



Introduction
Mitochondria are acknowledged as critical organelles responsible for the production of the energetic currency, Adenosine Triphosphate (ATP). Although often denoted as the “powerhouses of the cell” for their efficient metabolic capacity, mitochondria also influence the overall health of skeletal muscle, among other organs [1, 2]. Skeletal muscle, essential for overall motor function and whole-body metabolic control, relies on the oxidative capacity of mitochondria to adapt and maintain optimal health and function [3]. In energetically stimulating conditions, such as exercise training, the mitochondrial network undergoes beneficial adaptations, which in turn contributes to the improved endurance capacity of skeletal muscle [4]. On the other hand, the lack of physical activity often observed during prolonged bedrest and aging, dictates deleterious aberrations in the mitochondrial reticulum, such as reductions in content, and elevations in oxidative stress, thereby giving rise to a decline in oxidative capacity and consequently muscle health [5–9]. With the natural aging process, a hallmark event that typically occurs is the progressional loss of skeletal muscle mass, commonly denoted as sarcopenia [9]. Many decades of research have encapsulated the dramatic physiological changes that occur during aging; however, a common point of concern is the decline in mitochondrial health [10–13]. Since skeletal muscle is an integral facet in maintaining essential whole-body functioning, such age-related declines are therefore associated with deficits in the overall quality of life [14]. With that being said, there is an evolving need to preserve muscle health with age and given the central role of mitochondria in contributing to this process, fully elucidating how these organelles can be conserved with age is of critical significance.
Given the central importance of mitochondria in physiology and thus aging, a number of signaling cascades, termed mitochondrial quality control (MQC) processes, have been identified as crucial first responders to mitochondrial insults/dysfunction to thereby promote organelle maintenance and recovery, whilst undergoing metabolic adaptations [14]. Such MQC processes can be broken down into distinct pathways, such as biogenesis (synthesis of new mitochondria), mitophagy (degradation of dysfunctional mitochondria), and the transient activation of stress response mechanisms, notably the Integrated Stress Response (ISR) and the mitochondrial Unfolded Protein Response (UPRmt) [14]. Although a large focus has been placed on improving both biogenesis and mitophagy with age, very little work has assessed the overarching importance of stress response mechanisms in this context.
The ISR is an adaptive program that is activated in the face of various mitochondrial stressors, such as oxidative stress and aggregation of misfolded proteins [15]. This homeostatic mechanism, in mammalians cells, is responsible for coordinating the specific induction of the UPRmt, a highly specialized retrograde signaling pathway that enhances the expression of several mitochondrial chaperones, proteases and antioxidants [16–18]. This mitohormetic pathway was fully characterized in the nematode C. elegans, driven by the transcription factor ATFS-1 [19]. During the initial foray into unravelling the precise mechanisms of the UPRmt in C. elegans, it was reported that the activation of this mitochondrial regulatory process was beneficial and enhanced nematode longevity and health, therefore serving a greater role in cellular homeostasis within aging [20–22]. In mammalian cells, an ATFS-1 homologue, Activating Transcription Factor 5 (ATF5), has been shown to be required for the activation of this response during mitochondrial insults [23]. However, in the context of mammalian aging, it is not known whether this program and thus ATF5 is required to maintain optimal muscle health and function via the preservation of mitochondrial homeostasis. Although recent evidence from our laboratory has suggested the role of ATF5 in maintaining mitochondrial and skeletal muscle health basally in young mice [24], no work has focused on elucidating the importance of this transcription factor during the process of aging.
Therefore, to investigate the importance and relationship of this transcription factor, and thus mitochondrial stress response regulation in mediating muscle and mitochondrial health with age, we utilized an ATF5 KO model with both young (4-6 months) and middle-aged (14-16 months) mice, which will be denoted as the “aged” group in the study, representative of early phase sarcopenia. We assessed various muscle physiology parameters via the use of an acute in situ contractile paradigm. Our findings indicate an intricate importance of ATF5 in maintaining skeletal muscle health with age, as well as the altered regulation of mitochondrial quality control processes.
Results
ATF5 contributes to the age-related decline in muscle mass but is required to maintain muscle endurance performance
Parallel to previous findings [24], ATF5 KO mice displayed a significantly lower in body weight (g) (Figure 1A, P<0.05), a phenotype that is amplified with age, primarily reflected in the reduction in epidydimal fat mass (Supplementary Figure 1A). In particular, there was a 40% lower body weight in aged ATF5 KO mice compared to controls (Figure 1A, P<0.0001). Both hindlimb and cardiac muscle weight did not differ in either the presence or absence of ATF5 in young mice (Figure 1B–1E). Interestingly, with age, the lack of ATF5 prevented the age-related decline in hindlimb muscle weight. Tibalis Anterior (TA), Gastrocnemius (Gastroc), and Soleus muscle weights (mg/g) were significantly greater by approximately 22% in aged ATF5 KO mice, relative to WT controls (Figure 1B–1D, P<0.05, P<0.01). Moreover, heart weight was also elevated in aged ATF5 KO mice (Figure 1E, P<0.05), potentially emphasizing the role of ATF5 in mediating both skeletal and cardiac muscle mass regulation. To ascertain whether ATF5 was required to maintain muscle endurance capacity in both young and aged mice, we performed an acute contractile activity protocol in which the lower hindlimb muscles underwent a fatigue protocol for 9-minutes at increasing intensities. Young ATF5 KO mice did not display any differences in muscle fatiguability relative to WT controls (Figure 1F). However, with age, the absence of ATF5 negatively altered endurance capacity, as aged ATF5 KO mice displayed a significant reduction of muscle force and greater muscle fatigue by ~20% in comparison to aged WT controls over the course of the protocol (Figure 1G, P<0.01, P<0.001). Thus, from these data, it is evident that ATF5 negatively regulates skeletal muscle mass and is required to maintain overall muscle endurance capacity with increasing age.
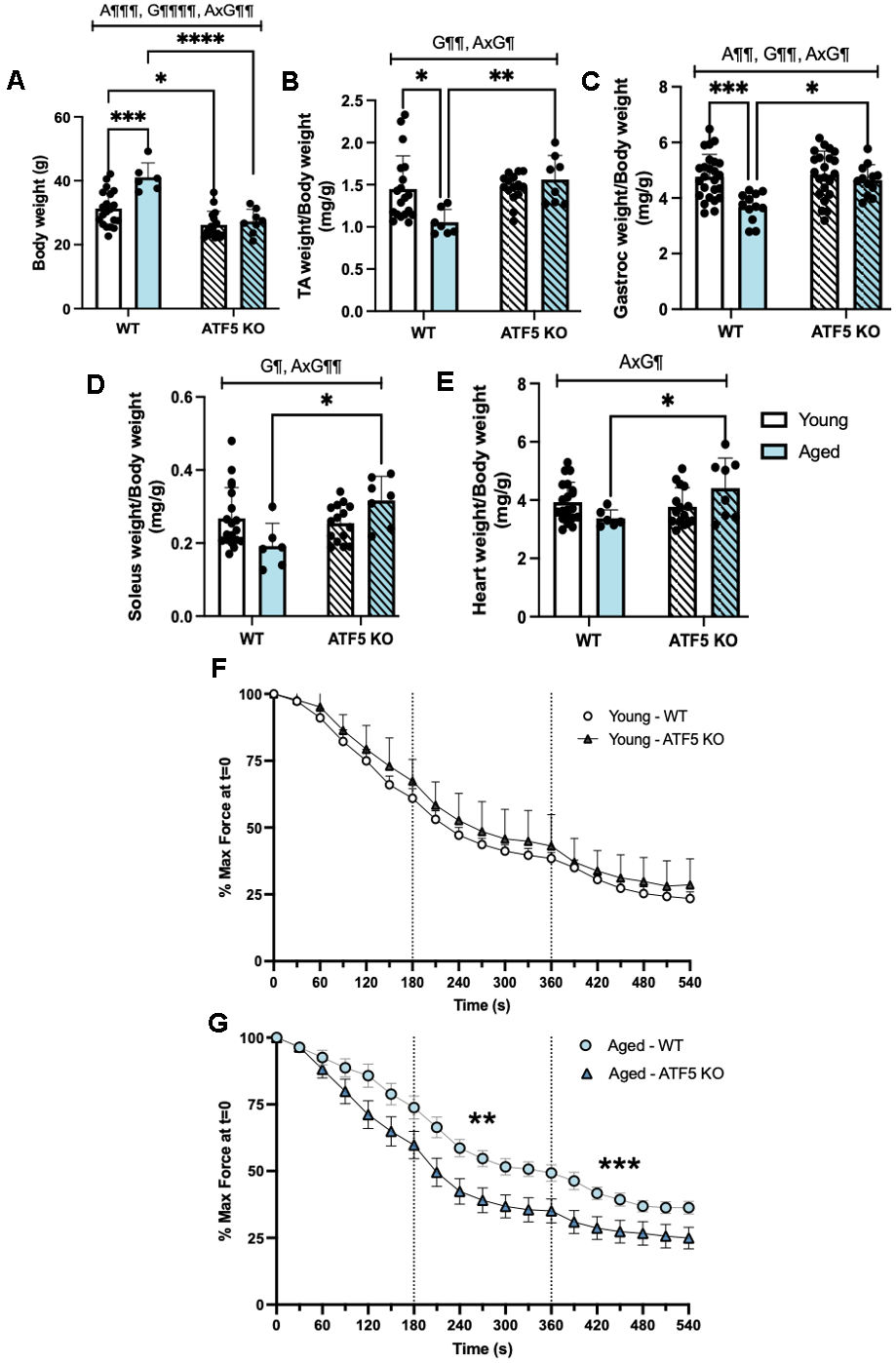
Figure 1. The lack of ATF5 rescues the age-related decline in muscle mass but reduces overall endurance performance. Phenotypic characteristics between genotypes and age were assessed, including (A) body weight (g), and muscle weights, (B) TA, (C) Gastrocnemius, (D) Soleus, (E) Heart weight, normalized for body weight (mg/g). Muscle endurance was assessed over the course of an acute contractile activity 9-minute in situ protocol, where the gastrocnemius muscle was stimulated at 0.25, 0.5 and 1 tetanic contractions per second (TPS). Muscle endurance, expressed as the percentage of max/initial force (t=0) in both (F) young and (G) aged mice, comparing phenotypic differences between genotypes. A¶¶ P<0.01, A¶¶¶ P<0.001, main effect of age; G¶ P<0.05, G¶¶ P<0.01, G¶¶¶¶ P<0.0001, main effect of genotype; AxG¶ P<0.05, AxG¶¶ P<0.01, interaction between age and genotype, Two-way ANOVA (n=6-20/group); Tukey’s post-hoc analyses, Young WT vs. Young ATF5 KO, Aged WT vs. Aged ATF5 KO, Young WT vs. Aged WT, Young ATF5 KO vs. Aged ATF5 KO, Young ATF5 KO vs. Aged WT, Young WT vs. Aged ATF5 KO. Tukey’s post-hoc: *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Data are means ± SD.
ATF5 regulates the expression of atrophy-mediating pathways, contributing to the altered regulation of muscle mass with age
The preservation of muscle weight observed in our aged (14-16 months) ATF5 KO mice prompted further exploration on the relationship between ATF5 and muscle atrophy programs. Thus, we assessed the protein expression of various Ubiquitin-Proteasome System (UPS) and other known age-related regulators of muscle atrophy in our model. We first evaluated the expression of p53, a well-known mediator of muscle atrophy and indicator of a senescent phenotype [25]. We observed a 4-fold increase in p53 expression in aged WT mice compared with young WT mice (Figure 2B, P<0.01). In the absence of ATF5, however, this upregulation was not observed, owing to an interaction between age and genotype (P=0.06). In addition, direct markers of UPS system activation, the E3 ubiquitin ligases, Atrogin-1 and MuRF1, displayed similar trends. There was a 2-fold elevation in these proteins with age, and this augmentation was blunted in the absence of ATF5 (Figure 2C, 2D, P<0.0001, P=0.05, P=0.07). To further fortify the regulatory capacity of ATF5 and the UPS during aging, we also assessed the expression of the 20S Proteosome Subunit PSMB4 which is required for the assembly and catalytic capacity of the 26S Proteasome [26]. The main effect of genotype on the expression of this subunit was observed, regardless of age (Figure 2E, P<0.05). This finding solidifies the role of ATF5 in dictating the capacity of the proteasomal system, an event that becomes more critical with age. We next assessed markers of muscle atrophy known to be regulated by ATF4 and also recognized as muscle senescent markers [27]. Both Gadd45α and p21 were upregulated with age (Figure 2F, 2G, P<0.01, P<0.05) but were not altered in ATF5 KO mice. In addition to the proteins involved in protein degradation, we assessed the overall drive for muscle protein synthesis via the expression of P/T-4E-BP1 levels, as an indirect indicator of mTORC1 activity. The level of P/T-4E-BP1 was differentially altered with age and was improved in the absence of ATF5 (Figure 2H, P<0.05). Thus, these data combined suggest that the absence of ATF5 alters the ratio between muscle protein degradation and synthesis with increasing age.
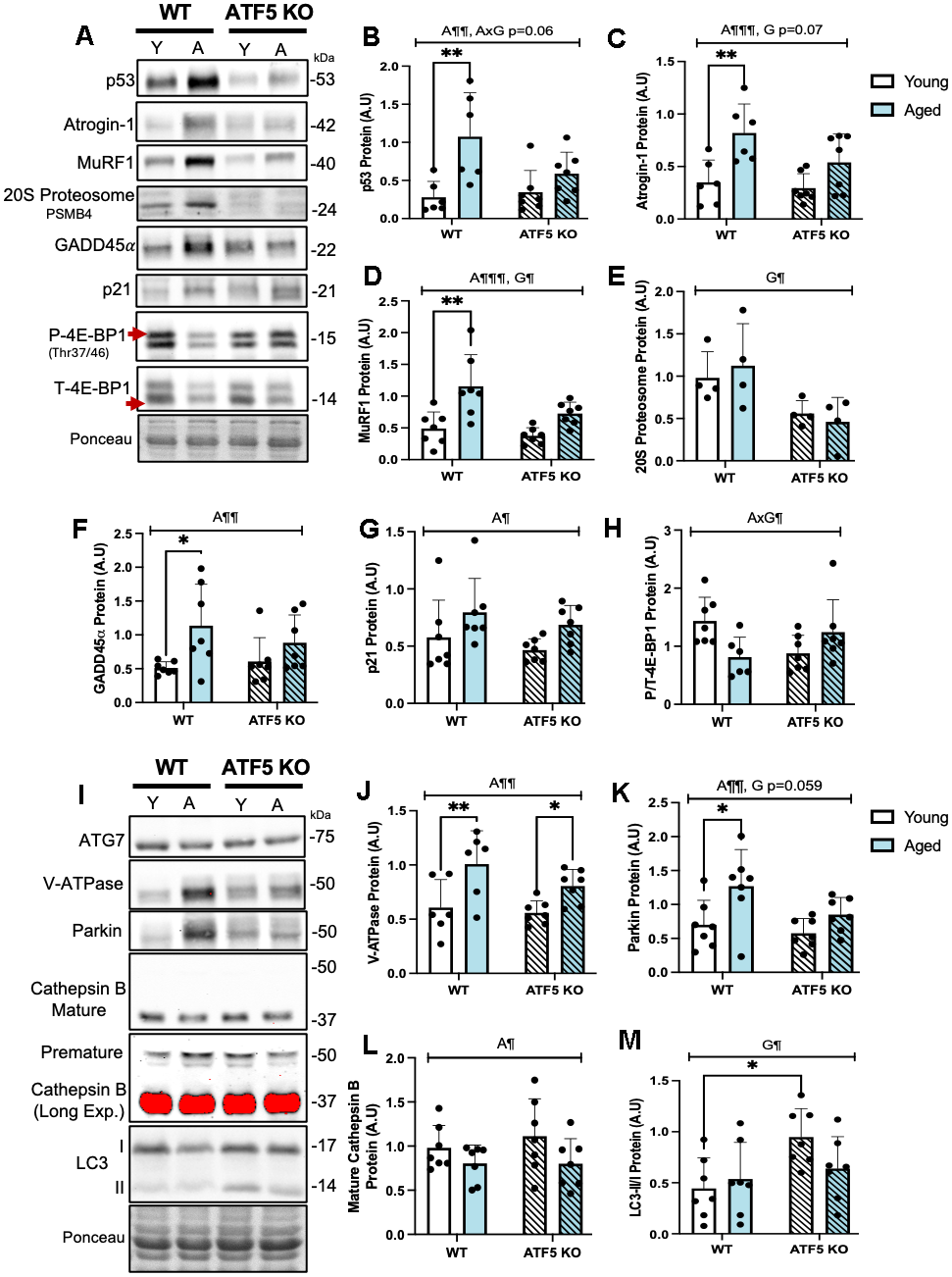
Figure 2. ATF5 regulates the expression of UPS, Autophagy-Lysosome System and other atrophy markers basally and with age. (A) Immunoblots of p53, Atrogin-1, MuRF1, 20S Proteasome subunit PSMB4, GADD45α, p21, Phosphorylated 4E-BP1 and Total 4E-BP1, corrected to Ponceau, with respective quantifications (B–H). Immunoblots of ATG7, V-ATPase, Parkin, Cathepsin B (mature (37kDa) and immature (50 kDa) and LC3I/II, with respective quantifications, corrected to Ponceau (I–M). Mature Cathepsin B was overexposed (long exposure: red saturation) in order to detect the levels of immature Cathepsin B. Mature Cathepsin B was quantified from the unsaturated/normal exposure immunoblot. The approximate molecular weights of proteins are indicated with a hash bar in kilodaltons (kDa). (n=4-7/group). A¶ P<0.05, A¶¶ P<0.01, A¶¶¶ P<0.001, main effect of age; G¶ P<0.05, main effect of genotype; AxG¶ P<0.05, interaction between age and genotype, Two-way ANOVA; Tukey’s post-hoc analyses, Young WT vs. Young ATF5 KO, Aged WT vs. Aged ATF5 KO, Young WT vs. Aged WT, Young ATF5 KO vs. Aged ATF5 KO, Young ATF5 KO vs. Aged WT, Young WT vs. Aged ATF5 KO. Tukey’s post-hoc: *P<0.05, **P<0.01. Y, Young; A, Aged. Data are means ± SD.
We next examined the role of ATF5 in maintaining the Autophagy-Lysosomal System (ALS) during aging, as another pathway potentially involved in the atrophy process. The protein V-ATPase, required for the acidification of the lysosomal lumen, was elevated with age and this elevation was slightly influenced by the absence of ATF5 (Figure 2J, P<0.01). ATG7, required for the initiation of autophagosome formation, remained unchanged (Figure 2I). Conversely, the mitophagy protein Parkin was elevated 2-fold with age, but this increase was blunted in ATF5 KO mice (Figure 2K, P<0.01, P=0.059). The mature: immature ratio of the lysosomal protease Cathepsin B was downregulated with age in both genotypes (Figure 2L, P<0.05). Interestingly, the ratio of LC3II/I was approximately 2-fold greater in ATF5 KO mice (Figure 2M, P<0.05), potentially illustrating a level of autophagy-lysosomal dysregulation. Thus, the lack of ATF5 induces a discoordination between muscle protein homeostasis mechanisms with age, suggesting an important role of ATF5 in regulating muscle proteostasis, with implications for the maintenance of muscle mass with age.
The lack of ATF5 influences the normal expression of ISR/UPRmt and antioxidant markers
As ATF5 is denoted as a regulator of the Integrated Stress Response (ISR) and the mitochondrial Unfolded Protein Response (UPRmt) in conjunction with ATF4 and CHOP, we sought to determine the extent of ATF5 involvement in these pathways with age. The expression of the mitochondrial protease LonP, corrected for mitochondrial content, showed a stark reduction in the absence of ATF5 (Figure 3B, P<0.05), however, we did not observe any changes in ClpP or the mitochondrial heat shock proteins, mtHSP70 and HSP60. This suggests a differential role of ATF5 in regulating HSP expression and perhaps indicates that ATF5 may not solely be required for stress response. Since the ISR/UPRmt pathways are regulated in concert by ATF5, ATF4 and CHOP, we determined whether the absence of ATF5 altered the expression of the other transcription factors as a potential compensatory mechanism. We did not detect any differences in ATF4 and CHOP protein expression in the absence of ATF5, however, with age, the expression of these factors was elevated by 1.5-2-fold (Figure 3E, 3F, P<0.05). In addition, since ATF5 can also coordinate the antioxidant response in response to elevations in ROS, various antioxidant markers were evaluated. The expression of Catalase and MnSOD2 remained unchanged with age or genotype (Figure 3H, 3I), however, NQO1 increased by 2-fold with age (Figure 3J, P<0.01), and this elevation was further enhanced in the absence of ATF5 (Figure 3J, P<0.05). In sum, these data point to an alternative function of ATF5 on regulating ISR/UPRmt and antioxidant markers outside of its canonical role.
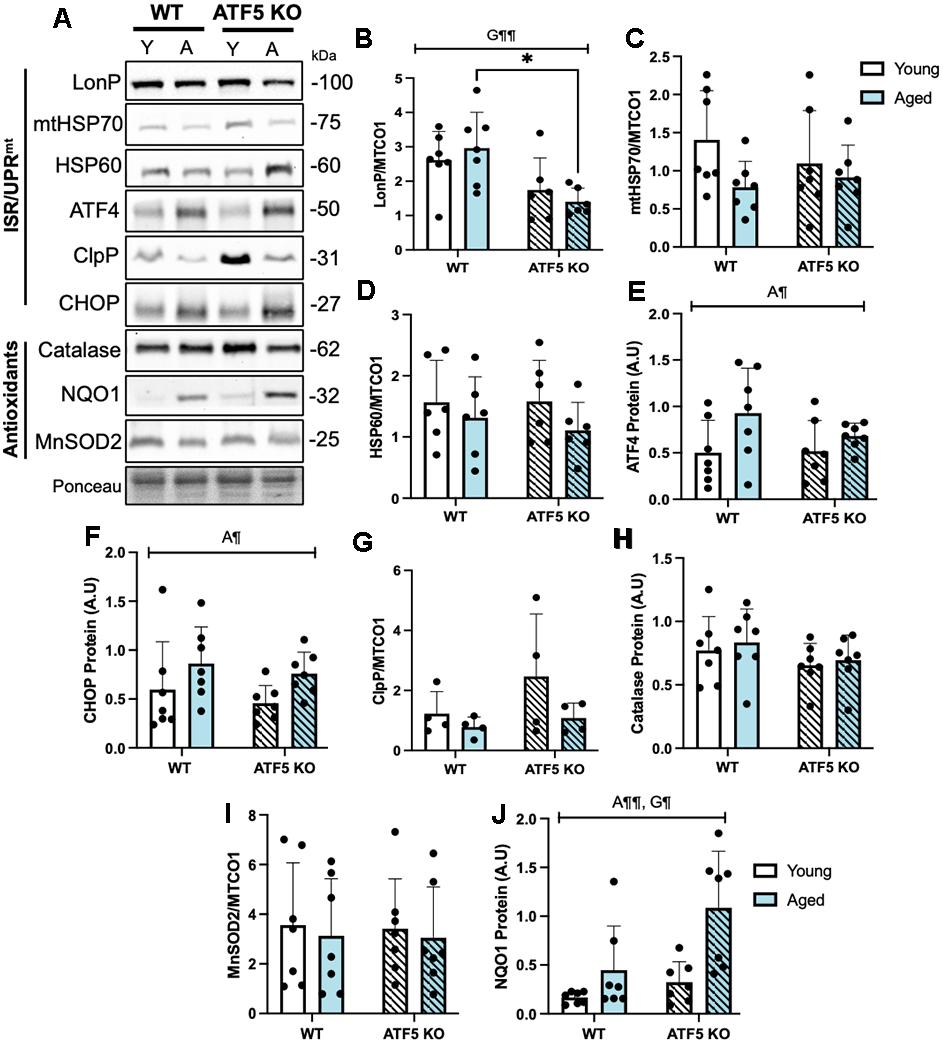
Figure 3. The normal expression of ISR/UPRmt and antioxidant markers are partially blunted in the absence of ATF5. (A) Representative immunoblots of LonP, mtHSP70, HSP60, corrected to mitochondrial content (MTCO1, blot from Figure 5A), and ATF4, ClpP, CHOP, Catalase, NQO1, MnSOD2 corrected to Ponceau, with respective quantifications (B–J). The approximate molecular weights of proteins are indicated with a hash bar in kilodaltons (kDa). (n=4-7/group). A¶ P<0.05, A¶¶ P<0.01, main effect of age; G¶ P<0.05, G¶¶ P<0.01 main effect of genotype, Two-way ANOVA. Y, Young; A, Aged. Data are means ± SD.
The normal induction of related ISR/UPRmt transcription factors following acute contractile activity requires ATF5
Previous evidence has demonstrated that acute in situ contractile activity elicits mitochondrial stress and the activation of the ISR/UPRmt [28]. To determine whether ATF5 is required for such responses, we measured established stress-responsive markers and the ISR transcription factors, ATF4 and CHOP. Contractile activity activated the ISR, evident from significant increases in eIF2α and JNK2 phosphorylation (Figure 4B, 4C, P<0.05). Similar activation levels were observed with age and genotype. Interestingly, both ISR transcription factors ATF4 and CHOP were markedly elevated with age and following stimulation in WT mice (Figure 4D, 4E, P<0.05 P<0.01, P<0.001), however these increases were blunted in the absence of ATF5 with age (Figure 4D, 4E, P<0.05 P<0.01), suggesting that the activation of these proteins following contractile activity requires a level of ATF5 regulation that is dependent on age.
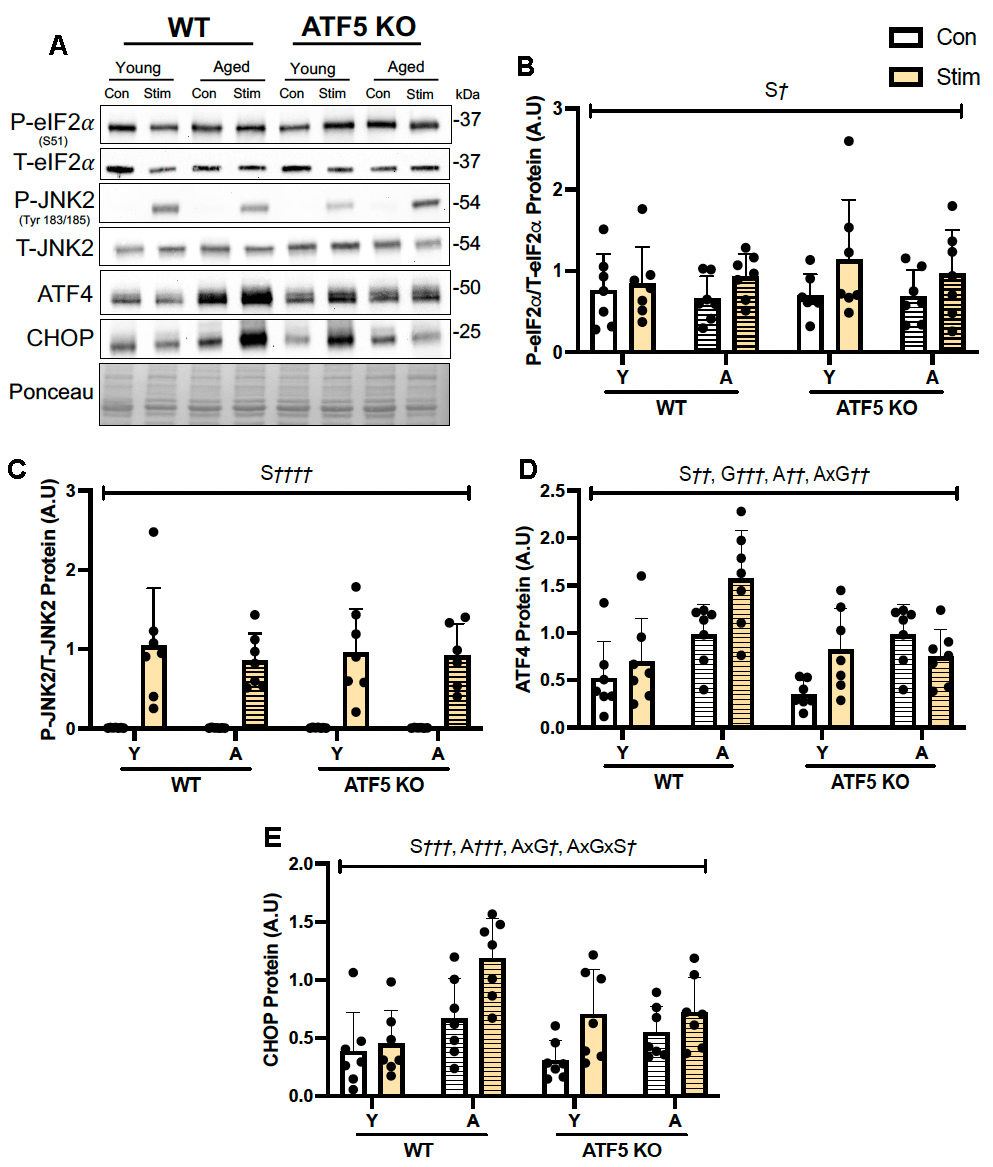
Figure 4. ATF5 is necessary for the normal induction of related ISR/UPRmt transcription factors following acute contractile activity with age. (A) Representative immunoblots of Total eIF2α, Phosphorylated eIF2α, Total JNK2, Phosphorylated JNK2, ATF4 and CHOP corrected to Ponceau, with respective quantifications (B–E). The approximate molecular weights of proteins are indicated with a hash bar in kilodaltons (kDa). (n=4-7/group). S† P<0.05, S†† P<0.01, S††† P<0.001, S†††† P<0.0001, main effect of acute contractile stimulation; A†† P<0.01, A††† P<0.001, main effect of age; G††† P<0.001, main effect of genotype; AxG† P<0.05, AxG†† P<0.01, interaction between age and genotype. AxGxS† P<0.05, interaction between age, genotype and stimulation; Three-way ANOVA. Y, Young; A, Aged; Con, Control; Stim, acute in situ stimulation. Data are means ± SD.
ATF5 is required to maintain a healthy pool of muscle mitochondria in both young and aged mice
The greater fatiguability observed in aged ATF5 KO mice prompted us to investigate whole muscle mitochondrial content and the function of mitochondrial subpopulations. Consistent with prior evidence [24], ATF5 KO animals displayed an elevation in mitochondrial content, as assessed via a Total OXPHOS blot. The expression of UQCRC2, NDUFB8, and SDHB was ~1.5-fold greater in ATF5 KO mice, regardless of age (Figure 5C–5F, P=0.06, P<0.05). However, no changes were observed for the expression of PGC-1α, a main regulator of mitochondrial biogenesis, nor were there any evident changes in the levels of mitochondrial dynamics regulators such as Opa1 and DRP1 (Supplementary Figure 2A–2D). The ratio of MTCO1:SDHB, an indicator of mitonuclear imbalance, which is the relative proportion of mitochondrial- and nuclear-derived proteins that exist in the mitochondria, was elevated ATF5 KO mice irrespective of age (Figure 5G, P<0.05). These data suggest that the absence of ATF5 leads to an elevation in mitochondrial content but imposes an imbalance in genomic regulation, potentially leading to mitochondrial dysregulation. This was supported by our investigation of mitochondrial respiration and ROS emission in both SS and IMF subpopulations. A 50% decline in the basal rate of respiration was observed in SS mitochondria in young ATF5 KO mice, relative to WT animals (Figure 5H, P<0.001). With age, this decline was further amplified, regardless of genotype (Figure 5H, P<0.0001). However, respiration was not worsened in aged ATF5 KO relative to WT controls. In concert with this, State IV SS ROS emission was 2.5-fold greater in aged ATF5 KO mice relative to controls and young mice (Figure 5I, P<0.001, P<0.0001). Moreover, there were no significant differences between genotypes in SS State IV ROS emission in the young cohort. In IMF mitochondria similar respiration trends were observed, with young ATF5 KO mice displaying a ~40% reduction in mitochondrial function (Figure 5J, P<0.01). Aged mice, regardless of genotype, also showed similar functional reductions compared to young WT mice (Figure 5J, P<0.0001, P<0.001). In SS mitochondria, respiration rates were similar between the aged ATF5 KO and their aged WT counterparts. State IV ROS emission did not differ between genotypes in young animals (Figure 5K), however aged mice produced a 4-fold increase in ROS emission, relative to young WT and ATF5 KO mice (Figure 5K, P<0.0001). It was interesting to observe that aged ATF5 KO mice showed a reduction in this elevation (Figure 5K, P<0.01). These data signify a critical importance for ATF5 regulating mitochondrial quality dichotomously in the mitochondrial sub-fractions, an event that becomes more important with age.
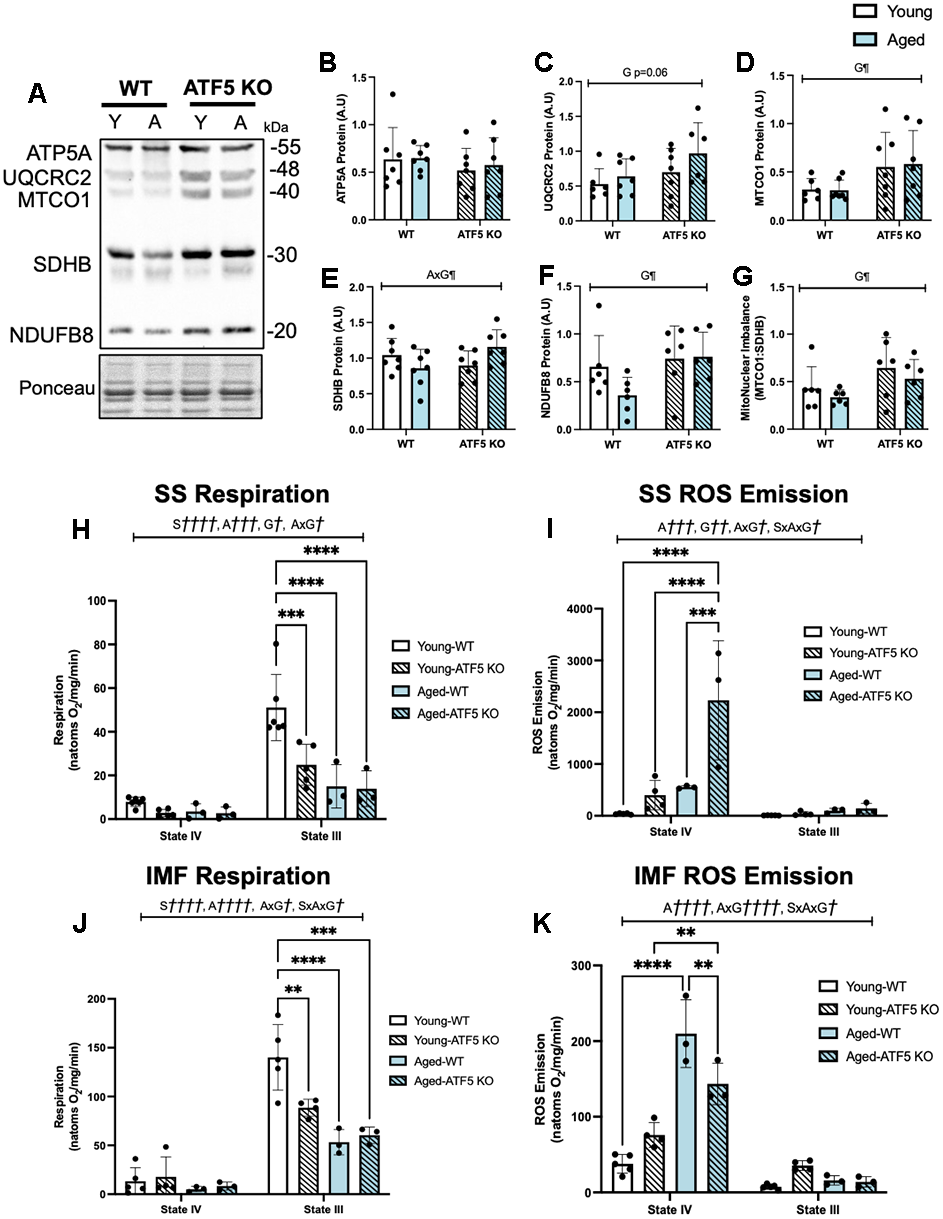
Figure 5. ATF5 KO mice contain a greater pool of poorly functioning skeletal muscle mitochondria, which is exacerbated with age. (A) Representative total OXPHOS blot (ATP5A, UQCRC2, MTCO1, SDHB, and NDUFB8), corrected to Ponceau, with respective quantifications (B–G). The approximate molecular weights of proteins are indicated with a hash bar in kilodaltons (kDa) (n=4-7/group). (H) Mitochondrial respiration, expressed in natoms O2/mg/min in both passive (State IV) and active (State III) respiratory conditions in SS mitochondria alongside (I) ROS emission. (J) Mitochondrial respiration and (K) ROS emission in IMF mitochondrial subfractions. (n=3-6/group). A¶¶¶ P<0.001, A¶¶¶¶ P<0.0001, main effect of age; G¶ P<0.05, G¶¶ P<0.01, main effect of genotype; AxG¶ P<0.05, AxG¶¶¶¶ P<0.0001, interaction between age and genotype; Two-way ANOVA (n=4-8/group). S†††† P<0.0001, main effect of respiratory state;A††† P<0.001, A†††† P<0.0001 main effect of age; G† P<0.05, G†† P<0.01, main effect of genotype; AxG† P<0.05, AxG†††† P<0.0001, interaction between age and genotype, SxAxG P<0.05, interaction between state, age and genotype; Three-way ANOVA (n=3-6/group), Tukey’s post-hoc analyses, Young WT vs. Young ATF5 KO, Aged WT vs. Aged ATF5 KO, Young WT vs. Aged WT, Young ATF5 KO vs. Aged ATF5 KO, Young ATF5 KO vs. Aged WT, Young WT vs. Aged ATF5 KO in both states. Tukey’s post-hoc: **P<0.01, ***P<0.001, ****P<0.0001. Data are means SD.
RNA-Seq analysis of young ATF5 KO mice relative to WT counterparts
To fortify the differences between ATF5 KO and WT mice, we performed RNAseq analysis on RNA isolated from gastrocnemius muscle (n=3/genotype) of young mice (4-6 months). ATF5 was downregulated in KO muscle confirming our model. We identified 116 upregulated, and 37 downregulated genes in KO muscle relative to WT (Figure 6A). Of the upregulated genes, we found 10 genes (Myl3, Tnni1, Tnnc1, Tnrt1, Myh7b, Sln, Myh7, Myl2, Ldhb-ps, Ldhb) that were of particular significance for muscle contraction and function. Through Gene Ontology assessment, we categorized the 116 upregulated genes and formulated the three different enrichment classes based on the Biological Process (BP), Cell Component (CC) and Molecular Function (MF) (Figure 6B). Of interest, we detected a large enrichment of genes associated with the “transition between fast and slow fibers”, indicating a potential fiber switch in the ATF5 KO muscle vs. WT. As well, genes associated with “skeletal muscle contraction” were also enriched. To further analyze the specific genes upregulated within the ATF5 KO mice, we formulated a heat map identifying the degree of change (Log2FC). The identified genes (Myl3, Tnni1, Tnnc1, Tnrt1, Myh7b, Sln, Myh7, Myl2, Ldhb-ps, Ldhb) remain the most relevant in terms of muscle health and regulation (Figure 6C). In that regard, a major finding from the RNA-Seq analysis, was that we did not discover any genes related to changes in mitochondrial health or stress response activation with the absence of ATF5 (Figure 6C). In addition, a heat map of downregulated genes was also formulated, and no specific genes related to muscle or mitochondrial health were identified (Figure 6D).
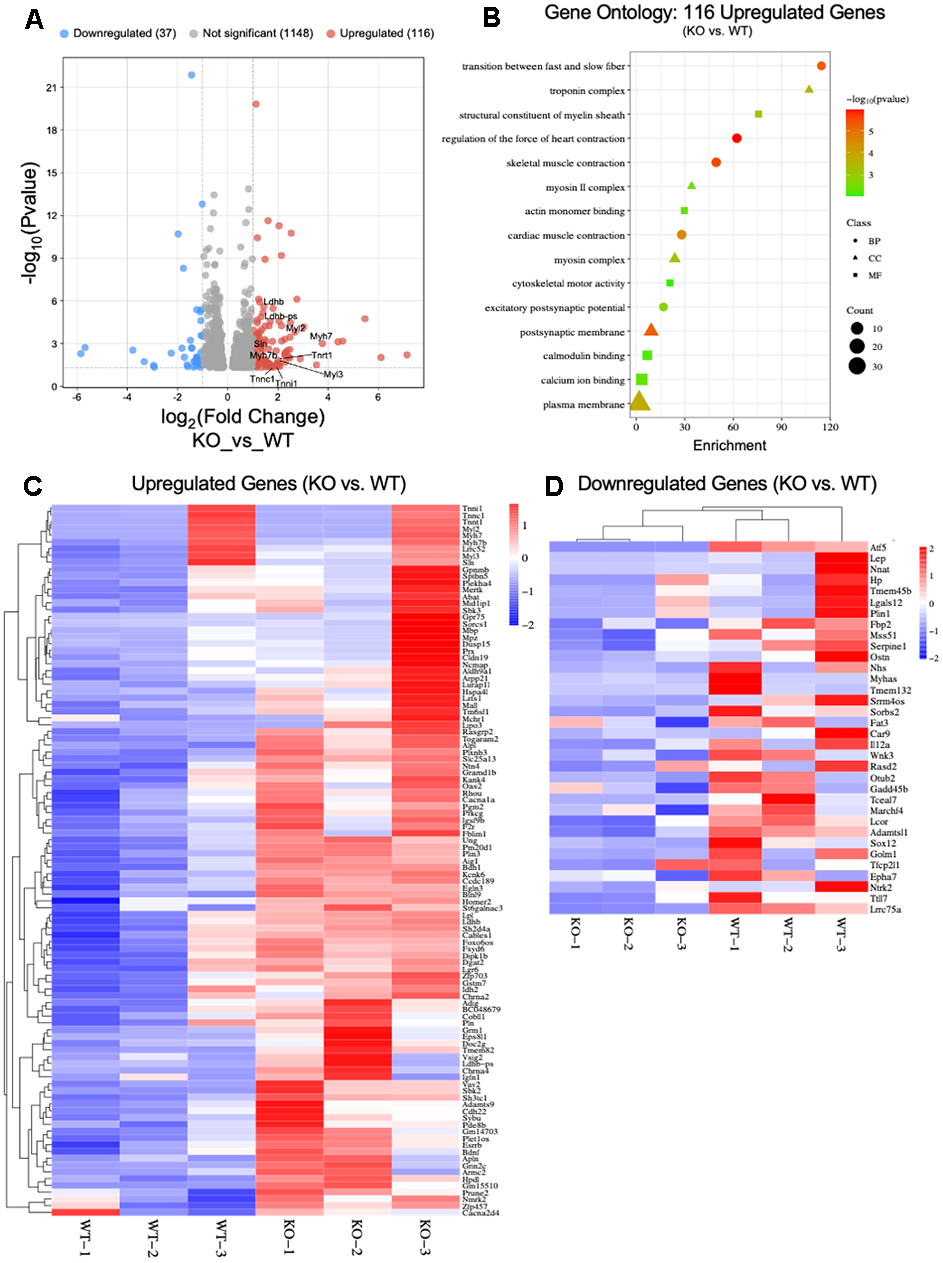
Figure 6. RNA-Seq analysis of young ATF5 KO mice relative to WT counterparts. (A) Volcano plot of significantly differential genes in WT vs. ATF5 KO mice. Genes which had abs(Log2FC) > 1 (=2x FC) and padj (FDR) < 0.05, where colour red or blue depending on the directional fold-change. (B) Gene Ontology (GO) Enrichment Analysis of Upregulated genes in ATF5 KO’s relative to WT’s. Classifications of genes were based of sorting into biological process (BP), Cellular Component (CC) and Molecular Function) with gene count size and enchainment score displayed. Heat map of DESeq2 RNA-Seq data showing differential expression of (C) Upregulated and (D) Downregulated genes in ATF5 KO vs. WT mice. Mitochondrial stress-related genes were not differentially altered. The data are expressed as log2 fold change from ATF5 KO vs. WT (enhanced gene expression, red; reduced gene abundance, blue; and no change in gene expression, white; FDR < 0.05 and padj <0.05 see Supplementary File 1 for expanded DESeq2 analysis data table.
Discussion
With age, a reduction in metabolic plasticity contributes to the onset of the sarcopenic phenotype in skeletal muscle [9, 29]. This decline is largely attributed to the deleterious changes that occur within the mitochondrial network, ranging from reduced content, respiration, and elevations in ROS production [12, 13]. Various mitochondrial quality control processes exist to maintain optimal organellar health, which encapsulates mitophagy, biogenesis, and the mitochondrial stress response (ISR/UPRmt). Although multiple lines of evidence have suggested reductions in the overall drive towards both efficient mitophagy and biogenesis during age in skeletal muscle [14], less is known on how the mitochondrial stress response (ISR/UPRmt) is altered. To this end, the present study explored whether a conserved regulator of the UPRmt, ATF5, is necessary for the normal induction of stress response regulation, mitochondrial quality, and thus skeletal muscle health with age. Furthermore, we examined the extent to which ATF5 was critical for the normal activation of the UPRmt during the induction of a mitochondrial stressor such as acute, progressively intense contractile activity, and whether the effect was altered with age.
To address these aims, along with the potential regulation of muscle mass, we utilized young (4-6 months) and aged/middle-aged (14-16 months) male ATF5 KO or WT mice. We acknowledge that males and females differ in muscle mass regulation, metabolism and other factors with age, and thereby recognise that future experiments should also be conducted on female ATF5 KO mice to confirm and address the following hypothesis. The absence of the female cohort limits generalizability and presents a limitation of this work.
As the age-related decline in muscle mass begins at around 40-50 years in the human population, we felt it necessary to assess the early decline/phase of sarcopenia in our model through utilizing middle-aged mice (denoted as aged within this study) to further emphasize and explore whether the lack of ATF5 would show a deleterious phenotype during such phase. Although our aged cohort of mice displayed an early sarcopenic phenotype, similar experiments should be conducted in a more exaggerated age group, such as in 24-month-old mice, to further explore the importance of ATF5 in very aged mice, thereby highlighting a limitation to this study. Nonetheless, we confirmed the relevant/notable age-related decline in muscle mass in our WT mice; however, this reduction was not detected in the absence of ATF5. These observations point to the role of ATF5 in regulating muscle atrophy. Indeed, our results indicate that in the absence of ATF5, the global expression of protein atrophy markers, in both the ALS, UPS and other related pathways were blunted with age. This suggests a more direct role of ATF5 in regulating the induction of these genes, however, future work confirming such dependency via a ChIP analysis is warranted. Through brief STRING analyses, it was revealed that various atrophy related transcription factors could, hypothetically, heterodimerize with ATF5, including CEBPβ, ATF4, FOXO3α, and TRB3 [30]. Yet no concrete evidence exists to date in literature, especially in skeletal muscle, regarding the involvement of ATF5 and such dimer partners. Therefore, the interaction between ATF5 and muscle atrophy-related transcription factors must be elucidated to confirm such a hypothesis, which undoubtably would help underpin the precise mechanisms in which ATF5 can modulate muscle atrophy. Nonetheless, our data shed light on a new role for ATF5 in modulating muscle protein turnover with age, at least in male mice.
While the current study evaluates the consequences of an ATF5 KO in skeletal muscle, a recent in vivo ATF5 overexpression study demonstrated a similar relationship to what was observed in our aged WT mice [31]. For example, there was an increased capacity for the hyperactivation of the ISR/UPRmt, elevations in protein catabolism-related metabolites, as well as reductions in muscle mass [31]. Moreover, the prolonged upregulation and activation of ATF5, as canonically observed in conditions of nutrient deprivation or aging, can elicit a prolonged ISR/UPRmt activation, which although is protective for the mitochondrial reticulum, can impose decrements in muscle regulation. Although it was not assessed in aforementioned study how the mitochondrial network was altered with the overexpression of ATF5, in our work, the absence of this transcription factor proved deleterious, regardless of age. These similar findings confirm that ATF5, when hyperactivated/expressed beyond normal levels, as observed with aging, promotes muscle atrophy potentially through chronic ISR/UPRmt activation, but protects the mitochondrial network.
In addition, the normal elevation in the Autophagy-Lysosomal System (ALS) with age in skeletal muscle was blunted in the absence of ATF5. This was illustrated by the reduced expression of Parkin, as well as the heightened LC3-II/I ratio, suggesting decreases in the capacity for mitochondrial targeting toward lysosomal degradation via the PINK1/Parkin pathway. This would undoubtedly contribute to the greater abundance of dysfunctional organelles that we observed in ATF5 KO muscle. Such regulation may serve as an important link between the ISR/UPRmt and the ALS in during elevations in muscle protein stress evident with age. Prior evidence from our laboratory in C2C12 myotubes and other work in mice have demonstrated the regulation of Parkin expression via ATF4 [32, 33]; however, it is unknown how ATF5 cooperates in such relationship. Further assessment of this interaction and potential dependence/heterodimerization of ATF4 and ATF5 must be evaluated.
We also observed an elevation in non-canonical muscle atrophy markers p53, p21 and Gadd45α with age. These proteins have been recently associated with ISR/UPRmt activation and the propagation of muscle atrophy primarily through ATF4 regulation during aging [25]. Previous evidence has suggested the capacity for ATF5 to also regulate these markers when overexpressed, alongside associated muscle mass consequences, similar to what we observed in aging WT mice [31]. Interestingly, p53, p21 and Gadd45α are also known mediators of cellular senescence [34–37]. Characterized by irreversible cell cycle arrest influenced by age-related cellular alterations such as DNA damage, ROS emission and proteome instability, the activation of cellular senescence has now become a common denominator in skeletal muscle aging [34, 38]. The reduced expression of p53 in aged ATF5 KO animals supports the current knowledge regarding the ISR/UPRmt and muscle atrophy and suggests a more regulatory role for ATF5 in propagating muscle atrophy potentially through a senescence-associated lens. Although speculative, this interconnection must be further explored in order to determine how senescence and the ISR/UPRmt are connected, particularly in the context of aging.
As ATF5 has been reported to regulate the ISR/UPRmt in multiple tissues, we evaluated the expression of several stress response markers in WT and KO animals. It was interesting to observe that both ATF4 and CHOP, known co-regulatory transcription factors of these pathways, were not altered in the absence of ATF5 in control muscle, and were both markedly elevated with age. This suggests that ATF5 may not be critical for the expression of these partner proteins, at least in skeletal muscle. The expression of the mitochondrial protease LonP, a downstream gene involved in the ISR/UPRmt adaptive response and required to maintain mitochondrial proteostasis, was significantly blunted in the absence of ATF5, corroborating recent findings during denervation-induced mitochondrial dysfunction in ATF5 KO mice [39]. This was not a surprising finding, considering that ATF5 is predominantly regarded as a regulator of mitochondrial protein homeostasis. In addition, a recent study suggested an importance of ATF5 in promoting LonP expression in MEF cells during mitochondrial protein handling stress [40]. With regards to other downstream targets, surprisingly, we did not observe any significant changes in the expression of mtHSP70 as well as ClpP in the absence of ATF5, suggesting that this transcription factor may not be a critical component in solely regulating these components of the UPRmt in young and aged skeletal muscle, a finding further solidified in denervation-induced mitochondrial dysfunction [39].
We also measured the relative balance in nuclear and mitochondrial encoded gene products in WT and KO muscle via the ratio of MTCO1:SDHB (Mito:Nuclear). Although there was no apparent effect of age, ATF5 KO mice showed a greater imbalance in this ratio relative to WT mice, indicating the over-abundance of mtDNA-encoded subunits relative to nuclear-encoded proteins. This could be a result of proteostatic failure and capacity to control the coordinated stoichiometry of the electron transport chain components or related to the decline in nuclear-encoded transcriptional activity relative to mitochondrial gene expression [41]. The lack of compensation in LonP expression likely further supports the inability to respond to genomic imbalance/proteotoxicity within the mitochondria in the absence of ATF5. Previous reports have also shown this alteration in Mito:Nuclear imbalance following exercise in aged muscle [42], and therefore our results indirectly suggest that the absence of ATF5 imposes a mitochondrial genomic stress similar to exercise. Thus, ATF5 is required to maintain optimal nuclear-mitochondrial proteomic balance in skeletal muscle, although the mechanism responsible remains elusive.
The interaction and expression of antioxidant-related proteins was also assessed as prior evidence suggested that ATF5 is highly responsive to mitochondrial derived oxidative stress and could coordinate the antioxidant stress response along with Nrf-2 [24]. Interestingly, NQO1, was the only antioxidant marker that was elevated in the absence of ATF5, particularly with age. This is likely a response to the higher level of oxidative stress evident in aged ATF5 KO muscle, in an attempt to sequester harmful ROS molecules. However, this observed response also represents a negative regulation of NQO1 by ATF5, a non-canonical regulatory relationship that has not been reported previously.
Indicators of muscle fatigue in the presence and absence of ATF5 were also evaluated. While no differences were observed in young animals, aged muscle from ATF5 KO mice displayed greater rates of fatigue compared to their WT counterparts. We determined whether these changes were a product of reduced mitochondrial content and/or function. Similar to previous findings from our laboratory [24], ATF5 KO mice showed greater mitochondrial content, and in our study this elevation was preserved with age. These findings support previous observations which measured mitochondrial content in the absence of ATF5 in a mammalian cell model [23], indicating a larger yet more dysfunctional mitochondrial pool. The dysfunction was not evident by significant reductions in respiratory capacity, assessed using standard Clark Electrode measures of oxygen consumption, but rather by the exaggerated rate of ROS emission in the absence of ATF5, particularly within the SS mitochondrial fraction. These large levels of ROS emission may influence the rate of peripheral muscle fatiguability via various putative pathways, including modifications of calcium release from the sarcoplasmic reticulum, or via the desensitization of the calcium-troponin C interaction, thereby impacting muscle force production [43–45]. In addition, the heightened and persistent ROS levels observed in aged ATF5 KO mice may also influence the activity of various glycolytic enzymes or increase the level of lipid peroxidation which could impact force production [46].
The decline in endurance capacity, coupled with preserved muscle mass and elevated ROS emission observed in the aged ATF5 KO mice represents a fascinating trade-off that becomes more relevant particularly during the process of aging. Our results suggest that although muscle mass seems to be preserved in the absence of ATF5 with age, the quality and mitochondrial health of these muscle fibers is compromised. The physiological implications for aging muscles therefore necessitates ATF5 as a critical regulator of skeletal muscle health.
The contractile activity protocol employed to investigate muscle fatigue imposes substantial mitochondrial stress, evident from the activation of the ISR markers such as eIF2α and JNK-P [28]. Since ATF4, CHOP, and ATF5 are involved in regulating this pathway, we sought to evaluate whether the expression of these alternative transcription factors was impacted by the absence of ATF5. Interestingly, our observations indicate that the changes in ATF4 and CHOP produced by contractile activity were not reliant on ATF5 in muscle of young animals, but that the response became clearly evident with age, a chronic mitochondrial stressor. The canonical mechanism of ATF5 expression in mammalian cells relies on the coordinated involvement of ATF4 and CHOP as heterodimerization partners [47], however, it was fascinating to observe that the lack of ATF5 can blunt these regulators and their responsiveness to acute activity/mitochondrial stress, suggesting a greater dependence on ATF5 in regulating both ATF4 and CHOP (and thus the ISR/UPRmt) in aging muscle. Indeed, previous evidence in young ATF5 KO mice from our laboratory similarly demonstrated altered ISR/UPRmt signaling during an acute bout of exhaustive treadmill running [24] and also during denervation [39], thereby highlighting such regulatory network. Although in this study, the levels of ATF4 and CHOP remained elevated with age in ATF5 KO control muscle, it is during a cellular/mitochondrial stressor where the normal induction of these regulators is blunted. This suggests that ATF4 and CHOP cannot impart a compensatory response in the absence of ATF5 following mitochondrial stress, thereby positioning ATF5 as a regulator of the ISR/UPRmt in this context.
To get a glimpse of the overall importance of ATF5 in maintaining normal cellular levels of gene expression, we conducted a limited RNA sequencing analysis of muscle from young ATF5 KO and WT mice. A small pool of genes was differentially expressed (upregulated: 116, or downregulated: 37). To further characterize these changes, our Gene-Ontology reports indicated a shift in gene expression in the absence of ATF5 from fast to slow-fibers. Interestingly, these changes were of insufficient magnitude to affect muscle physiology, measured as time-to-peak tension and half-relaxation time in the young cohort (Supplementary Figure 1). Given this, we did not further assess potential changes in histochemically-defined muscle fiber types. These data indicate that changes measured at the mRNA level, depending on their magnitude, do not always necessarily translate into functional changes.
In sum, our results indicate that the skeletal muscle of aged (14-16 months) ATF5 KO animals exhibits 1) a preservation of muscle mass and corresponding reductions in atrophy-related markers, 2) greater muscle fatiguability, 3) a more abundant mitochondrial pool, with reduced organelle function, and 4) altered expression and regulation of the ISR/UPRmt both basally and with acute contractile activity. This work thereby extends the previously known roles of ATF5 in skeletal muscle and mitochondrial health to age-specific analyses in male mice.
Materials and Methods
Animals
Male ATF5 whole-body KO mice were generated and genotyped, as previously discussed [24, 39]. Animals were housed in a 12:12-hr light-dark cycle and given food and water ad libitum. Experiments were conducted after approval by the York University Animal Care Committee in accordance with Canadian Council of Animal Care guidelines. Experimental animals were used at 4-6 months of age for the young (Y) cohort, and 14-16 months for the middle-aged/aged (A) cohort (n=3-20/group depending on the experimental analyses).
Acute in-situ muscle contractile activity protocol
The in situ muscle stimulation protocol was performed as described previously [28] (n=6-20/group). Briefly, animals were anesthetized under isoflurane inhalation with oxygen, and the right hind limb gastrocnemius muscle and innervating sciatic nerve were exposed, isolated, and doused in 0.9% saline. The right Achilles tendon and gastrocnemius were attached to a force transducer (Grass FT 10; Grass Instruments, Quincy, MA, USA) and the innervating sciatic nerve was stimulated at 0.25, 0.5 and 1 tetanic contractions per second (TPS, 100ms, duration at 100Hz) for 3 mins each. The gastrocnemius, TA, EDL, and Soleus muscles of both the stimulated and contralateral control limb were harvested, weighed, flash-frozen in liquid nitrogen and stored at -80° C for subsequent use in further experiments. Heart weights were also measured following sacrifice. All animals were sacrificed by cervical dislocation immediately after tissue harvest.
Whole muscle protein extracts
A portion of the gastrocnemius muscle (~20-30mg) that was flash-frozen in liquid nitrogen was diluted 10-fold and homogenized in Sakamoto buffer (20mM HEPES, 2mM EGTA, 1% Triton X-100, 50% Glycerol, 50 mM B-Glycerophosphate), with the addition of phosphatase (MilliporeSigma) and protease (Roche Mississauga, ON, Canada) inhibitors, using the Qiagen Tissue Lyser II. Samples were centrifuged (14,000g) for 10 minutes at 4° C and the supernatant was collected and stored at -80° C until further analysis.
Immunoblotting
Whole muscle protein extracts prepared from a portion of the gastrocnemius (n=4-7/group) were loaded (25-30μg of protein) and separated via electrophoresis on 10-15% SDS-PAGE at 120V for ~90 mins, and subsequently transferred onto nitrocellulose membranes (Bio-Rad, Mississauga, ON, Canada). Following the transfer, the membranes were stained with Ponceau Red, cut at the appropriate molecular weights, and blocked at room temperature in 5% skim milk or 5% BSA for phosphorylated proteins in TBS-T solution (25 mM Tris–HCl, 1 mM NaCl, 0.1% Tween-20, pH 7.5) for 1 hour with gentle agitation. The membranes were then coated and incubated with the appropriate antibodies (Supplementary Table 1) overnight at 4° C. Subsequently, membranes were then washed 3x for 5 minutes with TBS-T, followed by incubation of the appropriate secondary antibody conjugated to HRP at room temperature (1 hr), and washed again 3x for 5 minutes each with TBS-T. Membranes were imaged using an ECL kit (170-5061; Bio-Rad) on the Invitrogen iBright 1500 imager and revealed via enhanced chemiluminescence. Quantifications were performed with ImageJ software (NIH, Bethesda, MD, USA), and values were normalized to Ponceau. The approximate molecular weights in kDa (kilodaltons) of proteins are indicated on each blot. For the normalization of mitochondrial content in related immunoblots (Figure 3), we utilized MTCO1 as a mitochondrial protein content marker and following the corresponding Ponceau correction, we normalized each mitochondrial protein to the associated MTCO1 value for each respective sample.
Mitochondrial isolations
Approximately 1200-1500mg of fresh skeletal muscle was harvested from anesthetized animals, placed in ice-cold buffer, and subsequently minced and homogenized for a total of 9 minutes (n=3-6/group). Isolation of SS and IMF mitochondrial subpopulations were performed as previously described [24]. SS and IMF fractions were separated via differential centrifugation at 800g for 10 min. The Nagarse protease from Bacillus licheniformis, (P5380, MilliporeSigma) was utilized briefly to liberate the IMF mitochondrial fraction. Following additional centrifugation steps, both SS and IMF pellets were suspended in mitochondrial resuspension buffer (100 mM KCl, 10 mM MOPS, 0.2% BSA pH of 7.4). The protein concentration of each fraction was determined using the Bradford method. Mitochondrial fractions were used immediately to measure both respiration and ROS emission.
Mitochondrial respiration
Oxygen consumption (O2/mg/min) in both SS and IMF mitochondria was measured via a Clark Electrode (Strathkelvin Instruments) (n=3-6/group). SS or IMF fractions (50 μl) were incubated and continuously stirred at 30° C with 250 μl of VO2 buffer (250 mM sucrose, 50 mM KCl, 25 mM Tris base, 10 mM K2HPO4, pH 7.4). The rate of respiration (nanoatoms O2/min/mg) in both State IV (passive) and State III (active) was determined via the addition of 10 mM glutamate and 0.44 mM ADP, respectively. Following this, NADH was added to test mitochondrial membrane integrity.
Mitochondrial ROS emission
SS and IMF mitochondria (50 μg) were incubated in a black 96-well plate with VO2 buffer and 50 mM H2DCF-DA (D399, Thermo Fisher Scientific) at 37° C for 30 min (n=3-6/group). ROS emission was assessed following the addition of 10mM of glutamate either with (State III) or without (State IV) 0.44mM ADP. The fluorescent emission of H2DCF-DA (480-520nm), indicative of the presence of ROS within the mitochondrial fraction, was measured in the BioTek Cytation5 Imaging Reader (Agilent Technologies, Santa Clara, CA, USA). ROS emission/fluorescent values were normalized to the corresponding respiration rates in each mitochondrial subpopulation alongside total protein content, as previously described.
RNA-Seq and pathway analysis
RNA library preparation, sequencing, and pathway analysis were performed by the Toronto Centre for Applied Genomics (The Hospital for Sick Children). Briefly, NEBNext Ultra II Directional RNA libraries using poly-A selection were prepared. All 6 samples (three young ATF5 KO mice with matched young WT controls; n=3) were multiplexed on one lane of an SP flow cell and sequenced (paired end reads) on the Illumina NovaSeq 6000. Prior to performing RNA-Seq, the resulting DNA-free RNA samples were analyzed for quality on a 2100 Bioanalyzer System. Gene expression analysis was performed with DESeq2 (Supplementary File 1). Genes which had and absolute Log2FoldChange > 1 (=2x FC) and padj (FDR) <0.05 were denoted as differentially expressed. To investigate the effects of ATF5 KO on a global scale, Gene Ontology enrichment/pathway analysis was performed using DAVID Bioinformatics Resources 6.8 for upregulated genes (116) [48, 49]. The GO terms in each category (BP, CC, MP) pertaining to skeletal muscle with p<0.05 were defined as significantly enriched gene clusters. Scatter plots of the DEGs were visualized using the SRplot software. Volcano plot and heat maps were constructed to visualize differential gene expression between the groups, generated using SRplot [50]. Volcano plot and heat maps cutoffs were the following: p-value <0.05 and fold-change cutoff at 2. Heat maps were analyzed with the following settings: Clustering method, complete linkage. Distance measurement method, Euclidian. Young mice were used for this experiment due to technical requirements and limitations.
Statistical analysis
Data were analyzed using the GraphPad Prism 9.0 software and values are reported as means ± SD. Data were analyzed by a Two-Way to compare between genotype and age. A Three-way ANOVA was used in the comparisons of acute in situ stimulation, genotype and age, or within mitochondrial respiration parameters. ANOVA testing was following a Tukey’s post-hoc test, where necessary. Statistical significance was achieved at P ≤ 0.05.
Supporting Information
Additional supporting information may be found in the Supplementary Material section.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Author Contributions
V.C.S. and D.A.H. conceived and designed research; V.C.S. performed experiments; V.C.S. and D.A.H. analyzed data; V.C.S. and D.A.H. interpreted results of experiments; V.C.S. prepared figures; V.C.S. drafted manuscript; V.C.S. and D.A.H. edited and revised manuscript; D.A.H. approved final version of manuscript.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Ethical Statement
All animal procedures were conducted in accordance with the standards set by the Canadian Council on Animal Care, with the approval (2017-08) of York University Animal Care Committee (YUACC).
Funding
This work was supported by funding from the Natural Sciences and Engineering Research Council (NSERC) of Canada (Grant No. 03623) to D.A. Hood. V. C. Sanfrancesco is a recipient of the Canada Graduate Scholarship-Doctoral (CGS-D) NSERC award.
References
- 1. Hipkiss AR. Mitochondrial dysfunction, proteotoxicity, and aging: causes or effects, and the possible impact of NAD+-controlled protein glycation. Adv Clin Chem. 2010; 50:123–50. [PubMed]
- 2. Conley KE, Jubrias SA, Esselman PC. Oxidative capacity and ageing in human muscle. J Physiol. 2000; 526:203–10. https://doi.org/10.1111/j.1469-7793.2000.t01-1-00203.x [PubMed]
- 3. Argilés JM, Campos N, Lopez-Pedrosa JM, Rueda R, Rodriguez-Mañas L. Skeletal Muscle Regulates Metabolism via Interorgan Crosstalk: Roles in Health and Disease. J Am Med Dir Assoc. 2016; 17:789–96. https://doi.org/10.1016/j.jamda.2016.04.019 [PubMed]
- 4. Holloszy JO. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem. 1967; 242:2278–82. [PubMed]
- 5. Paddon-Jones D, Sheffield-Moore M, Cree MG, Hewlings SJ, Aarsland A, Wolfe RR, Ferrando AA. Atrophy and impaired muscle protein synthesis during prolonged inactivity and stress. J Clin Endocrinol Metab. 2006; 91:4836–41. https://doi.org/10.1210/jc.2006-0651 [PubMed]
- 6. Smuder AJ, Sollanek KJ, Nelson WB, Min K, Talbert EE, Kavazis AN, Hudson MB, Sandri M, Szeto HH, Powers SK. Crosstalk between autophagy and oxidative stress regulates proteolysis in the diaphragm during mechanical ventilation. Free Radic Biol Med. 2018; 115:179–90. https://doi.org/10.1016/j.freeradbiomed.2017.11.025 [PubMed]
- 7. Phillips SM, Glover EI, Rennie MJ. Alterations of protein turnover underlying disuse atrophy in human skeletal muscle. J Appl Physiol (1985). 2009; 107:645–54. https://doi.org/10.1152/japplphysiol.00452.2009 [PubMed]
- 8. Thomason DB, Booth FW. Atrophy of the soleus muscle by hindlimb unweighting. J Appl Physiol (1985). 1990; 68:1–12. https://doi.org/10.1152/jappl.1990.68.1.1 [PubMed]
- 9. Figueiredo PA, Mota MP, Appell HJ, Duarte JA. The role of mitochondria in aging of skeletal muscle. Biogerontology. 2008; 9:67–84. https://doi.org/10.1007/s10522-007-9121-7 [PubMed]
- 10. Larsson L, Degens H, Li M, Salviati L, Lee YI, Thompson W, Kirkland JL, Sandri M. Sarcopenia: Aging-Related Loss of Muscle Mass and Function. Physiol Rev. 2019; 99:427–511. https://doi.org/10.1152/physrev.00061.2017 [PubMed]
- 11. Demontis F, Piccirillo R, Goldberg AL, Perrimon N. Mechanisms of skeletal muscle aging: insights from Drosophila and mammalian models. Dis Model Mech. 2013; 6:1339–52. https://doi.org/10.1242/dmm.012559 [PubMed]
- 12. Chabi B, Ljubicic V, Menzies KJ, Huang JH, Saleem A, Hood DA. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell. 2008; 7:2–12. https://doi.org/10.1111/j.1474-9726.2007.00347.x [PubMed]
- 13. Chistiakov DA, Sobenin IA, Revin VV, Orekhov AN, Bobryshev YV. Mitochondrial aging and age-related dysfunction of mitochondria. Biomed Res Int. 2014; 2014:238463. https://doi.org/10.1155/2014/238463 [PubMed]
- 14. Liu BH, Xu CZ, Liu Y, Lu ZL, Fu TL, Li GR, Deng Y, Luo GQ, Ding S, Li N, Geng Q. Mitochondrial quality control in human health and disease. Mil Med Res. 2024; 11:32. https://doi.org/10.1186/s40779-024-00536-5 [PubMed]
- 15. Costa-Mattioli M, Walter P. The integrated stress response: From mechanism to disease. Science. 2020; 368:eaat5314. https://doi.org/10.1126/science.aat5314 [PubMed]
- 16. Teske BF, Fusakio ME, Zhou D, Shan J, McClintick JN, Kilberg MS, Wek RC. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol Biol Cell. 2013; 24:2477–90. https://doi.org/10.1091/mbc.e13-01-0067 [PubMed]
- 17. Zhou D, Palam LR, Jiang L, Narasimhan J, Staschke KA, Wek RC. Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. J Biol Chem. 2008; 283:7064–73. https://doi.org/10.1074/jbc.m708530200 [PubMed]
- 18. Arnould T, Michel S, Renard P. Mitochondria Retrograde Signaling and the UPR mt: Where Are We in Mammals? Int J Mol Sci. 2015; 16:18224–51. https://doi.org/10.3390/ijms160818224 [PubMed]
- 19. Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell. 2010; 37:529–40. https://doi.org/10.1016/j.molcel.2010.01.015 [PubMed]
- 20. Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011; 144:79–91. https://doi.org/10.1016/j.cell.2010.12.016 [PubMed]
- 21. Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013; 497:451–7. https://doi.org/10.1038/nature12188 [PubMed]
- 22. Schieber M, Chandel NS. TOR signaling couples oxygen sensing to lifespan in C. elegans. Cell Rep. 2014; 9:9–15. https://doi.org/10.1016/j.celrep.2014.08.075 [PubMed]
- 23. Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, Haynes CM. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr Biol. 2016; 26:2037–43. https://doi.org/10.1016/j.cub.2016.06.002 [PubMed]
- 24. Slavin MB, Kumari R, Hood DA. ATF5 is a regulator of exercise-induced mitochondrial quality control in skeletal muscle. Mol Metab. 2022; 66:101623. https://doi.org/10.1016/j.molmet.2022.101623 [PubMed]
- 25. Ebert SM, Dierdorff JM, Meyerholz DK, Bullard SA, Al-Zougbi A, DeLau AD, Tomcheck KC, Skopec ZP, Marcotte GR, Bodine SC, Adams CM. An investigation of p53 in skeletal muscle aging. J Appl Physiol (1985). 2019; 127:1075–84. https://doi.org/10.1152/japplphysiol.00363.2019 [PubMed]
- 26. Mairinger FD, Walter RF, Theegarten D, Hager T, Vollbrecht C, Christoph DC, Worm K, Ting S, Werner R, Stamatis G, Mairinger T, Baba H, Zarogoulidis K, et al. Gene Expression Analysis of the 26S Proteasome Subunit PSMB4 Reveals Significant Upregulation, Different Expression and Association with Proliferation in Human Pulmonary Neuroendocrine Tumours. J Cancer. 2014; 5:646–54. https://doi.org/10.7150/jca.9955 [PubMed]
- 27. Ebert SM, Dyle MC, Kunkel SD, Bullard SA, Bongers KS, Fox DK, Dierdorff JM, Foster ED, Adams CM. Stress-induced skeletal muscle Gadd45a expression reprograms myonuclei and causes muscle atrophy. J Biol Chem. 2012; 287:27290–301. https://doi.org/10.1074/jbc.m112.374777 [PubMed]
- 28. Sanfrancesco VC, Hood DA. Acute contractile activity induces the activation of the mitochondrial integrated stress response and the transcription factor ATF4. J Appl Physiol (1985). 2025; 138:857–71. https://doi.org/10.1152/japplphysiol.00307.2024 [PubMed]
- 29. Ljubicic V, Joseph AM, Adhihetty PJ, Huang JH, Saleem A, Uguccioni G, Hood DA. Molecular basis for an attenuated mitochondrial adaptive plasticity in aged skeletal muscle. Aging (Albany NY). 2009; 1:818–30. https://doi.org/10.18632/aging.100083 [PubMed]
- 30. Wilkinson DJ, Piasecki M, Atherton PJ. The age-related loss of skeletal muscle mass and function: Measurement and physiology of muscle fibre atrophy and muscle fibre loss in humans. Ageing Res Rev. 2018; 47:123–32. https://doi.org/10.1016/j.arr.2018.07.005 [PubMed]
- 31. Brearley-Sholto MC, Loczenski-Brown DM, Jones S, Daniel ZC, Ebling FJ, Parr T, Brameld JM. Effect of AAV-mediated overexpression of ATF5 and downstream targets of an integrated stress response in murine skeletal muscle. Sci Rep. 2021; 11:19796. https://doi.org/10.1038/s41598-021-99432-4 [PubMed]
- 32. Memme JM, Sanfrancesco VC, Hood DA. Activating transcription factor 4 regulates mitochondrial content, morphology, and function in differentiating skeletal muscle myotubes. Am J Physiol Cell Physiol. 2023; 325:C224–42. https://doi.org/10.1152/ajpcell.00080.2023 [PubMed]
- 33. Bouman L, Schlierf A, Lutz AK, Shan J, Deinlein A, Kast J, Galehdar Z, Palmisano V, Patenge N, Berg D, Gasser T, Augustin R, Trümbach D, et al. Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 2011; 18:769–82. https://doi.org/10.1038/cdd.2010.142 [PubMed]
- 34. Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, Miwa S, Olijslagers S, Hallinan J, Wipat A, Saretzki G, Rudolph KL, Kirkwood TB, von Zglinicki T. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol. 2010; 6:347. https://doi.org/10.1038/msb.2010.5 [PubMed]
- 35. Edwards MG, Anderson RM, Yuan M, Kendziorski CM, Weindruch R, Prolla TA. Gene expression profiling of aging reveals activation of a p53-mediated transcriptional program. BMC Genomics. 2007; 8:80. https://doi.org/10.1186/1471-2164-8-80 [PubMed]
- 36. Welle S, Brooks AI, Delehanty JM, Needler N, Bhatt K, Shah B, Thornton CA. Skeletal muscle gene expression profiles in 20-29 year old and 65-71 year old women. Exp Gerontol. 2004; 39:369–77. https://doi.org/10.1016/j.exger.2003.11.011 [PubMed]
- 37. Venturelli M, Morgan GR, Donato AJ, Reese V, Bottura R, Tarperi C, Milanese C, Schena F, Reggiani C, Naro F, Cawthon RM, Richardson RS. Cellular aging of skeletal muscle: telomeric and free radical evidence that physical inactivity is responsible and not age. Clin Sci (Lond). 2014; 127:415–21. https://doi.org/10.1042/cs20140051 [PubMed]
- 38. Sabath N, Levy-Adam F, Younis A, Rozales K, Meller A, Hadar S, Soueid-Baumgarten S, Shalgi R. Cellular proteostasis decline in human senescence. Proc Natl Acad Sci USA. 2020; 117:31902–13. https://doi.org/10.1073/pnas.2018138117 [PubMed]
- 39. Kuthe, J, Hood DA. ATF5 is required for the normal stress response during skeletal muscle denervation, Sports Medicine and Health Science. 2025; 40:ISSN 2666–3376. https://doi.org/10.1016/j.smhs.2026.01.001
- 40. Katiyar A, Fujimoto M, Tan K, Kurashima A, Srivastava P, Okada M, Takii R, Nakai A. HSF1 is required for induction of mitochondrial chaperones during the mitochondrial unfolded protein response. FEBS Open Bio. 2020; 10:1135–48. https://doi.org/10.1002/2211-5463.12863 [PubMed]
- 41. Braga RR, Crisol BM, Brícola RS, Sant'ana MR, Nakandakari SC, Costa SO, Prada PO, da Silva AS, Moura LP, Pauli JR, Cintra DE, Ropelle ER. Exercise alters the mitochondrial proteostasis and induces the mitonuclear imbalance and UPRmt in the hypothalamus of mice. Sci Rep. 2021; 11:3813. https://doi.org/10.1038/s41598-021-82352-8 [PubMed]
- 42. Cordeiro AV, Peruca GF, Braga RR, Brícola RS, Lenhare L, Silva VR, Anaruma CP, Katashima CK, Crisol BM, Barbosa LT, Simabuco FM, da Silva AS, Cintra DE, et al. High-intensity exercise training induces mitonuclear imbalance and activates the mitochondrial unfolded protein response in the skeletal muscle of aged mice. Geroscience. 2021; 43:1513–8. https://doi.org/10.1007/s11357-020-00246-5 [PubMed]
- 43. Moopanar TR, Allen DG. The activity-induced reduction of myofibrillar Ca2+ sensitivity in mouse skeletal muscle is reversed by dithiothreitol. J Physiol. 2006; 571:191–200. https://doi.org/10.1113/jphysiol.2005.101105 [PubMed]
- 44. Reid MB. Free radicals and muscle fatigue: Of ROS, canaries, and the IOC. Free Radic Biol Med. 2008; 44:169–79. https://doi.org/10.1016/j.freeradbiomed.2007.03.002 [PubMed]
- 45. Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev. 2008; 88:1243–76. https://doi.org/10.1152/physrev.00031.2007 [PubMed]
- 46. Mullarky E, Cantley LC. Diverting Glycolysis to Combat Oxidative Stress. 2015. https://doi.org/10.1007/978-4-431-55651-0_1 [PubMed]
- 47. Su N, Kilberg MS. C/EBP homology protein (CHOP) interacts with activating transcription factor 4 (ATF4) and negatively regulates the stress-dependent induction of the asparagine synthetase gene. J Biol Chem. 2008; 283:35106–17. https://doi.org/10.1074/jbc.m806874200 [PubMed]
- 48. Sherman BT, Hao M, Qiu J, Jiao X, Baseler MW, Lane HC, Imamichi T, Chang W. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022; 50:W216–21. https://doi.org/10.1093/nar/gkac194 [PubMed]
- 49. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4:44–57. https://doi.org/10.1038/nprot.2008.211 [PubMed]
- 50. Tang D, Chen M, Huang X, Zhang G, Zeng L, Zhang G, Wu S, Wang Y. SRplot: A free online platform for data visualization and graphing. PLoS One. 2023; 18:e0294236. https://doi.org/10.1371/journal.pone.0294236 [PubMed]