



Introduction
Chronic thromboembolic pulmonary hypertension (CTEPH) is a rare and debilitating disorder characterized by pulmonary artery obstruction with unresolved, organized thromboemboli [1, 2]. Due to delayed symptoms or misdiagnosis, CTEPH is typically diagnosed at an advanced stage, and as a result, disease prognosis is poor, with a < 40% 5-year survival rate [3, 4]. The only curative treatment currently available is pulmonary endarterectomy (PEA) for improving quality of life and prolonging survival [5–7]. However, not all patients are eligible for surgery, and alternative treatments are still required. Moreover, the exact pathophysiology of CTEPH remains largely unknown. To improve clinical outcomes, improved understanding of the detailed mechanisms involved in CTEPH is crucial.
The occurrence and development of CTEPH are complex processes involving various genes, multiple cell types, and a large number of signal transduction systems [8]. In particular, the roles of multiple cell types in disease development are gaining increasing attention. It has been reported that accumulation of circulating inflammatory cells [9] and excessive proliferation and migration of smooth muscle and endothelial cells [10–12] are involved in pulmonary arterial remodeling during CTEPH. Moreover, crosstalk between endothelial-like and myofibroblast-like cells has been shown to play a key role in endothelial cell dysfunction, contributing to vascular lesion in CTEPH [13]. Despite these findings, the cell types contributing to CTEPH progression have not been fully elucidated. A clear and complete atlas of the cellular repertoire that participates in the occurrence and development of CTEPH is vital for the development of effective diagnostic and therapeutic strategies to prevent CTEPH and decrease mortality.
The recent development of single-cell RNA sequencing (scRNA-seq) technology has provided new perspectives on the complex biological systems and key functions associated with disease development [14–16]. In the present study, we applied scRNA-seq to comprehensively characterize the cell cluster composition in the pulmonary endarterectomized tissues of patients with CTEPH for the first time. Our results revealed that the cell populations in pulmonary endarterectomized tissues include fibroblast/smooth muscle cell, T cell/natural killer (NK) cell, macrophage, cysteine rich secretory protein LCCL domain containing 2 (CRISPLD2)+ cell, cancer stem cell, mast cell, endothelial cell, and undefined. This study, for the first time, provided a comprehensive and unique resource revealing CTEPH cellular typing at single-cell resolution. Uncovering the function of these cell populations will help us to better understand CTEPH pathophysiology.
Results
Quality control analysis
To construct an atlas of the cell repertoire in CTEPH, we generated scRNA-seq profiles of pulmonary endarterectomized tissues from five CTEPH patients undergoing PEA. Tissue samples were digested into single-cell suspensions and analyzed by droplet-based single-cell transcriptome profiling using the 10× Genomics Single Cell Gene Expression Chromium system (https://www.10xgenomics.com/products/single-cell-gene-expression) (Figure 1). DNA sequencing reads were mapped to the hg19 human reference genome. We generated five-feature barcode matrices for each of the five samples independently, and these matrices were then aggregated. Subsequently, we filtered and collected 27,140 cells. By visualizing histograms representing gene number and unique molecular identifier (UMI) count, individual cells and their gene expression in the sample could be evaluated. For species for which mitochondrial genome information is available, the percentage of mitochondrial genes in cells was calculated to filter cells with relatively high mitochondrial gene content. Gene numbers, UMI counts, and percentage of mitochondrial genes are represented using violin plots (Supplementary Figure 1).
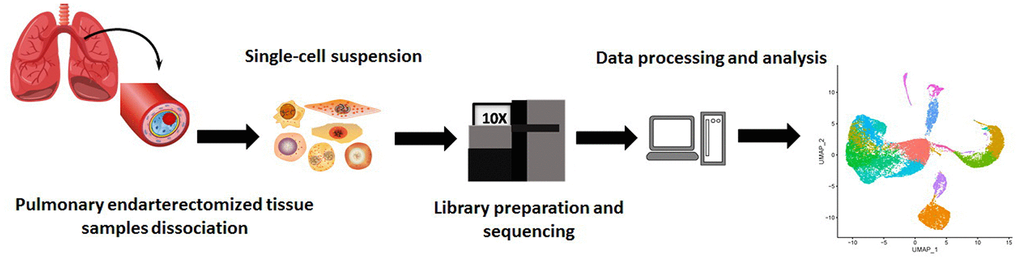
Figure 1. Schematic of the experimental design for single-cell RNA sequencing.
Overall characteristics of cell cluster composition in pulmonary tissues after endarterectomy of CTEPH patients
After quality filtering and normalization of gene expression, dimension reduction involving the top 20 components was conducted. As a result, a total of 17 cell clusters (clusters 0–16) was characterized (Figure 2A), corresponding to 10,518 marker genes. We then determined marker genes that were uniquely expressed in each cell cluster in comparison with all other clusters. The key marker genes for each cell cluster are presented in Figure 2B. Based on cell markers, fibroblast/smooth muscle cells were composed of clusters 3, 5, 6, 7, 9, and 15, and accounted for the largest proportion (37%) of total cells. Clusters 2 and 4 were classified as T-/NK cells, with a relative percentage of 18%. Clusters 1, 8, 10, 12, and 13 were classified as macrophages (11%), CRISPLD2+ cells (6%), cancer stem cells (4%), mast cells (2%), and endothelial cells (2%), respectively. Clusters 0, 11, 14, 16 were categorized as ‘undefined’ (20%) (Figure 2C, 2D).
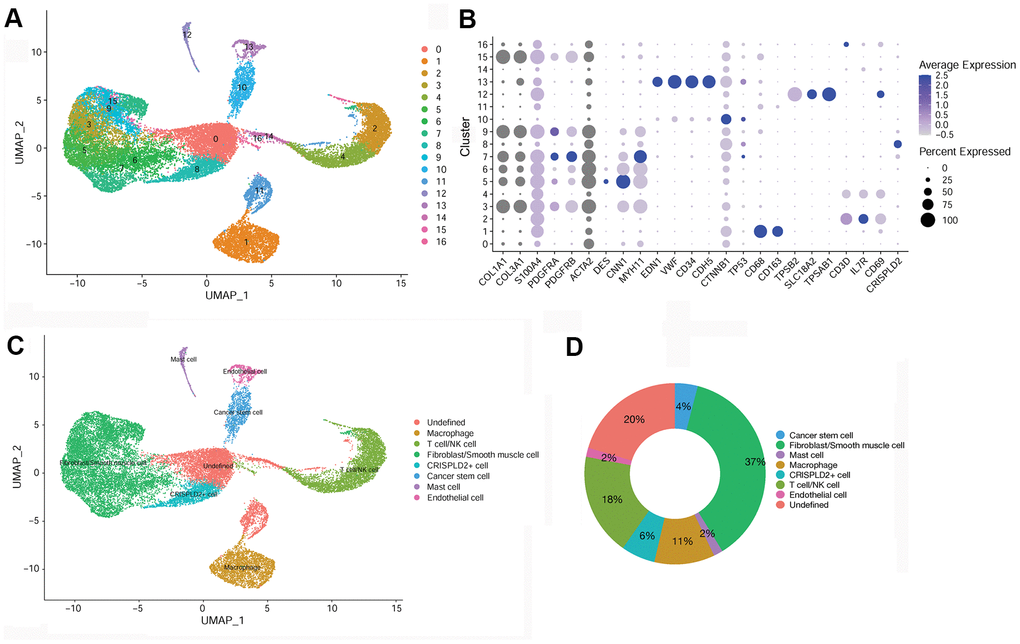
Figure 2. Identification of key marker genes and overall characteristics of cell cluster compositions in pulmonary endarterectomized tissues of CTEPH patients. (A) UMAP plot of 17 clusters. (B) Bubble diagram showing the key marker genes in 17 clusters. (C) UMAP plot of eight cell types. Based on cell markers, clusters 3, 5, 6, 7, 9, and 15 were annotated as fibroblasts/smooth muscle cells. Clusters 2 and 4 were classified as T-/NK cells. Clusters 1, 8, 10, 12, and 13 were classified as macrophages and CRISPLD2+, cancer stem, mast, and endothelial cells, respectively. Clusters 0, 11, 14, and 16 were classified as undefined. (D) Pie chart showing the proportion of each cell type among total cells. Abbreviations: CTEPH: chronic thromboembolic pulmonary hypertension; UMAP: Uniform Manifold Approximation and Projection; CRISPLD2: cysteine-rich secretory protein LCCL domain containing 2.
Characterization of fibroblast/smooth muscle cell and endothelial cell in the pulmonary endarterectomized tissues of CTEPH patients
To further examine fibroblast/smooth muscle cell clusters in CTEPH, Uniform Manifold Approximation and Projection (UMAP) were used to sub-cluster fibroblast/smooth muscle cells into three distinct subsets: fibroblasts, myofibroblasts, and smooth muscle cells (Figure 3A). Specific marker genes for the fibroblast/smooth muscle cell cluster were identified, including smooth muscle actin alpha 2 (ACTA2), platelet-derived growth factor receptor beta (PDGFRB), collagen type I alpha 1 chain (COL1A1), collagen type III alpha 1 chain (COL3A1), S100 calcium binding protein A4 (S100A4), platelet-derived growth factor receptor alpha (PDGFRA), desmin (DES), calponin 1 (CNN1), and myosin heavy chain 11 (MYH11) (Figure 3B). Moreover, these marker genes were significantly enriched for multiple functions associated with smooth muscle cell migration (Figure 3C).
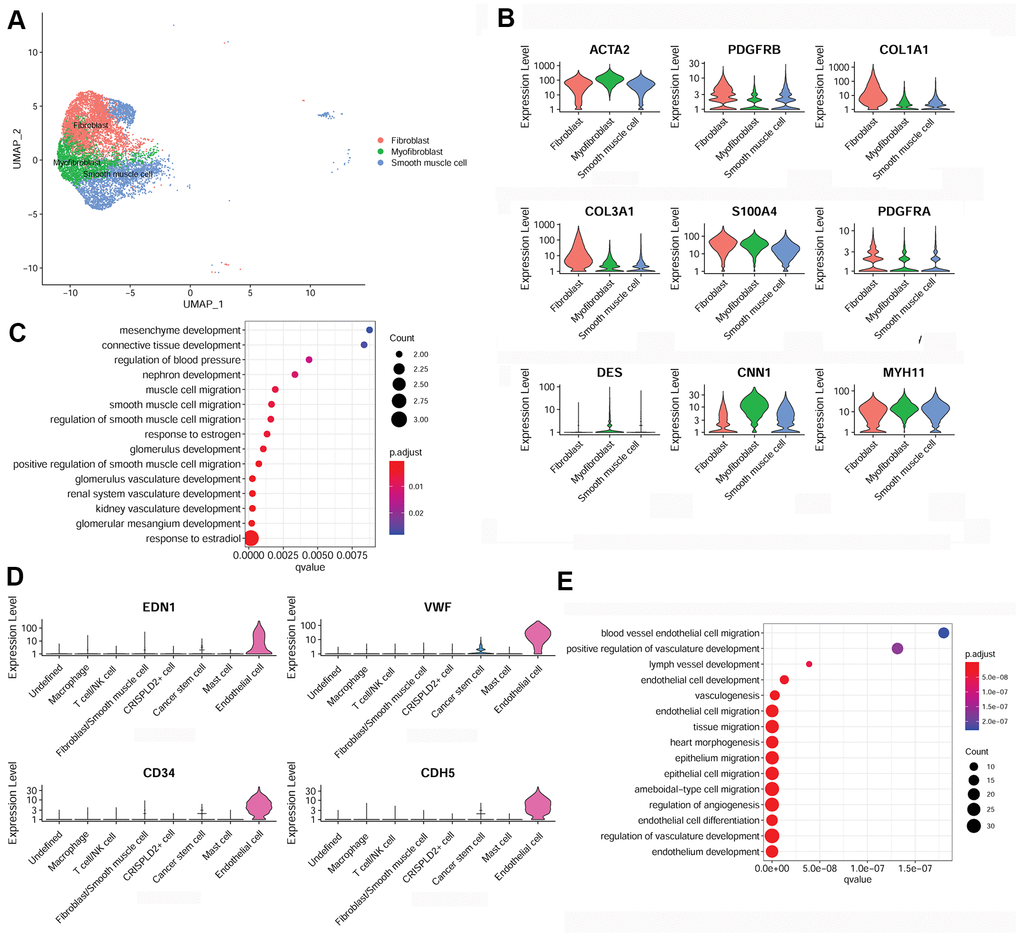
Figure 3. Characterization of fibroblasts/smooth muscle cells and endothelial cells in the pulmonary endarterectomized tissues of CTEPH patients. (A) UMAP plot of three subsets of fibroblasts/smooth muscle cells. (B) Violin plot illustrating fibroblast/smooth muscle cell/myofibroblast marker genes, including ACTA2, PDGFRB, COL1A1, COL3A1, S100A4, PDGFRA, DES, CNN1, and MYH11. (C) GO enrichment analysis of fibroblast/smooth muscle cell/myofibroblast markers. (D) Violin plot showing marker genes of endothelial cells, including END1, VWF, CD34, and CDH5. (E) GO enrichment analysis of endothelial cell marker genes. Abbreviations: CTEPH: chronic thromboembolic pulmonary hypertension; UMAP: Uniform Manifold Approximation and Projection; GO: Gene ontology.
In addition, specific marker genes of endothelial cells were determined to be endothelin 1 (END1), von Willebrand factor (VWF), CD34, and cadherin 5 (CDH5) (Figure 3D). These were mainly enriched for functions associated with cell migration, such as endothelial cell migration, epithelium migration, and ameboid-type cell migration (Figure 3E).
Characterization of T cell/NK cell, macrophage, and mast cell in the pulmonary endarterectomized tissues of CTEPH patients
To better understand the immune mechanisms underlying CTEPH, we further characterized T-/NK cells, macrophages, and mast cells in pulmonary endarterectomized tissues of CTEPH patients. The T-/NK cell cluster was also subdivided into T- and NK cells, as shown in the UMAP plot (Figure 4A). Figure 4B shows violin plots of the specific marker genes of the T-/NK cell cluster, including CD3D, interleukin 7 receptor (IL7R), CD69, and killer cell lectin-like receptor B1 (KLRB1). Further Gene Ontology (GO) enrichment analysis showed that T cell activation was an important function significantly enriched in the marker genes identified in T-/NK cells (Figure 4C). Specific macrophage marker genes were identified as CD68 and CD163 (Figure 5A), which were significantly involved in key functions in neutrophil activation, cell chemotaxis, and antigen processing and presentation via MHC Class II molecules (Figure 5B). In addition, the specific marker genes of mast cells were revealed to be tryptase beta 2 (TPSB2), solute carrier family 18 member A2 (SLC18A2), and tryptase alpha/beta 1 (TPSAB1) (Figure 5C). The significantly enriched GO functions of these mast cell marker genes included erythrocyte differentiation and homeostasis (Figure 5D).
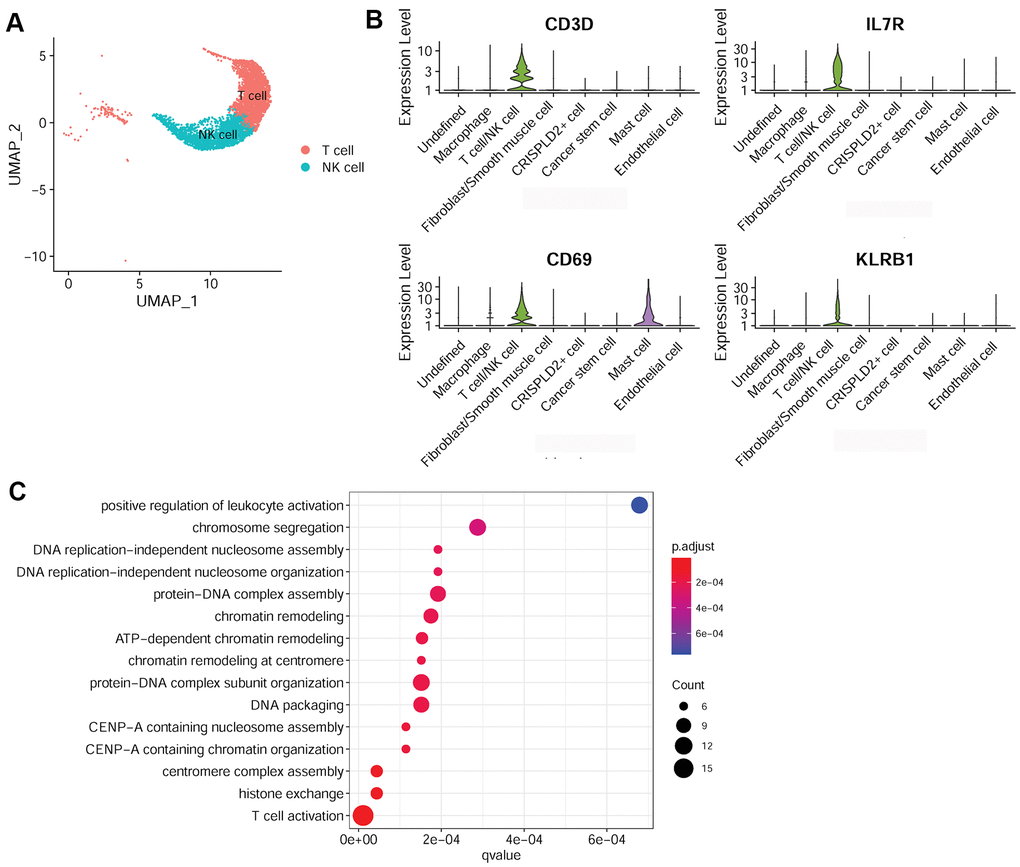
Figure 4. Characterization of T-/NK cells in pulmonary endarterectomized tissues of CTEPH patients. (A) UMAP plot of T and NK cells. (B) Violin plot showing the marker genes of T cell/NK cells, including CD3D, IL7R, CD69, and KLRB1. (C) GO enrichment analysis of marker genes of T-/NK cells. Abbreviations: CTEPH: chronic thromboembolic pulmonary hypertension; UMAP: Uniform Manifold Approximation and Projection; GO: Gene ontology.
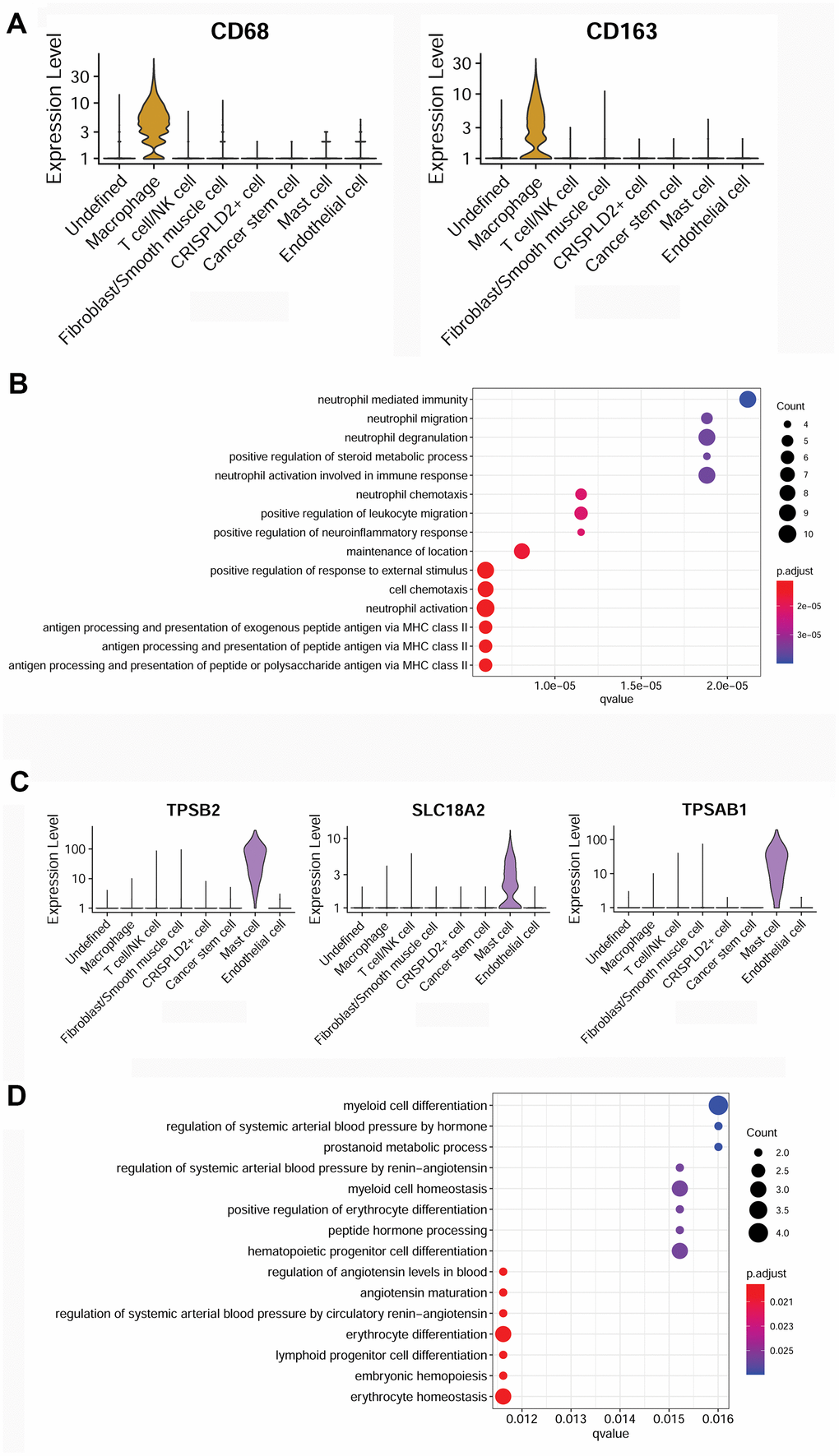
Figure 5. Characterization of macrophages and mast cells in pulmonary endarterectomized tissues of CTEPH patients. (A) Violin plot showing the marker genes of macrophages, including CD68 and CD163. (B) GO enrichment analysis of marker genes of macrophages. (C) Violin plot showing mast cell marker genes, including TPSB2, SLC18A2, and TPSAB1. (D) GO enrichment analysis of mast cell marker genes. Abbreviations: CTEPH: chronic thromboembolic pulmonary hypertension; GO: Gene ontology.
Characterization of other cell clusters in pulmonary endarterectomized tissues of CTEPH patients
Specific marker genes of cancer stem cells were determined to be catenin beta 1 (CTNNB1) and tumor protein P53 (TP53) (Figure 6A). The over-represented GO function in cancer stem cell marker genes was categorized as ‘tissue remodeling’ (Figure 6B). The specific marker gene of CRISPLD2+ cells was identified as CRISPLD2 (Figure 6C). However, no significant GO function was enriched for this gene.
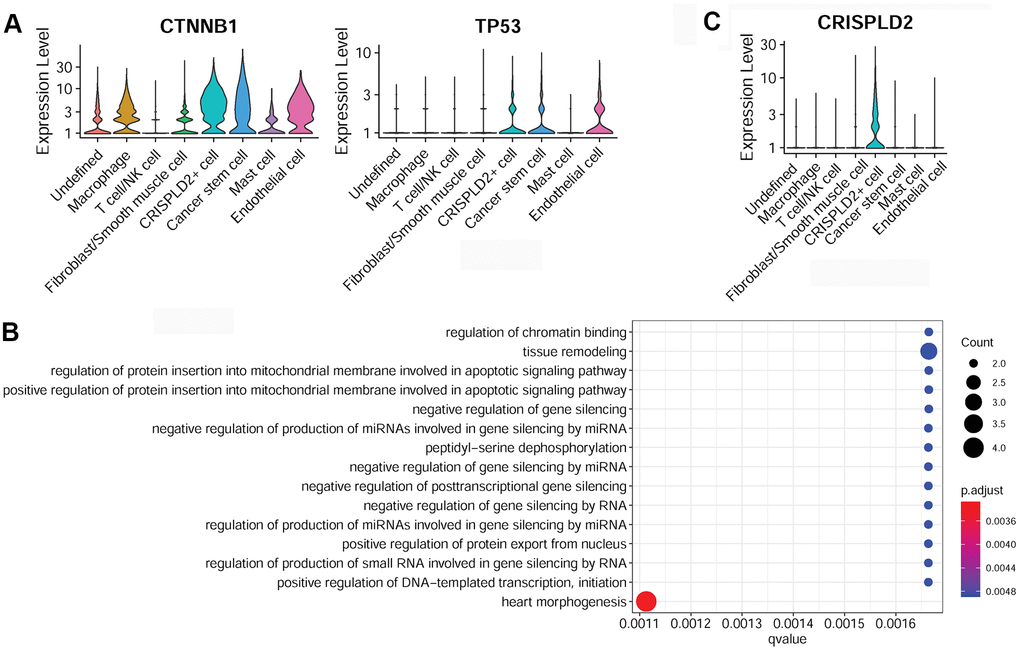
Figure 6. Characterization of cancer stem cells and CRISPLD2+ cells in pulmonary endarterectomized tissues of CTEPH patients. (A) Violin plot illustrating cancer stem cell marker genes, including CTNNB1 and TP53. (B) GO enrichment analysis of cancer stem cell marker genes. (C) Violin plot showing the CRISPLD2 marker of CRISPLD2+ cells. Abbreviations: CTEPH: chronic thromboembolic pulmonary hypertension; CRISPLD2: cysteine-rich secretory protein LCCL domain containing 2; GO: Gene ontology.
Discussion
CTEPH is a potentially curable disease, but its pathophysiology is not fully understood. There is limited research about the cells that make up the endarterectomized tissues. The present study, for the first time, investigated the cell landscape of pulmonary endarterectomized tissues of CTEPH patients using scRNA-seq. We found 17 cell clusters that could be divided into eight cell populations based on marker genes, as follows: including fibroblast/smooth muscle cell, T cell/NK cell, macrophage, CRISPLD2+ cell, cancer stem cell, mast cell, endothelial cell, and undefined. The functions of these cell populations with respect to CTEPH and their marker genes merit further discussion.
CTEPH is characterized by pulmonary vascular remodeling, in which fibroblasts, smooth muscle and endothelial cells have been shown to play important roles [17]. Vascular remodeling typically manifests as medial hypertrophy caused by increasing cell proliferation, or by attenuating apoptosis of vascular smooth muscle cells, as well as lumen obliteration, resulting from the over-proliferation of endothelial cells [17]. Moreover, lung fibroblasts have been reported to participate in developmental airway remodeling and tissue repair after inflammatory injury involving small airways [18]. In addition, pulmonary endothelial cells have been shown to be key regulators of pulmonary artery smooth muscle cell behavior in CTEPH [19]. In our study, fibroblast/smooth muscle cells/myofibroblasts accounted for the largest proportion (37%) of total cells, and endothelial cells accounted for only 2%. These data suggested an essential role for fibroblasts/smooth muscle cells/myofibroblasts and endothelial cell populations in CTEPH. Furthermore, specific marker genes of fibroblast/smooth muscle cells were identified, such as PDGFRB, COL1A1, and COL3A1, which were significantly enriched for multiple functions associated with smooth muscle cell migration. It has been reported that CTEPH endothelial cells can exhibit pro-fibrotic and pro-inflammatory phenotypes, as demonstrated by the expression of genes (COL1A1 and COL3A1) involved in extracellular matrix production and fibril organization [20]. In addition, PDGF is a potent mitogen for cells of mesenchymal origin, such as fibroblasts and smooth muscle cells [21]. Previous studies have revealed that smooth muscle 22 (SM22) and α-smooth muscle actin (α-SMA) are major marker genes of smooth muscle cells [22]. The PDGFRB pathway can inhibit the expression of SM22 and α-SMA and thus play crucial roles in phenotypic modulation of smooth muscle cells, facilitating the infiltration of inflammatory cells [23]. PDGFRB has been also identified to play important roles in CTEPH development [24]. Specific endothelial cell marker genes are END1 and CDH5, and these are mainly enriched for functions associated with endothelial cell migration. PKC-mediated END1 expression in endothelial cells has been shown to promote macrophage activation in atherogenesis [25]. A recent study has shown that END1 expression by endothelial cells may contribute to thrombofibrosis in the development of CTEPH [26]. CDH5, an endothelial marker gene, has been shown to promote endothelial–mesenchymal transition [27]. Sakao et al. showed that CDH5 is implicated in endothelial cell activation in CTEPH [13]. Based on our results, we speculated that fibroblast/smooth muscle cell/myofibroblast and endothelial cell populations may be involved in CTEPH development via the regulation of important functions associated with smooth muscle or endothelial cell migration.
Accumulating evidence has revealed the crucial role of dysregulated immune responses in CTEPH development [28], and such immune responses relate to either innate or adaptive immunity. Macrophages, NK cells, and mast cells participate in innate immunity, while T and B cells are implicated in adaptive immunity [28]. In CTEPH patients, macrophages and activated T and B lymphocytes are involved in thrombotic and atherosclerotic lesions [29]. Macrophages are first-line myeloid leucocytes in pulmonary lesions in patients with pulmonary hypertension [30]. Mast cells are tissue-resident immune cells that have key functions in the immune system through the production of inflammatory cytokines. Macrophages and mast cells play a pathological role in CTEPH via production of inflammatory cytokines, recruitment of other immune cells, and local inflammation and damage [28]. Moreover, CD68 and CD163 are two macrophage-associated markers, whose expression levels are associated with adverse outcomes in human atherosclerosis [31]. CD68- and CD163-positive tumor-associated macrophages are reported as key factors in tumor growth and metastasis through releasing chemokines such as inflammatory growth factors [32]. Our results showed that specific macrophage marker genes (such as CD68 and CD163) are significantly involved in key functions related to neutrophil activation, cellular chemotaxis, and antigen processing and presentation via MHC Class II molecules. Neutrophils are early responders and are recruited to respond to chemokines produced by tissue-resident immune cells such as macrophages and mast cells. In addition, it has been reported that CD3+ T cells accumulate in atherosclerotic and thrombotic lesions of CTEPH patients [29]. CD69 has been shown to be involved in immune cell homeostasis, which can activate T cell-mediated immune responses via regulating Th17 cell differentiation [33]. CD69 has also been identified as being inhibitory to cardiac inflammation and heart failure progression in autoimmune myocarditis [34]. Our analysis showed that specific T-/NK cell marker genes (such as CD3D and CD69) were remarkably enriched for T cell activation. It can be speculated that during CTEPH, the accumulation of macrophages and mast cells promotes the production of inflammatory cytokines and chemokines and subsequently induces T cell activation.
Notably, CRISPLD2, which encodes a secreted protein, has been shown to increase the anti-inflammatory effects of glucocorticoids to mediate immune responses in airway smooth muscle cells [35]. CRISPLD2 also functions as an endogenous anti-inflammatory gene in lung fibroblasts, and is an inhibitor of pro-inflammatory signaling in lung epithelial cells [36]. Based on our results, we speculated that CRISPLD2+ cells may be involved in CTEPH via the regulation of anti-inflammatory processes and subsequent immune responses. In addition to these, cancer stem cell clusters were also identified as a relatively newly recognized population in pulmonary endarterectomized tissues in CTEPH patients. Specific cancer stem cell marker genes are CTNNB1 and TP53. CTNNB1 expression is elevated in lung tissues from patients with idiopathic pulmonary arterial hypertension and CTNNB1 is involved in vascular remodeling via the Wnt/β-catenin pathway [37]. It has been shown that, in pulmonary artery smooth muscle cells, inhibition of TP53 results in upregulation of glycolysis and downregulation of mitochondrial respiration, indicating a proliferative phenotype resembling that of cancer cells [38]. Feng et al. demonstrated that miR-31a-5p targets TP53 to promote primary hypertension through accelerating proliferation and inhibiting apoptosis of arterial smooth muscle cells, suggesting a potential role of TP53 in pulmonary arterial hypertension [39]. In our study, specific cancer stem cell marker genes were enriched for tissue remodeling functions. We hypothesized that cancer stem cells may be involved in CTEPH by participating in tissue remodeling.
However, one of the limitations of our study was the relatively small sample size, which might be a consequence of the single-center study as here, the short duration of patient inclusion, and the low incidence of disease. A multicenter research involving a larger sample size may, in the future, provide additional strong evidence to support our findings reported here. Moreover, unidentified cells accounted for 20% of all cells. Elucidation of the fate of this large fraction of cells may provide new perspectives to better reveal the pathological mechanisms involved in CTEPH. Lastly, this study, for the first time, conducted scRNA-seq to reveal the cellular composition involved in CTEPH—essentially at single-cell resolution—and represents an exploratory study that provides a valuable resource facilitating researchers in future investigations of the possible mechanisms underlying CTEPH. However, CTEPH patients are rare and there are fewer CTEPH patients who eligible to receive surgical interventions. Coupled with the fact that this was a single-center study, it is difficult to assemble sufficient patient numbers and samples to carry out subsequent experiments in a short period of time. Therefore, we did not examine the pathology of cell types in CTEPH patients via histological analysis by immunohistochemistry to confirm our findings obtained from scRNA-seq, nor did we perform functional examinations of these cells. Further studies are still needed to combine clinical characterization of patients with experimental validation of existing clusters to explore possible mechanisms underpinning the disease.
In conclusion, our scRNA-seq analysis revealed that fibroblast/smooth muscle cells/myofibroblasts and endothelial cell populations may be involved in CTEPH development by participating in important functions associated with muscle or endothelial cell migration involving pulmonary vascular remodeling. Macrophage, mast cell, T cell/NK cell, and CRISPLD2+ cell are implicated in CTEPH via their involvement in inflammatory and immune responses. Cancer stem cells may be involved in CTEPH via participation in tissue remodeling. Collectively, our study provided the first atlas detailing the cellular landscape in pulmonary endarterectomized tissues of CTEPH patients, which will help us to better understand the pathological mechanisms involved in this disease.
Materials and Methods
Requirements for ethical approval and written informed consent were waived for this study because discarded pulmonary endarterectomized tissues of CTEPH patients were used here.
Handling of pulmonary endarterectomized tissue samples and dissociation to a single-cell suspension
Five consecutive patients were enrolled from October 2019 to June 2020 and definitively diagnosed with CTEPH. Pulmonary endarterectomized tissues from five CTEPH patients undergoing PEA were collected and stored in a tissue preservation solution. The baseline characteristics of these CTEPH patients are shown in Table 1.
Table 1. The baseline characteristics of CTEPH patients used for single-cell RNA sequencing.
| Features | CTEPH patients (N = 5) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Female/male | 1/4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age, years | 45.00 ± 13.34 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BMI, (kg/m2) | 25.18 ± 1.18 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| mPAP, mmHg | 54.40±3.21 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| WHO FC (I-II; III-IV) | 1/4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CI, L/(min·m2) | 1.78±0.13 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Family history of blood clots | No | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Smoking | 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Long period of inactivity | No | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Other CTEPH risk factors | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Pulmonary embolism | 3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Venous thromboembolism | 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviations: CTEPH: chronic thromboembolic pulmonary hypertension; BMI: body mass index; mPAP: mean pulmonary arterial pressure; WHO FC: World Health Organization function classification; CI: cardiac index. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The tissue samples were treated with enzymatic hydrolysate 1 at 30–40° C. Enzymatic hydrolysate 1 was prepared by adding a final concentration of 2.5 mg/mL collagenase I and 2.5 mM CaCl2 to RPMI 1640 cell culture medium. Then, 50 U/mL DNAseI was added to the enzymatic hydrolysate 1 to fully digest the tissue samples. After filtering, the samples were further digested using enzymatic hydrolysate 2, which was prepared by adding a final concentration of 1 mg/mL collagenase I, 1 mg/mL collagenase II, 0.2 mg/mL collagenase XI, 1.8 U/mL elastase, 50 U/mL DNAseI and 2.5 mM CaCl2 to RPMI 1640 medium. Digested tissue was macerated by passage through a 100 μM cell strainer, and red blood cell lysis buffer (Life Technologies, Ghent, Belgium) was added to eliminate potential interference from red blood cells. After terminating the reaction, cell pellets were collected by centrifugation. The cells were resuspended with deactivated magnetic beads, incubated at room temperature, added to the rinsed magnetic beads, and then collected by centrifugation. The cells were then resuspended again for preparation of single-cell suspensions.
scRNA-seq
The single-cell suspensions were loaded onto a Chromium™ Single-Cell Instrument (10× Genomics, Pleasanton, CA, USA) to create barcoded single-cell gel beads in emulsion (GEM), followed by reverse transcription and cDNA amplification. scRNA-seq libraries were then generated. Briefly, cDNA was sheared to 200–300 bp and then subjected to end repair and A-tailing, adapter ligation and sample index labeling for polymerase chain reaction, and sample cleanup. After quality checks, scRNA-seq was performed on the Illumina NovaSeq platform to obtain DNA sequencing data.
Data processing
All scRNA-seq data for the five samples were processed and quantified using Cell Ranger Software Suite v.3.1.0 software tools (https://support.10xgenomics.com). First, the sequenced reads were mapped to the hg19 human reference genome under the Cell Ranger ‘count’ module. We generated five-feature barcode matrices for each of the five samples independently. These matrices were then aggregated using the Cell Ranger ‘aggr’ module for subsequent analyses. Second, cells with gene numbers < 200 or > 7,500, mitochondrial gene expression ratio > 0.2, and at least one gene expressed in three cells were filtered using Seurat v.3.0 (https://satijalab.org/seurat/) [40]. Data were then normalized using the NormalizeData function (pbmc, normalization.method = "LogNormalize," scale.factor = 10000). The UMI count matrix was then converted to Seurat objects using Seurat v.3.0.
Identification of cluster marker genes and cell types
After data normalization and scaling, and dimensionality reduction of the top-20 components, cell clusters were identified using the ‘FindClusters’ function in Seurat v.3.0 and then visualized using the UMAP method after running ‘RunUMAP.’ Marker genes corresponding to each cell cluster were identified based on differential analysis using the ‘FindMakers’ function of the Seurat package and Wilcoxon rank-sum tests. The differential expression thresholds were set as log-fold change > 1 and P value < 0.05. The marker genes were then used to annotate cell types using the SCSA tool [41]. Violin plots of key marker genes were constructed using ggplot2 [42].
Supplementary Materials
Author Contributions
Conception and design of the research: RM and YY; acquisition of data: RM, XD, JG, YL, XG, JW, QH and YW; analysis and interpretation of data: RM, XD, JG, YL, XG, JW, QH, YW, JL, SY, TK, JW and ML; statistical analysis: RM, XD, JG, YL, XG, JW and QH; obtaining funding: RM, YL, SY, ML, ZZ, JZ and YY; drafting the manuscript: RM, XD and JG; revision of manuscript for important intellectual content: RM, JW, ZZ, JZ and YY. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This study was supported by National Natural Science Foundation of China (Project number: 81300044, 31670928, 81871356, 81770253, 81370362, 81871328, 81900047), Beijing Natural Science Foundation (Project number: 7162069, 7182149), Beijing Municipal Administration of Hospitals’ Youth Programme (Project number: QML20160301), the Open Foundation from Beijing Key Laboratory of Hypertension Research(2018GXY-KFKT-02), the National Key Research and Development Program of China (Project number: 2016YFC0905600), the National Major Research Plan Training Program of China (91849111) and Chinese Academy of Medical Sciences Central Public-interest Scientific Institution Basal Research Fund Young Medical Talents Award Project(Project number: 2018RC320013).
Editorial Note
This corresponding author has a verified history of publications using a personal email address for correspondence
References
- 1. Ranka S, Mohananey D, Agarwal N, Verma BR, Villablanca P, Mewhort HE, Ramakrishna H. Chronic Thromboembolic Pulmonary Hypertension-Management Strategies and Outcomes. J Cardiothorac Vasc Anesth. 2020; 34:2513–23. https://doi.org/10.1053/j.jvca.2019.11.019 [PubMed]
- 2. Hoeper MM, Madani MM, Nakanishi N, Meyer B, Cebotari S, Rubin LJ. Chronic thromboembolic pulmonary hypertension. Lancet Respir Med. 2014; 2:573–82. https://doi.org/10.1016/S2213-2600(14)70089-X [PubMed]
- 3. Mulchrone A, Kellihan HB, Forouzan O, Hacker TA, Bates ML, Francois CJ, Chesler NC. A Large Animal Model of Right Ventricular Failure due to Chronic Thromboembolic Pulmonary Hypertension: A Focus on Function. Front Cardiovasc Med. 2019; 5:189. https://doi.org/10.3389/fcvm.2018.00189 [PubMed]
- 4. Nishimura R, Tanabe N, Sugiura T, Shigeta A, Jujo T, Sekine A, Sakao S, Kasahara Y, Tatsumi K. Improved survival in medically treated chronic thromboembolic pulmonary hypertension. Circ J. 2013; 77:2110–17. https://doi.org/10.1253/circj.cj-12-1391 [PubMed]
- 5. Opitz I, Ulrich S. Chronic thromboembolic pulmonary hypertension. Swiss Med Wkly. 2018; 148:w14702. https://doi.org/10.4414/smw.2018.14702 [PubMed]
- 6. Jenkins D, Madani M, Fadel E, D’Armini AM, Mayer E. Pulmonary endarterectomy in the management of chronic thromboembolic pulmonary hypertension. Eur Respir Rev. 2017; 26:160111. https://doi.org/10.1183/16000617.0111-2016 [PubMed]
- 7. Delcroix M, Lang I, Pepke-Zaba J, Jansa P, D’Armini AM, Snijder R, Bresser P, Torbicki A, Mellemkjaer S, Lewczuk J, Simkova I, Barberà JA, de Perrot M, et al. Long-Term Outcome of Patients With Chronic Thromboembolic Pulmonary Hypertension: Results From an International Prospective Registry. Circulation. 2016; 133:859–71. https://doi.org/10.1161/CIRCULATIONAHA.115.016522 [PubMed]
- 8. Opitz I, Kirschner MB. Molecular Research in Chronic Thromboembolic Pulmonary Hypertension. Int J Mol Sci. 2019; 20:784. https://doi.org/10.3390/ijms20030784 [PubMed]
- 9. Lang IM, Dorfmüller P, Vonk Noordegraaf A. The Pathobiology of Chronic Thromboembolic Pulmonary Hypertension. Ann Am Thorac Soc. 2016 (Suppl 3); 13:S215–21. https://doi.org/10.1513/AnnalsATS.201509-620AS [PubMed]
- 10. Zhang YX, Li JF, Yang YH, Zhai ZG, Gu S, Liu Y, Miao R, Zhong PP, Wang Y, Huang XX, Wang C. Renin-angiotensin system regulates pulmonary arterial smooth muscle cell migration in chronic thromboembolic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2018; 314:L276–86. https://doi.org/10.1152/ajplung.00515.2016 [PubMed]
- 11. Simonneau G, Torbicki A, Dorfmüller P, Kim N. The pathophysiology of chronic thromboembolic pulmonary hypertension. Eur Respir Rev. 2017; 26:160112. https://doi.org/10.1183/16000617.0112-2016 [PubMed]
- 12. Deng C, Zhong Z, Wu D, Chen Y, Lian N, Ding H, Zhang Q, Lin Q, Wu S. Role of FoxO1 and apoptosis in pulmonary vascular remolding in a rat model of chronic thromboembolic pulmonary hypertension. Sci Rep. 2017; 7:2270. https://doi.org/10.1038/s41598-017-02007-5 [PubMed]
- 13. Sakao S, Hao H, Tanabe N, Kasahara Y, Kurosu K, Tatsumi K. Endothelial-like cells in chronic thromboembolic pulmonary hypertension: crosstalk with myofibroblast-like cells. Respir Res. 2011; 12:109. https://doi.org/10.1186/1465-9921-12-109 [PubMed]
- 14. Potter SS. Single-cell RNA sequencing for the study of development, physiology and disease. Nat Rev Nephrol. 2018; 14:479–92. https://doi.org/10.1038/s41581-018-0021-7 [PubMed]
- 15. Stubbington MJ, Rozenblatt-Rosen O, Regev A, Teichmann SA. Single-cell transcriptomics to explore the immune system in health and disease. Science. 2017; 358:58–63. https://doi.org/10.1126/science.aan6828 [PubMed]
- 16. Valenzi E, Bulik M, Tabib T, Morse C, Sembrat J, Trejo Bittar H, Rojas M, Lafyatis R. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Ann Rheum Dis. 2019; 78:1379–87. https://doi.org/10.1136/annrheumdis-2018-214865 [PubMed]
- 17. Mandegar M, Fung YC, Huang W, Remillard CV, Rubin LJ, Yuan JX. Cellular and molecular mechanisms of pulmonary vascular remodeling: role in the development of pulmonary hypertension. Microvasc Res. 2004; 68:75–103. https://doi.org/10.1016/j.mvr.2004.06.001 [PubMed]
- 18. Chapman HA. Epithelial-mesenchymal interactions in pulmonary fibrosis. Annu Rev Physiol. 2011; 73:413–35. https://doi.org/10.1146/annurev-physiol-012110-142225 [PubMed]
- 19. Mercier O, Arthur Ataam J, Langer NB, Dorfmüller P, Lamrani L, Lecerf F, Decante B, Dartevelle P, Eddahibi S, Fadel E. Abnormal pulmonary endothelial cells may underlie the enigmatic pathogenesis of chronic thromboembolic pulmonary hypertension. J Heart Lung Transplant. 2017; 36:305–14. https://doi.org/10.1016/j.healun.2016.08.012 [PubMed]
- 20. Bochenek M, Saar K, Marini F, Gerhold-Ay A, Huebner N, Muenzel T, Mayer E, Konstantinides S, Schaefer K. Phenotypic specification of endothelial cells in chronic thromboembolic pulmonary hypertension. Eur Heart J. 2021; 42:2137–40. https://doi.org/10.1093/eurheartj/ehab295 [PubMed]
- 21. Zheng X, Hu X, Zhang W. The phenotype of vascular smooth muscle cells co-cultured with endothelial cells is modulated by PDGFR-β/IQGAP1 signaling in LPS-induced intravascular injury. Int J Med Sci. 2019; 16:1149–56. https://doi.org/10.7150/ijms.34749 [PubMed]
- 22. Millette E, Rauch BH, Kenagy RD, Daum G, Clowes AW. Platelet-derived growth factor-BB transactivates the fibroblast growth factor receptor to induce proliferation in human smooth muscle cells. Trends Cardiovasc Med. 2006; 16:25–28. https://doi.org/10.1016/j.tcm.2005.11.003 [PubMed]
- 23. Rensen SS, Doevendans PA, van Eys GJ. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J. 2007; 15:100–08. https://doi.org/10.1007/BF03085963 [PubMed]
- 24. Wang M, Gu S, Liu Y, Yang Y, Yan J, Zhang X, An X, Gao J, Hu X, Su P. miRNA-PDGFRB/HIF1A-lncRNA CTEPHA1 Network Plays Important Roles in the Mechanism of Chronic Thromboembolic Pulmonary Hypertension. Int Heart J. 2019; 60:924–37. https://doi.org/10.1536/ihj.18-479 [PubMed]
- 25. Zhang J, Wang YJ, Wang X, Xu L, Yang XC, Zhao WS. PKC-Mediated Endothelin-1 Expression in Endothelial Cell Promotes Macrophage Activation in Atherogenesis. Am J Hypertens. 2019; 32:880–89. https://doi.org/10.1093/ajh/hpz069 [PubMed]
- 26. Bochenek ML, Leidinger C, Rosinus NS, Gogiraju R, Guth S, Hobohm L, Jurk K, Mayer E, Münzel T, Lankeit M, Bosmann M, Konstantinides S, Schäfer K. Activated Endothelial TGFβ1 Signaling Promotes Venous Thrombus Nonresolution in Mice Via Endothelin-1: Potential Role for Chronic Thromboembolic Pulmonary Hypertension. Circ Res. 2020; 126:162–81. https://doi.org/10.1161/CIRCRESAHA.119.315259 [PubMed]
- 27. Li Z, Kong X, Zhang Y, Zhang Y, Yu L, Guo J, Xu Y. Dual roles of chromatin remodeling protein BRG1 in angiotensin II-induced endothelial-mesenchymal transition. Cell Death Dis. 2020; 11:549. https://doi.org/10.1038/s41419-020-02744-y [PubMed]
- 28. Koudstaal T, Boomars KA, Kool M. Pulmonary Arterial Hypertension and Chronic Thromboembolic Pulmonary Hypertension: An Immunological Perspective. J Clin Med. 2020; 9:561. https://doi.org/10.3390/jcm9020561 [PubMed]
- 29. Quarck R, Wynants M, Verbeken E, Meyns B, Delcroix M. Contribution of inflammation and impaired angiogenesis to the pathobiology of chronic thromboembolic pulmonary hypertension. Eur Respir J. 2015; 46:431–43. https://doi.org/10.1183/09031936.00009914 [PubMed]
- 30. El Kasmi KC, Pugliese SC, Riddle SR, Poth JM, Anderson AL, Frid MG, Li M, Pullamsetti SS, Savai R, Nagel MA, Fini MA, Graham BB, Tuder RM, et al. Adventitial fibroblasts induce a distinct proinflammatory/profibrotic macrophage phenotype in pulmonary hypertension. J Immunol. 2014; 193:597–609. https://doi.org/10.4049/jimmunol.1303048 [PubMed]
- 31. de Gaetano M, Crean D, Barry M, Belton O. M1- and M2-Type Macrophage Responses Are Predictive of Adverse Outcomes in Human Atherosclerosis. Front Immunol. 2016; 7:275. https://doi.org/10.3389/fimmu.2016.00275 [PubMed]
- 32. Jamiyan T, Kuroda H, Yamaguchi R, Abe A, Hayashi M. CD68- and CD163-positive tumor-associated macrophages in triple negative cancer of the breast. Virchows Arch. 2020; 477:767–75. https://doi.org/10.1007/s00428-020-02855-z [PubMed]
- 33. de la Fuente H, Cruz-Adalia A, Martinez Del Hoyo G, Cibrián-Vera D, Bonay P, Pérez-Hernández D, Vázquez J, Navarro P, Gutierrez-Gallego R, Ramirez-Huesca M, Martín P, Sánchez-Madrid F. The leukocyte activation receptor CD69 controls T cell differentiation through its interaction with galectin-1. Mol Cell Biol. 2014; 34:2479–87. https://doi.org/10.1128/MCB.00348-14 [PubMed]
- 34. Cruz-Adalia A, Jiménez-Borreguero LJ, Ramírez-Huesca M, Chico-Calero I, Barreiro O, López-Conesa E, Fresno M, Sánchez-Madrid F, Martín P. CD69 limits the severity of cardiomyopathy after autoimmune myocarditis. Circulation. 2010; 122:1396–404. https://doi.org/10.1161/CIRCULATIONAHA.110.952820 [PubMed]
- 35. Himes BE, Jiang X, Wagner P, Hu R, Wang Q, Klanderman B, Whitaker RM, Duan Q, Lasky-Su J, Nikolos C, Jester W, Johnson M, Panettieri RA
Jr , et al. RNA-Seq transcriptome profiling identifies CRISPLD2 as a glucocorticoid responsive gene that modulates cytokine function in airway smooth muscle cells. PLoS One. 2014; 9:e99625. https://doi.org/10.1371/journal.pone.0099625 [PubMed] - 36. Zhang H, Kho AT, Wu Q, Halayko AJ, Limbert Rempel K, Chase RP, Sweezey NB, Weiss ST, Kaplan F. CRISPLD2 (LGL1) inhibits proinflammatory mediators in human fetal, adult, and COPD lung fibroblasts and epithelial cells. Physiol Rep. 2016; 4:e12942. https://doi.org/10.14814/phy2.12942 [PubMed]
- 37. Wu D, Talbot CC
Jr , Liu Q, Jing ZC, Damico RL, Tuder R, Barnes KC, Hassoun PM, Gao L. Identifying microRNAs targeting Wnt/β-catenin pathway in end-stage idiopathic pulmonary arterial hypertension. J Mol Med (Berl). 2016; 94:875–85. https://doi.org/10.1007/s00109-016-1426-z [PubMed] - 38. Wakasugi T, Shimizu I, Yoshida Y, Hayashi Y, Ikegami R, Suda M, Katsuumi G, Nakao M, Hoyano M, Kashimura T, Nakamura K, Ito H, Nojiri T, et al. Role of smooth muscle cell p53 in pulmonary arterial hypertension. PLoS One. 2019; 14:e0212889. https://doi.org/10.1371/journal.pone.0212889 [PubMed]
- 39. Feng Q, Tian T, Liu J, Zhang L, Qi J, Lin X. Deregulation of microRNA-31a-5p is involved in the development of primary hypertension by suppressing apoptosis of pulmonary artery smooth muscle cells via targeting TP53. Int J Mol Med. 2018; 42:290–98. https://doi.org/10.3892/ijmm.2018.3597 [PubMed]
- 40. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019; 177:1888–902.e21. https://doi.org/10.1016/j.cell.2019.05.031 [PubMed]
- 41. Cao Y, Wang X, Peng G. SCSA: A Cell Type Annotation Tool for Single-Cell RNA-seq Data. Front Genet. 2020; 11:490. https://doi.org/10.3389/fgene.2020.00490 [PubMed]
- 42. Ginestet C. ggplot2: elegant graphics for data analysis. J R Stat Soc Ser A Stat Soc. 2011; 174:245–46. https://doi.org/10.1111/j.1467-985X.2010.00676_9.x
- 43. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, et al, and The Gene Ontology Consortium. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000; 25:25–29. https://doi.org/10.1038/75556 [PubMed]
- 44. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012; 16:284–87. https://doi.org/10.1089/omi.2011.0118 [PubMed]