




Introduction
Pulmonary artery hypertension (PAH) is a life-threatening disease characterized by progressive pulmonary vascular remodeling, resulting in a sustained increase in Pulmonary artery pressure and eventually right heart failure [1–3]. The main histopathological features of PAH are vascular wall remodeling, which includes intimal hyperplasia, inner and outer membrane hypertrophy and extracellular matrix deposition. PAH involves complex structural and functional changes [4, 5]. Multiple cell types and growth factors are involved in the development of PAH. Sustained pulmonary vasoconstriction and vascular remodeling, characterized by concentric wall thickening and lumen occlusion of the small and medium pulmonary arteries (PAs), has convincing evidence that pulmonary smooth muscle cells (PASMCs) proliferation is a major feature of pulmonary artery remodeling and is the basis for the development of PAH [6, 7]. The constitutive proliferative phenotype of PASMCs is considered to be a key characteristic of PAH patients or animals that induce PAH [4, 8–10]. Thus, although current treatment for PAH can improve clinical symptoms, the progression of the disease is inevitable in most patients, and the mortality rate remains unacceptably high. The pathogenesis of PAH is complex and has not been fully elucidated. Therefore, a better understanding of the pathogenesis of the disease is crucial to identify new therapeutic targets.
Epigenetic regulation plays a key role in the evolution of PAH [11]. Among these factors, histone methyltransferases (HMTs) are tempting targets for disease intervention [12–14] because they are often maladjusted in the development of PAH and their enzyme activity can be used for treatment [15]. In mammalian cells, SETD2 is the main enzyme that catalyzes the trimethylation of lysine 36 on histone 3 (H3K36me3), a histone marker associated with an active transcriptional region [16]. Inactivation of SETD2 in the germ line of mice leads to defects in vascular remodeling, indicating that SETD2 plays a non-redundant role in development [17]. Cell culture-based studies have shown that there are defects such as chromosome separation, DNA repair and nucleotide pool reduction in SETD2 - deficient cells. Mechanism characteristics indicate that SETD2 mediated regulation of H3K36me3 and recruitment of DNMT3b ensure the fidelity of gene transcription initiation in embryonic stem cells [18]. In addition, SETD2 and H3K36me3 are involved in RNA splicing during gene transcription. SETD2 mediated H3K36me3 affects splicing site selection by recruiting morf-related gene 15 (MRG15) and polypyrimidine bundle binding protein 1 (PTB) [19]. However, in many cases, it is not clear whether SETD2 mediated changes in H3K36me3 are the driving force for PAH.
N6-methyl adenine (m6A), the most common internal modification of mRNA, is an important post-transcriptional gene regulation mechanism and plays an important role in various normal and pathological biological processes [20–23]. M6A modification is reversible and is added by methyltransferase complex and removed by m6A demethylase. M6A methyltransferase complex is composed of METTL3, METTL4, METTL14, WTAP, VIRMA, etc [24]. In contrast, FTO and AlkBH5 induced m6A demethylase. The effect of m6A on RNA stability depends on the m6A reader, including YTHDF1, YTHDF2, YTHDF3, and YTHDC1 [21, 22]. M6A modification was reported to be enriched near the H3K36me3 peak, and decreased in the overall range when the cell H3K36me3 was depleted [25]. Mechanistically, H3K36me3 is recognized as a key component of direct binding by METTL14 to the m6A methyltransferase complex (MTC), which promotes binding to m6A MTC adjacent to RNA polymerase II, thereby facilitating transcription of target genes [25]. However, the role of METTL14-mediated m6A modification in PAH is completely unknown.
Here, we constructed a mouse model with SETD2 specific knockout of smooth muscle cells (SETD2SM22α-cre). Our results suggest that the lack of SETD2 in SMCs protected mice from hypoxia-induced PAH and significantly reduced RVSP, [RV/(LV+S)], and pulmonary median width. Moreover, the deletion of SETD2 in SMCs significantly reduced the level of METTL14 and the methylation of m6A RNA induced by PAH. These results suggest that strategies that target the inhibition of SETD2/METTL14 activity may be a viable treatment for PAH in a clinical setting.
Results
SMCs specific SETD2 deficient ameliorates pulmonary pressure in a hypoxia-induced mouse model of PAH
To explore the role of SETD2 in the vascular remodeling during PAH, we constructed a C57BL/6 mouse with specific knockout of SETD2 in SMCs (SETD2SM22a-Cre C57BL/6 mice) (Figure 1A). The deleted expression of SETD2 in SMCs was confirmed by Western blotting (Figure 1B). Next, SETD2SM22a-Cre mice and SETD2fl/fl mice were subjected to induction of PAH under normobaric normoxic or hypoxic conditions (10% O2) for 28 days, respectively. After 28 days of hypoxic exposure, hypoxia significantly increased RVMP, RVSP, mPAP, TPR, and PAMT, compared with normoxic group, whereas RVCO in hypoxia treated groups was dramatically reduced (Figure 2). There were no significant differences in SBP and mSAP between hypoxic and normoxic groups (Figure 2). However, the mean of RVMP, RVSP, mPAP, TPR and PAMT were significantly lower in SETD2SM22a-Cre mice than that in SETD2fl/fl mice after hypoxic exposure (Figure 2). Furthermore, SETD2 deficiency in SMCs led to significant increases in RVCO in SETD2SM22a-Cre mice compared with SETD2fl/fl mice (Figure 2). Collectively, these data suggest that SETD2 deficiency in SMCs ameliorates PAH in hypoxia-induced mouse PAH model.
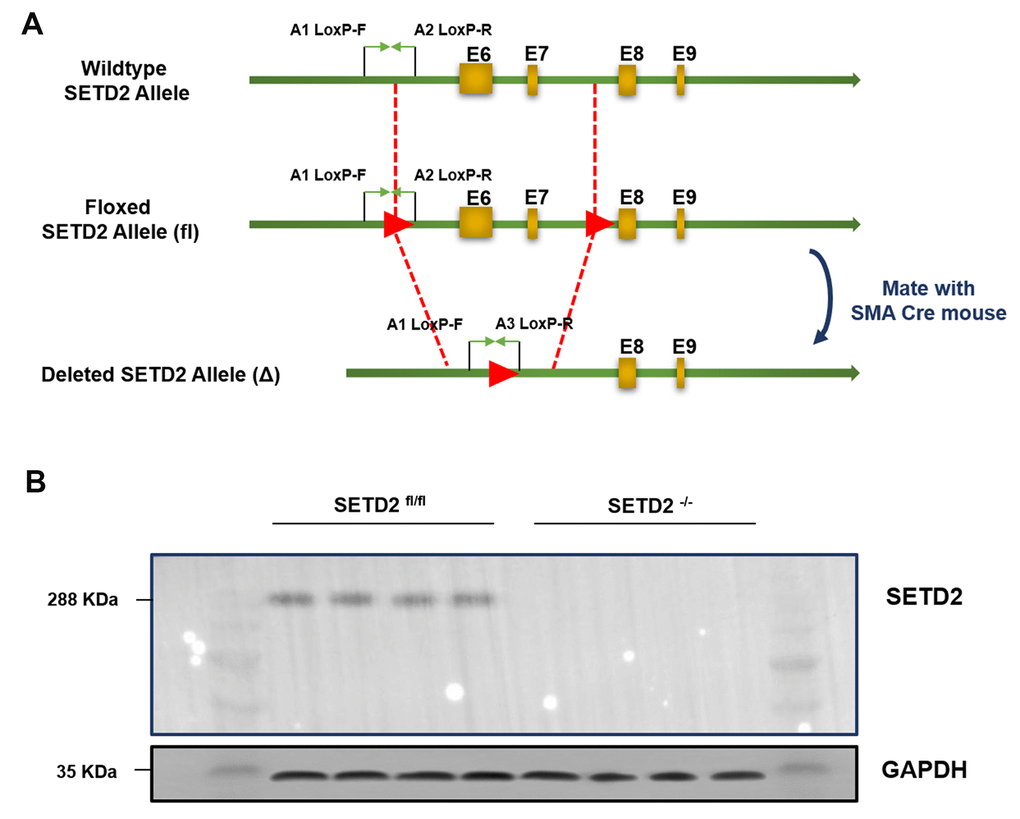
Figure 1. Generation and characterization of mice with smooth muscle cells specific knockout of SETD2. (A) The targeting strategy of mice with specific knockout of SETD2 in smooth muscle cells. (B) Western blotting was used to confirm the expression of SETD2 in smooth muscle cells from normal mice and SMCs SETD2 specific knockout mice.
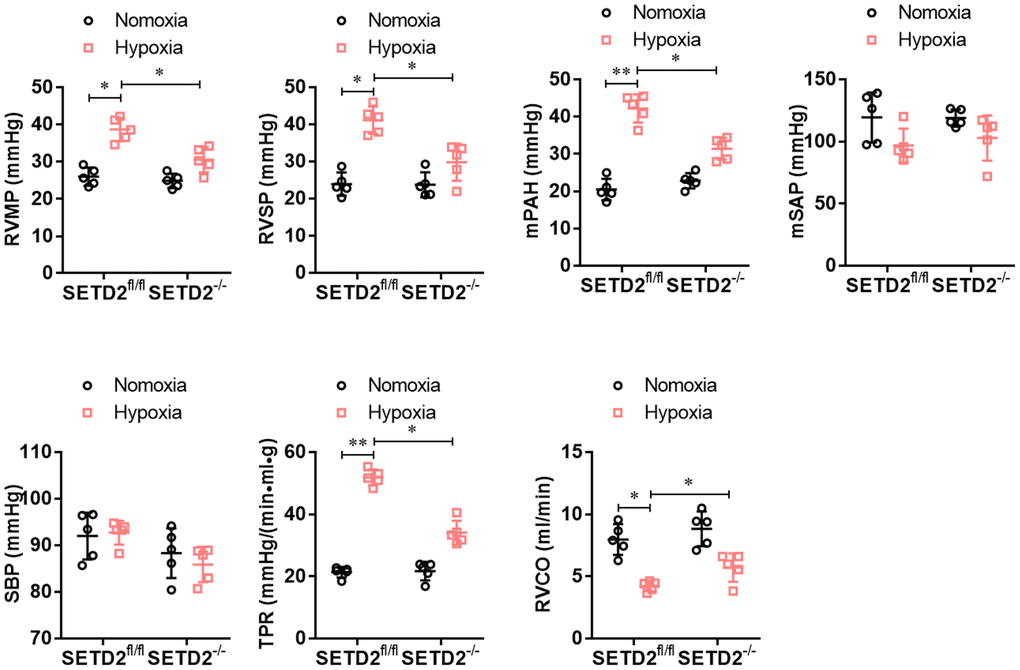
Figure 2. SMCs specific SETD2 deficient ameliorates pulmonary pressure in a hypoxia-induced mouse model of PAH. The hemodynamic measurements were performed to measure the right ventricular mean pressure (RVMP), right ventricular systolic pressure (RVSP) and mean pulmonary arterial pressure (mPAP). The left carotid artery cannulation was performed for systemic arterial pressure (mSAP) and systolic body pressure (SBP) measurement. Total pulmonary resistance (TPR) was calculated by the ratio of mPAP/RVCO. All the calculations were done with a LabChart8 (AD Instruments). All data were presented as Mean±SD (n=5). *P<0.05, **P<0.01 vs. corresponding group.
SMCs specific SETD2 deficient ameliorates right ventricular function in a hypoxia-induced mouse model of PAH
To explore whether SMCs specific SETD2 deficient ameliorates right ventricular dysfunction in mouse PAH model, we performed echocardiography. As shown in Figure 3, hypoxia significantly increased RVEDD, RVEDV, RVESD, RVESV, RVPWT and IVST and significantly decreased RVEF, RVFS and PAAT/CL compared with normoxic group. Consistently, SMCs specific SETD2 deficient obviously reduced RVEDD, RVEDV, RVESD, RVESV, RVPWT and IVST, and enhanced RVEF, RVFS and PAAT/CL compared with SETD2fl/fl mice in hypoxia group. These data indicate that hypoxia-induced PAH mice develop significant right ventricular dysfunction, which could be ameliorated by SMCs specific SETD2 deficient.
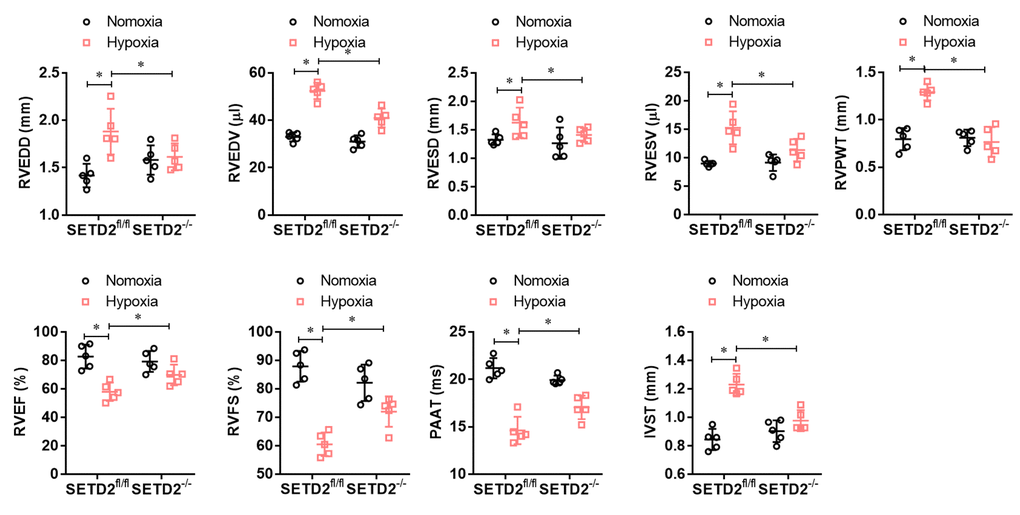
Figure 3. SMCs specific SETD2 deficient ameliorates right ventricular function in a hypoxia-induced mouse model of PAH. Echocardiography was used to measure the normalized cardiac cycle length measuring pulmonary acceleration time (PAAT/CL), RV end diastolic dimension (RVEDD), RV diastolic contraction size (RVESD), RV end diastolic volume (RVEDV), RV diastolic volume contraction (RVESV), RV cardiac output (RVCO), RV wall thickness (RVWT), the thickness of interventricular septum (IVS), RV ejection fraction (RVEF) and RV shortening fraction (RVFS). All data were presented as Mean±SD (n=5). *P<0.05, **P<0.01 vs. corresponding group.
SMCs specific SETD2 deficient attenuates pathological remodeling of pulmonary artery and right ventricular hypertrophy
To further evaluate the effect of SMCs specific SETD2 deficient on pathological remodeling of pulmonary artery and right ventricular hypertrophy, we firstly evaluated the RV/BW ratio (the ratio of the right ventricular mass to the sum of body masses) and RV/(L+S) ratio (the ratio of the right ventricular mass to the sum of the left ventricular and septal masses), a marker of hypertrophy in the right ventricle due to elevated right ventricular pressure and afterload. As shown in Figure 4A, RV/BW and RV/(LV+S) in hypoxia treated mice significantly increased compared with normoxic mice, but SMCs specific SETD2 deficient significantly decreased RV/BW and RV/(LV+S). Histological studies were next conducted to demonstrate the effect of SETD2 on pulmonary blood vessel remodeling during the course of PAH development. Remarkably, staining with haematoxylin and eosin (HE) (Figure 4B) demonstrated that loss of SMCs SETD2 significantly attenuated the PAMT ration of both small and medium pulmonary arteries, indicating the alleviated blood vessel remodeling (Figure 4B). Histochemical staining of SETD2 immunostaining revealed that the SETD2 expression in blood vessels of SETD2fl/fl mice were profoundly enhanced in response to chronic hypoxic exposure. In sharp contrast, the pulmonary arteries originated from SETD2SM22a-Cre mice were remodeled to a significantly less degree and harbored much lower SETD2 expressions (Figure 4C). These data indicate that SMCs specific SETD2 deficient obviously reverses pathological remodeling of pulmonary artery and right ventricular hypertrophy in PAH mice model induced by hypoxia.
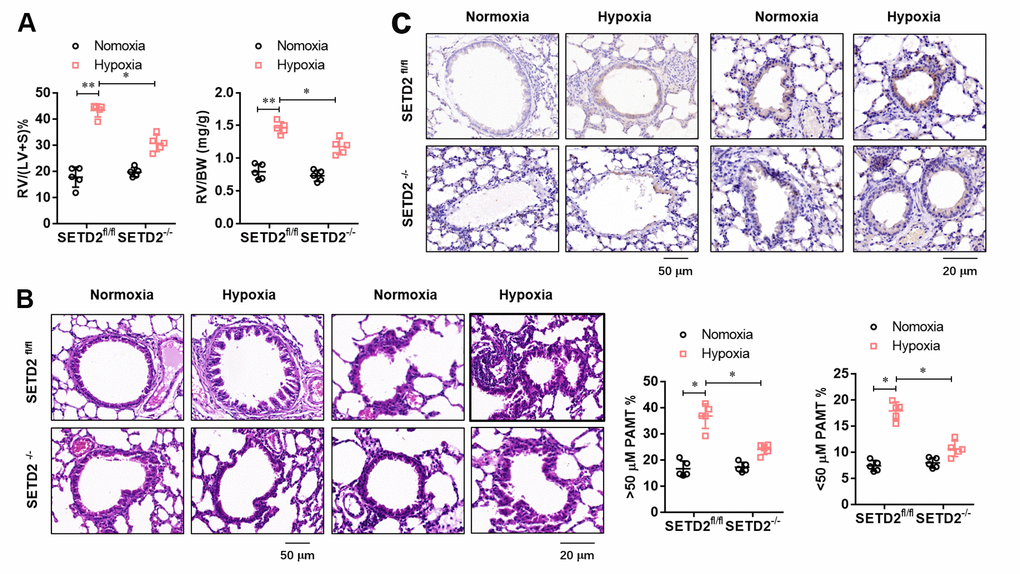
Figure 4. SMCs specific SETD2 deficient attenuates pathological remodeling of pulmonary artery and right ventricular hypertrophy. (A) The right ventricular hypertrophy was evaluated by the RV/BW ratio (the ratio of the right ventricular mass to the sum of body masses) and RV/(L+S) ratio (the ratio of the right ventricular mass to the sum of the left ventricular and septal masses). (B) Haematoxylin and eosin (HE) staining and the analyzed PAMT ration of both small and medium pulmonary arteries were used to evaluate the pulmonary blood vessel remodeling during the course of PAH development. (C) Histochemical staining of SETD2 immunostaining was used to detect the SETD2 expression in lung blood vessels. All data were presented as Mean±SD (n=5). *P<0.05, **P<0.01 vs. corresponding group.
SETD2 mediated H3K36me3 and METTL14 mediated m6A RNA modification are involved in hypoxia-induced PAH
To investigate whether SETD2 mediates H3K36me3 is involved in PAH, we evaluated the protein level of SETD2 and H3K36me3. Western blot analysis showed that hypoxia markedly elevated the protein level of SETD2 and H3K36me3 in SMCs (Figure 5A), but SMCs specific SETD2 deficient impaired the level of the protein level of SETD2 (Figure 5A, 5B) and H3K36me3 (Figure 5A, 5C). These data suggest that SETD2 mediates H3K36me3 is involved in PAH. As H3K36me3 is recognized and bound directly by METTL3/14 and deliver the m6A modification. We then evaluated the protein level of METTL3, METTL14 and the m6A modificated RNA level. Western blot analysis also showed that hypoxia markedly elevated the protein level of METTL14 in SMCs (Figure 5A, 5E), but SMCs specific SETD2 deficient impaired the level of the protein level of METTL14 (Figure 5A, 5E). However, the protein level of METTL3 was changed neither by hypoxia nor SMCs specific SETD2 deficient (Figure 5A, 5D). Moreover, the total m6A modificated RNA level, indicated by dot blot (Figure 6A) and m6A RNA methylation quantification (Figure 6B), were significantly enhanced in hypoxia induced PAH group, whereas diminished by SMCs specific SETD2 deficient. Collectively, these results demonstrated that SETD2 mediates H3K36me3 and METTL14 mediated m6A RNA modification contribute to hypoxia-induced pulmonary arterial hypertension in mice.
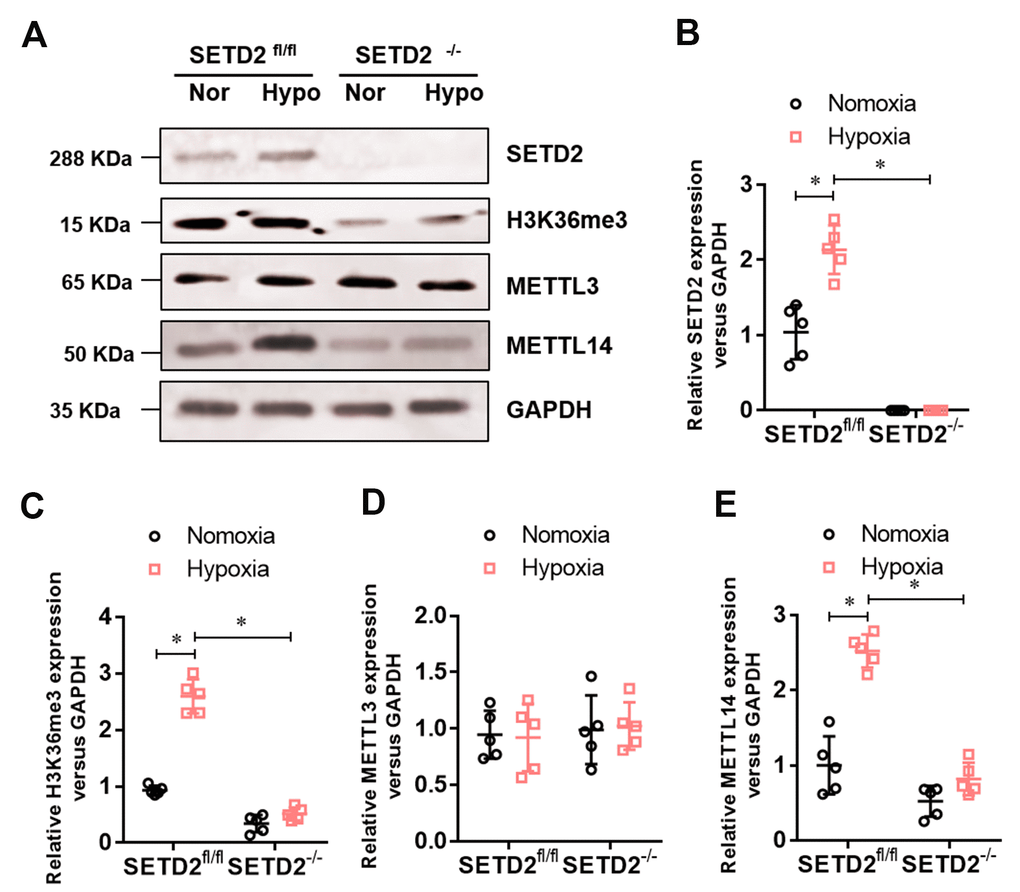
Figure 5. The expression of SETD2 mediates H3K36me3 and METTL14 are enhanced in hypoxia-induced PAH, whereas impaired by SMCs specific SETD2 deficient. (A) Represent images of Western blotting analysis. The statistical data showed that hypoxia markedly elevated the protein level of SETD2 (B), H3K36me3 (C) and METTL14 (E) in SMCs, but SMCs specific SETD2 deficient impaired the level of the protein level of H3K36me3 and METTL14. However, the protein level of METTL3 (D) was changed neither by hypoxia nor SMCs specific SETD2 deficient. All data were presented as Mean±SD (n=5). *P<0.05 vs. corresponding group.
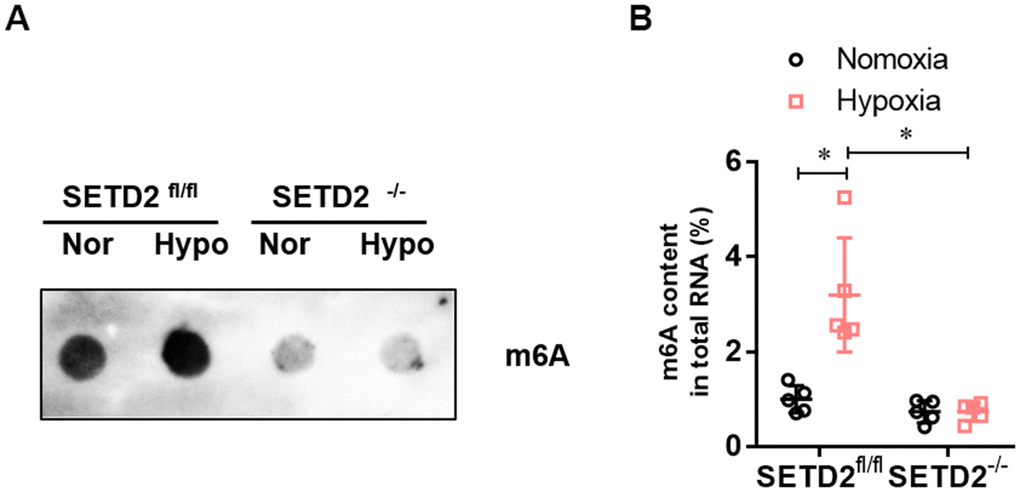
Figure 6. The level of m6A RNA modification is elevated in hypoxia-induced PAH, whereas impaired by SMCs specific SETD2 deficient. (A) The content of m6A was quantitatively determined by colorimetry. (B) The content of m6A was qualitatively evaluated by Dot blot. All data were presented as Mean±SD (n=5). *P<0.05 vs. corresponding group.
Discussion
Epigenetic changes caused by hypoxia can lead to permanent changes in early fetal development, increasing the risk of PAH in adults [11]. Unlike genetic mutations, epigenetic changes are pharmacologically reversible, making them an attractive target for the treatment of PAH. Data from this study showed that SMCs-specific SETD2 deletion resulted in the inactivation of H3K36me3 modification, which reversed hypoxia-induced PAH. Chronic hypoxia exposure can lead to significant increase in RVSP, increase in RV/(LV+S), and significant thickening of the middle membrane of pulmonary arterioles. However, the specific knockout of SETD2 in SMCs inhibits these increases. Furthermore, we showed that METTL14 mediated m6A RNA modification was elevated in the SMCs from hypoxia-induced PAH mice, which was similarly diminished by specific knockout of SETD2 in SMC. This novel information partially clarified that SETD2 mediates H3K36me3 and METTL14 mediated m6A RNA modification contribute to hypoxia-induced pulmonary arterial hypertension in mice.
In the development of PAH, the Warburg effect (glycolytic shift) and mitochondrial fission are critical factors that determine the phenotypic characteristics of the disease, such as proliferation, apoptotic resistance, migration, endothelial-mesenchymal transition, and extracellular matrix stiffness [3–5, 7]. Current therapies have focused on vasodilation and antithrombotic protection, but have not restored these abnormal phenotypes, suggesting the need to target other pathways. The multi-factor characteristics of PAH suggest that epigenetic changes are potential determinants of vascular remodeling [26, 27]. In recent years, studies on molecular markers for epigenetic changes in PAH have steadily increased [28, 29]. These experimental and clinical studies have shown that epigenetic processes, such as DNA methylation, ubiquitination, miRNA-dependent gene regulation, and HDACs are associated with PAH [26, 30]. Histone methylation is a process of transcriptional promotion or inhibition of post-translational modification based on specific methylated residues (lysine or arginine) and the number of methyl groups added [31]. However, histone methylation involved in PAH has been rarely reported. Our previous studies showed that NSD2, catalytic dimethylation of H3K36 (H3K36me2) changes in energy metabolism are involved in monocrotaline-induced PAH [32]. On the other hand, the SETD2fl/fl and SETD2-/- groups showed very similar pulmonary pressure and right ventricular function, which suggest that SETD2 mediated H3K36me3 may not be critical under normal physiological conditions. Moreover, SMCs specific SETD2 deficient did not alert the level of m6A and METTL14 under normal physiological conditions, whereas impaired the PAH induced elevation of m6A and METTL14. Hence, we think that METTL14 protein and m6A RNA are pathologically involved in the progression of PAH. Thus, this study further demonstrated that SETD2 mediated H3K36me3 is pathologically involved in hypoxia-induced PAH in mice.
Muscularization of vascular wall and phenotypic changes of endothelial cells lead to vascular remodeling, which is the main pathological feature of PAH [33, 34]. Arterioles obstructed by phenotypic changes and proliferation of cross-differentiated pulmonary artery endothelial cells (PAECs) are considered irreversible [35]. However, there is also some evidence that pulmonary artery SMCs can be transferred between proliferative and non-proliferative states, raising the possibility that this aspect of vascular reconstruction may be reversible [34, 36, 37]. The effect of m6A post-transcriptional modification on proliferation control has received extensive attention in recent years. METTL3, the main m6A methyltransferase, inhibits cell differentiation and apoptosis and promotes cell proliferation by increasing the translation of c-myc, bcl-2 and PTEN in leukemia [38]. WTAP, another member of m6A writer, enhances proliferation, migration, invasion, and tumorigenicity of glioblastoma cells in xenotransplantation by mediating the phosphorylation of epidermal growth factor receptor (EGFR) and AKT [39]. As far as we know, regulators of m6A play an important role in proliferation, especially in the development of cancer. In recent years, studies have found that total Panax notoginseng saponin regulate WTAP/p16 signal through m6A, and inhibit proliferation, migration and intimal hyperplasia of vascular smooth muscle cells [40]. However, the role of m6A post-transcriptional modification in the formation of PAH is unclear. In this study, we demonstrated that the total m6A modified RNA level was significantly increased in the hypoxia-induced PAH group, while the SMCs-specific SETD2 defect reduced the total m6A modified RNA level.
In summary, our study preliminarily found that SETD2 mediated H3K36me3 was involved in the occurrence and development of hypoxia-induced PAH through METTL14-mediated m6A RNA modification. Both SETD2 and METTL14 are promising targets for the prevention and treatment of PAH.
Materials and Methods
Generation and characterization of mice
All animals were maintained in a specific pathogen-free facility. SM22αCre transgenic mice were purchased from Beijing Biocytogen Co. Ltd. (Beijing, China) SETD2fl/fl C57BL/6 mice were generated by Shanghai Nanfang Research Center for Model Organisms, using conventional homologous recombination in embryonic stem cells, and the targeting strategy was shown in Figure 1A. All animal experiments were performed in accordance with the guidelines of NIH and under approved protocols of the Animal Care and Use Committee of Nanchang University.
Chronic hypoxia induced PAH mice model
At the age of 10–12 weeks, male SETD2SM22a-Cre mice and their WT littermates (SETD2fl/fl) were randomly assigned to the normoxia group (N) and hypoxia group (H). The normoxia group was exposed to room air, whereas the hypoxia-group mice were placed in a ventilated chamber with 10% O2 and CO2 < 0.5% environment. The environment was established by a mixture of room air and nitrogen with a detector (RCI Hudson, Anaheim, CA, USA) which monitored and controlled the fractional concentration of O2 automatically.
Antibodies
Antibodies against SETD2, H3K36me3, METTL3 and METTL14 were purchased from Cell Signaling Technologies (Danvers, MA, USA), GAPDH and m6A antibodies were purchased from Abcam (Cambridge, MA, USA).
Measurement of hemodynamics
After normal or hypoxic exposure, mice were anesthetized with sodium pentobarbital (60 mg/kg) and the hemodynamic measurements were performed as previously. In brief, the 25 g needle with a pressure sensor (FTH-1211B-0018, Primetech) were inserted through the jugular vein to measure the right ventricular mean pressure (RVMP), right ventricular systolic pressure (RVSP), mean pulmonary arterial pressure (mPAP), and the left carotid artery cannulation was performed for systemic arterial pressure (mSAP) and systolic body pressure (SBP) measurement. All the calculations were done with a LabChart8 (AD Instruments). Total pulmonary resistance (TPR) was calculated by the ratio of mPAP/RVCO.
Echocardiography
Echocardiography was measured using the Vevo 2100 high-resolution imaging system (Visual Sonics Inc.) as previously [32]. Normalized cardiac cycle length measuring pulmonary acceleration time (PAAT/CL), RV end diastolic dimension (RVEDD), RV diastolic contraction size (RVESD), RV end diastolic volume (RVEDV), RV diastolic volume contraction (RVESV), RV cardiac output (RVCO), RV wall thickness (RVWT), the thickness of interventricular septum (IVS), RV ejection fraction (RVEF) and RV shortening fraction (RVFS). All echocardiographic studies were analyzed by genotypic blind method.
Histological and immunohistochemical analysis
Histological and immunohistochemical analysis as described previously [32]. In brief, lung specimens were collected and stained with hematoxylin-eosin (HE). Immunohistochemical staining was performed on paraffin embedded equivalent slides (5 m) using a quick toolkit (Beyotime, Jiangsu, China) and stained with anti-SETD2 antibody (Abcam,1:1000 dilution).
Western blotting analysis
Western Blotting analysis were performed as described previously [32]. Briefly, the protein was isolated by RIPA lysis buffer (Beyotime) and the protein concentration was determined using the BCA protein detection kit (Beyotime), then transferred to PVDF membranes (Millipore, inc., USA) by SDS-PAGE. The membranes were blocked with 5% non-fat dry milk in TBS-T (20 mM Tris, 137 mM NaCl, 0.05% Tween-20, pH 7.4) at room temperature for 3 h and then incubated with primary antibodies at 4° C.
Dot blot
Total RNA was isolated by TriZol (Thermo Fisher) and loaded to an Amersham Hybond-N+ membrane (GE Healthcare, USA). After UV crosslinking and PBST washing, they were then stained with 0.02% methylene blue (Chinese worker) and the scans showed the total amount of incoming RNA. After incubation with 5% non-fat milk, the membrane was incubated overnight at 4° C with specific m6A antibody (1:1000, Millipore). After incubation with hrp-labeled anti-mouse immunoglobulin G (IgG) for 1 hour, the blobs were observed with an imaging system (Bio-Rad, USA).
Total RNA m6A quantification
Total RNA was extracted using the TriZol kit (Thermo Fisher), and polyadenylate mRNA was purified using the GenElute™mRNA Miniprep kit (Sigma-Aldrich, Germany) according to the manufacturer's protocol. EpiQuik™ m6A RNA methylation quantitative kit (colorimetry)(Epigentek, USA) was then used to determine the total level of m6A in the treated cells. Briefly, 500 ng RNA was added to each pore, followed by capture and detection antibodies. After multiple incubation, the content of m6A was quantitatively determined by colorimetry at the wavelength of 450nm and calculated according to the standard curve.
Statistical analysis
All data were expressed as mean ± standard deviation and analyzed by SPSS 11.0 statistical software. One-way ANOVA was used to compare the differences among the groups. P<0.05 was considered statistically significant.
Author Contributions
Xue-Liang Zhou performed molecular biology studies, Xue-Liang Zhou, Feng-Jian Huang, Yang Li and Huang Huang performed animal studies, Xue-Liang Zhou and Qi-Cai Wu designed the study and write the manuscript. All authors read and approved the manuscript.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 81970199, 81860045, 81860054), the Natural Science Foundation of Jiangxi Province (No. 20192ACB21030, 20192ACBL20036, 20202BAB206006) and the innovation of science and technology in Jiangxi Province outstanding young talent training plan (No. 20192BCBL23022).
References
- 1. Gerges C, Gerges M, Friewald R, Fesler P, Dorfmüller P, Sharma S, Karlocai K, Skoro-Sajer N, Jakowitsch J, Moser B, Taghavi S, Klepetko W, Lang IM. Microvascular disease in chronic thromboembolic pulmonary hypertension: hemodynamic phenotyping and histomorphometric assessment. Circulation. 2020; 141:376–86. https://doi.org/10.1161/CIRCULATIONAHA.119.041515 [PubMed]
- 2. Ruopp NF, Farber HW. The new world symposium on pulmonary hypertension guidelines: should Twenty-One be the new Twenty-Five? Circulation. 2019; 140:1134–36. https://doi.org/10.1161/CIRCULATIONAHA.119.040292 [PubMed]
- 3. White RJ, Wilkins MR. New therapeutic approaches in pulmonary arterial hypertension: the pantheon is getting crowded. Circulation. 2018; 137:2390–92. https://doi.org/10.1161/CIRCULATIONAHA.118.032700 [PubMed]
- 4. Gu S, Kumar R, Lee MH, Mickael C, Graham BB. Common genetic variants in pulmonary arterial hypertension. Lancet Respir Med. 2019; 7:190–91. https://doi.org/10.1016/S2213-2600(18)30448-X [PubMed]
- 5. Leopold JA. Pulmonary venous remodeling in pulmonary hypertension: the veins take center stage. Circulation. 2018; 137:1811–13. https://doi.org/10.1161/CIRCULATIONAHA.118.033013 [PubMed]
- 6. Hautefort A, Mendes-Ferreira P, Sabourin J, Manaud G, Bertero T, Rucker-Martin C, Riou M, Adão R, Manoury B, Lambert M, Boet A, Lecerf F, Domergue V, et al. Bmpr2 mutant rats develop pulmonary and cardiac characteristics of pulmonary arterial hypertension. Circulation. 2019; 139:932–48. https://doi.org/10.1161/CIRCULATIONAHA.118.033744 [PubMed]
- 7. Obokata M, Kane GC, Reddy YN, Melenovsky V, Olson TP, Jarolim P, Borlaug BA. The neurohormonal basis of pulmonary hypertension in heart failure with preserved ejection fraction. Eur Heart J. 2019; 40:3707–17. https://doi.org/10.1093/eurheartj/ehz626 [PubMed]
- 8. Lüscher TF. Arterial and pulmonary hypertension: risk assessment and current pharmacological and interventional management. Eur Heart J. 2018; 39:4127–31. https://doi.org/10.1093/eurheartj/ehy824 [PubMed]
- 9. Rhodes CJ, Batai K, Bleda M, Haimel M, Southgate L, Germain M, Pauciulo MW, Hadinnapola C, Aman J, Girerd B, Arora A, Knight J, Hanscombe KB, et al, and UK NIHR BioResource Rare Diseases Consortium, UK PAH Cohort Study Consortium, and US PAH Biobank Consortium. Genetic determinants of risk in pulmonary arterial hypertension: international genome-wide association studies and meta-analysis. Lancet Respir Med. 2019; 7:227–38. https://doi.org/10.1016/S2213-2600(18)30409-0 [PubMed]
- 10. Yuan K, Shamskhou EA, Orcholski ME, Nathan A, Reddy S, Honda H, Mani V, Zeng Y, Ozen MO, Wang L, Demirci U, Tian W, Nicolls MR, de Jesus Perez VA. Loss of endothelium-derived Wnt5a is associated with reduced pericyte recruitment and small vessel loss in pulmonary arterial hypertension. Circulation. 2019; 139:1710–24. https://doi.org/10.1161/CIRCULATIONAHA.118.037642 [PubMed]
- 11. Chen KH, Dasgupta A, Lin J, Potus F, Bonnet S, Iremonger J, Fu J, Mewburn J, Wu D, Dunham-Snary K, Theilmann AL, Jing ZC, Hindmarch C, et al. Epigenetic dysregulation of the dynamin-related protein 1 binding partners MiD49 and MiD51 increases mitotic mitochondrial fission and promotes pulmonary arterial hypertension: mechanistic and therapeutic implications. Circulation. 2018; 138:287–304. https://doi.org/10.1161/CIRCULATIONAHA.117.031258 [PubMed]
- 12. Sermer D, Pasqualucci L, Wendel HG, Melnick A, Younes A. Emerging epigenetic-modulating therapies in lymphoma. Nat Rev Clin Oncol. 2019; 16:494–507. https://doi.org/10.1038/s41571-019-0190-8 [PubMed]
- 13. Weinberg DN, Papillon-Cavanagh S, Chen H, Yue Y, Chen X, Rajagopalan KN, Horth C, McGuire JT, Xu X, Nikbakht H, Lemiesz AE, Marchione DM, Marunde MR, et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature. 2019; 573:281–86. https://doi.org/10.1038/s41586-019-1534-3 [PubMed]
- 14. Xue H, Yao T, Cao M, Zhu G, Li Y, Yuan G, Chen Y, Lei M, Huang J. Structural basis of nucleosome recognition and modification by MLL methyltransferases. Nature. 2019; 573:445–49. https://doi.org/10.1038/s41586-019-1528-1 [PubMed]
- 15. Legchenko E, Chouvarine P, Borchert P, Fernandez-Gonzalez A, Snay E, Meier M, Maegel L, Mitsialis SA, Rog-Zielinska EA, Kourembanas S, Jonigk D, Hansmann G. PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci Transl Med. 2018; 10:eaao0303. https://doi.org/10.1126/scitranslmed.aao0303 [PubMed]
- 16. Xu Q, Xiang Y, Wang Q, Wang L, Brind’Amour J, Bogutz AB, Zhang Y, Zhang B, Yu G, Xia W, Du Z, Huang C, Ma J, et al. SETD2 regulates the maternal epigenome, genomic imprinting and embryonic development. Nat Genet. 2019; 51:844–56. https://doi.org/10.1038/s41588-019-0398-7 [PubMed]
- 17. Diao YF, Lin T, Li X, Oqani RK, Lee JE, Kim SY, Jin DI. Dynamic changes of SETD2, a histone H3K36 methyltransferase, in porcine oocytes, IVF and SCNT embryos. PLoS One. 2018; 13:e0191816. https://doi.org/10.1371/journal.pone.0191816 [PubMed]
- 18. Zuo X, Rong B, Li L, Lv R, Lan F, Tong MH. The histone methyltransferase SETD2 is required for expression of acrosin-binding protein 1 and protamines and essential for spermiogenesis in mice. J Biol Chem. 2018; 293:9188–97. https://doi.org/10.1074/jbc.RA118.002851 [PubMed]
- 19. Bilokapic S, Halic M. Nucleosome and ubiquitin position Set2 to methylate H3K36. Nat Commun. 2019; 10:3795. https://doi.org/10.1038/s41467-019-11726-4 [PubMed]
- 20. Livneh I, Moshitch-Moshkovitz S, Amariglio N, Rechavi G, Dominissini D. The m6A epitranscriptome: transcriptome plasticity in brain development and function. Nat Rev Neurosci. 2020; 21:36–51. https://doi.org/10.1038/s41583-019-0244-z [PubMed]
- 21. Mathiyalagan P, Adamiak M, Mayourian J, Sassi Y, Liang Y, Agarwal N, Jha D, Zhang S, Kohlbrenner E, Chepurko E, Chen J, Trivieri MG, Singh R, et al. FTO-dependent N6-methyladenosine regulates cardiac function during remodeling and repair. Circulation. 2019; 139:518–32. https://doi.org/10.1161/CIRCULATIONAHA.118.033794 [PubMed]
- 22. Paris J, Morgan M, Campos J, Spencer GJ, Shmakova A, Ivanova I, Mapperley C, Lawson H, Wotherspoon DA, Sepulveda C, Vukovic M, Allen L, Sarapuu A, et al. Targeting the RNA m6A reader YTHDF2 selectively compromises cancer stem cells in acute myeloid leukemia. Cell Stem Cell. 2019; 25:137–48.e6. https://doi.org/10.1016/j.stem.2019.03.021 [PubMed]
- 23. Vu LP, Cheng Y, Kharas MG. The biology of m6A RNA methylation in normal and Malignant hematopoiesis. Cancer Discov. 2019; 9:25–33. https://doi.org/10.1158/2159-8290.CD-18-0959 [PubMed]
- 24. Dorn LE, Lasman L, Chen J, Xu X, Hund TJ, Medvedovic M, Hanna JH, van Berlo JH, Accornero F. The N6-methyladenosine mRNA methylase METTL3 controls cardiac homeostasis and hypertrophy. Circulation. 2019; 139:533–45. https://doi.org/10.1161/CIRCULATIONAHA.118.036146 [PubMed]
- 25. Garikipati VNS, Verma SK, Cheng Z, Liang D, Truongcao MM, Cimini M, Yue Y, Huang G, Wang C, Benedict C, Tang Y, Mallaredy V, Ibetti J, et al. Circular RNA CircFndc3b modulates cardiac repair after myocardial infarction via FUS/VEGF-A axis. Nat Commun. 2019; 10:4317. https://doi.org/10.1038/s41467-019-11777-7 [PubMed]
- 26. Napoli C, Benincasa G, Loscalzo J. Epigenetic inheritance underlying pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol. 2019; 39:653–64. https://doi.org/10.1161/ATVBAHA.118.312262 [PubMed]
- 27. Thenappan T, Ormiston ML, Ryan JJ, Archer SL. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ. 2018; 360:j5492. https://doi.org/10.1136/bmj.j5492 [PubMed]
- 28. Ryan J, Dasgupta A, Huston J, Chen KH, Archer SL. Mitochondrial dynamics in pulmonary arterial hypertension. J Mol Med (Berl). 2015; 93:229–42. https://doi.org/10.1007/s00109-015-1263-5 [PubMed]
- 29. Tuder RM, Archer SL, Dorfmüller P, Erzurum SC, Guignabert C, Michelakis E, Rabinovitch M, Schermuly R, Stenmark KR, Morrell NW. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol. 2013; 62:D4–12. https://doi.org/10.1016/j.jacc.2013.10.025 [PubMed]
- 30. Chelladurai P, Boucherat O, Stenmark K, Kracht M, Seeger W, Bauer UM, Bonnet S, Pullamsetti SS. Targeting histone acetylation in pulmonary hypertension and right ventricular hypertrophy. Br J Pharmacol. 2021; 178:54–71. https://doi.org/10.1111/bph.14932 [PubMed]
- 31. Li Y, Liu S, Zhang Y, Gao Q, Sun W, Fu L, Cao J. Histone demethylase JARID1B regulates proliferation and migration of pulmonary arterial smooth muscle cells in mice with chronic hypoxia-induced pulmonary hypertension via nuclear factor-kappa B (NFkB). Cardiovasc Pathol. 2018; 37:8–14. https://doi.org/10.1016/j.carpath.2018.07.004 [PubMed]
- 32. Zhou XL, Liu ZB, Zhu RR, Huang H, Xu QR, Xu H, Zeng L, Li YY, Huang CH, Wu QC, Liu JC. NSD2 silencing alleviates pulmonary arterial hypertension by inhibiting trehalose metabolism and autophagy. Clin Sci (Lond). 2019; 133:1085–96. https://doi.org/10.1042/CS20190142 [PubMed]
- 33. Leopold JA, Maron BA. Molecular mechanisms of pulmonary vascular remodeling in pulmonary arterial hypertension. Int J Mol Sci. 2016; 17:761. https://doi.org/10.3390/ijms17050761 [PubMed]
- 34. Dai Z, Zhu MM, Peng Y, Machireddy N, Evans CE, Machado R, Zhang X, Zhao YY. Therapeutic targeting of vascular remodeling and right heart failure in pulmonary arterial hypertension with a HIF-2α inhibitor. Am J Respir Crit Care Med. 2018; 198:1423–34. https://doi.org/10.1164/rccm.201710-2079OC [PubMed]
- 35. Thompson AA, Lawrie A. Targeting vascular remodeling to treat pulmonary arterial hypertension. Trends Mol Med. 2017; 23:31–45. https://doi.org/10.1016/j.molmed.2016.11.005 [PubMed]
- 36. Chen J, Sysol JR, Singla S, Zhao S, Yamamura A, Valdez-Jasso D, Abbasi T, Shioura KM, Sahni S, Reddy V, Sridhar A, Gao H, Torres J, et al. Nicotinamide phosphoribosyltransferase promotes pulmonary vascular remodeling and is a therapeutic target in pulmonary arterial hypertension. Circulation. 2017; 135:1532–46. https://doi.org/10.1161/CIRCULATIONAHA.116.024557 [PubMed]
- 37. Luo Y, Teng X, Zhang L, Chen J, Liu Z, Chen X, Zhao S, Yang S, Feng J, Yan X. CD146-HIF-1α hypoxic reprogramming drives vascular remodeling and pulmonary arterial hypertension. Nat Commun. 2019; 10:3551. https://doi.org/10.1038/s41467-019-11500-6 [PubMed]
- 38. Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, Chou T, Chow A, Saletore Y, MacKay M, Schulman J, Famulare C, Patel M, et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017; 23:1369–76. https://doi.org/10.1038/nm.4416 [PubMed]
- 39. Jin DI, Lee SW, Han ME, Kim HJ, Seo SA, Hur GY, Jung S, Kim BS, Oh SO. Expression and roles of Wilms’ tumor 1-associating protein in glioblastoma. Cancer Sci. 2012; 103:2102–09. https://doi.org/10.1111/cas.12022 [PubMed]
- 40. Zhu B, Gong Y, Shen L, Li J, Han J, Song B, Hu L, Wang Q, Wang Z. Total panax notoginseng saponin inhibits vascular smooth muscle cell proliferation and migration and intimal hyperplasia by regulating WTAP/p16 signals via m6A modulation. Biomed Pharmacother. 2020; 124:109935. https://doi.org/10.1016/j.biopha.2020.109935 [PubMed]