red), neurons with NeuN (yellow), and nuclei with DAPI (blue). The scale bar represents 50 μm. (A) Ctrl – control brain, (B) Isch – post-ischemic brain, (C) Quantification of the mean pixel intensities for Iba1 and GFAP signals of post-ischemic vs. control animals with 2 years survival. Values are presented as mean ± SEM. *** p<0.001. nCtrl = 16, nIsch = 16, n = number of analyzed cross sections. (D) Schematic representation at rat striatal level with STR-CP region indicated.
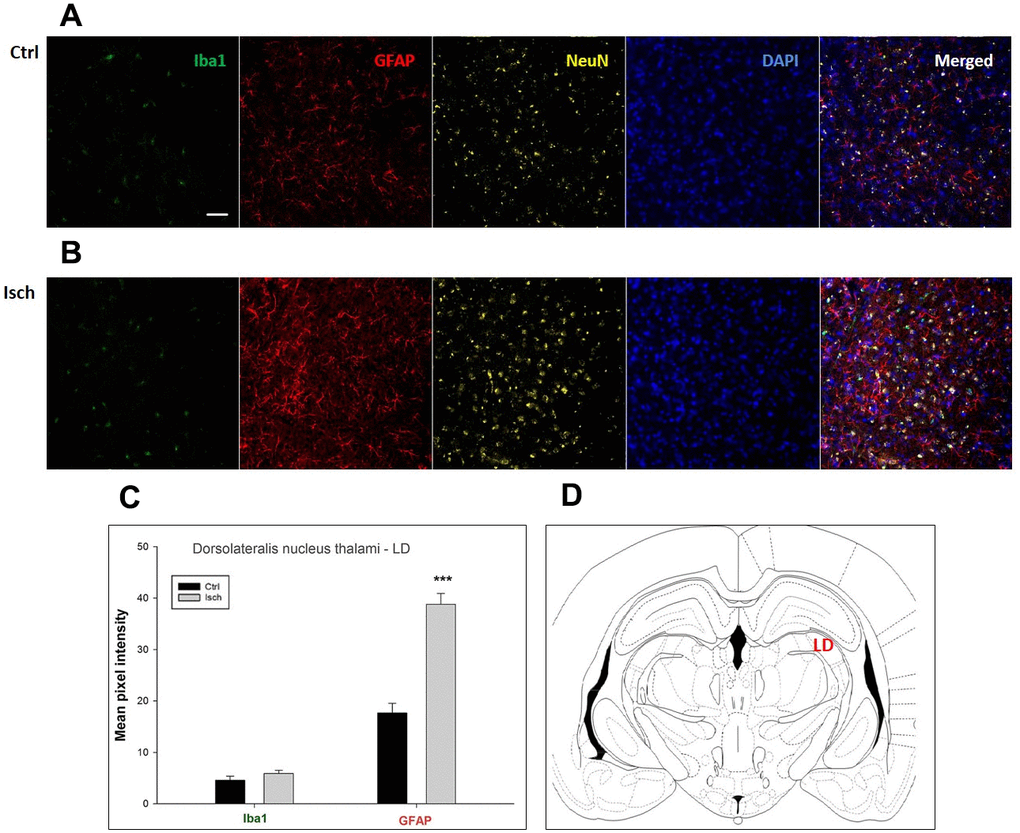
Figure 7. Confocal images of microglia and astrocytes in the post-ischemic dorso-lateral nucleus of thalami (LD) of the rat brain. Fourfold immunofluorescence labeling microglia with Iba1 (green), astrocytes with GFAP (red), neurons with NeuN (yellow), and nuclei with DAPI (blue). The scale bar represents 50 μm. (A) Ctrl – control brain, (B) Isch – post-ischemic brain, (C) Quantification of the mean pixel intensities for Iba1 and GFAP signals of post-ischemic vs. control animals with 2 years survival. Values are presented as mean ± SEM. *** p<0.001. nCtrl = 18, nIsch = 18, n = number of analyzed cross sections. (D) Schematic representation of the rat hippocampus level with LD region indicated.
In hippocampal CA1 and CA3 areas as well as in the motor cortex, GFAP and Iba1 staining was most prominent and significant (Figures 1, 2, 4). Qualitatively weaker signal from NeuN (Figures 1, 2, 4) indicated brain post-ischemic tissue damage in these areas. In terms of Iba1 marker, no statistical significance has been observed between the control and the post-ischemic dentate gyrus (Figure 3B, 3C), primary sensory cortex (Figure 5B, 5D), striatum-caudoputamen (Figure 6B, 6C), and dorso-lateral nucleus of thalamus (Figure 7B, 7C), however the strong GFAP staining indicated a significant increase of astrocytes activity in these brain regions.
The lack of colocalization of NeuN and DAPI signals in the striatum-caudoputamen indicated the presence of non-neuronal cells within the observed brain region. This DAPI signal may come from macrovacuoles that contribute to the sponge-like appearance of caudoputamen (Figure 6B). This indicates that during neuroinflammation after the induced ischemia macrovacuoles and caudoputamen fissures retain blood cells such as leukocytes, possibly also thrombocytes, which infiltrate the brain through the damaged blood-brain barrier [42].
Neurodegeneration response in the rat brain two years after ischemia
The differences in sensitivity to ischemia among seven different brain regions was studied by following the presence of Fluoro-Jade C-labeled neurons that undergo apoptosis in post-ischemic and control brains 2 years after the insult. A trend in increased coefficient of colocalization of Fluoro Jade C and NeuN signals in post-ischemic dentate gyrus, CA3 region, and pSS was found indicating an enhanced number of neurons still entering apoptotic death even 2 years after the ischemic episode (Figure 8). However, although neurodegeneration was found in all analyzed brain regions, a significant difference in coefficient of colocalization has not been observed (Figure 8).
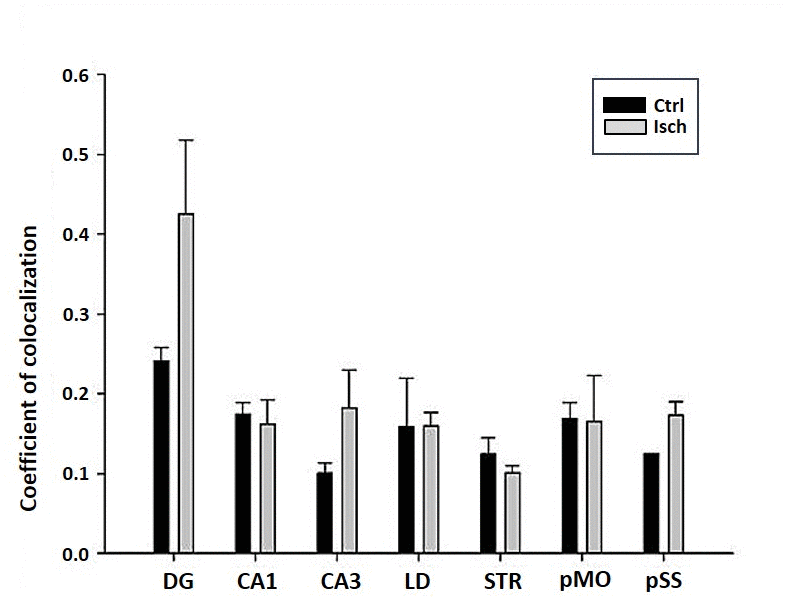
Figure 8. Post-ischemic neurodegeneration of neurons in seven investigated regions of the rat brain upon 2 years of survival. Neurons were immuno-labeled with NeuN, as a neuronal marker, and with Fluoro Jade C, as a marker of deteriorating neurons. The graph shows the quantitative analysis of the coefficient of colocalisation of the two markers in control (Ctrl) and ischemic (Isch) sections in various brain regions studied. DG - dentate gyrus, CA1 and CA3 regions of the hippocampus, LD - dorso-lateral nucleus of thalami, STR-CP - striatum-caudoputamen, pMO - primary motor cortex, pSS - primary somatosensory cortex. Data are presented as mean ± SEM.
Discussion
We are the first to present the heterogeneity in the distribution of microglial and astrocytes activity in seven different brain structures after a 10 min ischemia in the long-term survival rat model. We observed two new microglial and astrocyte activation patterns: in the first one activation was coincidental and statistically significant in both cell types, and in the second pattern, activation was statistically significant only for astrocytes. We have thus demonstrated that in the sensitive areas associated with cerebral ischemia i.e. in the hippocampal CA1 and CA3 areas and in the motor cortex, the co-activation of both microglia and astrocytes was statistically significant. On the other hand, in the resistant brain areas, i.e. in the dentate gyrus, sensory cortex, striatum, and dorso-lateral nucleus of the thalamus, a statistically significant activation was observed only for astrocytes. In general, our study has shed more light on the diversity of microglia and astrocytes in the brain neurodegeneration phenomena after ischemia, with the simultaneous pathology shown in previous studies of amyloid and tau protein [18, 19, 43]. Differences in activation correlate with behavioral deficits associated with ischemia since these depend on the integrity of the hippocampus as well as of the motor cortex [27, 28, 44–48].
Brain ischemia causes the activation of microglia and astrocytes, which triggers the production and secretion of inflammatory mediators, i.e. cytokines [49–51]. By way of these inflammatory cytokines activated microglia and astrocytes affected post-ischemic brain pathogenesis, resulting in increased hyper-phosphorylation of tau protein, amyloid production, transcription and translation of amyloid protein precursor and tau protein in neurons, amyloid-associated pathology, decreased amyloid clearance and activation of kinases CSK-3b and CDK5 [7, 8, 10, 13, 22–24, 52–64]. CDK5 is a kinase associated with hyperphosphorylation of the tau protein [24]. Hyperphosphorylated tau protein after ischemia aggregates into paired helical filaments [23] that eventually form neurofibrillary tangles [22, 24]. During pathological conditions astrocytes begin to express β-secretase, thus obtaining the ability to produce amyloid [32–34]. Neuroinflammation mediates the synergy between brain ischemia and amyloid, causing synaptic depression [65]. It is suggested that the elevated level of soluble β-amyloid peptide predisposes neurons to both hyperactivity and excitotoxicity [29], which is associated with an increase in focal microglial response [30, 31]. The neuroinflammation seen in post-ischemic brain injury presumably appears to play a leading role in increasing the amyloid burden and tau protein dysfunction, suggesting that this dual role may be the leading link between these seemingly different features of Alzheimer’s disease pathology [53]. On the other hand, mice overexpressing mutant tau protein with high aggregation ability demonstrated stimulated hyperactivation of microglia, as well as an extensive loss of neurons that could be dampened by immunosuppression [35]. Activation of microglia contributes to the spread of dysfunctional tau protein in the hippocampus and the motor cortex. It has been mechanistically assumed that microglia phagocytes the tau protein and releases it together with exosomes, thus contributing to the spread of the dysfunctional tau protein [36]. The continuous release of proinflammatory cytokines and neurotoxins from astrocytes and microglia serves to exacerbate the inflammation of the nervous system and contribute to neurodegeneration, leading to further activation of microglia and astrocytes [66].
Chronic opening of the blood-brain barrier has been shown in our model of ischemic rats with long-term survival also confirming that the hippocampus is most susceptible to neuroinflammation and accumulation of the β-amyloid peptide [19, 26, 67]. It has been shown that the blood-brain barrier is disrupted during the brain ischemia-reperfusion injury, in which neurovascular inflammation, characterized by an up-regulation of inflammatory mediators and proteases originating from endothelial and immune cells, plays a significant role [68]. Blood-brain barrier breach may also be strongly associated with the activation of microglia [68]. It has been shown that after an ischemic stroke, the blood-brain barrier integrity was influenced by microglia via an up-regulation of pro-inflammatory cytokines including IL-1b, TNF-a, and IL-6 [68]. It has also been reported that under stroke conditions, brain vessels become permissive to blood serum components, which leak into the parenchyma and thus promote microglial recruitment [69]. Post-ischemic blood-brain barrier disruption may occur by several mechanisms, including the physical alterations of astrocyte-endothelial junctions [41]. Digestion of blood-brain barrier matrix proteins by astrocyte matrix metalloprotease 2 and other matrix metalloproteases contributes to the physical disruption of the blood-brain barrier [41]. Thus, functional integrity of astrocytes proves to be essential for maintaining the blood-brain barrier integrity following the ischemia-reperfusion episode, particularly by reestablishing the astrocytic water channels, AQP4, which are essential for blood-brain barrier repair during post-stroke recovery [41]. Post-ischemic hippocampal and motor cortex neurons have shown the largest loss [19, 43], which correlates here with a significant inflammatory response of microglia and astrocytes in the same brain structures. Our previous study provides evidence of the role of neuroinflammation in post-ischemic cognitive impairments that are correlated with the atrophy of the hippocampus [26, 43, 70]. Microglia and astrocytes are strongly associated with inflammatory changes in the hippocampus and likely contribute to its neurodegeneration and related deterioration of cognitive functions [27, 28, 31]. In fact, some studies suggest that changes in the hippocampus may lead to persistent deterioration of memory in humans and animals, considered as the usual consequence of brain ischemia [44, 48, 71, 72]. In studies analyzing the post-mortem volume of the hippocampus in patients with dementia after ischemia, a decrease in the volume of CA1 and CA3 areas of the hippocampus was demonstrated by approximately 20% in each of the analyzed regions [71, 72]. Some studies, including ours, have shown that the appearance of characteristic features of neurodegeneration, amyloid, and tau protein dysfunction together with inflammatory changes, closely correlated with slow cognitive impairment and dementia after brain ischemia [28, 44].
The neuronal overproduction of the β-amyloid peptide, after cerebral ischemia, stimulates astrocytes to release complement C3, which binds to C3a receptors on neurons and microglia and causes impaired phagocytosis of microglia [73]. On the other hand, the cross-talk of astrocytes with microglia through the activation of complement affects the amyloid pathology [74]. During the development of inflammatory changes after cerebral ischemia, astrocytes either release inflammatory mediators [51], or communicate directly with microglia and/or neurons to modulate the inflammatory response. In focal cerebral ischemia, astrocytes and microglia show proliferative changes in the penumbra [75], indicating that both types of neuroglial cells are activated. Activated microglia has a huge impact on astrogliosis after brain damage, by affecting the development of the glial scar [76]. The glial scar acts as a barrier that prevents axonal ingrowth and reinervation, thus hindering regeneration [50]. It is thus possible that in the ischemic hippocampus, microglia contributes to the pathological changes not only through its direct action, but also indirectly by affecting astrocytes [77]. In fact, it was confirmed that the neurotoxicity of reactive astrocytes was induced by active microglia [77]. Astrocytes and microglia also cooperate in the phagocytosis of ectopic neurons [78]. In chronic, incomplete brain ischemia in rats, neurons with microglia and astrocytes work together creating ectopic and apoptotic neurons, as well as residual neurons. Astrocytes processes can then penetrate into the bodies of ectopic neurons, creating triads with activated microglia [78]. The formation of triads intensifies ischemic processes and leads to severe neurodegeneration. Throughout the progress of the pathological processes, astrocytes on the other hand, may also affect microglia by inhibiting its activity.
Our present study demonstrates that the neuroinflammatory response in the ischemic CA1 and CA3 areas and motor cortex clearly confirms a chronic character with powerful destructive influence. These findings reveal that the active post-ischemic neuroglial response in the hippocampus and the motor cortex lasts much longer than initially thought [79, 80] coinciding with the development of severe and progressive neurodegeneration and dementia after ischemia [28, 44, 46–48].
On the other hand, our study also shows that in ischemic areas of the brain, such as the dentate gyrus of the hippocampus, the sensory cortex, the striatum and the dorso-lateral nucleus of the thalamus, astrocytes alone may also play a role in the progression of cerebral ischemia. Previous studies in this brain ischemia model have shown activation of astrocytes with overexpressed cytokines IL-1β or IL-6 [51]. In the above tested structures under inflammatory changes, the effect of microglia and astrocytes on ischemic pathology is probably limited. We have in fact, shown a smaller effect of neuroinflammatory changes in resistant areas of the brain to ischemia.
Previous studies have been focused on the relationship between ischemic neurons and amyloid and tau protein pathology [7, 8, 10, 13, 18–24, 54–64] while neuroglial pathology has often been neglected. With the emerging new evidence, including the results of our study, it becomes evident that microglia and astrocytes are not only witnesses, but active and important participants in neurodegeneration in the post-ischemic brain, as well as in Alzheimer’s disease. Thus, we can foresee that the understanding of the relationship between neurons and neuroglial cells after brain ischemia will become more important in the future.
Conclusions
Our study, revealed the role of neuroinflammation in neurodegeneration throughout the post-ischemic brain. The role is quite complex and goes beyond the scope of only one research article. These findings show that the effect of brain ischemia on the activity of microglia and astrocytes is significantly different among the brain structures studied. This partly explains why in rats the extent to which neurodegeneration occurs in the brain after ischemia varies greatly depending on the area and does not develop at the same time [7–10, 13, 81–83].
Currently, the full temporal and spatial dynamics of the inflammatory response after ischemia is unknown. One way to broaden the scope of research is to move away from the biochemical theory of post-ischemic neurodegeneration towards cellular theory instead. We hope that the new experimental approaches, such as the study of gene expression changes in neuroglial cells in our model, will provide new knowledge about the complex spatial and temporal nature of the neuroinflammatory response after brain injury as a result of ischemia and reperfusion. Finally, the development of novel cell imaging tools in vivo can facilitate the development of new strategies for the treatment of stroke patients.
Materials and Methods
Animals
Two-month-old, female rats (Wistar, 160–180 g, n=16) were used for the study. Groups of four animals per cage, were housed in an air-conditioned room, at the temperature of 22 ± 2 °C, with 55 ± 5% humidity, and with lights 12 h/day (07.00 - 19.00). The animals were given commercial food and tap water ad libitum. All experimental procedures were performed during the light phase, between 9:00 and 15:00 under identical conditions. Animals used for procedures were treated in strict accordance with the European Communities Council Directive (86/609/EEC and 2010/63/EU) and with the approval of the local Ethical Committee. All efforts were made to minimize animal suffering and to reduce the number of animals used, in accordance with principles of good laboratory practice.
Brain ischemia model in rats with long-term survival
Our animal model of global cerebral ischemia clinically represents reversible cardiac arrest. Global cerebral ischemia was performed by cardiac arrest of 10 min duration [13, 84]. The animals were allowed to survive 2 years post-ischemia. Sham-operated rats were exposed to the same procedures as ischemic animals but without induced cardiac arrest and thus served as controls.
Immunocytochemistry
Immunocytochemistry was performed 2 years after the ischemic insult on 6 ischemic and 6 control rats. There was no mortality within 2 years after successful resuscitation (n = 6), but during cardiac arrest it reached 40% (n = 4/10). During brain autopsy of rats that died during cardiac arrest, no macroscopic lesions were observed. After transcardiac perfusion with 4 % paraformaldehyde the brains of resuscitated or sham controlled animals were postfixed in the same solution and cryoprotected in 30% sucrose. After freezing at -80oC brains were cut on a cryostat in 30 μm-thick coronal slices. Microglial cells were labeled with anti-Iba1 (1:250, Abcam), astrocytes with anti-GFAP (1:300, Daco), and neurons with anti-NeuN (1:100, Milipore) primary antibodies and visualized with the use of Alexa Fluor 488-conjugated donkey polyclonal anti-goat antibodies, Alexa Fluor 555-conjugated donkey polyclonal anti-rabbit, and Cy5 633-conjugated donkey polyclonal anti-mouse secondary antibodies, respectively (all 1:200, Molecular Probes). The slides were then washed with PBS and stained with the nuclear marker 4,6-diamidino-2-phenylindole (DAPI, 1:200, Molecular Probes). In parallel, double staining with anti-NeuN (1:100, Milipore) and Fluoro Jade C (1:100, Millipore) was used for visualization of neurodegenerated neurons. After washing the slides were dried and coverslipped using mounting medium (Mowiol, Sigma Aldrich). As a negative control primary antibodies were omitted. Studies of immunohistochemical reactions of different sections from all experimental and control groups were processed in parallel.
Image acquisition
Immunostained sections were imaged by a confocal laser scanning microscope (LSM 510, Carl Zeiss GmbH) with an argon laser (488 nm) utilized for the excitation of Alexa Fluor 488 and Fluoro Jade C, and helium-neon lasers (543 nm and 633 nm), for the excitation of Alexa Fluor 555 and Cy5, respectively. Objectives used were Plan - Neofluar 20x/0.5. Laser intensities, pinhole, scan speed, digital gain and offset were maintained constant throughout imaging.
Image analysis and quantification
Following acquisition with Zeiss LSM 510 confocal, images were processed using the Zeiss LSM 510 Basic software package v. 3.2. In order to investigate the activity of astrocytes and microglia within the specific brain regions, mean signal intensity of the Iba1 and the GFAP signal pixels were calculated for each image after thresholding for background fluorescence (six animals/group, 4–5 images/animal, in total 24-30 images/group). The number of analyzed brain slices was n=16-20 per group (control and ischemia).
In addition, we quantified by pixel analysis the colocalization of the green (Fluoro Jade C) over red (NeuN) signal. This was performed with confocal images taken from brain slices with the objective magnification of 20x, n=20 for each group (control or ischemia).
Statistics
Experimental values were statistically compared with the Student’s t-test using Sigma Plot 11.0 software package (Systat Software, Inc.).
Acknowledgments
The authors acknowledge the financial support from the following institutions: the Mossakowski Medical Research Centre, Polish Academy of Sciences, Warsaw, Poland (T3-RP), the Medical University of Lublin, Lublin, Poland (DS 475/19-SJC) and the Ministry of Education, Science and Technological Development Republic of Serbia (Grant No. 41005).
Conflicts of Interest
The authors declare that there are no conflicts of interest.
Funding
This research received no external funding.
References
- 1. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Després JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, et al, and Writing Group Members, American Heart Association Statistics Committee, and Stroke Statistics Subcommittee. Executive summary: heart disease and stroke statistics—2016 update: a report from the american heart association. Circulation. 2016; 133:447–54. https://doi.org/10.1161/CIR.0000000000000366 [PubMed]
- 2. Kim JH, Lee Y. Dementia and death after stroke in older adults during a 10-year follow-up: results from a competing risk model. J Nutr Health Aging. 2018; 22:297–301. https://doi.org/10.1007/s12603-017-0914-3 [PubMed]
- 3. Portegies ML, Wolters FJ, Hofman A, Ikram MK, Koudstaal PJ, Ikram MA. Prestroke vascular pathology and the risk of recurrent stroke and poststroke dementia. Stroke. 2016; 47:2119–22. https://doi.org/10.1161/STROKEAHA.116.014094 [PubMed]
- 4. Pendlebury ST, Rothwell PM. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: a systematic review and meta-analysis. Lancet Neurol. 2009; 8:1006–18. https://doi.org/10.1016/S1474-4422(09)70236-4 [PubMed]
- 5. Kokmen E, Whisnant JP, O’Fallon WM, Chu CP, Beard CM. Dementia after ischemic stroke: a population-based study in rochester, minnesota (1960-1984). Neurology. 1996; 46:154–9. https://doi.org/10.1212/wnl.46.1.154 [PubMed]
- 6. Cassella CR, Jagoda A. Ischemic stroke: advances in diagnosis and management. Emerg Med Clin North Am. 2017; 35:911–30. https://doi.org/10.1016/j.emc.2017.07.007 [PubMed]
- 7. Kocki J, Ułamek-Kozioł M, Bogucka-Kocka A, Januszewski S, Jabłoński M, Gil-Kulik P, Brzozowska J, Petniak A, Furmaga-Jabłońska W, Bogucki J, Czuczwar SJ, Pluta R. Dysregulation of amyloid-β protein precursor, β-secretase, presenilin 1 and 2 genes in the rat selectively vulnerable CA1 subfield of hippocampus following transient global brain ischemia. J Alzheimers Dis. 2015; 47:1047–56. https://doi.org/10.3233/JAD-150299 [PubMed]
- 8. Pluta R, Kocki J, Ułamek-Kozioł M, Petniak A, Gil-Kulik P, Januszewski S, Bogucki J, Jabłoński M, Brzozowska J, Furmaga-Jabłońska W, Bogucka-Kocka A, Czuczwar SJ. Discrepancy in expression of β-secretase and amyloid-β protein precursor in Alzheimer-related genes in the rat medial temporal lobe cortex following transient global brain ischemia. J Alzheimers Dis. 2016; 51:1023–31. https://doi.org/10.3233/JAD-151102 [PubMed]
- 9. Pluta R, Kocki J, Ułamek-Kozioł M, Bogucka-Kocka A, Gil-Kulik P, Januszewski S, Jabłoński M, Petniak A, Brzozowska J, Bogucki J, Furmaga-Jabłońska W, Czuczwar SJ. Alzheimer-associated presenilin 2 gene is dysregulated in rat medial temporal lobe cortex after complete brain ischemia due to cardiac arrest. Pharmacol Rep. 2016; 68:155–61. https://doi.org/10.1016/j.pharep.2015.08.002 [PubMed]
- 10. Pluta R, Bogucka-Kocka A, Ułamek-Kozioł M, Bogucki J, Januszewski S, Kocki J, Czuczwar SJ. Ischemic tau protein gene induction as an additional key factor driving development of Alzheimer’s phenotype changes in CA1 area of hippocampus in an ischemic model of Alzheimer’s disease. Pharmacol Rep. 2018; 70:881–84. https://doi.org/10.1016/j.pharep.2018.03.004 [PubMed]
- 11. Pluta R, Ułamek-Kozioł M, Januszewski S, Czuczwar SJ. Tau protein dysfunction after brain ischemia. J Alzheimers Dis. 2018; 66:429–37. https://doi.org/10.3233/JAD-180772 [PubMed]
- 12. Pluta R. Brain ischemia: Alzheimer’s disease mechanisms. Nova Science Publishers, Inc. New York. 2019.
- 13. Pluta R, Ułamek-Kozioł M, Kocki J, Bogucki J, Januszewski S, Bogucka-Kocka A, Czuczwar SJ. Expression of the tau protein and amyloid protein precursor processing genes in the CA3 area of the hippocampus in the ischemic model of Alzheimer’s disease in the rat. Mol Neurobiol. 2020; 57:1281–90. https://doi.org/10.1007/s12035-019-01799-z [PubMed]
- 14. Jendroska K, Poewe W, Daniel SE, Pluess J, Iwerssen-Schmidt H, Paulsen J, Barthel S, Schelosky L, Cervós-Navarro J, DeArmond SJ. Ischemic stress induces deposition of amyloid beta immunoreactivity in human brain. Acta Neuropathol. 1995; 90:461–6. https://doi.org/10.1007/BF00294806 [PubMed]
- 15. Jendroska K, Hoffmann OM, Patt S. Amyloid beta peptide and precursor protein (APP) in mild and severe brain ischemia. Ann N Y Acad Sci. 1997; 826:401–5. https://doi.org/10.1111/j.1749-6632.1997.tb48492.x [PubMed]
- 16. Qi JP, Wu H, Yang Y, Wang DD, Chen YX, Gu YH, Liu T. Cerebral ischemia and Alzheimer’s disease: the expression of amyloid-beta and apolipoprotein E in human hippocampus. J Alzheimers Dis. 2007; 12:335–41. https://doi.org/10.3233/jad-2007-12406 [PubMed]
- 17. Wiśniewski HM, Maślińska D. Beta-protein immunoreactivity in the human brain after cardiac arrest. Folia Neuropathol. 1996; 34:65–71. [PubMed]
- 18. Pluta R, Kida E, Lossinsky AS, Golabek AA, Mossakowski MJ, Wisniewski HM. Complete cerebral ischemia with short-term survival in rats induced by cardiac arrest. I. Extracellular accumulation of Alzheimer’s beta-amyloid protein precursor in the brain. Brain Res. 1994; 649:323–8. https://doi.org/10.1016/0006-8993(94)91081-2 [PubMed]
- 19. Pluta R, Ułamek M, Jabłoński M. Alzheimer’s mechanisms in ischemic brain degeneration. Anat Rec (Hoboken). 2009; 292:1863–81. https://doi.org/10.1002/ar.21018 [PubMed]
- 20. Pluta R, Ułamek-Kozioł M, Czuczwar SJ. Alzheimer’s disease-related proteins following ischemia-reperfusion brain injury. In Brain ischemia: Alzheimer’s disease mechanisms. Ed. Pluta R. Nova Science Publishers, Inc. New York. 2019:pp. 53–75.
- 21. van Groen T, Puurunen K, Mäki HM, Sivenius J, Jolkkonen J. Transformation of diffuse beta-amyloid precursor protein and beta-amyloid deposits to plaques in the thalamus after transient occlusion of the middle cerebral artery in rats. Stroke. 2005; 36:1551–6. https://doi.org/10.1161/01.STR.0000169933.88903.cf [PubMed]
- 22. Kato T, Hirano A, Katagiri T, Sasaki H, Yamada S. Neurofibrillary tangle formation in the nucleus basalis of meynert ipsilateral to a massive cerebral infarct. Ann Neurol. 1988; 23:620–3. https://doi.org/10.1002/ana.410230617 [PubMed]
- 23. Khan S, Yuldasheva NY, Batten TFC, Pickles AR, Kellett KAB, Saha S. Tau pathology and neurochemical changes associated with memory dysfunction in an optimized murine model of global cerebral ischaemia – A potential model for vascular dementia? Neurochem Int. 2018; 118:134–144. https://doi.org/10.1016/j.neuint.2018.04.004 [PubMed]
- 24. Wen Y, Yang SH, Liu R, Perez EJ, Brun-Zinkernagel AM, Koulen P, Simpkins JW. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim Biophys Acta. 2007; 1772:473–83. https://doi.org/10.1016/j.bbadis.2006.10.011 [PubMed]
- 25. Fu Y, Liu Q, Anrather J, Shi FD. Immune interventions in stroke. Nat Rev Neurol. 2015; 11:524–35. https://doi.org/10.1038/nrneurol.2015.144 [PubMed]
- 26. Sekeljic V, Bataveljic D, Stamenkovic S, Ułamek M, Jabłoński M, Radenovic L, Pluta R, Andjus PR. Cellular markers of neuroinflammation and neurogenesis after ischemic brain injury in the long-term survival rat model. Brain Struct Funct. 2012; 217:411–20. https://doi.org/10.1007/s00429-011-0336-7 [PubMed]
- 27. Thiel A, Cechetto DF, Heiss WD, Hachinski V, Whitehead SN. Amyloid burden, neuroinflammation, and links to cognitive decline after ischemic stroke. Stroke. 2014; 45:2825–9. https://doi.org/10.1161/STROKEAHA.114.004285 [PubMed]
- 28. Whitehead SN, Cheng G, Hachinski VC, Cechetto DF. Progressive increase in infarct size, neuroinflammation, and cognitive deficits in the presence of high levels of amyloid. Stroke. 2007; 38:3245–50. https://doi.org/10.1161/STROKEAHA.107.492660 [PubMed]
- 29. Pluta R, Salínska E, Puka M, Stafiej A, Lazarewicz JW. Early changes in extracellular amino acids and calcium concentrations in rabbit hippocampus following complete 15-min cerebral ischemia. Resuscitation. 1988; 16:193–210. https://doi.org/10.1016/0300-9572(88)90046-9 [PubMed]
- 30. Fan Z, Okello AA, Brooks DJ, Edison P. Longitudinal influence of microglial activation and amyloid on neuronal function in Alzheimer’s disease. Brain. 2015; 138:3685–98. https://doi.org/10.1093/brain/awv288 [PubMed]
- 31. Ferreira ST, Lourenco MV, Oliveira MM, De Felice FG. Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front Cell Neurosci. 2015; 9:191. https://doi.org/10.3389/fncel.2015.00191 [PubMed]
- 32. Carter SF, Herholz K, Rosa-Neto P, Pellerin L, Nordberg A, Zimmer ER. Astrocyte biomarkers in Alzheimer’s disease. Trends Mol Med. 2019; 25:77–95. https://doi.org/10.1016/j.molmed.2018.11.006 [PubMed]
- 33. Frost GR, Li YM. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017; 7:170228. https://doi.org/10.1098/rsob.170228 [PubMed]
- 34. Sastre M, Dewachter I, Landreth GE, Willson TM, Klockgether T, van Leuven F, Heneka MT. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J Neurosci. 2003; 23:9796–804. https://doi.org/10.1523/JNEUROSCI.23-30-09796.2003 [PubMed]
- 35. Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007; 53:337–51. https://doi.org/10.1016/j.neuron.2007.01.010 [PubMed]
- 36. Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, Lamb BT, Bhaskar K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain. 2015; 138:1738–55. https://doi.org/10.1093/brain/awv081 [PubMed]
- 37. Chamorro Á, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R. The immunology of acute stroke. Nat Rev Neurol. 2012; 8:401–10. https://doi.org/10.1038/nrneurol.2012.98 [PubMed]
- 38. Ma Y, Wang J, Wang Y, Yang GY. The biphasic function of microglia in ischemic stroke. Prog Neurobiol. 2017; 157:247–72. https://doi.org/10.1016/j.pneurobio.2016.01.005 [PubMed]
- 39. Hankey GJ. Stroke. Lancet. 2017; 389:641–54. https://doi.org/10.1016/S0140-6736(16)30962-X [PubMed]
- 40. Colombo E, Farina C. Astrocytes: key regulators of neuroinflammation. Trends Immunol. 2016; 37:608–20. https://doi.org/10.1016/j.it.2016.06.006 [PubMed]
- 41. Liu Z, Chopp M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog Neurobiol. 2016; 144:103–20. https://doi.org/10.1016/j.pneurobio.2015.09.008 [PubMed]
- 42. Pluta R, Lossinsky AS, Walski M, Wisniewski HM, Mossakowski MJ. Platelet occlusion phenomenon after short- and long-term survival following complete cerebral ischemia in rats produced by cardiac arrest. J Hirnforsch. 1994; 35:463–71. [PubMed]
- 43. Pluta R. The role of apolipoprotein E in the deposition of beta-amyloid peptide during ischemia-reperfusion brain injury. A model of early Alzheimer’s disease. Ann N Y Acad Sci. 2000; 903:324–34. https://doi.org/10.1111/j.1749-6632.2000.tb06383.x [PubMed]
- 44. Kiryk A, Pluta R, Figiel I, Mikosz M, Ulamek M, Niewiadomska G, Jablonski M, Kaczmarek L. Transient brain ischemia due to cardiac arrest causes irreversible long-lasting cognitive injury. Behav Brain Res. 2011; 219:1–7. https://doi.org/10.1016/j.bbr.2010.12.004 [PubMed]
- 45. Pluta R, Gajkowska B. Ultrastructural changes in the sensomotor cortex of the rabbit after complete 30-min brain ischemia. J Neurosci Res. 1984; 11:35–47. https://doi.org/10.1002/jnr.490110105 [PubMed]
- 46. de la Tremblaye PB, Plamondon H. Impaired conditioned emotional response and object recognition are concomitant to neuronal damage in the amygdala and perirhinal cortex in middle-aged ischemic rats. Behav Brain Res. 2011; 219:227–33. https://doi.org/10.1016/j.bbr.2011.01.009 [PubMed]
- 47. Li J, Wang YJ, Zhang M, Fang CQ, Zhou HD. Cerebral ischemia aggravates cognitive impairment in a rat model of Alzheimer’s disease. Life Sci. 2011; 89:86–92. https://doi.org/10.1016/j.lfs.2011.04.024 [PubMed]
- 48. Cohan CH, Neumann JT, Dave KR, Alekseyenko A, Binkert M, Stransky K, Lin HW, Barnes CA, Wright CB, Perez-Pinzon MA. Effect of cardiac arrest on cognitive impairment and hippocampal plasticity in middle-aged rats. PLoS One. 2015; 10:e0124918. https://doi.org/10.1371/journal.pone.0124918 [PubMed]
- 49. Kanazawa M, Ninomiya I, Hatakeyama M, Takahashi T, Shimohata T. Microglia and monocytes/macrophages polarization reveal novel therapeutic mechanism against stroke. Int J Mol Sci. 2017; 18:2135. https://doi.org/10.3390/ijms18102135 [PubMed]
- 50. Kawabori M, Yenari MA. Inflammatory responses in brain ischemia. Curr Med Chem. 2015; 22:1258–77. https://doi.org/10.2174/0929867322666150209154036 [PubMed]
- 51. Orzyłowska O, Oderfeld-Nowak B, Zaremba M, Januszewski S, Mossakowski M. Prolonged and concomitant induction of astroglial immunoreactivity of interleukin-1beta and interleukin-6 in the rat hippocampus after transient global ischemia. Neurosci Lett. 1999; 263:72–6. https://doi.org/10.1016/s0304-3940(99)00043-9 [PubMed]
- 52. Ahmad MH, Fatima M, Mondal AC. Influence of microglia and astrocyte activation in the neuroinflammatory pathogenesis of Alzheimer’s disease: rational insights for the therapeutic approaches. J Clin Neurosci. 2019; 59:6–11. https://doi.org/10.1016/j.jocn.2018.10.034 [PubMed]
- 53. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement (N Y). 2018; 4:575–90. https://doi.org/10.1016/j.trci.2018.06.014 [PubMed]
- 54. Banati RB, Gehrmann J, Wiessner C, Hossmann KA, Kreutzberg GW. Glial expression of the beta-amyloid precursor protein (APP) in global ischemia. J Cereb Blood Flow Metab. 1995; 15:647–54. https://doi.org/10.1038/jcbfm.1995.80 [PubMed]
- 55. Badan I, Platt D, Kessler C, Popa-Wagner A. Temporal dynamics of degenerative and regenerative events associated with cerebral ischemia in aged rats. Gerontology. 2003; 49:356–65. https://doi.org/10.1159/000073763 [PubMed]
- 56. Badan I, Dinca I, Buchhold B, Suofu Y, Walker L, Gratz M, Platt D, Kessler CH, Popa-Wagner A. Accelerated accumulation of N- and c-terminal beta APP fragments and delayed recovery of microtubule-associated protein 1B expression following stroke in aged rats. Eur J Neurosci. 2004; 19:2270–80. https://doi.org/10.1111/j.0953-816X.2004.03323.x [PubMed]
- 57. Majd S, Power JH, Koblar SA, Grantham HJ. Early glycogen synthase kinase-3β and protein phosphatase 2A independent tau dephosphorylation during global brain ischaemia and reperfusion following cardiac arrest and the role of the adenosine monophosphate kinase pathway. Eur J Neurosci. 2016; 44:1987–97. https://doi.org/10.1111/ejn.13277 [PubMed]
- 58. Fujii H, Takahashi T, Mukai T, Tanaka S, Hosomi N, Maruyama H, Sakai N, Matsumoto M. Modifications of tau protein after cerebral ischemia and reperfusion in rats are similar to those occurring in Alzheimer’s disease - hyperphosphorylation and cleavage of 4- and 3-repeat tau. J Cereb Blood Flow Metab. 2017; 37:2441–57. https://doi.org/10.1177/0271678X16668889 [PubMed]
- 59. Basurto-Islas G, Gu JH, Tung YC, Liu F, Iqbal K. Mechanism of tau hyperphosphorylation involving lysosomal enzyme asparagine endopeptidase in a mouse model of brain ischemia. J Alzheimers Dis. 2018; 63:821–33. https://doi.org/10.3233/JAD-170715 [PubMed]
- 60. Hall ED, Oostveen JA, Dunn E, Carter DB. Increased amyloid protein precursor and apolipoprotein E immunoreactivity in the selectively vulnerable hippocampus following transient forebrain ischemia in gerbils. Exp Neurol. 1995; 135:17–27. https://doi.org/10.1006/exnr.1995.1062 [PubMed]
- 61. Ishimaru H, Ishikawa K, Haga S, Shoji M, Ohe Y, Haga C, Sasaki A, Takashashi A, Maruyama Y. Accumulation of apolipoprotein E and beta-amyloid-like protein in a trace of the hippocampal CA1 pyramidal cell layer after ischaemic delayed neuronal death. Neuroreport. 1996; 7:3063–7. https://doi.org/10.1097/00001756-199611250-00054 [PubMed]
- 62. Lin B, Schmidt-Kastner R, Busto R, Ginsberg MD. Progressive parenchymal deposition of beta-amyloid precursor protein in rat brain following global cerebral ischemia. Acta Neuropathol. 1999; 97:359–68. https://doi.org/10.1007/s004010050999 [PubMed]
- 63. Lin B, Ginsberg MD, Busto R. Hyperglycemic but not normoglycemic global ischemia induces marked early intraneuronal expression of β-amyloid precursor protein. Brain Res. 2001; 888:107–116. https://doi.org/10.1016/S0006-8993(00)03023-7 [PubMed]
- 64. Sinigaglia-Coimbra R, Cavalheiro EA, Coimbra CG. Postischemic hyperthermia induces Alzheimer-like pathology in the rat brain. Acta Neuropathol. 2002; 103:444–52. https://doi.org/10.1007/s00401-001-0487-3 [PubMed]
- 65. Swardfager W, Lynch M, Dubey S, Nagy PM. Neuroinflammation mediates synergy between cerebral ischemia and amyloid-β to cause synaptic depression. J Neurosci. 2014; 34:13571–3. https://doi.org/10.1523/JNEUROSCI.3196-14.2014 [PubMed]
- 66. Arranz AM, De Strooper B. The role of astroglia in Alzheimer’s disease: pathophysiology and clinical implications. Lancet Neurol. 2019; 18:406–14. https://doi.org/10.1016/S1474-4422(18)30490-3 [PubMed]
- 67. Pluta R, Januszewski S, Jabłoński M, Ułamek M. Factors in creepy delayed neuronal death in hippocampus following brain ischemia-reperfusion injury with long-term survival. Acta Neurochir Suppl. 2010; 106:37–41. https://doi.org/10.1007/978-3-211-98811-4_5 [PubMed]
- 68. Qin C, Zhou LQ, Ma XT, Hu ZW, Yang S, Chen M, Bosco DB, Wu LJ, Tian DS. Dual functions of microglia in ischemic stroke. Neurosci Bull. 2019; 35:921–33. https://doi.org/10.1007/s12264-019-00388-3 [PubMed]
- 69. Jolivel V, Bicker F, Binamé F, Ploen R, Keller S, Gollan R, Jurek B, Birkenstock J, Poisa-Beiro L, Bruttger J, Opitz V, Thal SC, Waisman A, et al. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. 2015; 129:279–95. https://doi.org/10.1007/s00401-014-1372-1 [PubMed]
- 70. Hossmann KA, Schmidt-Kastner R, Grosse Ophoff B. Recovery of integrative central nervous function after one hour global cerebro-circulatory arrest in normothermic cat. J Neurol Sci. 1987; 77:305–20. https://doi.org/10.1016/0022-510x(87)90130-4 [PubMed]
- 71. Gemmell E, Bosomworth H, Allan L, Hall R, Khundakar A, Oakley AE, Deramecourt V, Polvikoski TM, O'Brien JT, Kalaria RN. Hippocampal neuronal atrophy and cognitive function in delayed poststroke and aging-related dementias. Stroke. 2012; 43:808–14. https://doi.org/10.1161/STROKEAHA.111.636498 [PubMed]
- 72. Gemmell E, Tam E, Allan L, Hall R, Khundakar A, Oakley AE, Thomas A, Deramecourt V, Kalaria RN. Neuron volumes in hippocampal subfields in delayed poststroke and aging-related dementias. J Neuropathol Exp Neurol. 2014; 73:305–11. https://doi.org/10.1097/NEN.0000000000000054 [PubMed]
- 73. Lian H, Yang L, Cole A, Sun L, Chiang AC, Fowler SW, Shim DJ, Rodriguez-Rivera J, Taglialatela G, Jankowsky JL, Lu HC, Zheng H. NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron. 2015; 85:101–15. https://doi.org/10.1016/j.neuron.2014.11.018 [PubMed]
- 74. Lian H, Litvinchuk A, Chiang AC, Aithmitti N, Jankowsky JL, Zheng H. Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J Neurosci. 2016; 36:577–89. https://doi.org/10.1523/JNEUROSCI.2117-15.2016 [PubMed]
- 75. Mu S, Liu B, Ouyang L, Zhan M, Chen S, Wu J, Chen J, Wei X, Wang W, Zhang J, Lei W. Characteristic changes of astrocyte and microglia in rat striatum induced by 3-NP and MCAO. Neurochem Res. 2016; 41:707–14. https://doi.org/10.1007/s11064-015-1739-2 [PubMed]
- 76. Röhl C, Lucius R, Sievers J. The effect of activated microglia on astrogliosis parameters in astrocyte cultures. Brain Res. 2007; 1129:43–52. https://doi.org/10.1016/j.brainres.2006.10.057 [PubMed]
- 77. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017; 541:481–87. https://doi.org/10.1038/nature21029 [PubMed]
- 78. Lana D, Melani A, Pugliese AM, Cipriani S, Nosi D, Pedata F, Giovannini MG. The neuron-astrocyte-microglia triad in a rat model of chronic cerebral hypoperfusion: protective effect of dipyridamole. Front Aging Neurosci. 2014; 6:322. https://doi.org/10.3389/fnagi.2014.00322 [PubMed]
- 79. Cordeau P
Jr , Lalancette-Hébert M, Weng YC, Kriz J. Live imaging of neuroinflammation reveals sex and estrogen effects on astrocyte response to ischemic injury. Stroke. 2008; 39:935–42. https://doi.org/10.1161/STROKEAHA.107.501460 [PubMed] - 80. Kriz J, Lalancette-Hébert M. Inflammation, plasticity and real-time imaging after cerebral ischemia. Acta Neuropathol. 2009; 117:497–509. https://doi.org/10.1007/s00401-009-0496-1 [PubMed]
- 81. Ułamek-Kozioł M, Kocki J, Bogucka-Kocka A, Petniak A, Gil-Kulik P, Januszewski S, Bogucki J, Jabłoński M, Furmaga-Jabłońska W, Brzozowska J, Czuczwar SJ, Pluta R. Dysregulation of autophagy, mitophagy, and apoptotic genes in the medial temporal lobe cortex in an ischemic model of Alzheimer’s disease. J Alzheimers Dis. 2016; 54:113–21. https://doi.org/10.3233/JAD-160387 [PubMed]
- 82. Ułamek-Kozioł M, Kocki J, Bogucka-Kocka A, Januszewski S, Bogucki J, Czuczwar SJ, Pluta R. Autophagy, mitophagy and apoptotic gene changes in the hippocampal CA1 area in a rat ischemic model of Alzheimer’s disease. Pharmacol Rep. 2017; 69:1289–94. https://doi.org/10.1016/j.pharep.2017.07.015 [PubMed]
- 83. Ułamek-Kozioł M, Czuczwar SJ, Kocki J, Januszewski S, Bogucki J, Bogucka-Kocka A, Pluta R. Dysregulation of autophagy, mitophagy, and apoptosis genes in the CA3 region of the hippocampus in the ischemic model of Alzheimer’s disease in the rat. J Alzheimers Dis. 2019; 72:1279–86. https://doi.org/10.3233/JAD-190966 [PubMed]
- 84. Pluta R, Lossinsky AS, Mossakowski MJ, Faso L, Wisniewski HM. Reassessment of a new model of complete cerebral ischemia in rats. Method of induction of clinical death, pathophysiology and cerebrovascular pathology. Acta Neuropathol. 1991; 83:1–11. https://doi.org/10.1007/BF00294424 [PubMed]