



Introduction
Colorectal cancer (CRC) is the third most frequently occurring cancer and the second leading cause of cancer-related deaths worldwide in 2018 [1]. The current therapeutic options for CRC include endoscopic and local surgical excision, downstaging preoperative radiotherapy and systemic therapy, extensive surgery, local ablative therapies for metastases, palliative chemotherapy, targeted therapy, and immunotherapy [2]. It’s a highly heterogeneous disease on account of accumulating mutations attributed to environmental and genetic factors for years, which makes prognostic prediction and treatment to be exceedingly challenging [3, 4]. Therefore, there is an urgent need to incorporate other important elements to guide personalized therapies for CRCs, thereby improving the survival and prognosis of CRCs.
In recent years, a myriad of publications have highlighted that the tumor immune microenvironment (TIME) is critically involved in cancer initiation and progression [5, 6]. For example, tumor-infiltrating lymphocytes (TILs) were in close interaction with relapse and mortality prediction in CRC [7–9]. Besides, immune checkpoint inhibitors (ICIs) targeted programmed cell death protein 1 (PD-1)/programmed Cell Death-Ligand 1 (PD-L1) have been proved effective in the treatment of CRC [10, 11], revolutionizing oncotherapy to a great extent. Michael J et al. have demonstrated that a combination of PD-1 inhibitor (nivolumab) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) inhibitor (ipilimumab) has comparatively better efficacy and is a promising new therapeutic option for metastatic DNA mismatch repair-deficient and microsatellite instability-high (dMMR–MSI-H) CRCs [12, 13]. As the most widely investigated marker, tumor PD-L1 expression might be useful as a predictive marker of response to anti-PD-1 treatment for non-small cell lung cancer (NSCLC), gastric cancer and gastroesophageal junction tumors [14, 15]. But in CRC, PD-L1 expression wasn’t tightly associated with the response or survival in the recent studies [16]. Thus far, several other biomarkers of potential response have been demonstrated, including high tumor mutation load [17, 18], high immunoscore [19, 20], and POLE mutation [21, 22]. However, these biomarkers that guided the use of ICIs in patients with CRC are not always consistent in clinical practice. For example, high immunoscore were also substantiated in pMMR–MSI-L CRCs, raising queries of whether single immunophenotype might robustly predict immunotherapy benefit [23]. Consequently, integrative immunogenic features of the TIME might be more precise in predicting immunotherapeutic response than either feature alone. In conclusion, developing a novel immune signature complementary for the currently established signatures is of great importance to optimize individual specialized immunotherapy for CRC patients.
Within the past decade, studies have aimed at elucidating the roles of immune-related genes (IRGs) in CRC. Li et al. have constructed an IRGs signature leveraging expression profiles and clinical characteristics from the GEO database and the TCGA database. Robust prognostic ability was demonstrated, meanwhile, the enrichment with cytotoxic immune cells as well as depletion of myeloid-derived suppressor cells (MDSC) and regulatory T cells (Tregs) were estimated in low-risk signature CRCs [24]. Lin et al. also comprehensively analyzed the role of IRGs in CRCs via the TCGA dataset, reporting a higher prognostic performance of 10 IRGs based signature in CRC and the infiltration degree of various immune cells [25]. Nevertheless, there has been no IRGs signature that comprehensively evaluates the TIME and predicts prognostic significance in conjunction with the response to chemotherapeutic and immunotherapeutic options of CRC.
In this study, we aimed at establishing a novel IRGs-based signature for CRC to investigate the interplay between colorectal immune activity profile and oncology genotype. Through systematic in silico analysis based on the constructed signature, we discovered that the IRGs risk score for CRC was associated with overall survival (OS), clinicopathological factors, and immunophenotypic characteristics. Moreover, we also assessed the efficiency of this IRGs signature in identifying chemotherapeutic compounds and immunotherapy with subtype-specific efficacy.
Materials and Methods
Data preparation
Processed RNA-Seq FPKM data and clinical information of CRC were collected from the TCGA database. The TCGA colon adenocarcinoma (COAD, n = 512) cohort and rectum adenocarcinoma (READ, n = 177) cohort were obtained from the GDC data portal (https://portal.gdc.cancer.gov/repository). For validation, the expression profiles and detailed clinical information of GSE39582 (including 562 CRC samples based on GPL570 platform) were retrieved from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). The immune gene lists were obtained from the ImmPort database (https://immport.niaid.nih.gov) [26] and overlapping genes from the TCGA dataset were defined as IRGs in the current study and extracted for the subsequent analysis.
To analyze the drug sensitivity in human CRCs, GSE17538 (including 232 CRC samples based on the GPL570 platform) was obtained from the GEO database. The expression profiles of human cancer cell lines (CCLs) were achieved from the Broad Institute Cancer Cell Line Encyclopedia (CCLE) project (https://portals.broadinstitute.org/ccle/) [27]. Drug sensitivity data of CCLs were extracted from the Cancer Therapeutics Response Portal (CTRP, https://portals.broadinstitute.org/ctrp) and PRISM Repurposing dataset (https://depmap.org/portal/prism/). The PRISM is composed of sensitivity data for 1448 compounds over 482 CCLs and the CTRP comprises of sensitivity data for 481 compounds over 835 CCLs. The area under the dose-response curve (area under the curve-AUC) values as a measure of drug sensitivity are presented in both two datasets, with lower AUC values indicating higher drug sensitivity. After the exclusion of compounds with more than 20% of missing data, the missing AUC values were imputed by K-nearest neighbor (k-NN) imputation.
To investigate the response to immunotherapy, tumor expression profiles of six immunotherapeutic cohorts were obtained. Roh et al. (2017) dataset contained melanoma patients receiving CTLA-4 or PD-1 blockade therapy was extracted from the supplementary files of reference [28]. Gene expression profiles and survival information of metastatic melanoma patients treated with CTLA-4 immuno-inhibitor were obtained from the work of Van Allen et al. (2015) [29]. The data of Ulloa Montoya et al. (2013) cohort with non–small-cell lung cancer (NSCLC) patients who were administered MAGE-A3 antigen-specific immunotherapy were downloaded from (GSE35640) [30]. The dataset of Hugo et al. (2016) included metastatic melanoma patients treated with anti-PD-1 agents was acquired from GSE78220 [31]. Moreover, patients with metastatic urothelial cancer treated with PD-L1 blockade therapy from the IMvigor210 cohort [32] and the dataset of Snyder et al. (2017) [33] were also enrolled.
Construction of the IRGs signature for CRC
Differentially expression genes (DEGs) between tumor and normal samples from CRC patients from TCGA-COAD and TCGA-READ cohorts were first screened by limma package [34] with a cutoff value of false discovery rate (FDR)-adjusted P-value < 0.01 and log2 | fold change (FC) | > 1. Then differentially expressed IRGs between the aforementioned CRC tumor and normal tissues were obtained using a strict criterion of FDR-adjusted P-value < 0.01 and log2 | fold change (FC) | > 2. Heatmaps were plotted utilizing pheatmap package and volcano plots were generated via R software.
The CRC tumor samples from the TCGA cohort were enrolled as the training cohort to construct the IRGs signature. Univariate Cox regression analysis of differentially expressed IRGs was performed by survival package in R. The prognosis-related IRGs (PRIRGs) were selected by a cutoff value of P < 0.01. To avoid the overfitting of IRGs signature and to delete highly correlated genes, dimensionality reduction analysis was conducted by the Least Absolute Shrinkage and Selection Operator (lasso) regression through survival and glmnet R packages using gene expression profiles and overall survival data. Lambda.min was set up as cutoff point to produce minimum mean cross-validated error and genes with the highest lambda values were selected for further analysis. Subsequently, multivariate Cox regression was harnessed to develop an IRGs signature based on the expression of these genes and to calculate the risk score for signature:
Survival analysis for high- and low-risk subgroups was then carried out using Kaplan-Meier methods and the log-rank test was used to determine the statistical significance of differences. Time-dependent receiver operating characteristic (ROC) curves were also generated leveraging survivalROC R package to validate the prognostic ability of the IRGs signature. The IRGs signature obtained from the training cohort were used to assign the validation cohort as well as datasets containing therapeutic information into high- and low- risk score subtypes. Furthermore, to assess the independence of the constructed signature’s predictive ability, we performed univariate analysis on the IRGs signature using all clinical factors in the training and validation cohort. The hazard ratio (HR) was measured by a Cox regression model using survival package in R and forest plots were drawn.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) in the CRC cohorts was carried out by clusterProfiler R package [35]. Fold change (FC) of each gene between subgroups was firstly produced by limma R package, and input genes were then ranked in descending order according to the logFC values. GSEA was subsequently applied to enrich 50 hallmark gene sets (h.all.v7.0.symbols) achieved from the Molecular Signatures Database (MSigDB) [36]. Enrichment significance was evaluated using default settings and FDR adjusted P-value < 0.05 was considered significantly enriched. The single sample gene set enrichment analysis (ssGSEA) [37] implemented in R package GSVA, was adopted to calculate the normalized enrichment score (NES) of immune-related signatures in the training and validation cohorts.
Estimation of drug response in clinical samples
Large-scale drug screening and molecular data across hundreds of cancer cell lines in pharmacogenomic databases of CTRP and PRISM make it possible for precise drug response prediction in clinical samples. Ridge regression model that located in the R package pRRophetic [45] was used to evaluate the drug responses in clinical samples, with a robust predictive power assessed by 10-fold cross-validation in default. The prediction model was merely employed on expression profiles and drug response data of solid CCLs, and the AUC value of each agent in each clinical sample was ultimately estimated. Agents with NAs in more than 20% of the samples and hematopoietic as well as lymphoid tissue-derived CCLs were excluded. Subclass mapping (SubMap) analysis (Gene Pattern modules, https://cloud.genepattern.org/), which can assess the similarity of molecular subtypes between independent patient cohorts based on mRNA expression matrix, was utilized to determine the potential immunotherapeutic benefit of distinct subtypes employing the available clinical response data and gene expression profiles from six immunotherapy datasets.
Statistical analysis
R statistical software (version 4.0.2) was implemented for all statistical analyses. The evaluation of normality distribution within continuous variables was performed by Shapiro-Wilk test. Comparison of a continuous variable in two or more than two groups was conducted by parametric test (Student’s t-test or analysis of variance, respectively) if the variable was normally distributed, otherwise, nonparametric test (Wilcoxon rank-sum test or Kruskal-Wallis test) was performed. Correlation between two continuous variables was evaluated by either Pearson’s r correlation or Spearman’s rank-order correlation. For all statistical analyses, unless otherwise noted, a two-tailed P-value < 0.05 was defined as statistically significant.
Results
Construction of IRGs signature in CRC cohorts
A total of 638 CRC and 51 adjacent normal tissues were acquired from the TCGA database. To establish a predictive IRGs signature, we performed differential expression analysis of genes and IRGs between tumor and normal tissues. A total of 3741 DEGs were identified, including 2,502 upregulated genes and 1,239 downregulated genes (Supplementary Figure 1A, 1B). 2,483 IRGs were also collected from the ImmPort database (Supplementary Table 1). Fulfilling the screening criteria, 294 differentially expressed IRGs were obtained, containing 99 upregulated IRGs and 195 downregulated IRGs (Supplementary Figure 1C, 1D). In total, 606 CRC samples with complete gene expression profiles and intact follow-up information from the TCGA database were enrolled for establishing IRGs signature in the training cohort.
To determine the IRGs related to tumorigenesis and development in CRC, univariate Cox regression analysis was implemented on the differentially expressed IRGs in the training cohort (P < 0.01), and 11 PRIRGs in all were obtained (Supplementary Figure 1E). Moreover, lasso regression was conducted to lessen the number of PRIRGs, and eight PRIRGs were thus filtered out (Supplementary Figure 1F, 1G and Supplementary Table 2). Through multivariate Cox regression analysis, seven-IRGs based signature was ultimately established, as depicted in Supplementary Table 3. The formula for calculating risk score is: Risk score = 0.139 х ExpFABP4 + 0.176 х ExpAMH + 0.207 х ExpGRP + 0.211 х ExpINHBB - 0.691 х ExpNRG1 + 0.274 х ExpUCN + 0.366 х ExpMC1R. Among these IRGs, NRG1 exhibited a negative coefficient, implying that it could be considered as a protective factor for CRCs; on the contrary, FABP4, AMH, GRP, INHBB, UCN, and MC1R possess positive coefficients, implying poor prognoses in CRCs with overexpression of these six genes.
According to the median value of the risk score (0.948), the 606 CRCs in the training cohort were divided into a high-risk group (n = 303) and a low-risk group (n = 303). The distribution of risk scores, survival status as well as the expression level of seven IRGs for the two subgroups in the training cohort were correspondingly displayed in Figure 1A. Kaplan-Meier survival analysis (Figure 1B) indicated dismal prognosis for patients in the high-risk group (P < 0.0001). To assess the predictive efficiency of the constructed seven IRGs signature, time-dependent ROC curves were plotted. As shown in Figure 1C, the AUCs for 1-, 3-, 5- year survival prediction was 0.692, 0.676, and 0.721, respectively.
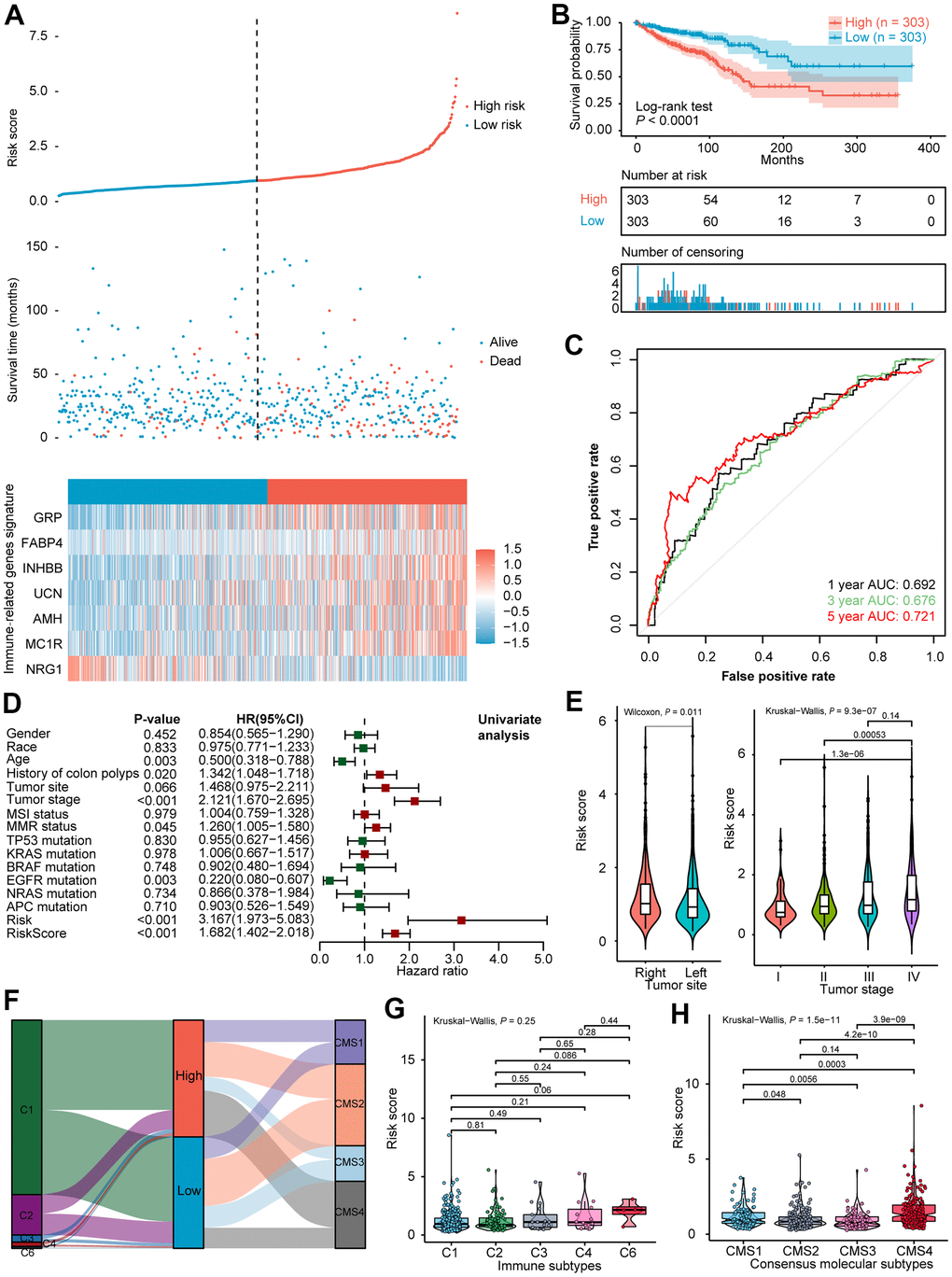
Figure 1. Exploration of the predictive power and clinical characteristics of seven IRGs signature in the training cohort. (A) Distribution of risk score, survival status, and expression of seven IRGs of CRCs. (B) Kaplan-Meier survival curve of the high- and low- risk subgroups. (C) ROC curve analysis of IRGs. (D) Univariate Cox analysis of prognostic factors and OS of CRCs. (E) Violin plot illustrated the correlation between risk score and tumor site as well as tumor stage. (F) Alluvial diagram for the two subtypes versus different immune subtypes and CMS. (G) Violin plot illustrated the correlation between risk score and immune subtypes, and (H) CMS. AUC, area under the curve; OS, overall survival; CRC, colorectal cancer; IRGs, immune-related genes; ROC, receiver operating characteristic; CMS, consensus molecular subtypes.
To evaluate the performance of the seven IRGs signature, the GSE39582 dataset (n=562) was used for validation. On the basis of signature information acquired from the training cohort, CRCs in the validation cohort were also classified into high-risk group (n = 281) and low-risk group (n = 281). The distribution of risk scores, survival status, and the expression level of seven IRGs for different subclasses in the validation cohort are correspondingly displayed in Supplementary Figure 2A. Kaplan-Meier survival analysis in Supplementary Figure 2B also revealed poor prognosis in patients of high-risk score (P < 0.0001). Similarly, time-dependent ROC curves were plotted. As exhibited in Supplementary Figure 2C, the AUC for 1-, 3-, 5- year survival prediction was 0.615, 0.616, and 0.662, respectively.
Evaluation of the IRGs signature in CRC cohorts
A detailed summary of the training and the validation cohorts selected for univariate analysis in this study is presented in Supplementary Table 4. Univariate analysis of the TCGA dataset suggested that age (P = 0.003), history of colon polyps (P = 0.020), tumor stage (P < 0.001), mismatch repair system (MMR) status (P = 0.045), EGFR mutation (P = 0.003), and risk score (P < 0.001) were significantly associated with OS (Figure 1D). Meanwhile, a high-risk score was correlated with increased age, history of colon polyps, right half of CRC, dMMR as well as EGFR mutation (Supplementary Table 4). Analogous analyses in the validation dataset showed that tumor stage (P < 0.001), KRAS mutation (P = 0.048), and risk score (P < 0.001) were closely connected with patient survival (Supplementary Figure 2D). As shown in Figure 1E, the risk score was significantly higher in right-side colorectal cancer than the left-side, and the risk score was significantly elevated as colorectal cancer progressed to an advanced stage.
The differences in the distribution of molecular subtypes within the IRGs risk score model were also investigated. In the TCGA cohort, there was no significant difference between the risk score and the immune subtypes (Figure 1F, 1G). Similar results were manifested in the validation dataset (Supplementary Figure 2E, 2F), probably because six immune subtypes were generated by immunogenomics analyses encompassing multiple cancer types [39]. With regards to CMS, the CMS4 subtype had significantly higher IRGs risk score than the other three molecular subtypes, whereas the CMS2 subtype held the lowest risk score (Figure 1F, 1H). A significantly difference was demonstrated among the four CMSs (P = 1.5e−11). In the GEO validation dataset, CMS was likewise found distributed between high- and low- risk subgroups (Supplementary Figure 2E), and allied results (P < 2.2e−16) were displayed in violin plot (Supplementary Figure 2G). Remarkably, the international CRC Subtyping Consortium proposed that superior survival was demonstrated in CMS2 patients while CMS4 patients displayed worse OS [46], consistent with our finding that a larger proportion of long-term survivors were identified in low-risk CRCs than the high-risk subset. The GSEA of 50 hallmark gene sets indicated that up-regulated genes of the high-risk group were enriched in multiple carcinogenesis related pathways, such as epithelial-mesenchymal transition (EMT), angiogenesis, Hedgehog signaling, myogenesis, transforming growth factor-beta (TGFβ) signaling, as well as hypoxia pathway targeted HIF1A (Figure 2A and Supplementary Figure 3A). GSEA analyses revealed the enrichment of tumor proliferation-associated signatures, such as E2F targets, MYC targets, and G2M checkpoint in IRGs low-risk subgroup. Several evidences suggested that they might denote dual role of regulating anti-tumor immunity and tumor cell proliferation. It’s indicated that the E2F1/SP3/STAT6 axis induced by IL-4 promoted EMT in CRC cells [47]. Activation of IL-6/p-STAT3/c-MYC signaling was demonstrated to enhance colorectal tumor growth in a TLR4-dependent manner [48]. In addition, MYC/PVT1 signaling induced immune surveillance via CD8+ TILs and peripheral blood mononuclear cells in CRCs [49]. As for G2M checkpoint, in-vitro co-culture assays of T cells and HCT-116 colorectal cancer cells reflected that immune checkpoint TIGIT blockade suppressed G2M transit [50]. Overall, these tumor proliferation-related pathways might also exert tumor immunity associated effects on the TIME of CRC, and the underlying mechanism deserved future investigation. Metabolism-related processes consisting of oxidative phosphorylation and fatty acid metabolism, as well as immune-related signaling involved in IL6/JAK/STAT3, were observed in the low-risk group.
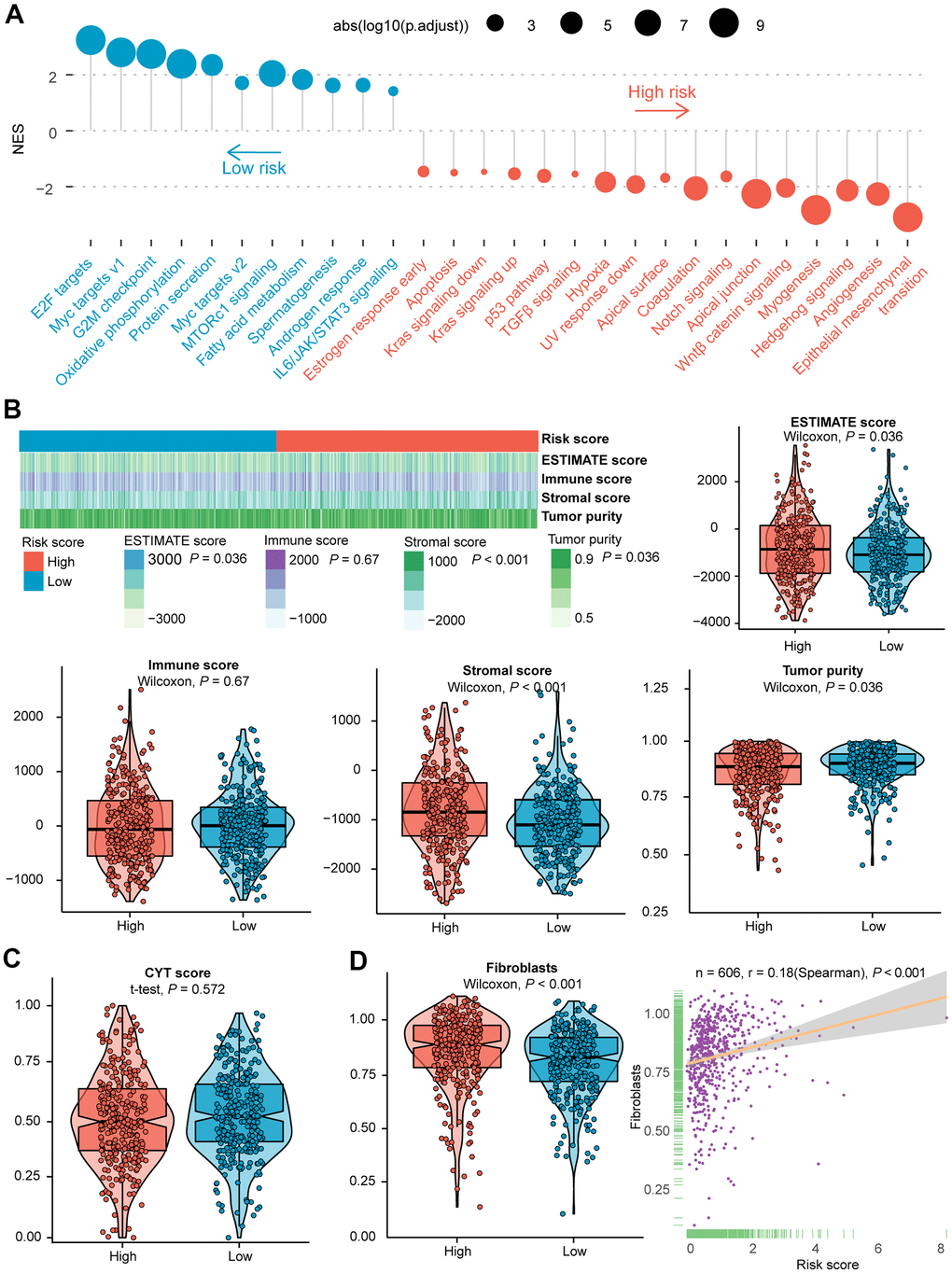
Figure 2. Evaluation of the role of IRGs-based risk score in the training cohort. (A) Results of GSEA of the high-risk group (red) compared with the low-risk group (blue). Color toward gray represents no statistical significance. (B) Heatmap and violin plots of the ESTIMATE score, immune score, stromal score, tumor purity between high- and low- risk subtypes. (C) Violin plot of the CYT score between high- and low- risk subtypes. (D) Violin plot of fibroblasts between two subtypes, and the association between risk score and the NES of fibroblasts. Statistical significance at the level of ns ≥ 0.05, * < 0.05, ** < 0.01 and *** < 0.001. GSEA, gene set enrichment analysis; CYT, cytolytic activity; NES, normalized enrichment score.
Besides, variation in the NES values of 10 common oncogenic pathways between the two subclasses were evaluated in the TCGA COAD and READ patients (Figure 3A). The Hippo-, Notch-, NRF2- and Wnt-related pathways exhibited significantly higher NES values in high-risk subtype than in low-risk subtype. The NES values of the PI3K, RAS, and TP53-related pathways were significantly higher in the immune risk score-low subtype than in the immune risk score-high subtype. Analogous effects in Hippo-, Notch- and Wnt-associated pathways were investigated in the GSE39582 validation cohort (Supplementary Figure 5C).
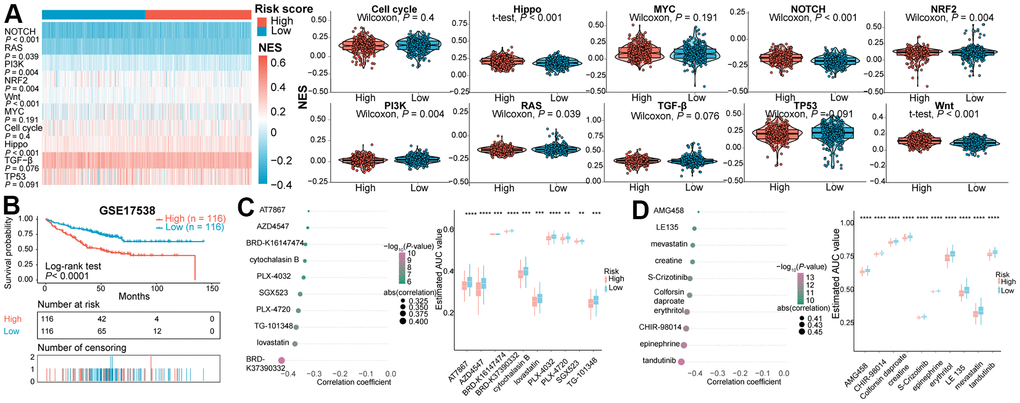
Figure 3. (A) Heatmap and violin plots of the NES of 10 oncogenic pathways between two subtypes in the TCGA cohort. (B) Kaplan-Meier survival curve of the high- and low- risk subgroups in GSE17538 dataset for identifying potential therapeutic agents. (C) Spearman’s correlation analysis and differential drug response analysis of 10 CTRP-derived compounds. (D) Spearman’s correlation analysis and differential drug response analysis of 10 PRISM-derived compounds. NES, normalized enrichment score.
The immune landscape of the microenvironment in CRC subclasses
To further evaluate the potential molecular mechanism, the connection between four types of score produced by the ESTIMATE algorithm and risk score was also examined. Among the training dataset, a higher risk score was unveiled with elevated ESTIMATE score and stromal score, nevertheless, with decreased tumor purity (Figure 2B). Analogous patterns were found in the validation dataset except for a significantly positive correlation between risk score and immune score (Supplementary Figure 3B). Besides, no statistical significance was shown in the CYT score between the two subclasses (Figure 2C and Supplementary Figure 3C). It has been documented that fibroblasts are critical in multiple immunologic responses and inflammatory responses to tumor tissue injury [51, 52]. In the training group, the risk score was markedly correlated with the NES of fibroblasts (Spearman’s r = 0.18, P-value < 0.001, Figure 2D). Likewise, we found increased NES in high-risk subgroup of the validation cohort (Supplementary Figure 3D).
Immunomodulators (IM) play a determinant role in clinical oncology and plenty of IM-related agonists and antagonists are being assessed [53]. To further figure out the underlying immune modules of the constructed IRGs model, the IM gene expression level between two subgroups in two CRC cohorts was compared. Among the IMs under investigation for cancer immunotherapy, certain of them were significantly related to the risk score (Figure 4A and Supplementary Figure 4A, 4B). In addition, we deeply investigated whether the risk score was associated with the expression level of T cell markers (CD4 and CD8A) and with six vital immune checkpoint genes (CD47, CTLA-4, LAG3, MAGE-A3, PD-1, and PD-L1). As shown in Figure 4B, the expression level of PD-1 was significantly higher in CRC of the high-risk subtype, while the risk score was negatively correlated with CD47 expression. Moreover, the differences in the expression level of CD47 and PD-1 between two subtypes of the TCGA dataset were statistically significant (Figure 4C). Even though the expression level of CD4 inclined to be elevated in high-risk subclass, no statistical difference was determined in the TCGA cohort. Statistical significance was verified in the validation cohort (Supplementary Figure 4C).
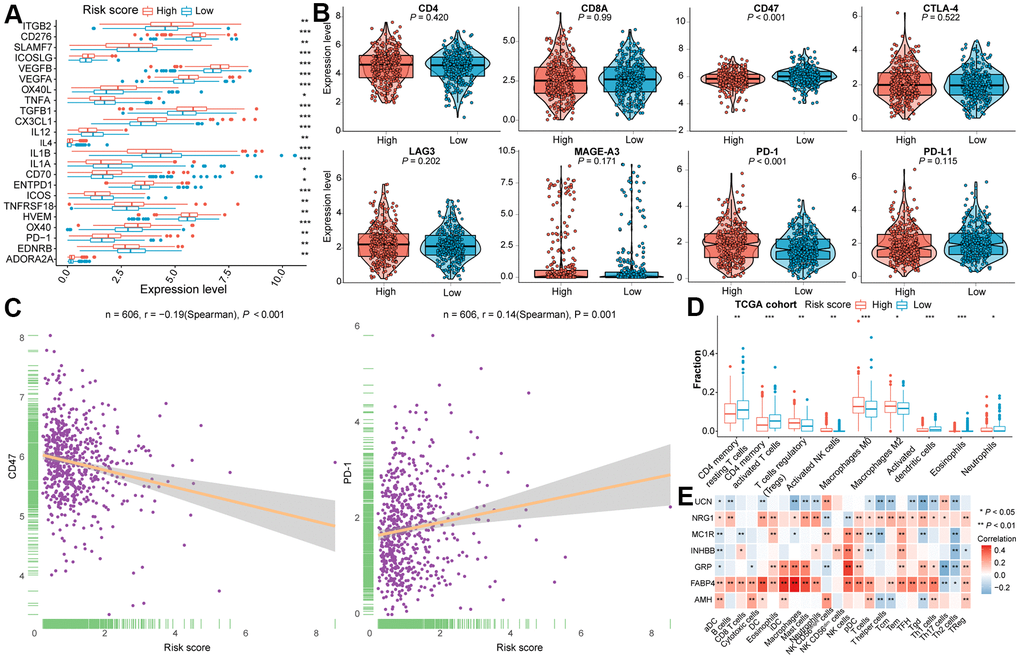
Figure 4. The immune landscape of two distinctive subclasses in the training cohort. (A) The differential expression level of immune checkpoint molecules between two subclasses with statistical significance. (B) Violin plots of the CD4, CD8A, CD47, CTLA4, LAG3, MAGE-A3, PD-1 and PD-L1 expression levels for two subtypes. (C) The association between risk score and CD47 as well as PD-1 expression levels. (D) Immune infiltration between high- and low- risk subtypes with statistical significance in the training cohort. (E) Correlation analysis between the expression of seven IRGs and the infiltration of immune cells. Statistical significance at the level of ns ≥ 0.05, * < 0.05, ** < 0.01 and *** < 0.001.
To investigate whether the immunophenotype may be shaped by immune cells, the relationship of immune infiltration with subtypes in both TCGA and GEO samples was examined in depth. We found that there was conspicuous heterogeneity in immune cell population among the established classifications, consistent with previous published TILs subpopulations in CRC [54]. As illustrated in Figure 4D and Supplementary Figure 5A, the infiltrated fractions of Tregs, activated NK cells, macrophage M0, and macrophage M2 was outstandingly augmented in the high-risk group. By contraries, markedly increased infiltration of CD4+ memory resting T cells, CD4+ memory activated T cells, activated dendritic cells (DCs), and neutrophils was observed in the low-risk group. Furthermore, we evaluate the correlation between the expression of seven IRGs and the infiltration of 24 types of immune cells by ssGSEA in CRC tissues. In the TCGA cohort (Figure 4E), there was a strong correlation of the FABP4 expression with the infiltration of NK cells (Spearman’s r = 0.34, P < 0.001), GRP with NK cells (Spearman’s r = 0.41, P < 0.001) or Th17 cells (Spearman’s r = -0.32, P < 0.001), INKBB with NK cells (Spearman’s r = 0.31, P < 0.001), as well as UCN with Tgd (Spearman’s r = -0.31, P < 0.001) (Supplementary Table 5). For the training dataset and validation dataset, strong connection was confirmed between the expression of FABP4 and the infiltration of DC (Spearman’s r = 0.38, P < 0.001), iDC (Spearman’s r = 0.44, P < 0.001), macrophages (Spearman’s r = 0.46, P < 0.001), and mast cells (Spearman’s r = 0.40, P < 0.001), the expression of GRP and infiltration of macrophages (Spearman’s r = 0.30, P < 0.001) included (Figure 4E and Supplementary Figure 5B and Supplementary Table 5).
Identification of potential therapeutic agents for CRCs with immune high-risk score
The CTRP and PRISM datasets shared 160 compounds, with 1770 compounds remained in total after removing duplication (Supplementary Table 6). Two approaches were utilized to screen candidate compounds with higher drug sensitivity in CRCs of high-risk score. By stratifying CRCs in GSE17538 dataset into high- and low- risk score subtypes based on seven IRGs (Figure 3B), the analyses were operated using CTRP and PRISM-derived drug response data, successively. First, differential drug response analysis between high- and low- risk groups was conducted to identify agents with differential estimated AUC values between subclasses (FDR < 0.05). Next, the Spearman correlation test between AUC value and risk score was adopted to identify drugs with negative correlation coefficient (Spearman’s r < −0.30 for CTRP or −0.40 for PRISM). Above analyses yielded 10 CTRP-derived agents (including AT7867, AZD4547, BRD-K37390332, Cytochalasin B, PLX-4720, SGX-523, PLX-4032, TG-101348, lovastatin, and BRD-K16147474) and 10 PRISM-derived agents (including AMG458, LE135, mevastatin, creatine, S-Crizotinib, Colforsin daproate, erythritol, CHIR-98014, epinephrine, and tandutinib). All these compounds presented lower estimated AUC values in the high-risk subgroup and a negative correlation with IRGs-based risk score (Figure 3C, 3D). Although the 20 candidate agents displayed a higher drug sensitivity in IRGs score-high patients, solely the analyses above could not draw to the conclusion that these compounds are promising treatment modality for the eradication of CRC. Therefore, an integrated literature retrieval was conducted in PubMed, DrugBank [55], and HERB [56] databases to search for the experimental and clinical evidence of candidate compounds for CRC (Supplementary Table 7). BRD-K16147474, SGX-523, BRD-K37390332, AMG458, LE135, creatine, colforsin daproate, erythritol, CHIR-98014, and epinephrine without supporting evidence for CRC were firstly excluded. Secondly, PLX-4032 [57, 58] and PLX-4720 [59] targeted B-rafV600E, AT7867 targeted Akt [60], tandutinib targeted Akt/mTOR pathway [61], TG-101348 targeted the JAK2/STAT3/PIM1 pathway [62], lovastatin [63] and mevastatin [64] inhibiting 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase weren’t considered as the potential compounds for risk score-high subclass. This is because these drugs functioned inconsistently with targets enriched in the immune score-high subclass through GSEA (Figure 2A and Supplementary Figure 3A). Collectively, AZD4547, Cytochalasin B and S-crizotinib, which held true in vitro and in silico evidence, were deemed the most promising therapeutic agents for CRCs with high IRG risk scores.
CRC subgroups have distinct responses to immunotherapy
Two different procedures were adopted in this study to identify subclass-specific candidate immunotherapies. Submap analysis was first used to find potential immunotherapeutic benefit of two subgroups through six immunotherapy datasets available with clinical response and gene expression information. As exhibited in Figure 5F, the high-risk subclass shared high similarity with anti-MAGE-A3 nonresponse group in Ulloa Montoya et al. (2013) dataset (P = 0.049) and anti-PD-1 nonresponse group in Hugo et al. (2016) dataset (P = 0.002), and the high-risk subgroup tended to be correlated with anti-PD-L1 nonresponse group in IMvigor210 cohort although no statistical significance was found (P = 0.08).
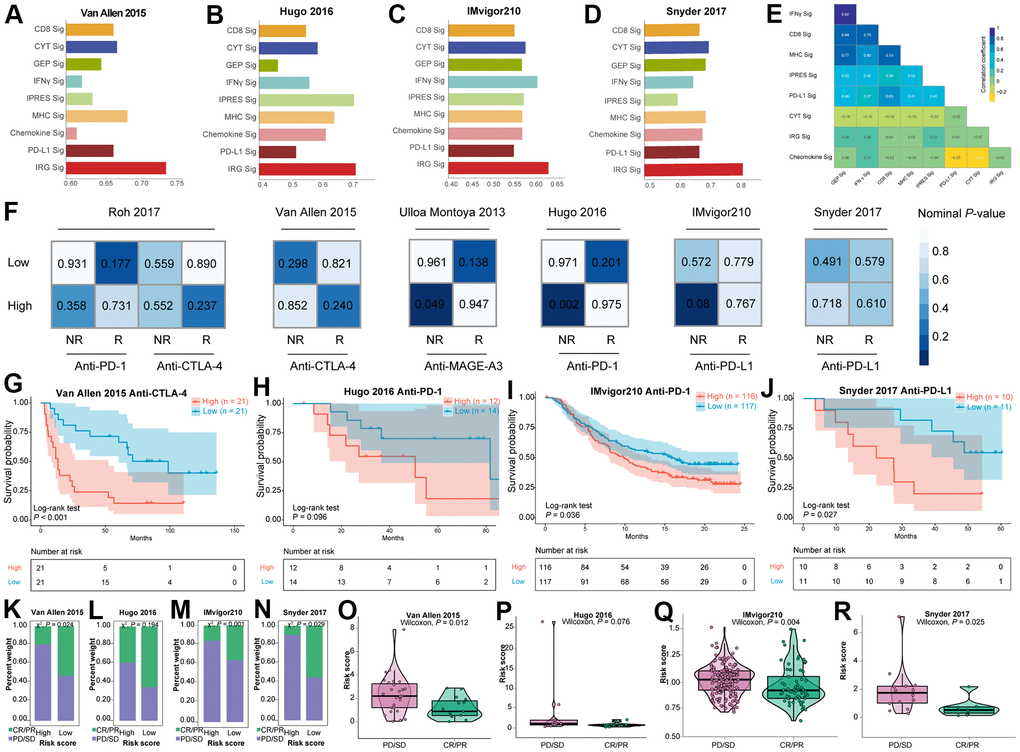
Figure 5. The immunotherapeutic benefit of the IRGs-based risk score in immunotherapeutic treatment cohorts. (A–D) Comparison of AUC values between IRGs-based signature and other eight previous published immune related signatures in four immunotherapeutic datasets. (E) Similarity comparison between IRGs-based signature and other seven previous signatures. (F) SubMap analysis utilizing six immunotherapy datasets. Kaplan-Meier survival curve of OS for patients with high- and low- risk score subtypes for (G) Van Allen et al. (2015) dataset, (H) Hugo et al. (2016) dataset, (I) IMvigor210 dataset, and (J) Snyder et al. (2017) dataset. Bar graph illustrated the treatment response to immunotherapy within high- and low- risk score subtypes in (K) Van Allen et al. (2015) dataset, (L) Hugo et al. (2016) dataset, (M) IMvigor210 dataset, and (N) Snyder et al. (2017) dataset. Violin plots illustrated the distribution of risk score for patients with different immunotherapy responses in (O) Van Allen et al. (2015) dataset, (P) Hugo et al. (2016) dataset, (Q) IMvigor210 dataset, and (R) Snyder et al. (2017) dataset.
Patients received immunotherapy in Van Allen et al. (2015) dataset, Hugo et al. (2016) dataset, IMvigor210 dataset, and Snyder et al. (2017) dataset were classified into high-risk subtype and low-risk subtype using the median IRGs-based risk score as the cutoff. Then, the AUC values for classifying the responder and non-responder cases of several previous signatures, including CD8 [65], CYT [17], T cell-inflamed GEP [66], IFNγ [66], IPRES [67], MHC [68], Chemokine [69], and PD-L1 [65] signatures as well as IRGs-based signature were calculated across all the four immunotherapeutic datasets with abundant gene expression profiles. Notably, IRGs signature outperformed the other eight signatures and the AUC values exceeded 0.7 in three out of four datasets (Figure 5A–5D). The results of performance comparison in four independent datasets suggested that the predictive power of IRGs signature ranked the highest. The association across these signatures indicated that five signatures, including IFNγ, CD8, MHC, IPRES, and PD-L1 signatures correlated closely with each other (Figure 5E). By contrast, IRGs signature displayed relatively weak correlation with other signatures, implying its complementary role rather than the alternative as an immunotherapeutic indicator. Patients in the low-risk subclass presented significant longer OS than those in the high-risk subclass of Van Allen et al. (2015) dataset (log-rank test P-value < 0.001, Figure 5G), IMvigor210 dataset (log-rank test P-value = 0.036, Figure 5I) and Snyder et al. (2017) dataset (log-rank test P-value = 0.027, Figure 5J), however, no statistical difference was observed in Hugo et al. (2016) dataset (log-rank test P-value = 0.096, Figure 5H). These findings demonstrated that the lower risk score was associated with better survival outcomes in tumor patients treated with immunotherapy. Collecting immunotherapeutic response data in four cohorts mentioned above, we determined the correlation between immunotherapeutic response and risk score. It’s shown that patients in the low-risk subtype had a dramatically higher response to immunotherapy than patients in the high-risk subtype among three datasets (P-value = 0.024 for Van Allen et al. anti-CTLA-4 cohort, P-value = 0.001 for IMvigor210 anti-PD-L1 cohort, and P-value = 0.029 for Snyder et al. anti-PD-L1 cohort; X2 test, Figure 5K, 5M, 5N), apart from (P-value = 0.194 for Hugo et al. anti-PD-1 cohort, X2 test, Figure 5L). According to Van Allen et al. (2015) anti-CTLA-4 cohort (Figure 5O), IMvigor210 anti-PD-L1 cohort (Figure 5Q), and Snyder et al. (2017) anti-PD-L1 cohort (Figure 5R), violin plots revealed that the risk score was significantly decreased in patients responsive to the immunotherapeutic invention, compared to non-responsive patients. Nonetheless, no statistical significance was observed in Hugo et al. (2016) anti-PD-1 cohort (Figure 5P).
Discussion
Despite the advances in treatment, CRC is a lethal disease of great heterogeneity, prompting therapeutic optimization to prolong survival outcomes and reduce mortality. Hence, it’s essential to acquire reliable prognostic biomarkers to stratify survival risk and to predict subclass-specific therapeutic strategies. Tailoring specialized management for patients depends on personalized clinical and molecular features. Gaining insight into IRGs involved in CRC enables scientists to recapitulate the underlying mechanism of carcinogenesis in CRC and identify patients who may benefit from adaptive therapy. In this study, by exploiting a compendium of IRGs, a robust prognostic immune-based signature was built using public CRC cohorts. CRC samples with intact expression profiles and clinical characteristics were downloaded from the TCGA and the GEO database. Multivariate Cox regression was utilized to calculate the risk score for each cohort based on the seven IRGs signature independently. Besides, bioinformatic analyses were separately performed in different CRC cohorts, the normalized process was thus unneeded.
The IRGs-based risk score was found to be significantly correlated with OS in CRCs and remained significant after adjustment for clinical and pathological parameters. To characterize the TIME immune infiltration, we explored the divergent immune cell subpopulation via the CIBERSORT algorithm between subgroups. The higher pro-tumor immunocytes encompassing Tregs, macrophage M0, and macrophage M2 were observed in the high-risk group, in contrast, immune cells orchestrating anti-tumor responses including CD4+ memory resting T cells, CD4+ memory activated T cells, activated DCs, and neutrophils accumulated in the low-risk group. Imbalances in immune cell components are associated with undesirable prognosis and inferior survival outcomes in cancer patients [70, 71]. Toor et al. documented the aggregation of CD4+ and FoxP3+ TILs in CRC tissues, compared to para-carcinoma normal tissues [72]. In humans, the accumulation of Tregs within TIME is regarded as a disadvantageous prognostic factor in a plethora of cancers [73]. However, Tregs infiltration in CRC tissues is incapable of predicting the prognosis [74, 75]. Elevated infiltration of Tregs could trigger low tumor differentiation and aggrandized involvement of lymph node [74]. In contrast, enhanced Tregs densities have also been correlated with better relapse-free survival (RFS) [76, 77]. Some heterogeneous subsets of Tregs facilitate CRC progression, covering CD8+ Tregs [78] and RORγt+ Tregs [79]. Macrophage polarization plays a prominent role in tumor pathogenesis. In response to distinct microenvironments, primary macrophages (M0) migrate out of vessels and could be polarized toward pro-inflammatory (M1) macrophages or anti-inflammatory (M2) macrophages, while resting macrophages undergo diverse functional alterations [80, 81]. To some extent, M2 macrophage infiltration is closely linked with increased involvement of CRC liver metastasis and malignant lesion in the liver [82]. Moreover, cancer-associated fibroblasts (CAFs) in CRC fuel tumor-associated macrophages (TAMs) infiltration and macrophages M2 polarization in TIME, subsequently impairing the function of NK cells [83]. The increased level of CD4+ TILs has been deemed as favorable clinical outcome in CRC [84], highlighting the crucial role of CD4+ cells in regulating immune system to exert anti-neoplastic activity. In CRCs, elevated expression of Th1 transcripts is correlated with beneficial prognosis, whereas the elevated expression of Th17 transcripts is correlated with poor clinical outcome [85]. Additionally, effector and memory Th1 CD4+ T cells are pivotal in effective anti-tumor immunity and that CD4+ T cells induce more durable immune responses than CD8+ T cells [86]. DCs act a key role in presenting tumor antigens and eliciting tumoricidal processes of T cells [87], and activated DCs might potentiate immunotherapeutic efficacy in advanced CRCs [88]. On the contrary, inhibited functions of DCs in cancer patients lead to the suppression of protective immune responses and facilitating disease progression [89]. An increased intra-tumoral abundance of neutrophil has been shown in CRC [90], and elevated neutrophil/lymphocyte ratio (NLR) in peripheral blood of advanced CRCs is related to unfavorable prognostic aspects [91]. By frequently colocalizing with CD8+ T cells, neutrophils could also irritate CD8+ T cell response to T cell receptor priming, thus reflecting that neutrophils might have notably anti-oncogenic efficacy [92]. Thus far, the roles of neutrophils and other immune cells in CRC progression have not been fully elucidated. The investigation in-depth, presented herein, opens new avenues for understanding the relationship between immune cells and the progression of CRC.
Among the seven IRGs in the classifier, NRG1 was considered as a protective factor for CRCs while FABP4, AMH, GRP, INHBB, UCN and MC1R were risk factors for CRCs. These IRGs have been previously reported to be involved in tumorigenesis. The growth factor neuregulin 1 (NRG1) comprises of an epidermal growth factor (EGF)-like domain that binds to human tyrosine kinases of the ErbB/HER receptor family, contributing to heterodimerization and activation of the ErbB-mediated downstream signaling pathways [93]. CRC is an NRG1 fusion-positive tumor [94, 95], in which the expression of NRG1 III is significantly upregulated and negatively correlated with lymph node metastasis [96], implying a satisfactory prognosis. Primarily expressed in the adipocytes and macrophages [97], fatty acid binding protein 4 (FABP4) is involved in lipid transfer between adipocytes and tumor cells, provoking the fatty acid oxidation to induce tumor growth [98, 99]. The elevated expression of FABP4 was confirmed as a robust risk factor for the progression of CRC in a Chinese cohort [100], while an in-silico study also uncovered that FABP4 imposed conceivably poor prognosis on CRCs [25]. Herein, FABP4 harbored detrimental effects on CRCs and the strong interaction between the FABP4 expression and macrophages was also manifested in our study, supporting FABP4’s crosstalk with macrophages in the TIME. As a corticotropin-releasing factor-related peptide, urocortin (UCN) participated in gastrointestinal motor and visceral pain during stress response [101]. In the current study, UCN was correlated with poor CRC prognosis, in tandem with anteriorly proposed CRC signature [25, 102, 103]. The melanocortin-1 receptor (MC1R) has been regarded as an adverse parameter for survival in CRC [102]. Nevertheless, the specific implication of MC1R in CRC is rarely known. Patients carrying the MC1R variants are presented with elevated melanoma risk, and MC1R had been a therapeutic target for melanoma [104, 105]. Consequently, preclinical studies on the importance of MC1R in the development of CRC are needed. Anti-Müllerian hormone (AMH) is a member of the TGFβ family that engages in cell proliferation, differentiation, and apoptosis in normal tissues [106]. AMH was positively related to the risk of breast cancer [107], and the downregulation of AMH lower the risk of CRC was forecasted in two bioinformatic analyses [108, 109]. The inhibin subunit beta B (INHBB) is a subunit of the activin B, a functional cytokine of the TGFβ superfamily [110, 111]. INHBB is upregulated and exerts tumorigenic activity in a variety of malignant tumors ranging from oral cancer [112] to endometrial cancer [113], prostate cancer [114], and thyroid cancer [115]. In our model, elevated INHBB expression predicted an adverse outcome. Analogously, Yuan et al. indicated that the expression of INHBB was enhanced in CRC tissue, bringing about worse OS and disease-free survival (DFS) [116]. As a subtribe of the bombesin (BN)-like peptide family, gastrin-releasing peptide (GRP) is principally served as gastrointestinal hormone and neurotransmitter [117, 118]. GRP modulates the growth and differentiation of numerous human tumors including CRC [119, 120]. The GRP receptor (GRPR) has been shown to be overexpressed in human CRCs, when compared to normal colonic epithelial cells [121, 122]. Moreover, GRP and the co-expression of GRPR acted in differentiation, with the highest levels observed in well-differentiated CRC cells [123]. BN/GRP antagonists, such as RC-3095 and RC-3940-II, have been reported to exert anti-tumor activities in in-vitro and in-vivo mouse xenografts [124, 125]. RC-3940-II also exerted potent anti-neoplastic activity on the human CRC cell lines both in vitro and in vivo [126]. Li et al. pointed out that GRP could predict the prognosis of DFS in CRC [127], uncovering its involvement in the prognosis and survival of CRC. Bedke et al. demonstrated that GRP and GRPR were mainly expressed by TAMs in renal cell carcinomas (RCC) [128], accordantly, the current study indicated that the expression of GRP was positively correlated with the degree of macrophage infiltration. Briefly, these compelling evidences for the significance of GRP show great potential at unmasking the malignancy-associated roles of TAMs in CRC.
Three drugs, including AZD4547, Cytochalasin B, and S-crizotinib, harbored more notable anti-neoplastic activity in the immune risk score-high group. Intriguingly, high-risk specific agents are all anti-tumor targeted compounds, and a striking consistency was shown between the mechanism of action (MOA) of these chemical entities and enriched signatures obtained from GSEA. As prominent segment in the TME composed of cancer cells and stromal or immune cells, CAFs crosstalk with tumor cells contributes to the progression of tumor [129]. Overexpression of the fibroblast growth factor receptor-1 (FGFR-1) has been correlated with liver metastasis in CRC [130]. The fibroblast growth factor 1 (FGF1)/FGFR-3 signaling mediates migration and invasion in CRC, and activated fibroblasts upregulate the expression of FGF1 [131]. AZD4547 is an orally potent and highly selective tyrosine kinase inhibitor (TKI) targeted FGFR 1-3 [132]. Preclinical data recapitulates that AZD4547 possesses anti-oncogenic activity against various tumors, such as gastric [133], lung [134], and pancreatic [135] cancers. Yao et al. reported that AZD4547 delayed CRC tumor growth in vitro, and its activity was in close interaction with the expression level of FGFR [136]. In our study, the infiltration of fibroblasts was apparently higher in high-risk score CRCs, compellingly argue for clinical investigations of AZD4547 for treating high-risk specific CRCs. Cytochalasin B is a common microfilament-disrupting compound that impacts various cellular physiological processes mediated by F-actin, encompassing cell motility, endocytosis and adherence [137–139]. Treating human CRC SW480 cells with cytochalasin B attenuated the downregulation of E-cadherin expression [140]. Indeed, the loss or dysregulation of E-cadherin expression expedites the growth, invasion, and drug resistance in CRC cells [141, 142]. EMT, a morphogenetic process whereby epithelial cells transform to the mesenchymal phenotype, critically engaged in tumorigenesis and cancer progression [143]. In tumor, the expression of epithelial markers, E-cadherin particularly, is downregulated during the process of EMT, ultimately destroying cell adhesion, promoting cell motility and stages of cancer [144, 145]. Conversely, inhibited EMT as evidenced by the elevated expression of E-cadherin exerts suppressive effects on the growth and invasion of human CRC via the Wnt/β-catenin signaling [146, 147]. c-MET/RON activation initiates many facets of cellular responses covering motility, proliferation, EMT, and angiogenesis [148, 149]. Typically, c-Met and RON signaling irritate angiogenesis through the interplay with vascular endothelial growth factor (VEGF) stimulated by hypoxia-inducible factor 1-alpha (HIF-1α). Crizotinib is an extensively functioning, small-molecule TKI clinically approved for treating non-small-cell lung cancer (NSCLC) patients [150]. In a three-dimensional CRC culture system, Li et al. found that crizotinib restored cetuximab sensitivity in the HCA-7 CRC cell line [151]. By inhibiting c-MET/RON/ALK/MTH1, S-crizotinib is an optical isomer of a clinical anticancer compound, R-crizotinib, with inhibited efficacy in suppressing MTH1 compared to S-crizotinib [152, 153]. Previous evidence suggests that MTH1 inhibition via S-crizotinib induced an increase in DNA single strand breaks as well as activated DNA repair in SW480 cells [153]. In human cells, acute MTH1 inhibition enables p53-dependent cellular senescence upon hyperoxia [154]. Moreover, MTH1 is pivotal in RAS-driven oncogenesis and its overexpression accelerates the spectrum of RAS-driven carcinogenic transformation [155]. Notably, elevated expression of MTH1 enhances the transformation of immortalized cells through RAS and maintains pro-oncogenic phenotype, EMT [156, 157]. Collectively, we postulate that AZD4547, Cytochalasin B and S-crizotinib are attractive compounds for further pre-clinical investigations and could be promising novel anti-cancer agents for IRGs risk score-high CRCs.
Immunotherapy, with special regard to ICIs, has attracted great interest in oncotherapy and has been applied in clinical practice for a variety of malignancies. Pembrolizumab and nivolumab that inhibited PD-1 and ipilimumab targeted CTLA-4 have been approved by the United States Food and Drug Administration (FDA) as second-line treatment in MSI-high and dMMR advanced CRCs. Focused on the findings from KEYNOTE 028 [13] and CheckMate 142 [11], solely a modest percentage of advanced CRCs harbored a persistent and stable response during the ICI therapy, with response rate at 30-55%. Therefore, it is of great clinical significance to develop a biomarker for predicting immunotherapeutic efficacy. In this study, we confirmed that CRCs with a low-risk immune signature were markedly related with enhanced response to ICIs targeted PD-1, PD-L1 and CTLA-4, while the immune score-high CRCs exhibited nonresponse to PD-1 inhibitor and MAGE-A3 based immunotherapy. These findings illustrated that the IRGs-based risk score could be served as a practical tool for assessing immunotherapeutic efficacy in CRC, in accordance with a recent study on the immune signature score for colon cancer [24]. Compared to the immune high-risk subclass, the low-risk subclass exhibited significantly higher infiltration of anti-tumor immune cells and expression of immune checkpoint genes, which may account for diverse responses between the two subclasses. Furthermore, the GSEA of hallmark gene sets indicated that the upregulated genes in the high-risk subgroup were enriched in Wnt/β-catenin signaling, consistent with previous findings that the activation of tumor-intrinsic β-catenin pathway could induce T-cell exclusion, thereby causing resistance to PD-L1 or CTLA-4 blockade immunotherapy [158]. Thus, altered Wnt/β-catenin signaling activation may be associated with immunotherapeutic resistance in CRC.
However, there are still some limitations in this study. Firstly, we attempted to obtain abundant CRC cohorts to achieve more reliable results with sufficient sample size. But the intra-tumor or intra-patient heterogeneity of the TIME in CRCs was not fully considered, which impacted the effect of chemotherapy and immunotherapy. Secondly, the median cutoff of IRGs risk score was utilized to stratify the CRC samples into high-risk subtype and low-risk subtype, and the optimal cutoff of the risk score is needed to best classify the CRCs. Thirdly, all the conclusions in this study were inferred from in-silico analyses, and further in-vitro or in-vivo experiments and clinical validations are needed to promote the clinical application of our findings. Finally, due to the paucity of CRC cohorts treated with immunotherapy, more prospective clinical studies are required to further verify this novel IRGs-based signature in CRCs.
Conclusions
The IRGs signature is valuable for its correlation with immune infiltration, and the association between the risk score and OS in the integrated analysis of CRC cohorts suggests that it is a robust prognostic biomarker for CRC. This IRGs model harbors crucial clinical practicality in both high- and low- risk CRCs who had failed first-line treatment or progressed. For immune low-risk score patients, clinicians could adopt ICIs targeted PD-1, PD-L1 and CTLA-4 as well as MAGE-A3 immunotherapy strategies to avoid excessive treatment, so these CRCs could acquire a better quality of life with a favorable prognosis. For immune high-risk score patients, AZD4547, Cytochalasin B and S-crizotinib might be used in cases of immunotherapeutic resistance. Generally, our finding provides new insights into determining the prognosis of CRCs, and sheds new light on tailoring CRCs with precise treatment.
Supplementary Materials
Author Contributions
WY, and JC designed and edited this study; YL and ZJX searched the databases and collected data; YL, YYL, ZJX, DZ, XMC, XYW, and JL analyzed the data and wrote the manuscript. WY and JC critically revised the article for essential intellectual content and administrative support. All authors read and approved the final version of the manuscript. All authors reviewed and revised the manuscript.
Acknowledgments
We would like to thank our researchers for their hard work and the reviewers for their valuable advice. We thank Home for Researchers editorial team (https://www.home-for-researchers.com) for language editing service.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Funding
This research was supported by the National Key R&D Program of China (2019YFA0110300, 2020YFA0509400); the National Natural Science Foundation of China (82071745); the Science and Technology Program of Guangzhou (202002030069); and the Guangdong project (2019QN01Y212).
References
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018; 68:394–424. https://doi.org/10.3322/caac.21492 [PubMed]
- 2. Dekker E, Tanis PJ, Vleugels JL, Kasi PM, Wallace MB. Colorectal cancer. Lancet. 2019; 394:1467–80. https://doi.org/10.1016/S0140-6736(19)32319-0 [PubMed]
- 3. Inamura K. Colorectal Cancers: An Update on Their Molecular Pathology. Cancers (Basel). 2018; 10:26. https://doi.org/10.3390/cancers10010026 [PubMed]
- 4. Schmoll HJ, Van Cutsem E, Stein A, Valentini V, Glimelius B, Haustermans K, Nordlinger B, van de Velde CJ, Balmana J, Regula J, Nagtegaal ID, Beets-Tan RG, Arnold D, et al. ESMO Consensus Guidelines for management of patients with colon and rectal cancer. a personalized approach to clinical decision making. Ann Oncol. 2012; 23:2479–516. https://doi.org/10.1093/annonc/mds236 [PubMed]
- 5. Condamine T, Ramachandran I, Youn JI, Gabrilovich DI. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu Rev Med. 2015; 66:97–110. https://doi.org/10.1146/annurev-med-051013-052304 [PubMed]
- 6. Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019; 79:4557–66. https://doi.org/10.1158/0008-5472.CAN-18-3962 [PubMed]
- 7. Mlecnik B, Tosolini M, Kirilovsky A, Berger A, Bindea G, Meatchi T, Bruneval P, Trajanoski Z, Fridman WH, Pagès F, Galon J. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J Clin Oncol. 2011; 29:610–8. https://doi.org/10.1200/JCO.2010.30.5425 [PubMed]
- 8. Pagès F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C, Lugli A, Zlobec I, Rau TT, Berger MD, Nagtegaal ID, Vink-Börger E, Hartmann A, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 2018; 391:2128–39. https://doi.org/10.1016/S0140-6736(18)30789-X [PubMed]
- 9. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoué F, Bruneval P, Cugnenc PH, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006; 313:1960–4. https://doi.org/10.1126/science.1129139 [PubMed]
- 10. Xiao Y, Freeman GJ. The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov. 2015; 5:16–8. https://doi.org/10.1158/2159-8290.CD-14-1397 [PubMed]
- 11. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, Wong F, Azad NS, Rucki AA, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017; 357:409–13. https://doi.org/10.1126/science.aan6733 [PubMed]
- 12. Overman MJ, Lonardi S, Wong KY, Lenz HJ, Gelsomino F, Aglietta M, Morse MA, Van Cutsem E, McDermott R, Hill A, Sawyer MB, Hendlisz A, Neyns B, et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol. 2018; 36:773–9. https://doi.org/10.1200/JCO.2017.76.9901 [PubMed]
- 13. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, Desai J, Hill A, Axelson M, Moss RA, Goldberg MV, Cao ZA, Ledeine JM, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017; 18:1182–91. https://doi.org/10.1016/S1470-2045(17)30422-9 [PubMed]
- 14. Hersom M, Jørgensen JT. Companion and Complementary Diagnostics-Focus on PD-L1 Expression Assays for PD-1/PD-L1 Checkpoint Inhibitors in Non-Small Cell Lung Cancer. Ther Drug Monit. 2018; 40:9–16. https://doi.org/10.1097/FTD.0000000000000460 [PubMed]
- 15. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn MJ, Felip E, et al, and KEYNOTE-001 Investigators. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015; 372:2018–28. https://doi.org/10.1056/NEJMoa1501824 [PubMed]
- 16. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015; 372:2509–20. https://doi.org/10.1056/NEJMoa1500596 [PubMed]
- 17. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015; 160:48–61. https://doi.org/10.1016/j.cell.2014.12.033 [PubMed]
- 18. Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC, Tadmor AD, Schoenberger SP, Huber C, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015; 520:692–6. https://doi.org/10.1038/nature14426 [PubMed]
- 19. Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D, Church SE, Lafontaine L, Fischer M, Fredriksen T, Sasso M, Bilocq AM, Kirilovsky A, et al. Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity. 2016; 44:698–711. https://doi.org/10.1016/j.immuni.2016.02.025 [PubMed]
- 20. Bruni D, Angell HK, Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer. 2020; 20:662–80. https://doi.org/10.1038/s41568-020-0285-7 [PubMed]
- 21. Jansen AM, van Wezel T, van den Akker BE, Ventayol Garcia M, Ruano D, Tops CM, Wagner A, Letteboer TG, Gómez-García EB, Devilee P, Wijnen JT, Hes FJ, Morreau H. Combined mismatch repair and POLE/POLD1 defects explain unresolved suspected Lynch syndrome cancers. Eur J Hum Genet. 2016; 24:1089–92. https://doi.org/10.1038/ejhg.2015.252 [PubMed]
- 22. Domingo E, Freeman-Mills L, Rayner E, Glaire M, Briggs S, Vermeulen L, Fessler E, Medema JP, Boot A, Morreau H, van Wezel T, Liefers GJ, Lothe RA, et al, and Epicolon consortium. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: a retrospective, pooled biomarker study. Lancet Gastroenterol Hepatol. 2016; 1:207–16. https://doi.org/10.1016/S2468-1253(16)30014-0 [PubMed]
- 23. Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, Diaz LA
Jr . Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol. 2019; 16:361–75. https://doi.org/10.1038/s41575-019-0126-x [PubMed] - 24. Li X, Wen D, Li X, Yao C, Chong W, Chen H. Identification of an Immune Signature Predicting Prognosis Risk and Lymphocyte Infiltration in Colon Cancer. Front Immunol. 2020; 11:1678. https://doi.org/10.3389/fimmu.2020.01678 [PubMed]
- 25. Lin K, Huang J, Luo H, Luo C, Zhu X, Bu F, Xiao H, Xiao L, Zhu Z. Development of a prognostic index and screening of potential biomarkers based on immunogenomic landscape analysis of colorectal cancer. Aging (Albany NY). 2020; 12:5832–57. https://doi.org/10.18632/aging.102979 [PubMed]
- 26. Bhattacharya S, Andorf S, Gomes L, Dunn P, Schaefer H, Pontius J, Berger P, Desborough V, Smith T, Campbell J, Thomson E, Monteiro R, Guimaraes P, et al. ImmPort: disseminating data to the public for the future of immunology. Immunol Res. 2014; 58:234–9. https://doi.org/10.1007/s12026-014-8516-1 [PubMed]
- 27. Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, Barretina J, Gelfand ET, Bielski CM, Li H, Hu K, Andreev-Drakhlin AY, Kim J, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019; 569:503–8. https://doi.org/10.1038/s41586-019-1186-3 [PubMed]
- 28. Roh W, Chen PL, Reuben A, Spencer CN, Prieto PA, Miller JP, Gopalakrishnan V, Wang F, Cooper ZA, Reddy SM, Gumbs C, Little L, Chang Q, et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Transl Med. 2017; 9:eaah3560. https://doi.org/10.1126/scitranslmed.aah3560 [PubMed]
- 29. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MH, Goldinger SM, Utikal J, Hassel JC, Weide B, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015; 350:207–11. https://doi.org/10.1126/science.aad0095 [PubMed]
- 30. Ulloa-Montoya F, Louahed J, Dizier B, Gruselle O, Spiessens B, Lehmann FF, Suciu S, Kruit WH, Eggermont AM, Vansteenkiste J, Brichard VG. Predictive gene signature in MAGE-A3 antigen-specific cancer immunotherapy. J Clin Oncol. 2013; 31:2388–95. https://doi.org/10.1200/JCO.2012.44.3762 [PubMed]
- 31. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, Seja E, Lomeli S, Kong X, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016; 165:35–44. https://doi.org/10.1016/j.cell.2016.02.065 [PubMed]
- 32. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE II, Koeppen H, Astarita JL, Cubas R, Jhunjhunwala S, Banchereau R, Yang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018; 554:544–8. https://doi.org/10.1038/nature25501 [PubMed]
- 33. Snyder A, Nathanson T, Funt SA, Ahuja A, Buros Novik J, Hellmann MD, Chang E, Aksoy BA, Al-Ahmadie H, Yusko E, Vignali M, Benzeno S, Boyd M, et al. Contribution of systemic and somatic factors to clinical response and resistance to PD-L1 blockade in urothelial cancer: An exploratory multi-omic analysis. PLoS Med. 2017; 14:e1002309. https://doi.org/10.1371/journal.pmed.1002309 [PubMed]
- 34. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. https://doi.org/10.1093/nar/gkv007 [PubMed]
- 35. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012; 16:284–7. https://doi.org/10.1089/omi.2011.0118 [PubMed]
- 36. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015; 1:417–25. https://doi.org/10.1016/j.cels.2015.12.004 [PubMed]
- 37. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013; 14:7. https://doi.org/10.1186/1471-2105-14-7 [PubMed]
- 38. Eide PW, Bruun J, Lothe RA, Sveen A. CMScaller: an R package for consensus molecular subtyping of colorectal cancer pre-clinical models. Sci Rep. 2017; 7:16618. https://doi.org/10.1038/s41598-017-16747-x [PubMed]
- 39. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, Ziv E, Culhane AC, Paull EO, et al, and Cancer Genome Atlas Research Network. The Immune Landscape of Cancer. Immunity. 2018; 48:812–30.e14. https://doi.org/10.1016/j.immuni.2018.03.023 [PubMed]
- 40. Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, Carter SL, Getz G, Stemke-Hale K, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013; 4:2612. https://doi.org/10.1038/ncomms3612 [PubMed]
- 41. Xiao Y, Ma D, Zhao S, Suo C, Shi J, Xue MZ, Ruan M, Wang H, Zhao J, Li Q, Wang P, Shi L, Yang WT, et al, and AME Breast Cancer Collaborative Group. Multi-Omics Profiling Reveals Distinct Microenvironment Characterization and Suggests Immune Escape Mechanisms of Triple-Negative Breast Cancer. Clin Cancer Res. 2019; 25:5002–14. https://doi.org/10.1158/1078-0432.CCR-18-3524 [PubMed]
- 42. Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, Chakravarty D, Daian F, Gao Q, et al, and Cancer Genome Atlas Research Network. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell. 2018; 173:321–37.e10. https://doi.org/10.1016/j.cell.2018.03.035 [PubMed]
- 43. Barros L, Pretti MA, Chicaybam L, Abdo L, Boroni M, Bonamino MH. Immunological-based approaches for cancer therapy. Clinics (Sao Paulo). 2018; 73:e429s. https://doi.org/10.6061/clinics/2018/e429s [PubMed]
- 44. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, Bruneval P, Fridman WH, Becker C, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013; 39:782–95. https://doi.org/10.1016/j.immuni.2013.10.003 [PubMed]
- 45. Geeleher P, Cox N, Huang RS. pRRophetic: an R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS One. 2014; 9:e107468. https://doi.org/10.1371/journal.pone.0107468 [PubMed]
- 46. Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, Bot BM, Morris JS, Simon IM, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015; 21:1350–6. https://doi.org/10.1038/nm.3967 [PubMed]
- 47. Chen J, Gong C, Mao H, Li Z, Fang Z, Chen Q, Lin M, Jiang X, Hu Y, Wang W, Zhang X, Chen X, Li H. E2F1/SP3/STAT6 axis is required for IL-4-induced epithelial-mesenchymal transition of colorectal cancer cells. Int J Oncol. 2018; 53:567–78. https://doi.org/10.3892/ijo.2018.4429 [PubMed]
- 48. Chen T, Li Q, Wu J, Wu Y, Peng W, Li H, Wang J, Tang X, Peng Y, Fu X. Fusobacterium nucleatum promotes M2 polarization of macrophages in the microenvironment of colorectal tumours via a TLR4-dependent mechanism. Cancer Immunol Immunother. 2018; 67:1635–46. https://doi.org/10.1007/s00262-018-2233-x [PubMed]
- 49. Kikuchi Y, Tokita S, Hirama T, Kochin V, Nakatsugawa M, Shinkawa T, Hirohashi Y, Tsukahara T, Hata F, Takemasa I, Sato N, Kanaseki T, Torigoe T. CD8+ T-cell Immune Surveillance against a Tumor Antigen Encoded by the Oncogenic Long Noncoding RNA PVT1. Cancer Immunol Res. 2021; 9:1342–53. https://doi.org/10.1158/2326-6066.CIR-20-0964 [PubMed]
- 50. Shao Q, Wang L, Yuan M, Jin X, Chen Z, Wu C. TIGIT Induces (CD3+) T Cell Dysfunction in Colorectal Cancer by Inhibiting Glucose Metabolism. Front Immunol. 2021; 12:688961. https://doi.org/10.3389/fimmu.2021.688961 [PubMed]
- 51. Spranger S. Mechanisms of tumor escape in the context of the T-cell-inflamed and the non-T-cell-inflamed tumor microenvironment. Int Immunol. 2016; 28:383–91. https://doi.org/10.1093/intimm/dxw014 [PubMed]
- 52. Hakelius M, Reyhani V, Rubin K, Gerdin B, Nowinski D. Normal Oral Keratinocytes and Head and Neck Squamous Carcinoma Cells Induce an Innate Response of Fibroblasts. Anticancer Res. 2016; 36:2131–7. [PubMed]
- 53. Tang J, Shalabi A, Hubbard-Lucey VM. Comprehensive analysis of the clinical immuno-oncology landscape. Ann Oncol. 2018; 29:84–91. https://doi.org/10.1093/annonc/mdx755 [PubMed]
- 54. Angelova M, Charoentong P, Hackl H, Fischer ML, Snajder R, Krogsdam AM, Waldner MJ, Bindea G, Mlecnik B, Galon J, Trajanoski Z. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015; 16:64. https://doi.org/10.1186/s13059-015-0620-6 [PubMed]
- 55. Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, Assempour N, Iynkkaran I, Liu Y, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018; 46:D1074–82. https://doi.org/10.1093/nar/gkx1037 [PubMed]
- 56. Fang S, Dong L, Liu L, Guo J, Zhao L, Zhang J, Bu D, Liu X, Huo P, Cao W, Dong Q, Wu J, Zeng X, et al. HERB: a high-throughput experiment- and reference-guided database of traditional Chinese medicine. Nucleic Acids Res. 2021; 49:D1197–206. https://doi.org/10.1093/nar/gkaa1063 [PubMed]
- 57. Herr R, Köhler M, Andrlová H, Weinberg F, Möller Y, Halbach S, Lutz L, Mastroianni J, Klose M, Bittermann N, Kowar S, Zeiser R, Olayioye MA, et al. B-Raf inhibitors induce epithelial differentiation in BRAF-mutant colorectal cancer cells. Cancer Res. 2015; 75:216–29. https://doi.org/10.1158/0008-5472.CAN-13-3686 [PubMed]
- 58. Whittaker SR, Cowley GS, Wagner S, Luo F, Root DE, Garraway LA. Combined Pan-RAF and MEK Inhibition Overcomes Multiple Resistance Mechanisms to Selective RAF Inhibitors. Mol Cancer Ther. 2015; 14:2700–11. https://doi.org/10.1158/1535-7163.MCT-15-0136-T [PubMed]
- 59. Herr R, Halbach S, Heizmann M, Busch H, Boerries M, Brummer T. BRAF inhibition upregulates a variety of receptor tyrosine kinases and their downstream effector Gab2 in colorectal cancer cell lines. Oncogene. 2018; 37:1576–93. https://doi.org/10.1038/s41388-017-0063-5 [PubMed]
- 60. Zhang S, Deng Z, Yao C, Huang P, Zhang Y, Cao S, Li X. AT7867 Inhibits Human Colorectal Cancer Cells via AKT-Dependent and AKT-Independent Mechanisms. PLoS One. 2017; 12:e0169585. https://doi.org/10.1371/journal.pone.0169585 [PubMed]
- 61. Ponnurangam S, Standing D, Rangarajan P, Subramaniam D. Tandutinib inhibits the Akt/mTOR signaling pathway to inhibit colon cancer growth. Mol Cancer Ther. 2013; 12:598–609. https://doi.org/10.1158/1535-7163.MCT-12-0907 [PubMed]
- 62. Liu Q, Li A, Wang L, He W, Zhao L, Wu C, Lu S, Ye X, Zhao H, Shen X, Xiao X, Liu Z. Stomatin-like Protein 2 Promotes Tumor Cell Survival by Activating the JAK2-STAT3-PIM1 Pathway, Suggesting a Novel Therapy in CRC. Mol Ther Oncolytics. 2020; 17:169–79. https://doi.org/10.1016/j.omto.2020.03.010 [PubMed]
- 63. Lee SJ, Lee I, Lee J, Park C, Kang WK. Statins, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors, potentiate the anti-angiogenic effects of bevacizumab by suppressing angiopoietin2, BiP, and Hsp90α in human colorectal cancer. Br J Cancer. 2014; 111:497–505. https://doi.org/10.1038/bjc.2014.283 [PubMed]
- 64. Wächtershäuser A, Akoglu B, Stein J. HMG-CoA reductase inhibitor mevastatin enhances the growth inhibitory effect of butyrate in the colorectal carcinoma cell line Caco-2. Carcinogenesis. 2001; 22:1061–7. https://doi.org/10.1093/carcin/22.7.1061 [PubMed]
- 65. Xiong D, Wang Y, You M. A gene expression signature of TREM2hi macrophages and γδ T cells predicts immunotherapy response. Nat Commun. 2020; 11:5084. https://doi.org/10.1038/s41467-020-18546-x [PubMed]
- 66. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, Piha-Paul SA, Yearley J, Seiwert TY, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017; 127:2930–40. https://doi.org/10.1172/JCI91190 [PubMed]
- 67. Thompson JC, Hwang WT, Davis C, Deshpande C, Jeffries S, Rajpurohit Y, Krishna V, Smirnov D, Verona R, Lorenzi MV, Langer CJ, Albelda SM. Gene signatures of tumor inflammation and epithelial-to-mesenchymal transition (EMT) predict responses to immune checkpoint blockade in lung cancer with high accuracy. Lung Cancer. 2020; 139:1–8. https://doi.org/10.1016/j.lungcan.2019.10.012 [PubMed]
- 68. Lauss M, Donia M, Harbst K, Andersen R, Mitra S, Rosengren F, Salim M, Vallon-Christersson J, Törngren T, Kvist A, Ringnér M, Svane IM, Jönsson G. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat Commun. 2017; 8:1738. https://doi.org/10.1038/s41467-017-01460-0 [PubMed]
- 69. Messina JL, Fenstermacher DA, Eschrich S, Qu X, Berglund AE, Lloyd MC, Schell MJ, Sondak VK, Weber JS, Mulé JJ. 12-Chemokine gene signature identifies lymph node-like structures in melanoma: potential for patient selection for immunotherapy? Sci Rep. 2012; 2:765. https://doi.org/10.1038/srep00765 [PubMed]
- 70. Ali HR, Chlon L, Pharoah PD, Markowetz F, Caldas C. Patterns of Immune Infiltration in Breast Cancer and Their Clinical Implications: A Gene-Expression-Based Retrospective Study. PLoS Med. 2016; 13:e1002194. https://doi.org/10.1371/journal.pmed.1002194 [PubMed]
- 71. Bense RD, Sotiriou C, Piccart-Gebhart MJ, Haanen JB, van Vugt MA, de Vries EG, Schröder CP, Fehrmann RS. Relevance of Tumor-Infiltrating Immune Cell Composition and Functionality for Disease Outcome in Breast Cancer. J Natl Cancer Inst. 2016; 109:djw192. https://doi.org/10.1093/jnci/djw192 [PubMed]
- 72. Toor SM, Murshed K, Al-Dhaheri M, Khawar M, Abu Nada M, Elkord E. Immune Checkpoints in Circulating and Tumor-Infiltrating CD4+ T Cell Subsets in Colorectal Cancer Patients. Front Immunol. 2019; 10:2936. https://doi.org/10.3389/fimmu.2019.02936 [PubMed]
- 73. Huang Y, Liao H, Zhang Y, Yuan R, Wang F, Gao Y, Wang P, Du Z. Prognostic value of tumor-infiltrating FoxP3+ T cells in gastrointestinal cancers: a meta analysis. PLoS One. 2014; 9:e94376. https://doi.org/10.1371/journal.pone.0094376 [PubMed]
- 74. Zhuo C, Xu Y, Ying M, Li Q, Huang L, Li D, Cai S, Li B. FOXP3+ Tregs: heterogeneous phenotypes and conflicting impacts on survival outcomes in patients with colorectal cancer. Immunol Res. 2015; 61:338–47. https://doi.org/10.1007/s12026-014-8616-y [PubMed]
- 75. Sideras K, Galjart B, Vasaturo A, Pedroza-Gonzalez A, Biermann K, Mancham S, Nigg AL, Hansen BE, Stoop HA, Zhou G, Verhoef C, Sleijfer S, Sprengers D, et al. Prognostic value of intra-tumoral CD8+ /FoxP3+ lymphocyte ratio in patients with resected colorectal cancer liver metastasis. J Surg Oncol. 2018; 118:68–76. https://doi.org/10.1002/jso.25091 [PubMed]
- 76. Kuwahara T, Hazama S, Suzuki N, Yoshida S, Tomochika S, Nakagami Y, Matsui H, Shindo Y, Kanekiyo S, Tokumitsu Y, Iida M, Tsunedomi R, Takeda S, et al. Intratumoural-infiltrating CD4 + and FOXP3 + T cells as strong positive predictive markers for the prognosis of resectable colorectal cancer. Br J Cancer. 2019; 121:659–65. https://doi.org/10.1038/s41416-019-0559-6 [PubMed]
- 77. Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, Platell C, Iacopetta B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol. 2009; 27:186–92. https://doi.org/10.1200/JCO.2008.18.7229 [PubMed]
- 78. Chaput N, Louafi S, Bardier A, Charlotte F, Vaillant JC, Ménégaux F, Rosenzwajg M, Lemoine F, Klatzmann D, Taieb J. Identification of CD8+CD25+Foxp3+ suppressive T cells in colorectal cancer tissue. Gut. 2009; 58:520–29. https://doi.org/10.1136/gut.2008.158824 [PubMed]
- 79. Blatner NR, Mulcahy MF, Dennis KL, Scholtens D, Bentrem DJ, Phillips JD, Ham S, Sandall BP, Khan MW, Mahvi DM, Halverson AL, Stryker SJ, Boller AM, et al. Expression of RORγt marks a pathogenic regulatory T cell subset in human colon cancer. Sci Transl Med. 2012; 4:164ra159. https://doi.org/10.1126/scitranslmed.3004566 [PubMed]
- 80. Reddy JP, Atkinson RL, Larson R, Burks JK, Smith D, Debeb BG, Ruffell B, Creighton CJ, Bambhroliya A, Reuben JM, Van Laere SJ, Krishnamurthy S, Symmans WF, et al. Mammary stem cell and macrophage markers are enriched in normal tissue adjacent to inflammatory breast cancer. Breast Cancer Res Treat. 2018; 171:283–93. https://doi.org/10.1007/s10549-018-4835-6 [PubMed]
- 81. Martinez FO. Regulators of macrophage activation. Eur J Immunol. 2011; 41:1531–4. https://doi.org/10.1002/eji.201141670 [PubMed]
- 82. Guo L, Wang C, Qiu X, Pu X, Chang P. Colorectal Cancer Immune Infiltrates: Significance in Patient Prognosis and Immunotherapeutic Efficacy. Front Immunol. 2020; 11:1052. https://doi.org/10.3389/fimmu.2020.01052 [PubMed]
- 83. Zhang R, Qi F, Zhao F, Li G, Shao S, Zhang X, Yuan L, Feng Y. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis. 2019; 10:273. https://doi.org/10.1038/s41419-019-1435-2 [PubMed]
- 84. Kuwahara T, Hazama S, Suzuki N, Yoshida S, Tomochika S, Nakagami Y, Matsui H, Shindo Y, Kanekiyo S, Tokumitsu Y, Iida M, Tsunedomi R, Takeda S, et al. Correction: Intratumoural-infiltrating CD4 + and FOXP3 + T cells as strong positive predictive markers for the prognosis of resectable colorectal cancer. Br J Cancer. 2019; 121:983–4. https://doi.org/10.1038/s41416-019-0605-4 [PubMed]
- 85. Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, Berger A, Bruneval P, Fridman WH, Pagès F, Galon J. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011; 71:1263–71. https://doi.org/10.1158/0008-5472.CAN-10-2907 [PubMed]
- 86. Melssen M, Slingluff CL
Jr . Vaccines targeting helper T cells for cancer immunotherapy. Curr Opin Immunol. 2017; 47:85–92. https://doi.org/10.1016/j.coi.2017.07.004 [PubMed] - 87. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012; 12:265–77. https://doi.org/10.1038/nrc3258 [PubMed]
- 88. Du XH, Liu HL, Li L, Xia SY, Ning N, Zou ZY, Teng D, Xiao CH, Li R, Xu YX. Clinical significance of immunotherapy with combined three kinds of cells for operable colorectal cancer. Tumour Biol. 2015; 36:5679–85. https://doi.org/10.1007/s13277-015-3242-4 [PubMed]
- 89. Hargadon KM. Strategies to Improve the Efficacy of Dendritic Cell-Based Immunotherapy for Melanoma. Front Immunol. 2017; 8:1594. https://doi.org/10.3389/fimmu.2017.01594 [PubMed]
- 90. Rao HL, Chen JW, Li M, Xiao YB, Fu J, Zeng YX, Cai MY, Xie D. Increased intratumoral neutrophil in colorectal carcinomas correlates closely with malignant phenotype and predicts patients’ adverse prognosis. PLoS One. 2012; 7:e30806. https://doi.org/10.1371/journal.pone.0030806 [PubMed]
- 91. Chua W, Charles KA, Baracos VE, Clarke SJ. Neutrophil/lymphocyte ratio predicts chemotherapy outcomes in patients with advanced colorectal cancer. Br J Cancer. 2011; 104:1288–95. https://doi.org/10.1038/bjc.2011.100 [PubMed]
- 92. Governa V, Trella E, Mele V, Tornillo L, Amicarella F, Cremonesi E, Muraro MG, Xu H, Droeser R, Däster SR, Bolli M, Rosso R, Oertli D, et al. The Interplay Between Neutrophils and CD8+ T Cells Improves Survival in Human Colorectal Cancer. Clin Cancer Res. 2017; 23:3847–58. https://doi.org/10.1158/1078-0432.CCR-16-2047 [PubMed]
- 93. Mei L, Xiong WC. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci. 2008; 9:437–52. https://doi.org/10.1038/nrn2392 [PubMed]
- 94. Laskin J, Liu SV, Tolba K, Heining C, Schlenk RF, Cheema P, Cadranel J, Jones MR, Drilon A, Cseh A, Gyorffy S, Solca F, Duruisseaux M. NRG1 fusion-driven tumors: biology, detection, and the therapeutic role of afatinib and other ErbB-targeting agents. Ann Oncol. 2020; 31:1693–703. https://doi.org/10.1016/j.annonc.2020.08.2335 [PubMed]
- 95. Cadranel J, Liu SV, Duruisseaux M, Branden E, Goto Y, Weinberg BA, Heining C, Schlenk RF, Cheema P, Jones MR, Drilon A, Trombetta D, Muscarella LA, et al. Therapeutic Potential of Afatinib in NRG1 Fusion-Driven Solid Tumors: A Case Series. Oncologist. 2021; 26:7–16. https://doi.org/10.1634/theoncologist.2020-0379 [PubMed]
- 96. Guo Y, Duan Z, Jia Y, Ren C, Lv J, Guo P, Zhao W, Wang B, Zhang S, Li Y, Li Z. HER4 isoform CYT2 and its ligand NRG1III are expressed at high levels in human colorectal cancer. Oncol Lett. 2018; 15:6629–35. https://doi.org/10.3892/ol.2018.8124 [PubMed]
- 97. Fujii K, Kondo T, Yokoo H, Yamada T, Iwatsuki K, Hirohashi S. Proteomic study of human hepatocellular carcinoma using two-dimensional difference gel electrophoresis with saturation cysteine dye. Proteomics. 2005; 5:1411–22. https://doi.org/10.1002/pmic.200401004 [PubMed]
- 98. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS, Yamada SD, Peter ME, Gwin K, Lengyel E. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011; 17:1498–503. https://doi.org/10.1038/nm.2492 [PubMed]
- 99. Nieman KM, Romero IL, Van Houten B, Lengyel E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim Biophys Acta. 2013; 1831:1533–41. https://doi.org/10.1016/j.bbalip.2013.02.010 [PubMed]
- 100. Zhang Y, Zhao X, Deng L, Li X, Wang G, Li Y, Chen M. High expression of FABP4 and FABP6 in patients with colorectal cancer. World J Surg Oncol. 2019; 17:171. https://doi.org/10.1186/s12957-019-1714-5 [PubMed]
- 101. Martinez V, Wang L, Million M, Rivier J, Taché Y. Urocortins and the regulation of gastrointestinal motor function and visceral pain. Peptides. 2004; 25:1733–44. https://doi.org/10.1016/j.peptides.2004.05.025 [PubMed]
- 102. Chen H, Luo J, Guo J. Development and validation of a five-immune gene prognostic risk model in colon cancer. BMC Cancer. 2020; 20:395. https://doi.org/10.1186/s12885-020-06799-0 [PubMed]
- 103. Wang J, Yu S, Chen G, Kang M, Jin X, Huang Y, Lin L, Wu D, Wang L, Chen J. A novel prognostic signature of immune-related genes for patients with colorectal cancer. J Cell Mol Med. 2020; 24:8491–504. https://doi.org/10.1111/jcmm.15443 [PubMed]
- 104. Scherer D, Nagore E, Bermejo JL, Figl A, Botella-Estrada R, Thirumaran RK, Angelini S, Hemminki K, Schadendorf D, Kumar R. Melanocortin receptor 1 variants and melanoma risk: a study of 2 European populations. Int J Cancer. 2009; 125:1868–75. https://doi.org/10.1002/ijc.24548 [PubMed]
- 105. Chen S, Zhu B, Yin C, Liu W, Han C, Chen B, Liu T, Li X, Chen X, Li C, Hu L, Zhou J, Xu ZX, et al. Palmitoylation-dependent activation of MC1R prevents melanomagenesis. Nature. 2017; 549:399–403. https://doi.org/10.1038/nature23887 [PubMed]
- 106. MacLaughlin DT, Donahoe PK. Müllerian inhibiting substance/anti-Müllerian hormone: a potential therapeutic agent for human ovarian and other cancers. Future Oncol. 2010; 6:391–405. https://doi.org/10.2217/fon.09.172 [PubMed]
- 107. Clendenen TV, Ge W, Koenig KL, Afanasyeva Y, Agnoli C, Brinton LA, Darvishian F, Dorgan JF, Eliassen AH, Falk RT, Hallmans G, Hankinson SE, Hoffman-Bolton J, et al. Breast cancer risk prediction in women aged 35-50 years: impact of including sex hormone concentrations in the Gail model. Breast Cancer Res. 2019; 21:42. https://doi.org/10.1186/s13058-019-1126-z [PubMed]
- 108. Gong B, Kao Y, Zhang C, Sun F, Gong Z, Chen J. Identification of Hub Genes Related to Carcinogenesis and Prognosis in Colorectal Cancer Based on Integrated Bioinformatics. Mediators Inflamm. 2020; 2020:5934821. https://doi.org/10.1155/2020/5934821 [PubMed]
- 109. Pang B, Xu X, Lu Y, Jin H, Yang R, Jiang C, Shao D, Liu Y, Shi J. Prediction of new targets and mechanisms for quercetin in the treatment of pancreatic cancer, colon cancer, and rectal cancer. Food Funct. 2019; 10:5339–49. https://doi.org/10.1039/c9fo01168d [PubMed]
- 110. Namwanje M, Brown CW. Activins and Inhibins: Roles in Development, Physiology, and Disease. Cold Spring Harb Perspect Biol. 2016; 8:a021881. https://doi.org/10.1101/cshperspect.a021881 [PubMed]
- 111. Wijayarathna R, de Kretser DM. Activins in reproductive biology and beyond. Hum Reprod Update. 2016; 22:342–57. https://doi.org/10.1093/humupd/dmv058 [PubMed]
- 112. Kita A, Kasamatsu A, Nakashima D, Endo-Sakamoto Y, Ishida S, Shimizu T, Kimura Y, Miyamoto I, Yoshimura S, Shiiba M, Tanzawa H, Uzawa K. Activin B Regulates Adhesion, Invasiveness, and Migratory Activities in Oral Cancer: a Potential Biomarker for Metastasis. J Cancer. 2017; 8:2033–41. https://doi.org/10.7150/jca.18714 [PubMed]
- 113. Mylonas I. Inhibin-alpha, -betaA and -betaB subunits in uterine non-endometrioid carcinomas: prognostic significance and clinical implications. Eur J Cancer. 2010; 46:2485–93. https://doi.org/10.1016/j.ejca.2010.06.001 [PubMed]
- 114. Hofland J, van Weerden WM, Steenbergen J, Dits NF, Jenster G, de Jong FH. Activin A stimulates AKR1C3 expression and growth in human prostate cancer. Endocrinology. 2012; 153:5726–34. https://doi.org/10.1210/en.2011-2065 [PubMed]
- 115. Matsuo SE, Ebina KN, Kulcsar MA, Friguglietti CU, Kimura ET. Activin betaB expression in rat experimental goiter and human thyroid tumors. Thyroid. 2003; 13:239–47. https://doi.org/10.1089/105072503321582033 [PubMed]
- 116. Yuan J, Xie A, Cao Q, Li X, Chen J. INHBB Is a Novel Prognostic Biomarker Associated with Cancer-Promoting Pathways in Colorectal Cancer. Biomed Res Int. 2020; 2020:6909672. https://doi.org/10.1155/2020/6909672 [PubMed]
- 117. Pansky A, DE Weerth A, Fasler-Kan E, Boulay JL, Schulz M, Ketterer S, Selck C, Beglinger C, VON Schrenck T, Hildebrand P. Gastrin releasing peptide-preferring bombesin receptors mediate growth of human renal cell carcinoma. J Am Soc Nephrol. 2000; 11:1409–18. https://doi.org/10.1681/ASN.V1181409 [PubMed]
- 118. Thomas RP, Hellmich MR, Townsend CM
Jr , Evers BM. Role of gastrointestinal hormones in the proliferation of normal and neoplastic tissues. Endocr Rev. 2003; 24:571–99. https://doi.org/10.1210/er.2002-0028 [PubMed] - 119. Glover SC, Tretiakova MS, Carroll RE, Benya RV. Increased frequency of gastrin-releasing peptide receptor gene mutations during colon-adenocarcinoma progression. Mol Carcinog. 2003; 37:5–15. https://doi.org/10.1002/mc.10117 [PubMed]
- 120. Schally AV, Szepeshazi K, Nagy A, Comaru-Schally AM, Halmos G. New approaches to therapy of cancers of the stomach, colon and pancreas based on peptide analogs. Cell Mol Life Sci. 2004; 61:1042–68. https://doi.org/10.1007/s00018-004-3434-3 [PubMed]
- 121. Chave HS, Gough AC, Palmer K, Preston SR, Primrose JN. Bombesin family receptor and ligand gene expression in human colorectal cancer and normal mucosa. Br J Cancer. 2000; 82:124–30. https://doi.org/10.1054/bjoc.1998.0888 [PubMed]
- 122. Patel O, Dumesny C, Giraud AS, Baldwin GS, Shulkes A. Stimulation of proliferation and migration of a colorectal cancer cell line by amidated and glycine-extended gastrin-releasing peptide via the same receptor. Biochem Pharmacol. 2004; 68:2129–42. https://doi.org/10.1016/j.bcp.2004.08.009 [PubMed]
- 123. Matkowskyj KA, Keller K, Glover S, Kornberg L, Tran-Son-Tay R, Benya RV. Expression of GRP and its receptor in well-differentiated colon cancer cells correlates with the presence of focal adhesion kinase phosphorylated at tyrosines 397 and 407. J Histochem Cytochem. 2003; 51:1041–8. https://doi.org/10.1177/002215540305100807 [PubMed]
- 124. Rick FG, Schally AV, Block NL, Nadji M, Szepeshazi K, Zarandi M, Vidaurre I, Perez R, Halmos G, Szalontay L. Antagonists of growth hormone-releasing hormone (GHRH) reduce prostate size in experimental benign prostatic hyperplasia. Proc Natl Acad Sci USA. 2011; 108:3755–60. https://doi.org/10.1073/pnas.1018086108 [PubMed]
- 125. Schally AV, Comaru-Schally AM, Nagy A, Kovacs M, Szepeshazi K, Plonowski A, Varga JL, Halmos G. Hypothalamic hormones and cancer. Front Neuroendocrinol. 2001; 22:248–91. https://doi.org/10.1006/frne.2001.0217 [PubMed]
- 126. Rick FG, Buchholz S, Schally AV, Szalontay L, Krishan A, Datz C, Stadlmayr A, Aigner E, Perez R, Seitz S, Block NL, Hohla F. Combination of gastrin-releasing peptide antagonist with cytotoxic agents produces synergistic inhibition of growth of human experimental colon cancers. Cell Cycle. 2012; 11:2518–25. https://doi.org/10.4161/cc.20900 [PubMed]
- 127. Li C, Shen Z, Zhou Y, Yu W. Independent prognostic genes and mechanism investigation for colon cancer. Biol Res. 2018; 51:10. https://doi.org/10.1186/s40659-018-0158-7 [PubMed]
- 128. Bedke J, Hemmerlein B, Perske C, Gross A, Heuser M. Tumor-associated macrophages in clear cell renal cell carcinoma express both gastrin-releasing peptide and its receptor: a possible modulatory role of immune effectors cells. World J Urol. 2010; 28:335–41. https://doi.org/10.1007/s00345-009-0492-z [PubMed]
- 129. Ribatti D, Vacca A, Rusnati M, Presta M. The discovery of basic fibroblast growth factor/fibroblast growth factor-2 and its role in haematological malignancies. Cytokine Growth Factor Rev. 2007; 18:327–34. https://doi.org/10.1016/j.cytogfr.2007.04.011 [PubMed]
- 130. Sato T, Oshima T, Yoshihara K, Yamamoto N, Yamada R, Nagano Y, Fujii S, Kunisaki C, Shiozawa M, Akaike M, Rino Y, Tanaka K, Masuda M, Imada T. Overexpression of the fibroblast growth factor receptor-1 gene correlates with liver metastasis in colorectal cancer. Oncol Rep. 2009; 21:211–16. [PubMed]
- 131. Henriksson ML, Edin S, Dahlin AM, Oldenborg PA, Öberg Å, Van Guelpen B, Rutegård J, Stenling R, Palmqvist R. Colorectal cancer cells activate adjacent fibroblasts resulting in FGF1/FGFR3 signaling and increased invasion. Am J Pathol. 2011; 178:1387–94. https://doi.org/10.1016/j.ajpath.2010.12.008 [PubMed]
- 132. Gavine PR, Mooney L, Kilgour E, Thomas AP, Al-Kadhimi K, Beck S, Rooney C, Coleman T, Baker D, Mellor MJ, Brooks AN, Klinowska T. AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. 2012; 72:2045–56. https://doi.org/10.1158/0008-5472.CAN-11-3034 [PubMed]
- 133. Jang J, Kim HK, Bang H, Kim ST, Kim SY, Park SH, Lim HY, Kang WK, Lee J, Kim KM. Antitumor Effect of AZD4547 in a Fibroblast Growth Factor Receptor 2-Amplified Gastric Cancer Patient-Derived Cell Model. Transl Oncol. 2017; 10:469–75. https://doi.org/10.1016/j.tranon.2017.03.001 [PubMed]
- 134. Paik PK, Shen R, Berger MF, Ferry D, Soria JC, Mathewson A, Rooney C, Smith NR, Cullberg M, Kilgour E, Landers D, Frewer P, Brooks N, André F. A Phase Ib Open-Label Multicenter Study of AZD4547 in Patients with Advanced Squamous Cell Lung Cancers. Clin Cancer Res. 2017; 23:5366–73. https://doi.org/10.1158/1078-0432.CCR-17-0645 [PubMed]
- 135. Guan Z, Lan H, Sun D, Wang X, Jin K. A potential novel therapy for FGFR1-amplified pancreatic cancer with bone metastasis, screened by next-generation sequencing and a patient-derived xenograft model. Oncol Lett. 2019; 17:2303–7. https://doi.org/10.3892/ol.2018.9876 [PubMed]
- 136. Yao TJ, Zhu JH, Peng DF, Cui Z, Zhang C, Lu PH. AZD-4547 exerts potent cytostatic and cytotoxic activities against fibroblast growth factor receptor (FGFR)-expressing colorectal cancer cells. Tumour Biol. 2015; 36:5641–8. https://doi.org/10.1007/s13277-015-3237-1 [PubMed]
- 137. Scherlach K, Boettger D, Remme N, Hertweck C. The chemistry and biology of cytochalasans. Nat Prod Rep. 2010; 27:869–86. https://doi.org/10.1039/b903913a [PubMed]
- 138. Van Goietsenoven G, Mathieu V, Andolfi A, Cimmino A, Lefranc F, Kiss R, Evidente A. In vitro growth inhibitory effects of cytochalasins and derivatives in cancer cells. Planta Med. 2011; 77:711–7. https://doi.org/10.1055/s-0030-1250523 [PubMed]
- 139. MacLean-Fletcher S, Pollard TD. Mechanism of action of cytochalasin B on actin. Cell. 1980; 20:329–41. https://doi.org/10.1016/0092-8674(80)90619-4 [PubMed]
- 140. Foran E, McWilliam P, Kelleher D, Croke DT, Long A. The leukocyte protein L-plastin induces proliferation, invasion and loss of E-cadherin expression in colon cancer cells. Int J Cancer. 2006; 118:2098–104. https://doi.org/10.1002/ijc.21593 [PubMed]
- 141. Karatzas G, Karayiannakis AJ, Syrigos KN, Chatzigianni E, Papanikolaou S, Riza F, Papanikolaou D. E-cadherin expression correlates with tumor differentiation in colorectal cancer. Hepatogastroenterology. 1999; 46:232–5. [PubMed]
- 142. Chen X, Wang Y, Xia H, Wang Q, Jiang X, Lin Z, Ma Y, Yang Y, Hu M. Loss of E-cadherin promotes the growth, invasion and drug resistance of colorectal cancer cells and is associated with liver metastasis. Mol Biol Rep. 2012; 39:6707–14. https://doi.org/10.1007/s11033-012-1494-2 [PubMed]
- 143. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014; 15:178–96. https://doi.org/10.1038/nrm3758 [PubMed]
- 144. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009; 119:1420–8. https://doi.org/10.1172/JCI39104 [PubMed]
- 145. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009; 139:871–90. https://doi.org/10.1016/j.cell.2009.11.007 [PubMed]
- 146. Gu J, Cui CF, Yang L, Wang L, Jiang XH. Emodin Inhibits Colon Cancer Cell Invasion and Migration by Suppressing Epithelial-Mesenchymal Transition via the Wnt/β-Catenin Pathway. Oncol Res. 2019; 27:193–202. https://doi.org/10.3727/096504018X15150662230295 [PubMed]
- 147. Marjaneh RM, Rahmani F, Hassanian SM, Rezaei N, Hashemzehi M, Bahrami A, Ariakia F, Fiuji H, Sahebkar A, Avan A, Khazaei M. Phytosomal curcumin inhibits tumor growth in colitis-associated colorectal cancer. J Cell Physiol. 2018; 233:6785–98. https://doi.org/10.1002/jcp.26538 [PubMed]
- 148. Zhang J, Jiang X, Jiang Y, Guo M, Zhang S, Li J, He J, Liu J, Wang J, Ouyang L. Recent advances in the development of dual VEGFR and c-Met small molecule inhibitors as anticancer drugs. Eur J Med Chem. 2016; 108:495–504. https://doi.org/10.1016/j.ejmech.2015.12.016 [PubMed]
- 149. Chang K, Karnad A, Zhao S, Freeman JW. Roles of c-Met and RON kinases in tumor progression and their potential as therapeutic targets. Oncotarget. 2015; 6:3507–18. https://doi.org/10.18632/oncotarget.3420 [PubMed]
- 150. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Jänne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010; 363:1693–703. https://doi.org/10.1056/NEJMoa1006448 [PubMed]
- 151. Li C, Singh B, Graves-Deal R, Ma H, Starchenko A, Fry WH, Lu Y, Wang Y, Bogatcheva G, Khan MP, Milne GL, Zhao S, Ayers GD, et al. Three-dimensional culture system identifies a new mode of cetuximab resistance and disease-relevant genes in colorectal cancer. Proc Natl Acad Sci USA. 2017; 114:E2852–61. https://doi.org/10.1073/pnas.1618297114 [PubMed]
- 152. Niu Y, Pan D, Shi D, Bai Q, Liu H, Yao X. Influence of Chirality of Crizotinib on Its MTH1 Protein Inhibitory Activity: Insight from Molecular Dynamics Simulations and Binding Free Energy Calculations. PLoS One. 2015; 10:e0145219. https://doi.org/10.1371/journal.pone.0145219 [PubMed]
- 153. Huber KV, Salah E, Radic B, Gridling M, Elkins JM, Stukalov A, Jemth AS, Göktürk C, Sanjiv K, Strömberg K, Pham T, Berglund UW, Colinge J, et al. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature. 2014; 508:222–7. https://doi.org/10.1038/nature13194 [PubMed]
- 154. Rai P, Onder TT, Young JJ, McFaline JL, Pang B, Dedon PC, Weinberg RA. Continuous elimination of oxidized nucleotides is necessary to prevent rapid onset of cellular senescence. Proc Natl Acad Sci USA. 2009; 106:169–74. https://doi.org/10.1073/pnas.0809834106 [PubMed]
- 155. Rai P, Young JJ, Burton DG, Giribaldi MG, Onder TT, Weinberg RA. Enhanced elimination of oxidized guanine nucleotides inhibits oncogenic RAS-induced DNA damage and premature senescence. Oncogene. 2011; 30:1489–96. https://doi.org/10.1038/onc.2010.520 [PubMed]
- 156. Rai P. Human Mut T Homolog 1 (MTH1): a roadblock for the tumor-suppressive effects of oncogenic RAS-induced ROS. Small GTPases. 2012; 3:120–5. https://doi.org/10.4161/sgtp.19556 [PubMed]
- 157. Giribaldi MG, Munoz A, Halvorsen K, Patel A, Rai P. MTH1 expression is required for effective transformation by oncogenic HRAS. Oncotarget. 2015; 6:11519–29. https://doi.org/10.18632/oncotarget.3447 [PubMed]
- 158. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015; 523:231–5. https://doi.org/10.1038/nature14404 [PubMed]