



Introduction
Respiratory infection is one of the most common respiratory diseases in which pulmonary infection is one of the principal causes of death in the elderly and children. [1]. The main symptoms of the first onset of the disease are dyspnea and shortness of breath, and some patients will have a loss of appetite, lethargy, disturbance of consciousness, and dehydration [2]. At present, many researchers have conducted in-depth research and discussion on the pathogenesis of pneumonia. Therefore, effective anti-inflammatory therapy is one of the critical strategies for the prevention and treatment of pneumonia.
Long non-coding RNAs(LncRNA) is a class of transcripts with a length of more than 200 nucleotides. LncRNA has a variety of biological functions. It regulates the expression of specific genes in the process of recruitment, expression, post-transcriptional modification and epigenetics of necessary transcription, and participates in the regulation of myeloid determination, innate and adaptive immune function by interacting with DNA, protein, and RNA [3]. Under physiological and pathological conditions, it plays a crucial role in maintaining tissue homeostasis, lipid homeostasis, and epithelial, and smooth muscle cell homeostasis [4, 5]. LncRNA is closely related to the occurrence and development of malignant tumors and has certain advantages as a marker of tumor diagnosis and prognosis [6]. There are many reports on the cancer, but there are few studies on lung injury. LncRNA in acute pneumonia is explored to provide new methods and ideas for the diagnosis and treatment of severe pneumonia. It has been found that lncRNA FOXD3-AS1 was significantly upregulated in human alveolar epithelial cells after oxidative stress. Further studies have shown that lncRNA FOXD3-AS1 as a molecular sponge of miR-150 or competition for endogenous non-coding RNA, limits the ability of miR-150 to promote cell growth, thus aggravating the death of pulmonary epithelial cells [7]. In the experiment of ALI mice and A549 cells induced by LPS, it was reported that lncRNA CASC2 reduced the apoptosis of pulmonary epithelial cells by regulating the miR-144-3p/AQP1 axis, thus improving acute lung injury [8]. High expression of lncRNA HAGLROS was found in the serum of patients with severe pneumonia. Further experiments showed that the down-regulation of HAGLROS could alleviate LPS-induced inflammatory damage of WI-38 cells by regulating the miR-100/NF-kB axis [9]. Through the lncRNA sequencing study, the expression of nine lncRNAs in pneumonia was significantly upregulated compared with the control group, such as RP11-248E9.5 and RP11456D7. RP11-248E9.5 could regulate QRFP and epidermal growth factor receptor pathway substrate 8 (EPS8) gene expression, in which QRFP encodes pyroglutamate aminopeptidase, participates in the degradation of many proteins, and is related to G protein-coupled receptor signal pathway, while the lack of G protein-coupled receptor kinase 5 can regulate inflammation. EPS8 encodes the epidermal growth factor receptor (EG-FR), and its action mode is similar to that of EGFR. EGFR is involved in the production of IL-8 in mycoplasma pneumonia [10].
Our data demonstrated that FGD5-AS1 controlled TNF-α and alleviated inflammation in pneumonia by targeting miR-301a-3p, which would provide a new basis for clinical research.
Results
The increased expression of FGD5-AS-1 in LPS-induced acute pneumonia mice
In order to explore the possible effect of lncRNA on lung function, we established a mouse model of severe pneumonia and determined the expression profile of previously annotated lncRNAs by microarray analysis. In the LPS group, 119 lncRNAs were upregulated, and 81 lncRNAs were downregulated (Figure 1A, 1B). Among them, the change of FGD5-AS1 is relatively significant. Then we detected the expression of FGD5-AS1 in lung tissue of LPS-induced acute pneumonia and sham mice. The qRT-PCR assay showed that FGD5-AS1 was significantly upregulated in the LPS group (Figure 1C). As shown in Figure 1D, 1E, we found that FGD5-AS1 expression, predominantly in the cytoplasm, was dramatically raised in lung tissue of LPS mice compared with the sham group.
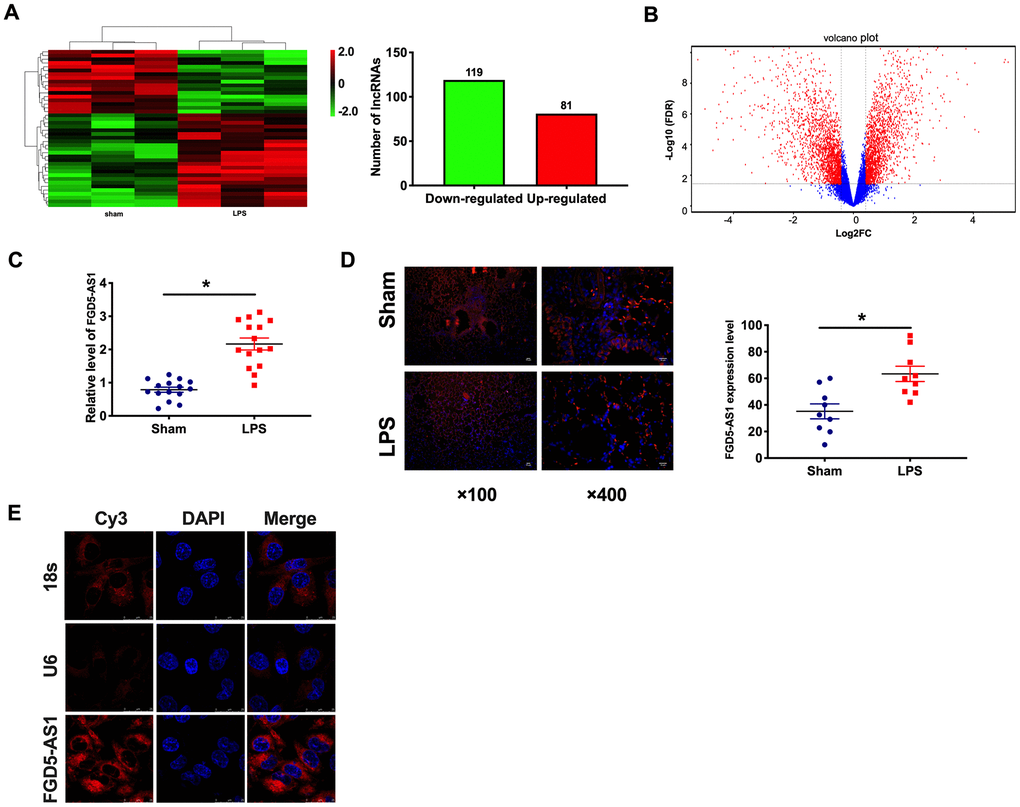
Figure 1. The upregulated of FGD5-AS1 in acute pneumonia. (A) Heat map of lncRNA profile comparison between LPS group and normal lung samples. n=3. (B) A cluster heat map was used to show the expression variations of these lncRNA transcripts in the LPS group and normal lung samples. (C) The expression of FGD5-AS1 in the LPS group and normal lung samples. n=15. (D) FISH analysis of the localization and expression levels of FGD5-AS1 in lung tissues. n=9. (E) The FISH assay was performed to explore the location of FGD5-AS1. *P < 0.05.
Knockdown of FGD5-AS1 alleviates LPS-induced mouse lung injury
For further research, we constructed lentiviral plasmid for knockdown the expression of FGD5-AS1 (LV-sh-FGD5-AS1, LV-sh-NC was indicated as a control group). The survival curve showed that LPS significantly decreased the survival rate of mice, while the mortality of LPS +LV-sh-FGD5-AS1 mice decreased. Compared with LPS mice, the survival rate of LPS + LV-sh-NC mice had no significant difference (Figure 2A). Meanwhile, LPS induced weight loss in mice, knockdown of FGD5-AS1 restored the bodyweight of mice to some extent (Figure 2B). Histological examination showed that LPS caused structural damage of lung tissue in mice, accompanied by apparent inflammatory cell infiltration and alveolar hemorrhage. LV-sh-FGD5-AS1 could significantly alleviate the above pathological changes induced by LPS (Figure 2C). Meanwhile, these changes were evaluated by calculating the lung injury score (Figure 2D). E-cadherin (E-cad) is a transmembrane protein of epithelial cells with high specificity and is often used as a marker to express epithelial cells [11]. We found that LPS inhibited the expression of E-cad, and FGD5-AS1 restored the expression of E-cad (Figure 2E, 2F). LPS induces actin stress fiber formation and cell contraction, accompanied by the appearance of paracellular spaces. Knocking down FGD5-AS1 can mitigate these changes (Figure 2G).
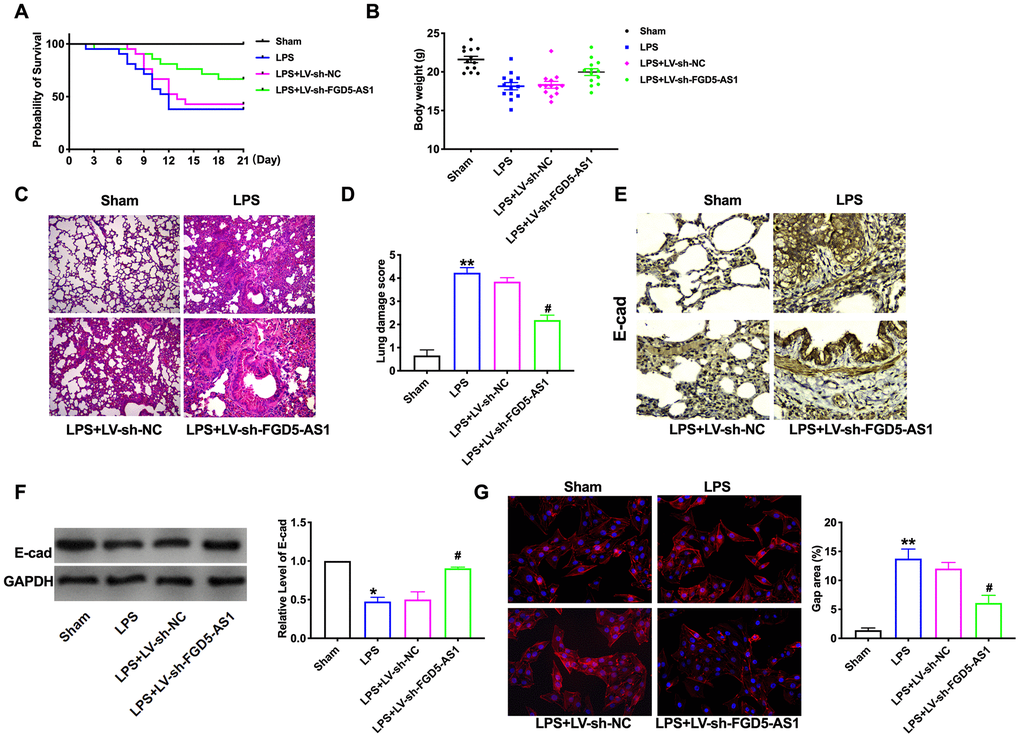
Figure 2. Knockdown of FGD5-AS1 alleviates LPS-induced mouse lung injury. (A) The survival curves of mice in different groups. n=13. (B) The body weight of mice in different groups. n=13. (C) H&E analysis of the morphological changes for lung tissue. (D) The lung damage score of mice in different groups. n=6. (E) Immunohistochemistry analysis of the expression levels of E-cad in lung tissue. (F) The protein level of E-cad in lung tissue. n=6. (G) Immunofluorescence assay of F-Actin in lung tissue. n=4. *P < 0.05, #P<0.05.
Knockdown of FGD5-AS1 decreases inflammatory cell in the bronchoalveolar lavage fluid
To further verify the effect of FGD5-AS1 on pulmonary inflammation, The number of inflammatory cells in bronchoalveolar lavage fluid (BALF) was measured. LPS significantly increased the levels of macrophages and neutrophils in BALF. Compared with LPS alone, knockdown of FGD5-AS1 significantly reduced the infiltration of macrophages and neutrophils (Figure 3A–3C). Pro-inflammatory cytokines related to neutrophil recruitment, such as TNFα, IL1β, and IL6, are involved in the pathogenesis of ALI induced by lipopolysaccharide. Therefore, we also measured the levels of these three inflammatory mediators in BALF. TNFα, IL1β, and IL6 were significantly increased in the LPS treatment group. Knockdown of FGD5-AS1 reduced the level of these pro-inflammatory cytokines, which in turn improved tissue damage (Figure 3D). These results suggested that the down-regulation of FGD5-AS1 improves lung injury by reducing the expression of pro-inflammatory cytokines in acute pneumonia.
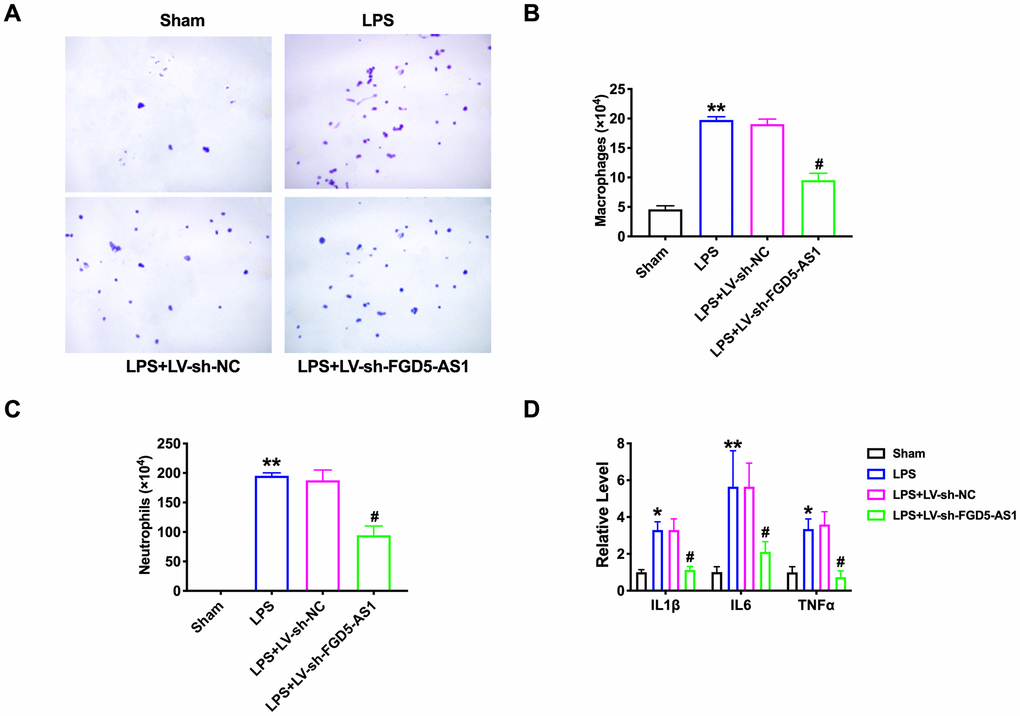
Figure 3. FGD5-AS1 regulates inflammatory cells in the bronchoalveolar lavage fluid. (A) The cells isolated from BALF were counted by blood cell count and represented by Diff-Quick staining. Large and blue indicated macrophages, small and purple indicated neutrophils. (B) The statistical column of macrophage count. n=4. (C) The statistical column of neutrophils count. n=4. (D) The level of cytokines was assessed by ELISA. n=4. *P < 0.05, **P < 0.01, #P < 0.05.
FGD5-AS1 regulates the level of TNFα in acute pneumonia
Then, we detected the level of TNFα in lung tissue by immunofluorescence. The expression of TNFα in the LPS group increased significantly, while knockdown of FGD5-AS1 remitted the level of TNFα (Figure 4A). Then we transfected sh-FGD5-AS1/sh-NC into TC-1 cells after LPS treatment. The protein level of TNFα was assessed by western blot. Sh-FGD5-AS1 abolished the increased level of TNFα induced by LPS (Figure 4B). Further, we determined the activity of TNFα in TC-1 cells. The results showed that LPS produced the event of TNFα, while sh-FGD5-AS1 remitted the LPS function (Figure 4C). TNFα, IL1β, and IL6 were significantly increased in the LPS treatment group. The knockdown of FGD5-AS1 reduced the level of these pro-inflammatory cytokines (Figure 4D). Meanwhile, ROS production induced by LPS was alleviated by knockdown of FGD-AS1 (Figure 4E).
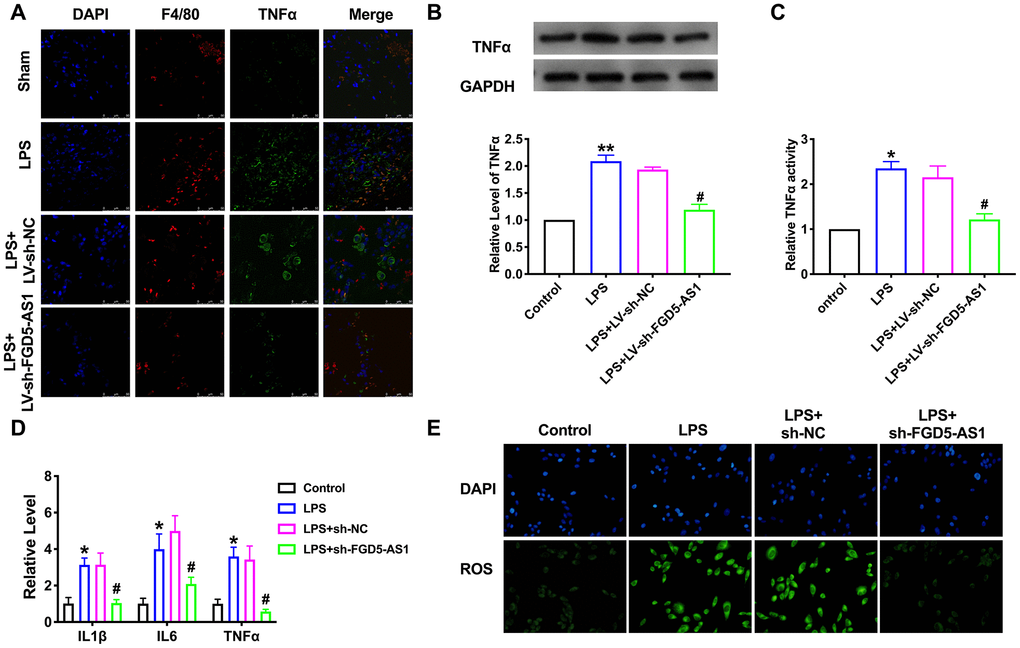
Figure 4. FGD5-AS1 regulates the expression of TNFα in acute pneumonia. (A) Immunofluorescence assay of F4/80 and TNFα in lung tissue. (B) The protein level of TNFα in TC-1 cells. n=5. (C) The activity of TNFα was determined by the TNFα activity kit. n=4. (D) The level of cytokines was assessed by ELISA. n=4. (E) The production of ROS in TC-1 cells. *P < 0.05, **P < 0.01, #P < 0.05.
TNFα neutralizing antibody prevented LPS induced acute pneumonia
Next, we treated the mice TNFα neutralizing antibody (anti-TNFα) by tail vein injection. Histological results showed that there was no difference between WT mice and anti-TNFα treatment WT mice (Figure 5A). LPS caused structural damage to lung tissue in mice, which was remitted by anti-TNFα treatment (Figure 5B, 5C). Immunohistochemical assay performed that LPS inhibited the expression of E-cad, and anti-TNFα restored the expression of E-cad (Figure 5D), and western blot showed similar results (Figure 5E). As Figure 5F shown, anti-TNFα decreased the actin stress fiber formation and cell contraction (Figure 5F). Anti-TNFα also reduced the number of macrophages and neutrophils in BALF (Figure 5G–5I).
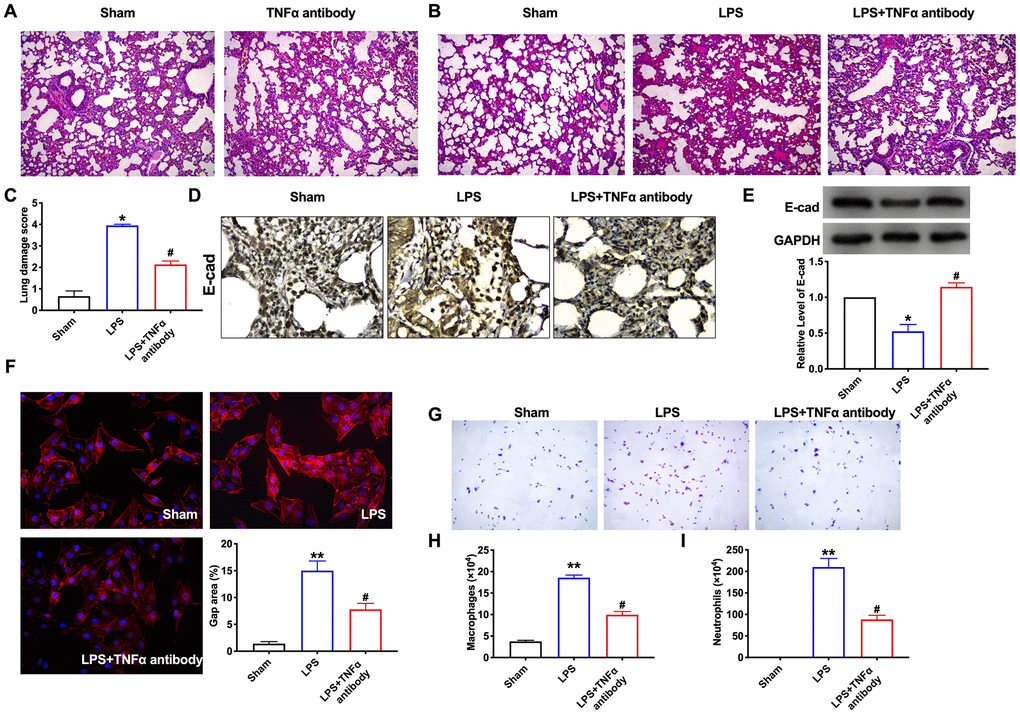
Figure 5. TNFα neutralizing antibody prevented LPS induced acute pneumonia. (A) H&E analysis of the morphological for lung tissue. (B) H&E analysis of the morphological changes for lung tissue. (C) The lung damage score of mice in different groups. n=5. (D) Immunohistochemistry analysis of the expression levels of E-cad in lung tissue. n=5. (E) The protein level of E-cad in lung tissue. (F) Immunofluorescence assay of F-Actin in lung tissue. n=4. (G) The cells isolated from BALF were counted by blood cell count and represented by Diff-Quick staining. (H) Statistical column of macrophage count. n=4. (I) The statistical column of neutrophils count. n=4. *P < 0.05, **P < 0.01, #P < 0.05.
FGD5-AS1 acts as a sponge of miR-301a-3p
Microarray analysis showed the expression profile of previously annotated miRNAs, which including miR-301a-3p (Figure 6A). By performing FISH, we observed that miR-301a-3p, localized in the cytoplasm, was down-regulated and had a negative correlation with FGD5-AS1 expression in lung tissues (Figure 6B–6D). Bioinformatics website predicts the existence of binding sites between FGD5-AS1 and miR-301a-3p. Next, we performed luciferase assays to confirm whether FGD5-AS1 binds to miR-301a-3p. The assay report showed that FGD5-AS1 could bind with miR-301a-3p (Figure 6E). Further, we carried out anti-AGO2 RIP in TC-1 cells. FGD5-AS1 pull-down by AGO2 was enriched in cells transfected with miR-301a-3p, revealing the direct binding of FGD5-AS1 with miR-301a-3p (Figure 6F).
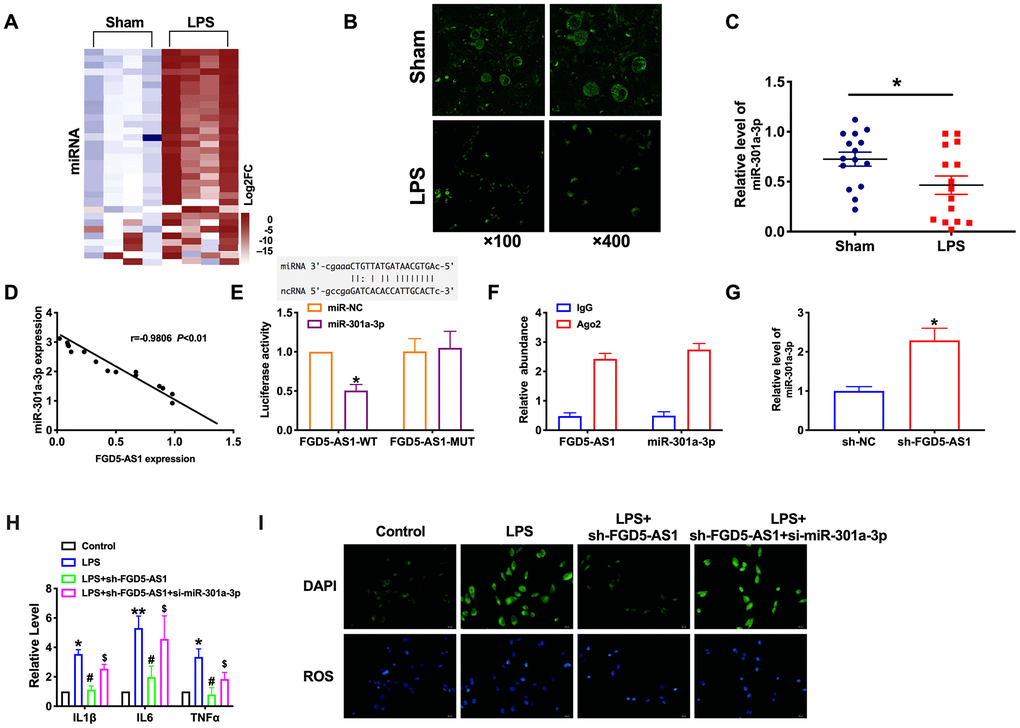
Figure 6. FGD5-AS1 acts as a sponge of miR-301a-3p. (A) Heat map of miRNA profile comparison between LPS group and normal lung samples. (B–D) FISH analysis of the localization and expression levels of miR-301a-3p and validation of the correlation of FGD5-AS1 with miR-301a-3p expression in lung tissues. n=15. (E) Luciferase activity of WT or Mut FGD5-AS1 after the co-transfection with the miR-301a-3p mimic. n=4, (F) RIP analysis of the amount of FGD5-AS1 and miR-301a-3p pulled down from the Ago2 protein. n=4. (G) The expression level of miR-301a-3p was measured. n=6. (H) The level of cytokines was assessed by ELISA. n=4. (I) The production of ROS in TC-1 cells. *P < 0.05, **P < 0.01, #P < 0.05, $P<0.05.
Moreover, knockdown FGD5-AS1 induced the expression of miR-301a-3p (Figure 6G). Further, we found that the silencing of miR-301a-3p would prevent the inhibition function of FGD5-AS1 on cytokines and ROS production (Figure 6H, 6I). In summary, miR-301a-3p would be a target of FGD5-AS1, which involved in inflammation progression.
MiR-301a-3p reversed FGD5-AS1 induced TNFα expression in TC-1 cells
Different databases were used to predict targets of miR-301a-3p (Figure 7A). We found that TNFα had an increased expression in lung tissue of LPS treated mice compared with sham mice. (Figure 7B). Luciferase assays showed that miR-301a-3p could bind with TNFα (Figure 7C). RT-PCR assay results performed that forced expression of miR-301-3p could inhibit the expression of TNFα (Figure 7D). The decreased level of TNFα induced by sh-FGD5-AS1 could restore by co-transfection si-miR-301a-3p (Figure 7E).
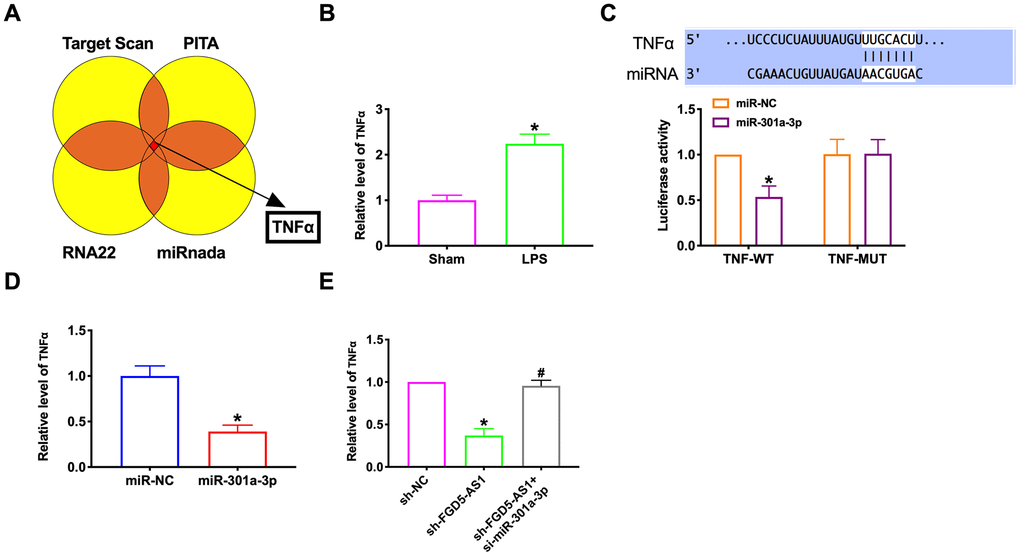
Figure 7. MiR-301a-3p reversed FGD5-AS1 induced TNFα expression in TC-1 cells. (A) Different databases were used to predict targets of miR-301a-3p. (B) TNFα expression was determined by RT-PCR. (C) Luciferase activity of WT or Mut TNFα after the co-transfection with the miR-301a-3p mimic. (D, E) TNFα expression was determined by RT-PCR. *P < 0.05.
Discussion
Cumulative data showed that the down-regulation of FGD5-AS1 could reduce LPS-induced pulmonary inflammation by decreasing the activity of TNFα. Meanwhile, the interaction between FGD5-AS1 and miR-301a-3p regulates TNFα, the target of miR-301a-3p, and established a new signal pathway, which laid a foundation for the future study of acute lung injury and severe pneumonia (Figure 8).
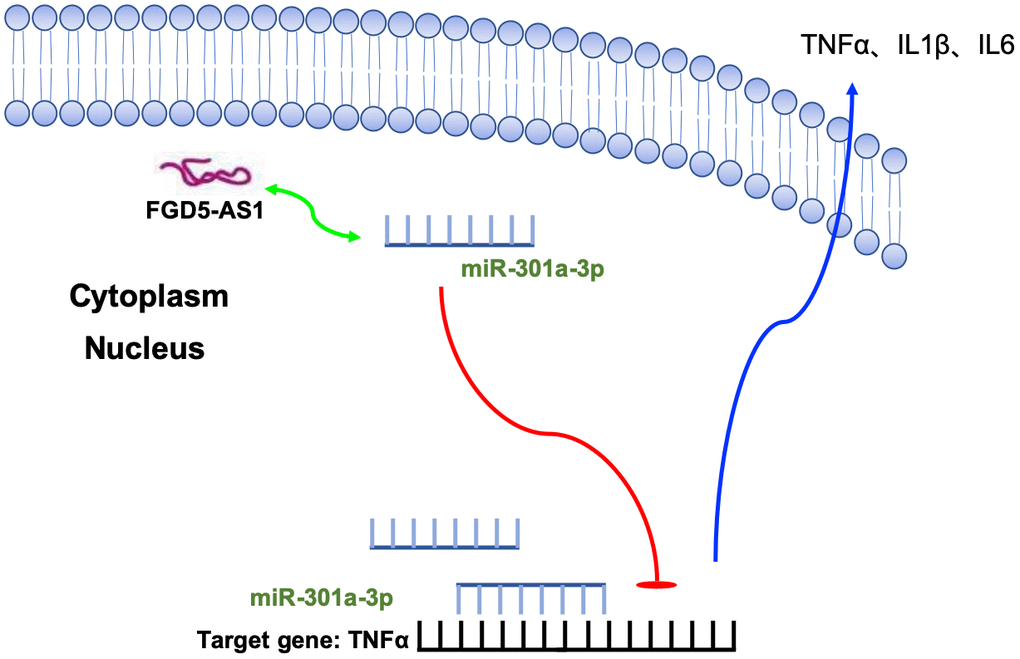
Figure 8. Schematic model describing the effect of FGD5-AS1 on homeostasis and function in TC-1 cells. FGD5-AS1 interacted with miR-301a-3p and induced the expression of TNFα, which would aggravate inflammation.
The severity of pneumonia depends on the degree of local inflammation, the spread of pulmonary inflammation, and the degree of systemic inflammation. The inflammation of lung tissue caused by different etiology, different pathogens, and various occasions has similar or the same pathophysiological process. When it develops to a particular stage of the disease, it can worsen and aggravate into severe pneumonia [12, 13].
Non-coding RNA is a research hotspot in recent years; only a small number of non-coding RNA contains a short open reading frame, can encode functional peptides, most non-coding RNA does not encode proteins but can regulate gene expression and participate in a variety of critical physiological processes. So far, there are few studies on lncRNAs compared with miRNA. Like miRNA, lncRNAs have abundant functions, including participation in messenger RNA splicing, degradation, and chromatin modification. Pulmonary fibrosis is an essential pathological stage in the development of ALI. In recent years, studies have focused on lncRNAs and pneumonia. In the mouse models of peritonitis and pneumonia, Neat1 deficiency significantly reduced inflammatory responses. These results revealed a previously unrecognized role of lncRNAs in innate immunity and suggest that Neat1 was a common mediator for inflammasome stimuli [14]. Zhou Z et al. Found that SNHG16 effected LPS-induced pneumonia in WI-38 cells through competitively interacting miR-146a-5p with CCL5 by controlling JNK and NF-κB pathways [15]. Liu M. et al. revealed that knockdown of HAGLROS could remit LPS-induced pneumonia by regulating miR-100/NF-κB axis [9]. LncRNA MIAT2 could also decrease LPS-induced inflammatory damage by sponging miRNA-15 [16]. Here, we firstly explore the mechanism of FGD5-AS1 in acute pneumonia. The abnormal expression of FGD5-AS1 was found in LPS induced lung tissues. FGD5-AS1 would sponge miR-301a-3p and regulated the inflammation level.
One of the pathological manifestations of acute pneumonia is diffuse alveolar injury. The histological characterization of this process occurs in the early stage of infiltration or damage, goes through the stage of proliferation or histology, and finally enters the stage of healing or regression. The proliferative phase is characterized by interstitial edema and thickening of the alveolar septum caused by mild inflammation. This stage is characterized by necrosis of AT epithelial cells, injury of endothelial cells, and exfoliation of the alveolar basement membrane. This loss of alveolar integrity causes fibrin-rich protein fluid to leak into the alveolar cavity while inflammatory cells recruited to the area. Neutrophils attach to the injured capillary endothelium and enter the alveolar hole filled with protein-rich edematous fluid through the interstitial [17, 18]. In the alveolar cavity, alveolar macrophages secrete cytokines, IL1, IL6, IL8 and TNFα, which locally stimulate neutrophil chemotaxis and activation. Macrophages also secrete other cytokines, including IL1, IL6, and IL 10 [19–21]. IL1 can also stimulate extracellular matrix production through fibroblasts. Neutrophils release oxidants, proteases, leukotrienes, and other pro-inflammatory factors, such as platelet-activating factor (PAF). There are also many anti-inflammatory mediators in the alveolar environment, including IL1 receptor antagonists, soluble tumor necrosis factor receptors, autoantibodies to IL8, and cytokines such as IL10 and IL11. The flow of protein edematous fluid into the alveoli can lead to the inactivation of surfactants, make the alveolar surface lose the protective layer, and then destroy the surface cell structure. Alveolar epithelial cell damage plays a vital role in the development and recovery of the disease. In our research, Our experimental results once again confirmed the part of inflammatory cell infiltration and the release of cytokines in pneumonia. Knockout of FGD5-AS1 reduced macrophage and neutrophil infiltration, inhibited the release of TNFα, IL1β, and IL6, which effectively alleviated lung injury caused by acute pneumonia.
When pneumonia occurs, it is necessary to seek medical treatment in an effective way. Although modern medicine has explored a variety of drugs that can effectively treat pneumonia, it still cannot meet the treatment needs of patients. New treatment strategies need to be studied in depth. Our study is expected to be a theoretical basis for the clinical treatment of pneumonia.
Materials and Methods
Establishment of mouse model of acute pneumonia by LPS nasal inhalation
After intraperitoneal injection of 22 mg/mL pentobarbital sodium (diluted with normal saline), the caudal root, hindlimb, and eyelash reflexes disappeared after 10 min, and slow breathing was considered as deep anesthesia [22]. Nasopharyngeal drip: LPS saline solution was prepared according to 167um g/mL. After anesthesia, the mouse head was tilted downward, and the tongue was pulled out with tweezers. The 60 μL LPS saline solution (about 10 μg LPS) was absorbed by a fluid transfer gun and dropped into the oral cavity through the posterior wall of the pharynx. The nostril was quickly pinched and maintained for 30 seconds, and the model was successful when all the liquid was absorbed into the nasal cavity, and slight tracheal rales appeared. 150 μL lentivirus containing FGD5-AS1-shRNA/NC-shRNA was injected in the tail vein of mice. 21 days after the establishment of the model, mice were intraperitoneally injected with 3% pentobarbital sodium and were euthanatized by excessive anesthesia with a dose of 90 mL/kg, and the organs and tissues were removed for follow-up study. The research protocol of this study was approved by the Animal Care and Use Committee of the Linyi People's Hospital.
Cell culture
The TC-1 cell lines (Mouse alveolar epithelial cells) were purchased from the Science Cell Laboratory. Cell lines were cultured in DMEM (Thermo-life, United States) with 10 % FBS (Thermo Fisher, USA) and 100 μL/mL penicillin and streptomycin (Beyotime, China) and placed at 37° C with 5% CO2.
Quantitative real-time PCR
RNA isolation, reverse transcription, and quantitative expression were carried out according to manufacturer’s instructions. All the kits were purchased from Vazyme, and gene expression was calculated using 2-ΔΔCt method.
Dual-Luciferase reporter assay
20 mmol/L miRNA mimic or negative control (NC) and FGD5-AS1/TNFα were co-transfected into HEK293T cells. Luciferase activity was detected with Luciferase Reporter Assay Kit (Biovision, China) on a luminometer (Berthold, Germany) 48 hr after the transfection.
Western blot analysis
Total protein was isolated from tissues or cells with RIPA lysis Mix. Western blotting assay was performed as previously described. Briefly, 50-80 μg protein extraction was loaded via SDS-PAGE and transferred onto nitrocellulose membranes (absin, China), then incubated with primary antibodies for 2 hrs at temperature, then plated at 4° C overnight, the membranes were incubated in 5% non-fat milk blocking buffer for horizontal mode 3h. After incubation with secondary antibodies, the membranes were scanned using an Odyssey, and data were analyzed with Odyssey software (LI-COR, USA).
Histological analysis
The tissues were gathered and fixed in 4% paraformaldehyde for 24 hrs. Then the fixed tissues were embedded in paraffin. Next, Paraffin slicer machine was used to cut slices (5-mm cross-sectional). H&E staining was used to evaluate cardiac morphology. The pathological grades were scored according to the degree of lung pathological changes at different time points.
Fish
The sample was grown or adhered to or sliced on the cover slide and permeated with 70% ethanol. Hybridization can be done in a traditional laboratory incubator at 37° C within 4 hours. After hybridization, the washing buffer was incubated briefly to remove the excess probe. The total time is 1-1.5 hours. The sample can be imaged using a standard fluorescence microscope.
Immunohistochemical staining
Paraffin sections of lung tissue were dewaxing to water in xylene and descending series of ethanol. We penetrated parts using 0.5% Triton X-100. After 3 times wash, we blocked sections with 50% goat serum. Then, sections were incubated with an E-cadherin antibody overnight. We produced the parts using a secondary antibody followed by DAPI staining. The segments were photographed by light scope under an IX73 fluorescence microscope (Olympus, Valley, PA) and analyzed by Image J software.
Collection of bronchoalveolar lavage fluid
After anesthesia, the mice were fixed in supine position, disinfected neck and chest with ethanol, opened chest cavity, separated cervical trachea, ligated right main bronchus, injected human 0.5ml PBS solution into bronchoalveolar lavage for 3 times. Collect bronchoalveolar lavage fluid [23].
Statistical analysis
All data is statistical as a mean ± S.E.M. We performed Student's t-test or a one-way ANOVA for statistical analysis. P < 0.05 was described as statistically significant.
Author Contributions
Yi-Yu He designed and coordinated the research; Qing-Min Liu performed the majority of experiments, collected and analyzed the data; Li-Li Liu performed the molecular investigations; Li-Kun Wang wrote the paper.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding
This study was supported by Natural Science Foundation of Shandong Province, No. ZR2014HM081; Shandong Province Medical and Health Science and Technology Development Plan, No. 2015WS0378; National Natural Science Foundation of China, No. 81800444; Natural Science Foundation of Hubei Province, No. 2018CFB415.
References
- 1. Quinton LJ, Walkey AJ, Mizgerd JP. Integrative physiology of pneumonia. Physiol Rev. 2018; 98:1417–64. https://doi.org/10.1152/physrev.00032.2017 [PubMed]
- 2. Wunderink RG, Waterer G. Advances in the causes and management of community acquired pneumonia in adults. BMJ. 2017; 358:j2471. https://doi.org/10.1136/bmj.j2471 [PubMed]
- 3. Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013; 152:1298–307. https://doi.org/10.1016/j.cell.2013.02.012 [PubMed]
- 4. Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018; 172:393–407. https://doi.org/10.1016/j.cell.2018.01.011 [PubMed]
- 5. Satpathy AT, Chang HY. Long noncoding RNA in hematopoiesis and immunity. Immunity. 2015; 42:792–804. https://doi.org/10.1016/j.immuni.2015.05.004 [PubMed]
- 6. Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016; 29:452–63. https://doi.org/10.1016/j.ccell.2016.03.010 [PubMed]
- 7. Zhang D, Lee H, Haspel JA, Jin Y. Long noncoding RNA FOXD3-AS1 regulates oxidative stress-induced apoptosis via sponging microRNA-150. FASEB J. 2017; 31:4472–81. https://doi.org/10.1096/fj.201700091R [PubMed]
- 8. Li H, Shi H, Gao M, Ma N, Sun R. Long non-coding RNA CASC2 improved acute lung injury by regulating miR-144-3p/AQP1 axis to reduce lung epithelial cell apoptosis. Cell Biosci. 2018; 8:15. https://doi.org/10.1186/s13578-018-0205-7 [PubMed]
- 9. Liu M, Han T, Shi S, Chen E. Long noncoding RNA HAGLROS regulates cell apoptosis and autophagy in lipopolysaccharides-induced WI-38 cells via modulating miR-100/NF-κB axis. Biochem Biophys Res Commun. 2018; 500:589–96. https://doi.org/10.1016/j.bbrc.2018.04.109 [PubMed]
- 10. Huang S, Feng C, Chen L, Huang Z, Zhou X, Li B, Wang LL, Chen W, Lv FQ, Li TS. Identification of Potential Key Long Non-Coding RNAs and Target Genes Associated with Pneumonia Using Long Non-Coding RNA Sequencing (lncRNA-Seq): A Preliminary Study. Med Sci Monit. 2016; 22:3394–3408. https://doi.org/10.12659/msm.900783 [PubMed]
- 11. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, Herridge M, Randolph AG, Calfee CS. Acute respiratory distress syndrome. Nat Rev Dis Primers. 2019; 5:18. https://doi.org/10.1038/s41572-019-0069-0 [PubMed]
- 12. Todd NW, Marciniak ET, Sachdeva A, Kligerman SJ, Galvin JR, Luzina IG, Atamas SP, Burke AP. Organizing pneumonia/non-specific interstitial pneumonia overlap is associated with unfavorable lung disease progression. Respir Med. 2015; 109:1460–68. https://doi.org/10.1016/j.rmed.2015.09.015 [PubMed]
- 13. Brown SM, Jones JP, Aronsky D, Jones BE, Lanspa MJ, Dean NC. Relationships among initial hospital triage, disease progression and mortality in community-acquired pneumonia. Respirology. 2012; 17:1207–13. https://doi.org/10.1111/j.1440-1843.2012.02225.x [PubMed]
- 14. Zhang P, Cao L, Zhou R, Yang X, Wu M. The lncRNA Neat1 promotes activation of inflammasomes in macrophages. Nat Commun. 2019; 10:1495. https://doi.org/10.1038/s41467-019-09482-6 [PubMed]
- 15. Zhou Z, Zhu Y, Gao G, Zhang Y. Long noncoding RNA SNHG16 targets miR-146a-5p/CCL5 to regulate LPS-induced WI-38 cell apoptosis and inflammation in acute pneumonia. Life Sci. 2019; 228:189–97. https://doi.org/10.1016/j.lfs.2019.05.008 [PubMed]
- 16. Zhang H, Zhao J, Shao P. Long noncoding RNA MIAT2 alleviates lipopolysaccharide-induced inflammatory damage in WI-38 cells by sponging microRNA-15. J Cell Physiol. 2020; 235:3690–03697. https://doi.org/10.1002/jcp.29263 [PubMed]
- 17. Bordon J, Aliberti S, Fernandez-Botran R, Uriarte SM, Rane MJ, Duvvuri P, Peyrani P, Morlacchi LC, Blasi F, Ramirez JA. Understanding the roles of cytokines and neutrophil activity and neutrophil apoptosis in the protective versus deleterious inflammatory response in pneumonia. Int J Infect Dis. 2013; 17:e76–83. https://doi.org/10.1016/j.ijid.2012.06.006 [PubMed]
- 18. Pechous RD. With friends like these: the complex role of neutrophils in the progression of severe pneumonia. Front Cell Infect Microbiol. 2017; 7:160. https://doi.org/10.3389/fcimb.2017.00160 [PubMed]
- 19. Mizgerd JP. Inflammation and pneumonia: why are some more susceptible than others? Clin Chest Med. 2018; 39:669–76. https://doi.org/10.1016/j.ccm.2018.07.002 [PubMed]
- 20. Divangahi M, King IL, Pernet E. Alveolar macrophages and type I IFN in airway homeostasis and immunity. Trends Immunol. 2015; 36:307–14. https://doi.org/10.1016/j.it.2015.03.005 [PubMed]
- 21. Bhattacharya J, Westphalen K. Macrophage-epithelial interactions in pulmonary alveoli. Semin Immunopathol. 2016; 38:461–69. https://doi.org/10.1007/s00281-016-0569-x [PubMed]
- 22. Pera T, Zuidhof A, Valadas J, Smit M, Schoemaker RG, Gosens R, Maarsingh H, Zaagsma J, Meurs H. Tiotropium inhibits pulmonary inflammation and remodelling in a Guinea pig model of COPD. Eur Respir J. 2011; 38:789–96. https://doi.org/10.1183/09031936.00146610 [PubMed]
- 23. McLachlan A, Borchers M, Velayutham P, Wagner M, Limbach PA. Characterizing the reproducibility of a protein profiling method for the analysis of mouse bronchoalveolar lavage fluid. J Proteome Res. 2006; 5:3059–65. https://doi.org/10.1021/pr060241r [PubMed]