



Introduction
Atrial fibrillation (AF) is a common arrhythmia during clinical diagnosis and treatment in cardiology departments. Due to the rapid advancement of surgical instruments and cardiac drugs, significant progress has been achieved in the treatment of AF and its complications (tachyarrhythmia, thrombosis, heart failure, etc.) [1]. According to a systematic review including population studies worldwide, the number of AF patients globally was estimated to be 33.5 million in 2010 [2–4]. However, our understanding of the mechanism of AF occurrence and persistence is still inadequate.
Many case–control studies have demonstrated that the levels of inflammatory markers such as C-reactive protein (CRP), interleukin-6 (IL-6), IL-8, and tumor necrosis factor-α (TNF-α) in the AF patient population are significantly higher than those in the sinus rhythm population [5–10]. Additionally, the CRP level can be used to predict new-onset AF [11–13]. These results indicate a certain correlation between the inflammatory response and the occurrence of AF. Notably, in a canine sterile pericarditis model, anti-inflammatory treatment significantly reduced the incidence of aseptic inflammation-induced AF [14], which may suggest that the inflammatory response is the initiating factor in AF.
AF induced by inflammation can stimulate the body to generate new inflammatory responses and initiate atrial remodeling, resulting in AF maintenance [1]. AF patients have higher levels of inflammatory markers than previous AF patients who have recovered sinus rhythm [15, 16]. After continuous fast atrial pacing in dogs (pacing time >1 week), peripheral blood CRP levels were significantly increased, the atrial effective refractory period was shortened, and AF susceptibility was increased [17], indicating that AF may trigger new inflammatory responses, which likely result in AF persistence [17].
In summary, although the inflammatory response and AF are strongly correlated, the underlying mechanisms remain poorly understood. With the widespread application of second-generation sequencing technology and immune infiltration analysis technology, more powerful research tools are available to solve this problem. Therefore, this study (Supplementary Figure 2) was conducted to screen AF/immune infiltration–related differentially expressed genes (DEGs) based on existing gene expression data from the Gene Expression Omnibus (GEO) database. On the basis of these DEGs, the weighted gene co-expression network analysis (WGCNA) algorithm was used to identify key modules. Finally, hub genes associated with the AF/inflammatory response were further screened, and their possible role in the development of AF is discussed.
Materials and Methods
Data download
GEOquery [18] was used to download four sets of data: GSE115574, GSE41177, GSE79768, and GSE2240. GSE2240 was used as the verification dataset. Probes corresponded to genes, and the median value of the probes for the same gene was used for analysis. The GSE115574, GSE41177, and GSE79768 datasets were pooled. Batch differences (Supplementary Figure 1) were eliminated using the ComBat function of the sva package [19], and then the gene expression levels were homogenized using the limma package [20].
Immune cell infiltration
The gene expression data of the samples were uploaded to CIBERSORTx (https://cibersortx.stanford.edu/)). Bulk-mode batch correction and absolute mode were selected to analyze the level of immune infiltration, and the absolute score indicating the level of infiltration was obtained for each sample. The median absolute score of all samples was calculated. An absolute score greater than or equal to the median value indicated high infiltration, and an absolute score less than the median indicated low infiltration.
Analysis of DEGs
The limma [20] package was used to analyze DEGs in a high-infiltration group, a low-infiltration group, an AF group, and a sinus rhythm (SR) group, and the intersections of the DEGs of pairs of groups were obtained. P<0.01 was considered indicative of a significant difference.
Enrichment analysis
The clusterProfiler [21] package of R was used for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of DEGs.
WGCNA
We used WGCNA to construct a weighted co-expression network of DEGs. Based on traits of interest, the module with the smallest P value and the highest correlation was selected as the key module.
Screening of hub genes
Genes that were most relevant to a trait (gene significance (GS) ≥0.3) and a module (module membership (MM) ≥0.7) among the key WGCNA modules were selected as the hub genes in the modules. All the genes in the key modules were input into the STRING website (https://string-db.org/). The protein–protein interaction (PPI) network was obtained and then plotted using Cytoscape software. Genes with a high degree of gene connectivity (≥30) were selected as key genes in the PPI network. The final set of hub genes was obtained by taking the intersection of the WGCNA key module genes and PPI network key genes.
Analysis of clinical characteristics of hub genes
The age and sex information of each sample was used for grouping. The expression levels of the hub genes in different groups were analyzed. The median age was set as the dichotomizing line.
Hub gene validation
The expression levels of the hub genes were verified in GSE165838.
Unsupervised clustering analysis
Based on the expression levels of the hub genes, the best number of classifications was determined by determining the best sum of squared error (SSE) inflection point, and the samples were classified using k-means clustering combined with t-distributed stochastic neighbor embedding dimensionality reduction. Differences in immune infiltration and the expression levels of hub genes between different types were investigated.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, and further inquiries can be directed to the corresponding author/s.
Results
Data set selection and content introduction
The AF and SR datasets of GSE115574, GSE41177, GSE79768, and GSE2240 were downloaded from the GEO database (Table 1). GSE2240 was used as the verification dataset. The other three datasets were combined to obtain the expression data of 21,502 genes in 123 samples, including 74 AF and 49 SR samples.
Table 1. AF and SR datasets from the GEO database.
| Dataset | Platform | Atrial fibrillation | Sinus rhythm |
| GSE115574 | GPL570 | 28 | 31 |
| GSE41177 | GPL570 | 32 | 6 |
| GSE79768 | GPL570 | 14 | 12 |
| GSE2240 | GPL96 | 10 | 20 |
The difference of immune cell infiltration is shown
According to the gene expression data, the immune cell infiltration level was analyzed using CIBERSORTx. We marked the differences of immune cells between the two groups, and the results are shown in the Figure 1A. Each square represents the infiltration level of immune cells in the sample.
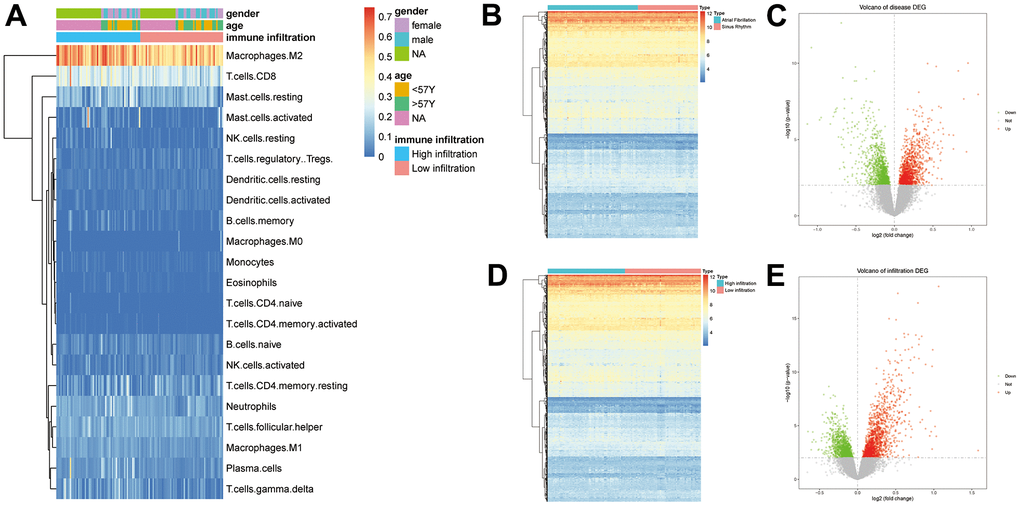
Figure 1. (A) Results of the immune infiltration analysis (p>0.05, *: p<0.05, **: p<0.01, ***: p<0.001, ****: p<0.0001). (B) Volcano plot of DEGs in AF and SR. (C) Heat map for the expression of the 2379 DEGs in AF. (D) Volcano plot of DEGs in immune infiltration. (E) Heat map for the expression of the 2607 DEGs in immune infiltration.
Differential analysis strategy and differential analysis results
P<0.01 was used as the threshold to define a DEG. A total of 2379 DEGs were identified in the 74 AF samples and the 49 SR samples, 1213 of which were upregulated, while 1167 were downregulated (Figure 1B, 1C). A total of 2607 DEGs were related to immune infiltration, 1432 of which were upregulated, while 1184 were downregulated (Figure 1D, 1E). The intersection of these two groups included 586 DEGs.
GO and KEGG enrichment analysis
Pathway enrichment analysis of the 586 shared DEGs was performed (Figure 2, P_FDR<0.05). As shown in Figure 2, the GO categories of biological process (BP) and cell component (CC) and the KEGG pathways are shown only with the 20 most significant results. Most were related to immune responses (e.g., the regulation of the innate immune response), immune cells (e.g., T cell activation, leukocyte migration), and immune activities (e.g., chemokine signaling, immunoglobulin binding).
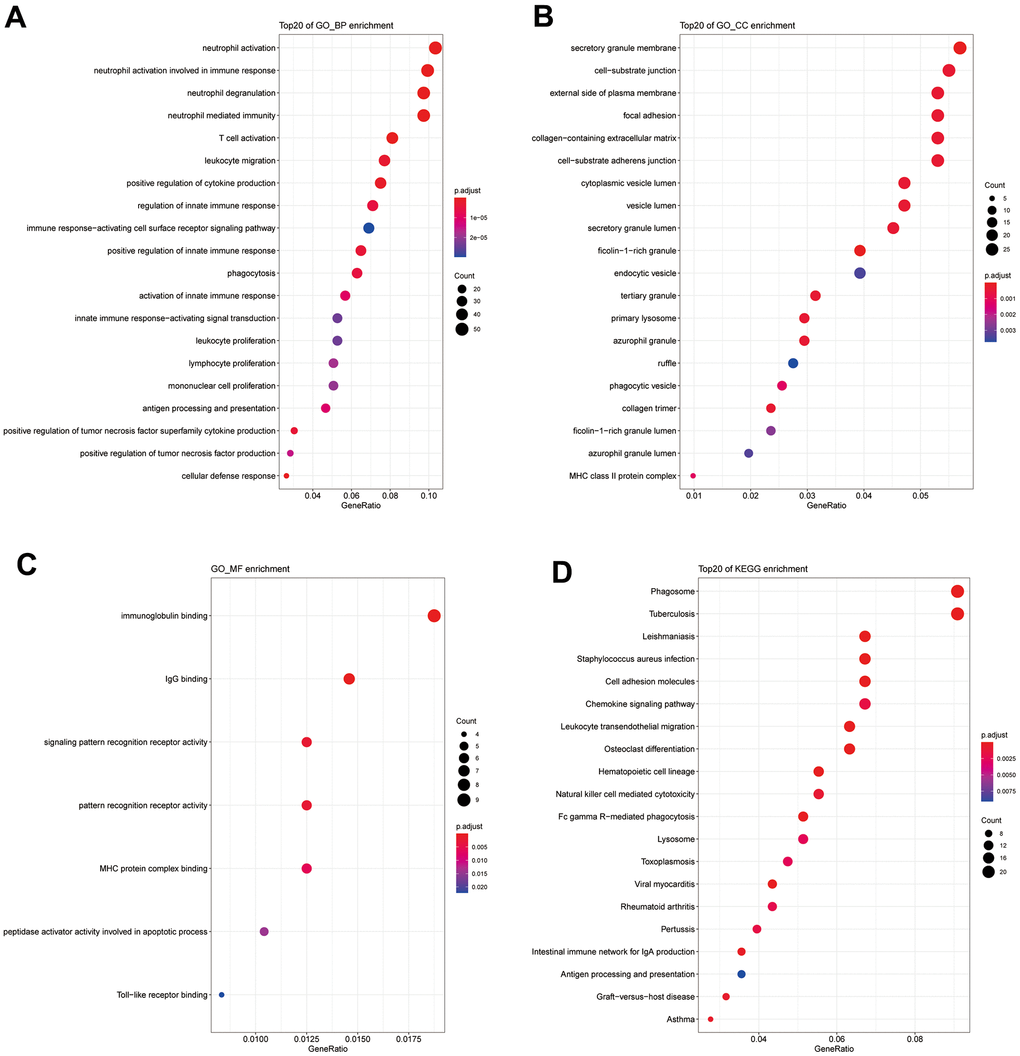
Figure 2. (A) GO_BP enrichment results for DEGs. (B) GO_CC enrichment results for DEGs. (C) GO_MF enrichment results for DEGs. (D) KEGG enrichment results for DEGs.
Construction of WGCNA co-expression network
WGCNA was performed on the 586 shared DEGs (The soft-threshold β=6. The minimum module size is 10. We merged similar modules, mergeCutHeight=0.25. We use Person correlation when we associate features with modules.). A scale-free network was constructed (R2=0.8850) (Figure 3A). Hierarchical clustering was performed to divide the network into modules. A total of 10 modules were obtained. In the figure, gray indicates no modules included (Figure 3B). The numbers of genes in each module are shown in Table 2. For several key T cell-related immune modules (P<0.05 with a correlation) (Figure 3C), gamma delta cells had a strong correlation with each module. The turquoise module, representing the smallest P value and the highest correlation, was selected as the key module and contained a total of 172 genes.
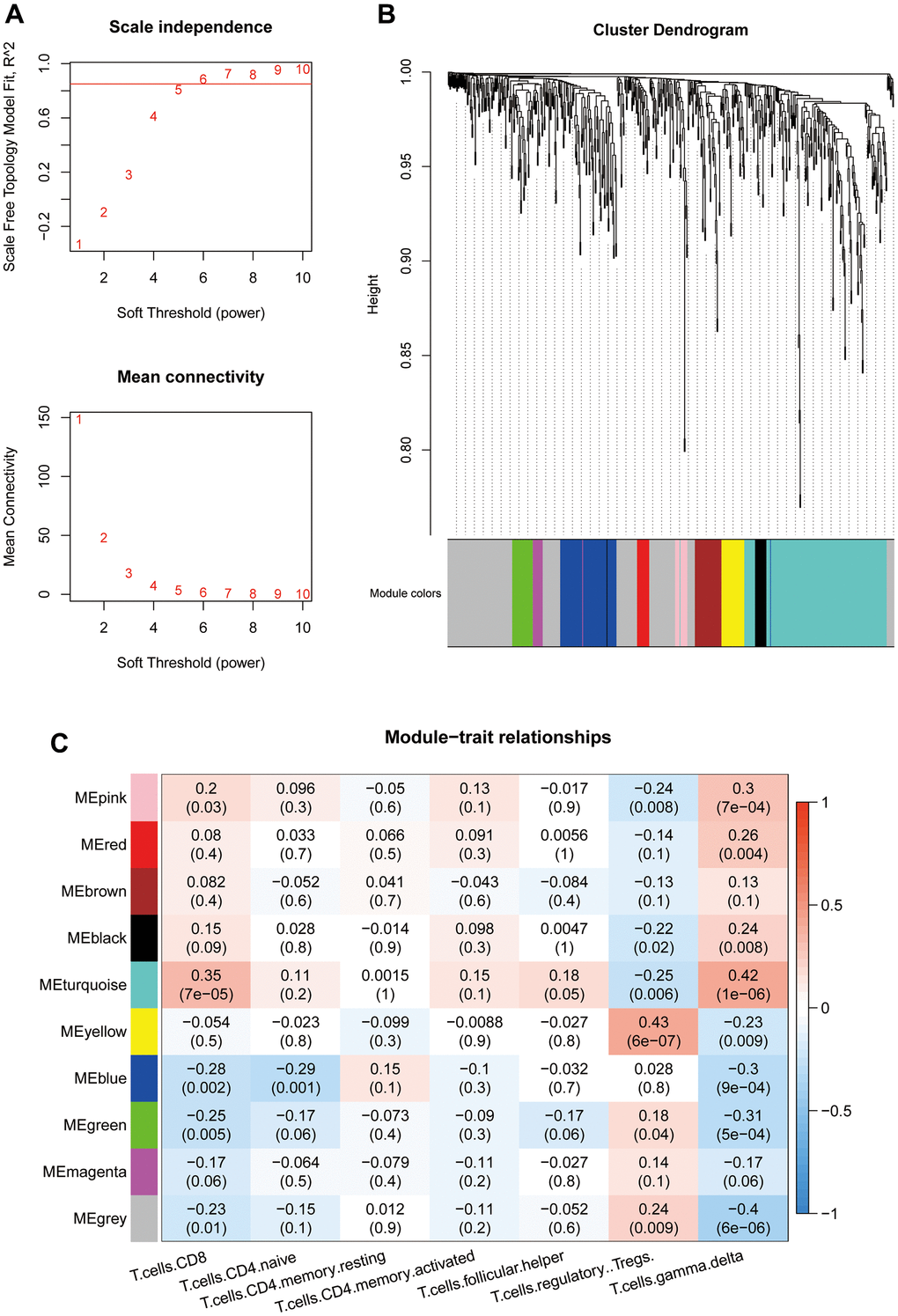
Figure 3. (A) Threshold selection for WGCNA network construction (β=6). (B) WGCNA network module classification (mergeCutHeight=0.25. The minimum module size is 10.). (C) Association between module eigenvectors and T cells. The first rows in each block are the correlation coefficients. Red indicates a positive correlation, and blue indicates a negative correlation. P values are provided in parentheses in the second row.
Table 2. WGCNA module statistics.
| Module | Number of genes |
| Pink | 15 |
| Red | 16 |
| Brown | 35 |
| Black | 16 |
| Turquoise | 172 |
| Yellow | 30 |
| Blue | 73 |
| Green | 27 |
| Magenta | 14 |
| Gray | 188 |
Screening methods and results display of hub genes
The 172 genes in the turquoise module were input into the STRING website to construct a PPI network. A total of 29 genes with a connectivity of ≥30 were selected as hub genes (Figure 4A). A total of 31 genes with MM ≥0.7 and GS ≥0.3 were selected as hub genes in the turquoise module (Figure 4B). Ten key genes were obtained after the intersection of key genes of the PPI and key genes of the turquoise module: CTSS, NCF2, MNDA, CCR2, TYROBP, LAPTM5, IGSF6, PTPRC, AIF1, and ITGAL (Figure 4C).
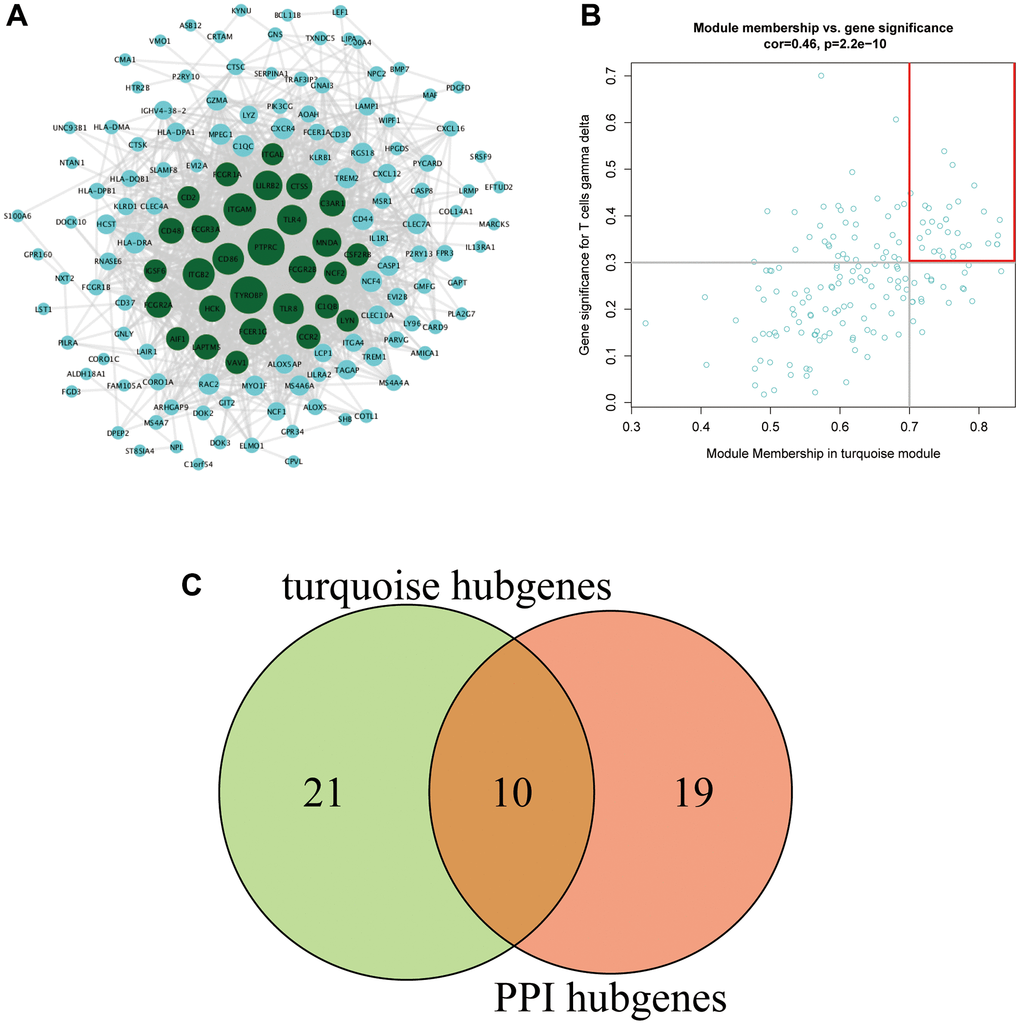
Figure 4. (A) PPI network of the turquoise module. The gene node size in the network reflects the degree of nodes, with a higher degree of gene connectivity corresponding to a larger font with which the gene is written. Green represents genes with a connectivity degree ≥30 (a total of 29 genes). (B) The key genes in the turquoise module. (C) Venn diagram of the key genes.
Correlation analysis between hub genes and clinical features
Among the 123 samples, 64 samples from GSE41177 and GSE79768 contained sex and age information. The median age (57 years) was used as the dichotomizing line for age grouping. Figure 5A shows that hub genes had no significant differences in expression between age groups. Figure 5B shows the expression of hub genes in different sexes. The expression levels of CTSS, IGSF6, CCR2, and PPTRC were significantly higher in females than in males.
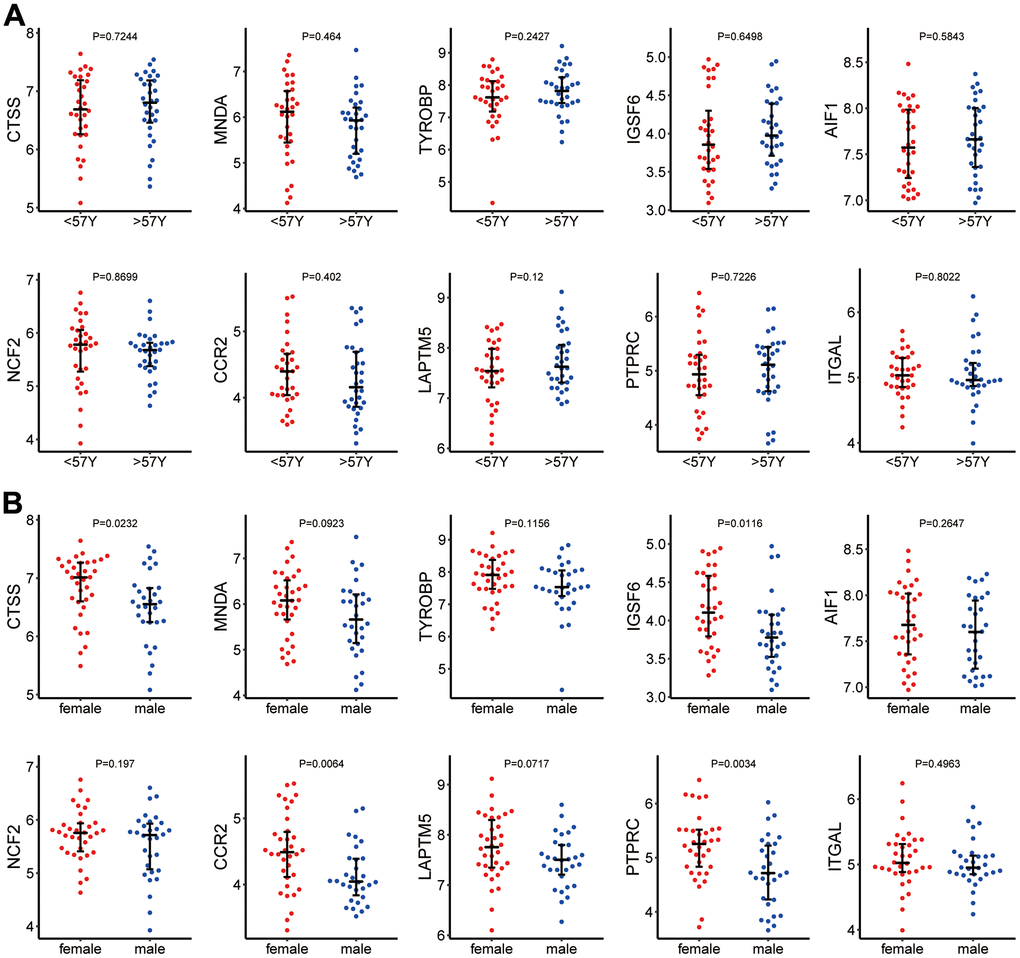
Figure 5. (A) Hub gene expression in different age groups. (B) Hub gene expression in different sexes.
Hub genes validation
The Seurat function was used to analyze GSE165838, and the expression distribution of 10 hub genes in the scRNA dataset was visualized (Figure 6).
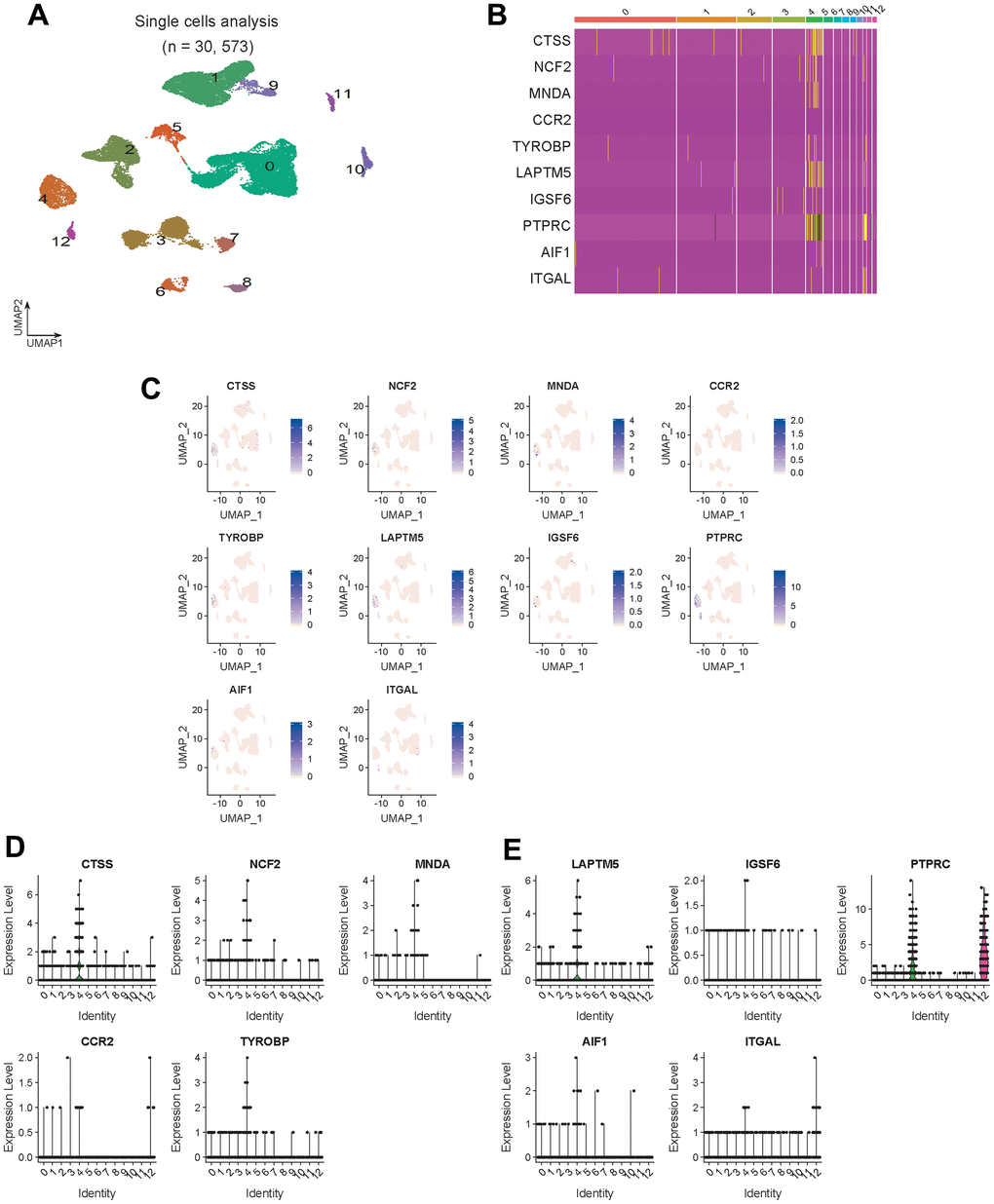
Figure 6. Hub gene expression validation. (A) Visualized cell cluster map of 30,573 single cells. (B) Heat map of 10 hub genes. (C) Expression distribution of 10 hub genes in the visual cell cluster. (D, E) Violin map of 10 hub genes showing the expression distribution of genes in different cell types.
Unsupervised clustering of hub genes
As shown in Figure 7A, we first determined the inflection point of SSE (i.e., the sum of the squares of the distances of all the points to the cluster center to which the points belonged). We searched for the optimal number of clusters K. When K=4, the SSE declined slowly, and all 123 samples were therefore classified into four categories by k-means clustering. Cluster 1 (blue), cluster 2 (red), cluster 3 (green), and cluster 4 (gray) had 27, 53, 17, and 26 samples, respectively (Figure 7A). The expression of the hub genes differed between clusters (Figure 7B). Therefore, the hub genes had important significance in disease classification. We next looked at the difference in immune infiltration of different types (Figure 7B). Cluster 1 and cluster 2 were significantly different from cluster 3 and cluster 4. We also examined the expression of hub genes in different types (Figure 7D) and found that the expression levels of all hub genes were ranked as cluster 1 < cluster 2 < cluster 4 < cluster 3.
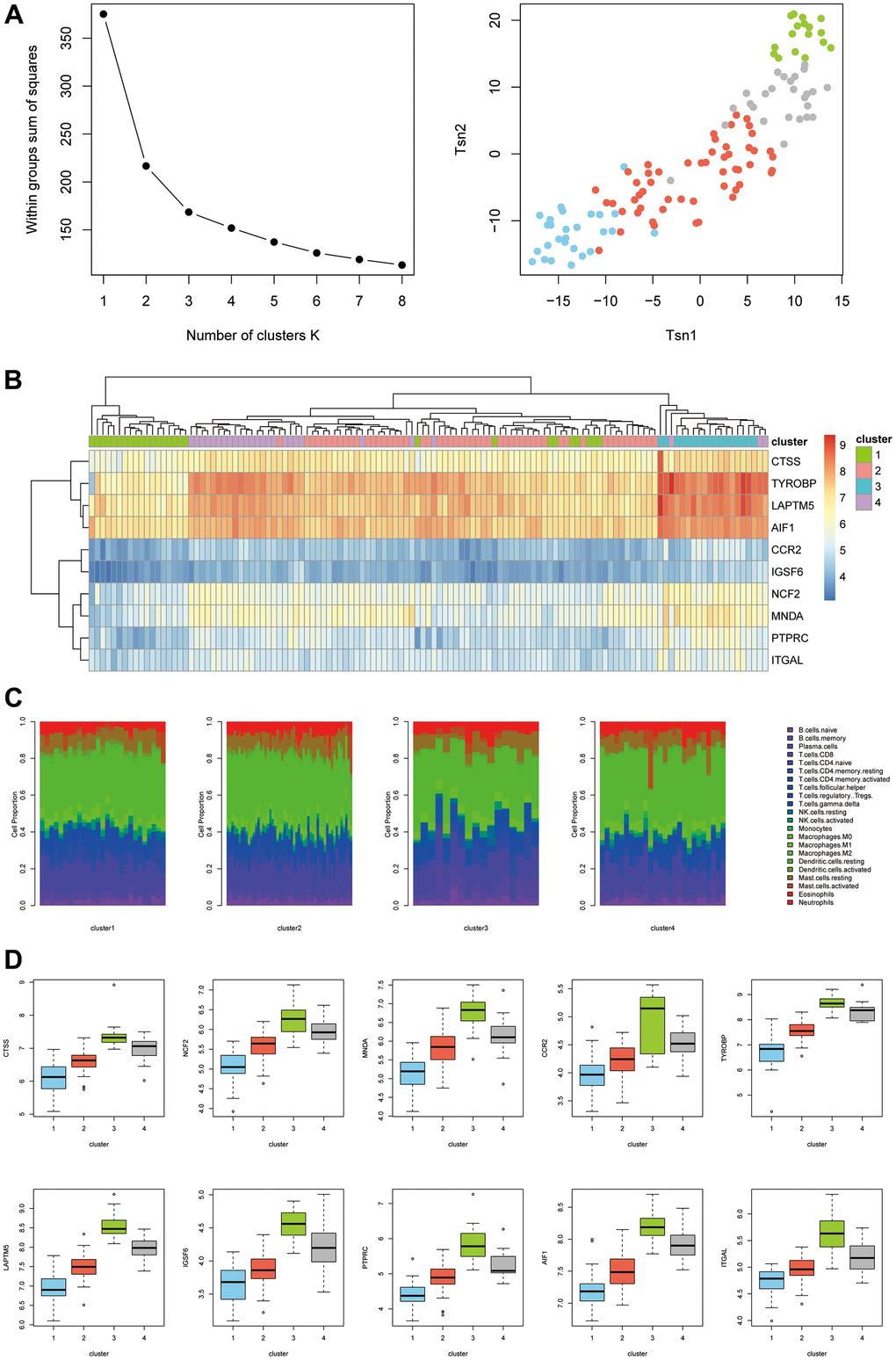
Figure 7. (A) K-means clustering classification. (B) Expression levels of different types of hub genes. (C) Differences in immune infiltration between different types. (D) Expression levels of hub genes in different types.
Discussion
T lymphocytes play a central role in cell-mediated immunity. A large number of studies have shown that T cells are closely related to the occurrence of many types of cardiovascular diseases [22, 23], but their roles in AF pathogenesis have not been well established. Therefore, this study focused on T cells. Notably, however, the results of our analysis show that most DEGs are not strongly associated with T cells. Therefore, the hub genes identified in this study not only affect T cell function but may also have other functions. Because of the complexity and diversity of inflammatory responses during the occurrence and development of AF, we believe that AF samples should be classified using hub genes because these genes can more accurately guide exploration of the pathogenesis of AF and the development of relevant immunotherapy methods.
Of the 10 hub genes discovered in this study, MNDA, TYROBP, LAPTM5, IGSF6, AIF1, and ITGAL have never been reported to be associated with AF. We have now elaborated the possible mechanisms by which these genes are associated with AF.
CTSS encodes cathepsin S (CTSS), which is mainly involved in the pathogenesis of a variety of cardiovascular diseases by mediating the degradation of extracellular matrix (ECM) proteins [24]. Under ischemic and high-fat diet (HFD) loads, CTSS participates in cardiovascular remodeling and the formation of atherosclerosis by mediating transforming growth factor-beta (TGF-β) and peroxisome proliferator- activated receptor gamma (PPAR-γ) or activating the p38 mitogen-activated protein kinase (MAPK) pathway [25–27]. Cardiovascular remodeling, especially atrial reconstruction, is an important basis for the formation and maintenance of AF. Therefore, CTSS likely promotes the proliferation and differentiation of fibroblasts by influencing TGF-β and other related pathways, resulting in atrial reconstruction. However, this hypothesis still requires further experimental verification.
Neutrophil cytosolic factor 2 (NCF2), also known as NOXA2, is a subunit of NADPH oxidase. The function of this oxidase is to produce O2- using NADPH as a substrate. The change in the NCF2 expression level significantly affects the level of reactive oxygen species (ROS), which is related to the occurrence and progression of cardiovascular diseases [28]. In addition, NCF2 is mainly expressed in neutrophils, an important site for ROS production. Existing evidence suggests a significant causal relationship between neutrophil activity and the onset of AF [29]. A mouse-based study showed that fibrosis in AF relies heavily on neutrophil activation [30]. The production of ROS can activate metalloproteinases, a key enzyme family involved in fibrosis [31]. Whether NCF2 participates in the occurrence and progression of AF relying on the aforementioned mechanisms remains to be further explored.
CCR2 encodes C-C motif chemokine receptor 2 (CCR2), which occurs ubiquitously on the surface of mononuclear macrophages. It binds CC motif chemokine ligand 2 (CCL2) and plays a major role in the recruitment of mononuclear macrophages to inflammatory sites [32]. The accumulation of macrophages is involved in TGF-β-mediated myocardial fibrosis. As a source of profibrotic-associated factors, macrophages can promote fibroblast proliferation and activation. Activated fibroblasts in turn promote structural remodeling of the atria by expressing fibrosis-related substances in large quantities [33, 34]. Macrophages also play an important role in electrical remodeling of the atria, which is mainly accomplished by their secretion of TNF-α. In summary, macrophages are involved in both the structural and electrical remodeling processes of the atria during the course of AF.
Myeloid nuclear differentiation antigen (MNDA) was first found to be expressed in macrophages and fibroblasts in inflammatory areas but not in noninflammatory areas. MNDA is highly expressed in macrophages in atherosclerotic plaques [35], but whether it is directly involved in the occurrence and progression of AF remains unknown. Given the role of macrophages in the occurrence and progression of AF, the inclusion of MNDA as a hub gene is presumably because MNDA is a characteristic marker of macrophages in inflammatory regions.
TYRO protein tyrosine kinase-binding protein (TYROBP) plays an important role in the pathogenesis of Alzheimer’s disease (AD). TYROBP seems to enhance the phagocytic activity of microglia, while timely clearing of apoptotic neurons and related metabolites is important for maintaining normal brain function. TYROBP can also mediate the anti-inflammatory response, thereby maintaining the immune balance function in the process of neuroinflammation. Triggering chronic inflammation results in neurodegenerative diseases, thereby increasing the risk of AD [36]. Interestingly, a series of clinical studies demonstrated that AF patients have a significantly increased risk of AD, but the cause of this phenomenon remains controversial [37–42]. Microglia in the brain rarely require peripheral supplementation in adulthood [43]. However, if microglial depletion occurs in the central nervous system under some special circumstances (e.g., inflammation, infection, injury, etc.), bone marrow–derived microglia are added [44]. AF patients may have chronic embolism or hemorrhage (macro- or micro), hypoperfusion, oxidative stress, and proinflammatory conditions [41]. Would this cause not only neuronal injury but also the depletion of microglia in the central nervous system? Is the change in the hub gene TYROBP also a hallmark of this pathological process? These questions remain to be further explored.
Allograft inflammatory factor 1 (AIF1) plays an important role in the occurrence and progression of atherosclerosis by stimulating the migration and proliferation of human smooth muscle cells and promoting the activation of macrophages [45]. AIF1 can induce the expression of fibrosis-related factors in normal fibroblasts [46]. AIF1 also induces monocytes to secrete IL-6 and enhance the chemotaxis of fibroblasts [47], thereby causing fibrosis. Given the important role of fibrosis in the occurrence and progression of AF, further exploring the potential mechanism of this gene during the occurrence and progression of AF would provide valuable data.
As a member of the protein tyrosine phosphatase (PTP) family, protein tyrosine phosphatase receptor type C (PTPRC) is also known as CD45 [48]. CD45 plays a role in regulating leukocyte adhesion, cytokine signaling, and immune receptor signaling (e.g., Fc, NK, Toll-like receptors) [48]. Accumulating evidence indicates that atrial tissue in AF patients is infiltrated with a large number of CD45+ cells [49–51]. Most of these CD45+ immune cells are CD68+ macrophages [52]. Given the important role of macrophages in the pathogenesis of AF, it makes sense that PPTRC would be a signature gene of AF. Integrin subunit alpha L (ITGAL), also known as CD11a, mainly functions through CD11a/CD18 integrins. It plays a role in the process of interleukocyte adhesion [53]. The role of CD11a in the pathogenesis of AF is unknown, but another integrin, CD11b/CD18, plays an important role in the pathogenesis of AF (ref?). The role of polymorphonuclear neutrophils (PMNs) in AF is mainly mediated by CD11b/CD18. The MPO released by PMNs directly activates more PMNs through the CD11b/CD18/MAPK pathway58, and the MPO released by PMNs furthermore acts directly on endothelial cells through cell junctions mediated by CD11b/CD18 integrins [54]. MPO can catalyze the oxidation of chloride to form hypochlorous acid, thereby activating MMP to participate in the atrial fibrosis process [29]. Whether CD11a/CD18 integrins play a similar role requires further investigation.
As a lysosome-related transmembrane receptor, lysosomal-associated protein transmembrane 5 (LAPTM5) is involved in the occurrence and progression of some neoplastic diseases [55]. Immunoglobulin superfamily member 6 (IGSF6) has been reported to be associated with inflammatory bowel disease [56]. Whether these two hub genes are involved in AF needs to be further explored.
Conclusions
In this study, we identified the hub genes associated with AF/immune infiltration through a series of analytical methods and tools, including CTSS, NCF2, MNDA, CCR2, TYROBP, LAPTM5, IGSF6, PTPRC, AIF1, and ITGAL. The expression levels of these genes did not differ between samples from AF patients at different ages. However, the mRNA levels of CTSS, IGSF6, CCR2, and PTPRC were significantly higher in males than in females. We subsequently verified the above hub genes in an external dataset and then confirmed differences in AF/immune infiltration based on the screened hub genes. This study is the first to classify AF into four types using immune infiltration differences. Due to the small amount of sample data that could be included in this study, our results may not represent the full clinical picture of AF/immune infiltration. In addition, due to the difficulty of acquiring human atrial specimens, we cannot conduct biological validation on the results. Taken together, the findings of this study provide new insight into the pathogenesis and progression of AF from the perspective of immune infiltration.
Supplementary Materials
Author Contributions
Tong Zou and Yuqing Tian designed the research; Yuqing Tian, Shiying Liu, Yanan Zhang and Xue Yu analyzed the data; Yuqing Tian, Peiyao Guo and Hongchao Zhang wrote the paper, which was revised by Tong Zou and Jiefu Yang. All authors have read and approved the final version of the manuscript.
Acknowledgments
We are grateful to Dr. Cheng-Gang Zou (Yunnan University, China) and Zhi-Gang Zhang (Yunnan University, China) for their critical reading of this manuscript.
Conflicts of Interest
The authors have no financial or nonfinancial conflicts of interest.
Funding
This work was supported by a grant from the Beijing Municipal Science and Technology Commission (D181100000218005).
Editorial Note
This corresponding author has a verified history of publications using a personal email address for correspondence.
References
- 1. Hu YF, Chen YJ, Lin YJ, Chen SA. Inflammation and the pathogenesis of atrial fibrillation. Nat Rev Cardiol. 2015; 12:230–43. https://doi.org/10.1038/nrcardio.2015.2 [PubMed]
- 2. Chugh SS, Havmoeller R, Narayanan K, Singh D, Rienstra M, Benjamin EJ, Gillum RF, Kim YH, McAnulty JH
Jr , Zheng ZJ, Forouzanfar MH, Naghavi M, Mensah GA, et al. Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 Study. Circulation. 2014; 129:837–47. https://doi.org/10.1161/CIRCULATIONAHA.113.005119 [PubMed] - 3. Ball J, Carrington MJ, McMurray JJ, Stewart S. Atrial fibrillation: profile and burden of an evolving epidemic in the 21st century. Int J Cardiol. 2013; 167:1807–24. https://doi.org/10.1016/j.ijcard.2012.12.093 [PubMed]
- 4. Lip GYH, Brechin CM, Lane DA. The global burden of atrial fibrillation and stroke: a systematic review of the epidemiology of atrial fibrillation in regions outside North America and Europe. Chest. 2012; 142:1489–98. https://doi.org/10.1378/chest.11-2888 [PubMed]
- 5. Guo Y, Lip GY, Apostolakis S. Inflammation in atrial fibrillation. J Am Coll Cardiol. 2012; 60:2263–70. https://doi.org/10.1016/j.jacc.2012.04.063 [PubMed]
- 6. Patel P, Dokainish H, Tsai P, Lakkis N. Update on the association of inflammation and atrial fibrillation. J Cardiovasc Electrophysiol. 2010; 21:1064–70. https://doi.org/10.1111/j.1540-8167.2010.01774.x [PubMed]
- 7. Boos CJ, Anderson RA, Lip GY. Is atrial fibrillation an inflammatory disorder? Eur Heart J. 2006; 27:136–49. https://doi.org/10.1093/eurheartj/ehi645 [PubMed]
- 8. Hu YF, Yeh HI, Tsao HM, Tai CT, Lin YJ, Chang SL, Lo LW, Tuan TC, Suenari K, Li CH, Chao TF, Chen SA. Electrophysiological correlation and prognostic impact of heat shock protein 27 in atrial fibrillation. Circ Arrhythm Electrophysiol. 2012; 5:334–40. https://doi.org/10.1161/CIRCEP.111.965996 [PubMed]
- 9. Jacob KA, Nathoe HM, Dieleman JM, van Osch D, Kluin J, van Dijk D. Inflammation in new-onset atrial fibrillation after cardiac surgery: a systematic review. Eur J Clin Invest. 2014; 44:402–28. https://doi.org/10.1111/eci.12237 [PubMed]
- 10. Smit MD, Maass AH, De Jong AM, Muller Kobold AC, Van Veldhuisen DJ, Van Gelder IC. Role of inflammation in early atrial fibrillation recurrence. Europace. 2012; 14:810–7. https://doi.org/10.1093/europace/eur402 [PubMed]
- 11. Aviles RJ, Martin DO, Apperson-Hansen C, Houghtaling PL, Rautaharju P, Kronmal RA, Tracy RP, Van Wagoner DR, Psaty BM, Lauer MS, Chung MK. Inflammation as a risk factor for atrial fibrillation. Circulation. 2003; 108:3006–10. https://doi.org/10.1161/01.CIR.0000103131.70301.4F [PubMed]
- 12. Marott SC, Nordestgaard BG, Zacho J, Friberg J, Jensen GB, Tybjaerg-Hansen A, Benn M. Does elevated C-reactive protein increase atrial fibrillation risk? A Mendelian randomization of 47,000 individuals from the general population. J Am Coll Cardiol. 2010; 56:789–95. https://doi.org/10.1016/j.jacc.2010.02.066 [PubMed]
- 13. Conen D, Ridker PM, Everett BM, Tedrow UB, Rose L, Cook NR, Buring JE, Albert CM. A multimarker approach to assess the influence of inflammation on the incidence of atrial fibrillation in women. Eur Heart J. 2010; 31:1730–6. https://doi.org/10.1093/eurheartj/ehq146 [PubMed]
- 14. Zhang Z, Zhang C, Wang H, Zhao J, Liu L, Lee J, He Y, Zheng Q. n-3 polyunsaturated fatty acids prevents atrial fibrillation by inhibiting inflammation in a canine sterile pericarditis model. Int J Cardiol. 2011; 153:14–20. https://doi.org/10.1016/j.ijcard.2010.08.024 [PubMed]
- 15. Marcus GM, Smith LM, Glidden DV, Wilson E, McCabe JM, Whiteman D, Tseng ZH, Badhwar N, Lee BK, Lee RJ, Scheinman MM, Olgin JE. Markers of inflammation before and after curative ablation of atrial flutter. Heart Rhythm. 2008; 5:215–21. https://doi.org/10.1016/j.hrthm.2007.10.007 [PubMed]
- 16. Chung MK, Martin DO, Sprecher D, Wazni O, Kanderian A, Carnes CA, Bauer JA, Tchou PJ, Niebauer MJ, Natale A, Van Wagoner DR. C-reactive protein elevation in patients with atrial arrhythmias: inflammatory mechanisms and persistence of atrial fibrillation. Circulation. 2001; 104:2886–91. https://doi.org/10.1161/hc4901.101760 [PubMed]
- 17. Shiroshita-Takeshita A, Brundel BJ, Lavoie J, Nattel S. Prednisone prevents atrial fibrillation promotion by atrial tachycardia remodeling in dogs. Cardiovasc Res. 2006; 69:865–75. https://doi.org/10.1016/j.cardiores.2005.11.028 [PubMed]
- 18. Davis S, Meltzer PS. GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics. 2007; 23:1846–7. https://doi.org/10.1093/bioinformatics/btm254 [PubMed]
- 19. Leek JT, Johnson WE, Parker HS, Fertig EJ, Jaffe AE, Zhang Y, Storey JD. Leonardo Collado Torres: sva: Surrogate Variable Analysis. In. 2019.
- 20. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. https://doi.org/10.1093/nar/gkv007 [PubMed]
- 21. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012; 16:284–7. https://doi.org/10.1089/omi.2011.0118 [PubMed]
- 22. Witztum JL, Lichtman AH. The influence of innate and adaptive immune responses on atherosclerosis. Annu Rev Pathol. 2014; 9:73–102. https://doi.org/10.1146/annurev-pathol-020712-163936 [PubMed]
- 23. Lazaros G, Karavidas A, Spyropoulou M, Tsiachris D, Halapas A, Zacharoulis A, Arapi S, Matzaraki V, Papadopoulos K, Korres D, Iniotaki A, Pyrgakis V, Stefanadis C. The role of the immunogenetic background in the development and recurrence of acute idiopathic pericarditis. Cardiology. 2011; 118:55–62. https://doi.org/10.1159/000324309 [PubMed]
- 24. Wu H, Du Q, Dai Q, Ge J, Cheng X. Cysteine Protease Cathepsins in Atherosclerotic Cardiovascular Diseases. J Atheroscler Thromb. 2018; 25:111–23. https://doi.org/10.5551/jat.RV17016 [PubMed]
- 25. Li X, Cheng XW, Hu L, Wu H, Guo-Ping, Hao CN, Jiang H, Zhu E, Huang Z, Inoue A, Sasaki T, Du Q, Takeshita K, et al. Cathepsin S activity controls ischemia-induced neovascularization in mice. Int J Cardiol. 2015; 183:198–208. https://doi.org/10.1016/j.ijcard.2015.01.058 [PubMed]
- 26. Chen H, Wang J, Xiang MX, Lin Y, He A, Jin CN, Guan J, Sukhova GK, Libby P, Wang JA, Shi GP. Cathepsin S-mediated fibroblast trans-differentiation contributes to left ventricular remodelling after myocardial infarction. Cardiovasc Res. 2013; 100:84–94. https://doi.org/10.1093/cvr/cvt158 [PubMed]
- 27. Wu H, Cheng XW, Hu L, Takeshita K, Hu C, Du Q, Li X, Zhu E, Huang Z, Yisireyili M, Zhao G, Piao L, Inoue A, et al. Cathepsin S Activity Controls Injury-Related Vascular Repair in Mice via the TLR2-Mediated p38MAPK and PI3K-Akt/p-HDAC6 Signaling Pathway. Arterioscler Thromb Vasc Biol. 2016; 36:1549–57. https://doi.org/10.1161/ATVBAHA.115.307110 [PubMed]
- 28. O’Neill S, Brault J, Stasia MJ, Knaus UG. Genetic disorders coupled to ROS deficiency. Redox Biol. 2015; 6:135–56. https://doi.org/10.1016/j.redox.2015.07.009 [PubMed]
- 29. Liu Y, Shi Q, Ma Y, Liu Q. The role of immune cells in atrial fibrillation. J Mol Cell Cardiol. 2018; 123:198–208. https://doi.org/10.1016/j.yjmcc.2018.09.007 [PubMed]
- 30. Rudolph V, Andrié RP, Rudolph TK, Friedrichs K, Klinke A, Hirsch-Hoffmann B, Schwoerer AP, Lau D, Fu X, Klingel K, Sydow K, Didié M, Seniuk A, et al. Myeloperoxidase acts as a profibrotic mediator of atrial fibrillation. Nat Med. 2010; 16:470–4. https://doi.org/10.1038/nm.2124 [PubMed]
- 31. Friedrichs K, Baldus S, Klinke A. Fibrosis in Atrial Fibrillation - Role of Reactive Species and MPO. Front Physiol. 2012; 3:214. https://doi.org/10.3389/fphys.2012.00214 [PubMed]
- 32. França CN, Izar MCO, Hortêncio MNS, do Amaral JB, Ferreira CES, Tuleta ID, Fonseca FAH. Monocyte subtypes and the CCR2 chemokine receptor in cardiovascular disease. Clin Sci (Lond). 2017; 131:1215–24. https://doi.org/10.1042/CS20170009 [PubMed]
- 33. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012; 18:1028–40. https://doi.org/10.1038/nm.2807 [PubMed]
- 34. Desmoulière A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993; 122:103–11. https://doi.org/10.1083/jcb.122.1.103 [PubMed]
- 35. Briggs RC, Atkinson JB, Miranda RN. Variable expression of human myeloid specific nuclear antigen MNDA in monocyte lineage cells in atherosclerosis. J Cell Biochem. 2005; 95:293–301. https://doi.org/10.1002/jcb.20435 [PubMed]
- 36. Ma J, Jiang T, Tan L, Yu JT. TYROBP in Alzheimer’s disease. Mol Neurobiol. 2015; 51:820–6. https://doi.org/10.1007/s12035-014-8811-9 [PubMed]
- 37. Bunch TJ, Weiss JP, Crandall BG, May HT, Bair TL, Osborn JS, Anderson JL, Muhlestein JB, Horne BD, Lappe DL, Day JD. Atrial fibrillation is independently associated with senile, vascular, and Alzheimer’s dementia. Heart Rhythm. 2010; 7:433–7. https://doi.org/10.1016/j.hrthm.2009.12.004 [PubMed]
- 38. de Bruijn RF, Heeringa J, Wolters FJ, Franco OH, Stricker BH, Hofman A, Koudstaal PJ, Ikram MA. Association Between Atrial Fibrillation and Dementia in the General Population. JAMA Neurol. 2015; 72:1288–94. https://doi.org/10.1001/jamaneurol.2015.2161 [PubMed]
- 39. Rollo J, Knight S, May HT, Anderson JL, Muhlestein JB, Bunch TJ, Carlquist J. Incidence of dementia in relation to genetic variants at PITX2, ZFHX3, and ApoE ε4 in atrial fibrillation patients. Pacing Clin Electrophysiol. 2015; 38:171–7. https://doi.org/10.1111/pace.12537 [PubMed]
- 40. Jacobs V, Woller SC, Stevens S, May HT, Bair TL, Anderson JL, Crandall BG, Day JD, Johanning K, Long Y, Mallender C, Olson JL, Osborn JS, et al. Time outside of therapeutic range in atrial fibrillation patients is associated with long-term risk of dementia. Heart Rhythm. 2014; 11:2206–13. https://doi.org/10.1016/j.hrthm.2014.08.013 [PubMed]
- 41. Poggesi A, Inzitari D, Pantoni L. Atrial Fibrillation and Cognition: Epidemiological Data and Possible Mechanisms. Stroke. 2015; 46:3316–21. https://doi.org/10.1161/STROKEAHA.115.008225 [PubMed]
- 42. Jacobs V, Woller SC, Stevens SM, May HT, Bair TL, Crandall BG, Cutler M, Day JD, Weiss JP, Osborn JS, Mallender C, Anderson JL, Bunch TJ. Percent Time With a Supratherapeutic INR in Atrial Fibrillation Patients Also Using an Antiplatelet Agent Is Associated With Long-Term Risk of Dementia. J Cardiovasc Electrophysiol. 2015; 26:1180–6. https://doi.org/10.1111/jce.12776 [PubMed]
- 43. Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007; 10:1538–43. https://doi.org/10.1038/nn2014 [PubMed]
- 44. Waisman A, Ginhoux F, Greter M, Bruttger J. Homeostasis of Microglia in the Adult Brain: Review of Novel Microglia Depletion Systems. Trends Immunol. 2015; 36:625–36. https://doi.org/10.1016/j.it.2015.08.005 [PubMed]
- 45. Tian Y, Kelemen SE, Autieri MV. Inhibition of AIF-1 expression by constitutive siRNA expression reduces macrophage migration, proliferation, and signal transduction initiated by atherogenic stimuli. Am J Physiol Cell Physiol. 2006; 290:C1083–91. https://doi.org/10.1152/ajpcell.00381.2005 [PubMed]
- 46. Del Galdo F, Jiménez SA. T cells expressing allograft inflammatory factor 1 display increased chemotaxis and induce a profibrotic phenotype in normal fibroblasts in vitro. Arthritis Rheum. 2007; 56:3478–88. https://doi.org/10.1002/art.22877 [PubMed]
- 47. Yamamoto A, Ashihara E, Nakagawa Y, Obayashi H, Ohta M, Hara H, Adachi T, Seno T, Kadoya M, Hamaguchi M, Ishino H, Kohno M, Maekawa T, Kawahito Y. Allograft inflammatory factor-1 is overexpressed and induces fibroblast chemotaxis in the skin of sclerodermatous GVHD in a murine model. Immunol Lett. 2011; 135:144–50. https://doi.org/10.1016/j.imlet.2010.10.015 [PubMed]
- 48. Johnson P, Samarakoon A, Saunders AE, Harder KW. CD45 (PTPRC). In: Encyclopedia of Signaling Molecules. Ed S.Choi. Springer International Publishing, Cham. 2018; 912–9. https://doi.org/10.1007/978-3-319-67199-4_34
- 49. Wu L, Emmens RW, van Wezenbeek J, Stooker W, Allaart CP, Vonk ABA, van Rossum AC, Niessen HWM, Krijnen PAJ. Atrial inflammation in different atrial fibrillation subtypes and its relation with clinical risk factors. Clin Res Cardiol. 2020; 109:1271–81. https://doi.org/10.1007/s00392-020-01619-8 [PubMed]
- 50. Mitrofanova LB, Orshanskaya V, Ho SY, Platonov PG. Histological evidence of inflammatory reaction associated with fibrosis in the atrial and ventricular walls in a case-control study of patients with history of atrial fibrillation. Europace. 2016; 18:iv156–62. https://doi.org/10.1093/europace/euw361 [PubMed]
- 51. Chen MC, Chang JP, Liu WH, Yang CH, Chen YL, Tsai TH, Wang YH, Pan KL. Increased inflammatory cell infiltration in the atrial myocardium of patients with atrial fibrillation. Am J Cardiol. 2008; 102:861–5. https://doi.org/10.1016/j.amjcard.2008.05.038 [PubMed]
- 52. Yamashita T, Sekiguchi A, Iwasaki YK, Date T, Sagara K, Tanabe H, Suma H, Sawada H, Aizawa T. Recruitment of immune cells across atrial endocardium in human atrial fibrillation. Circ J. 2010; 74:262–70. https://doi.org/10.1253/circj.cj-09-0644 [PubMed]
- 53. DesJardin LE, Kaufman TM, Potts B, Kutzbach B, Yi H, Schlesinger LS. Mycobacterium tuberculosis-infected human macrophages exhibit enhanced cellular adhesion with increased expression of LFA-1 and ICAM-1 and reduced expression and/or function of complement receptors, FcgammaRII and the mannose receptor. Microbiology (Reading). 2002; 148:3161–71. https://doi.org/10.1099/00221287-148-10-3161 [PubMed]
- 54. Jerke U, Rolle S, Purfürst B, Luft FC, Nauseef WM, Kettritz R. β2 integrin-mediated cell-cell contact transfers active myeloperoxidase from neutrophils to endothelial cells. J Biol Chem. 2013; 288:12910–9. https://doi.org/10.1074/jbc.M112.434613 [PubMed]
- 55. Nuylan M, Kawano T, Inazawa J, Inoue J. Down-regulation of LAPTM5 in human cancer cells. Oncotarget. 2016; 7:28320–8. https://doi.org/10.18632/oncotarget.8614 [PubMed]
- 56. Bates EE, Kissenpfennig A, Péronne C, Mattei MG, Fossiez F, Malissen B, Lebecque S. The mouse and human IGSF6 (DORA) genes map to the inflammatory bowel disease 1 locus and are embedded in an intron of a gene of unknown function. Immunogenetics. 2000; 52:112–20. https://doi.org/10.1007/s002510000259 [PubMed]