



Introduction
Hepatocellular carcinoma (HCC) is one of the five leading causes of cancer-related death worldwide. Recent developments in the treatment of HCC proved insufficient to cure unresectable disease and to substantially prevent HCC progression. The limitations of current therapeutics primarily reflect incomplete understanding of the complex molecular signaling processes contributing to the heterogeneous nature of the disease [1].
Liver carcinogenesis is a multistep, long process associated with multiple risk factors, e.g. viral hepatitis, alcohol abuse, metabolic disorders, and obesity [2]. Over the last two decades, studies based on genome-wide gene expression and functional profiling have revealed the great diversity of transcriptional alterations occurring in liver carcinogenesis. However, translating these findings into individualized treatments has proved to be difficult [2].
Transcription factors (TFs) drive gene expression programs that shape specific phenotypes [3], and are frequently dysregulated in cancer [4]. Correspondingly, most cancer signaling pathways seem to converge on one or more TFs, termed “master regulators” (MRs) [4], which direct tumor development, progression, and metastasis through hierarchical control of gene expression patterns. Thus, MRs comprise typically a small number of TF-encoding genes (and their products) that control a disproportionate level of gene expression, giving rise to distinct molecular phenotypes associated with a particular disease. Therefore, identification and functional characterization of MRs is critical to understand associated disease processes and design effective therapeutic options [5]. For instance, a recent study indicated that SOX4 acted as a MR of epithelial-to-mesenchymal transition (EMT) in HCC [6]. However, studies evaluating the presence of MRs in HCC are sparse and have yielded limited insight into cancer risk.
Since the expression of genes defining discrete phenotypes is highly coordinated, application of reverse engineering algorithms to transcriptome datasets allows interpreting transcriptional networks by defining MRs and their associated regulons and gene circuits. Focusing on computational and statistical aspects of MR discovery, the ARACNe-MRA (Algorithm for the Reconstruction of Accurate Cellular Networks-Master Regulator Analysis) method has shown competent performance in this regard [4]. Using ARACNe-MRA, researchers have successfully identified prognostically relevant MRs in glioma, ovarian, breast, and prostate cancer [4, 7].
In the present study, we model liver cancer through a TF-centered regulatory network derived from HCC transcriptional datasets, followed by MRA analysis of changes in transcriptional regulatory programs related to tumor phenotype. Our analysis provides insights into the gene regulatory circuits operating in HCC and has implications for the identification of novel therapeutic targets.
Results
Generation of a compendium of gene expression profiles in human HCC
To assemble well-annotated human HCC gene expression profiles, 21 liver-oriented datasets were retrieved, yielding 1,316 gene expression profiles (Figure 1A). A detailed characterization of the HCC datasets is provided in Supplementary Table 1.
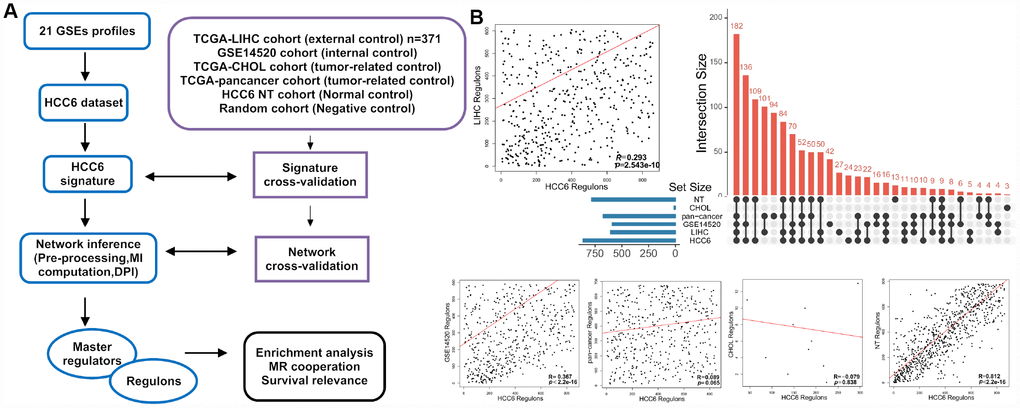
Figure 1. Schematic representation of MR discovery and validation in HCC datasets. (A) Different HCC-related gene expression datasets were analyzed in parallel with the ARACNe-MRA pipeline. (B) MRA agreement among different cohorts using unfiltered networks. The scatter plots show ranking agreement (by enrichment P-values) for all regulons between the different cohorts. Each dot represents one regulon (MR and all its targets) in the TN. The correlation coefficient R is given for each pairwise ranking. An UpSet plot of the intersection of identified regulons in different cohorts is presented in the right panel.
To ensure that the datasets generated from different types of arrays are comparable, the ComBat method [8], which is especially robust when handling multiple batches [9], was applied. For a more quantitative evaluation of the effects of dataset adjustment, principal variance component analysis (PVCA) [10] was performed to verify significant improvement after adjusting for intraplatform batch-to-batch differences (Supplementary Figure 1). In total, 12,100 annotated genes were present in the final dataset (named HCC6 hereafter).
Functional enrichment analysis of HCC signatures
Based on identical normalization, filtration, and statistical treatment of raw datasets, the HCC6 signature was defined as a set of genes differentially expressed between tumor tissues (PT) and adjacent non-tumor tissues (NT). It consisted of 483 genes (32% of them upregulated) and included well-known HCC biomarkers (e.g. GPC3, AFP, KPNA2; Supplementary Table 2). We carried out gene ontology and KEGG pathway enrichment analysis to uncover potential biological functions for genes in the HCC6 signature, which were consistent with reprogramming features characteristic of cancer cells (Supplementary Figure 2).
In line with previous research [2], overrepresentation of cell proliferation markers was the most prominent feature in our HCC6 signature. We observed upregulation of numerous cell cycling genes, including cyclins (e.g. CCNA2, CCNB1, CCNB2), cyclin-dependent kinases and inhibitors (e.g. CDK1, CDK4, CDKN3), and genes acting on cell cycle and cell division checkpoints (e.g. AURKA, NDC80). Also, the HCC6 signature highlighted deregulation of genes associated with DNA replication (e.g. MCM2-6, RFC4), DNA unwinding (e.g. TOP2A), and DNA repair (e.g. RRM2, FEN1). Increased expression of several genes associated with protein translation, i.e. ribosomal subunits (e.g. RPS5, RPL38), and translation initiation factors (e.g. EIF4G2) was also observed. In addition, genes acting at the epigenetic level such as regulators of chromatin assembly and remodeling (e.g. CHAF1A, HDAC1, HDAC5, HMGB2), and components of the polycomb-repressive complex 2 (e.g. EZH2, SUZ12) were also up-regulated.
Metabolic reprogramming was noticeable for downregulated genes involved in liver specific metabolism, including those encoding acute phase plasma proteins (e.g. A2M, ALB, CP), components of complement and coagulation cascades (e.g. C5-9, CFB, F2), and detoxication enzymes (e.g. ADH1A, CYP2E1). Key regulators of oxidation-reduction processes (e.g. STEAP3, CYP3A4, ALDH8A) were also repressed.
Regulon profiling in HCC
Next, a genome-wide transcriptional network (TN) centered on TFs and their predicted target genes was inferred using the RTN package [11, 12]. The computational pipeline is summarized in Figure 1A. Briefly, the TN was constructed by computing mutual information (MI) between annotated TFs [13] (n = 2,020 genes) and all potential targets in each cohort based on the ARACNe approach [14]. Then, the MRA algorithm was applied to compute the statistical significance of the overlap between the network and the molecular signature (differentially expressed genes). The groups of inferred target genes associated with each MR are hereinafter referred to as regulons.
To assess whether our results on the HCC6 dataset are consistent with other tumor-profiling datasets, we applied RTN on The Cancer Genome Atlas Liver Hepatocellular Carcinoma (TCGA-LIHC) data collection (external control), and on GSE14520 (internal control), etc, independently (Figure 1A). The enrichment P-value for each regulon was used to rank MRs identified in each network and the correlation statistic ® between these lists was compared.
We first ranked HCC6 regulons based on the enrichment score and found good agreement (R = 0.29-0.37) between HCC6, TCGA-LIHC, and GSE14520 (using its own differentially expressed genes as signature), suggesting that these HCC datasets share similar sets of regulated genes (Figure 1B). We then computed a TN for 990 tumor-matched, adjacent normal tissues from HCC6 patients. Enrichment for most MRs was found in this network, suggesting both PT and NT tissues share partial common regulatory network. Of note, after carrying out MRA on a network derived from a TCGA pan-cancer dataset, a lower level of correlation was observed (R = 0.09), while virtually no overlap was found between HCC6 and cholangiocarcinoma (TCGA-CHOL).
Landscape of master regulators of HCC
To preserve the dominant TF-target pairs in the filtered TN, the weakest interactions between any two TFs and a common target gene were removed applying the data processing inequality (DPI) method. The analysis yielded 120 MR candidates in the HCC6 dataset (Supplementary Table 3). Again, there was good agreement between regulons in the HCC6, LIHC, and GSE14520 cohorts (Figure 2A, 2B). The regulatory network graph in Figure 2C shows association patterns between MRs in the HCC6 dataset.
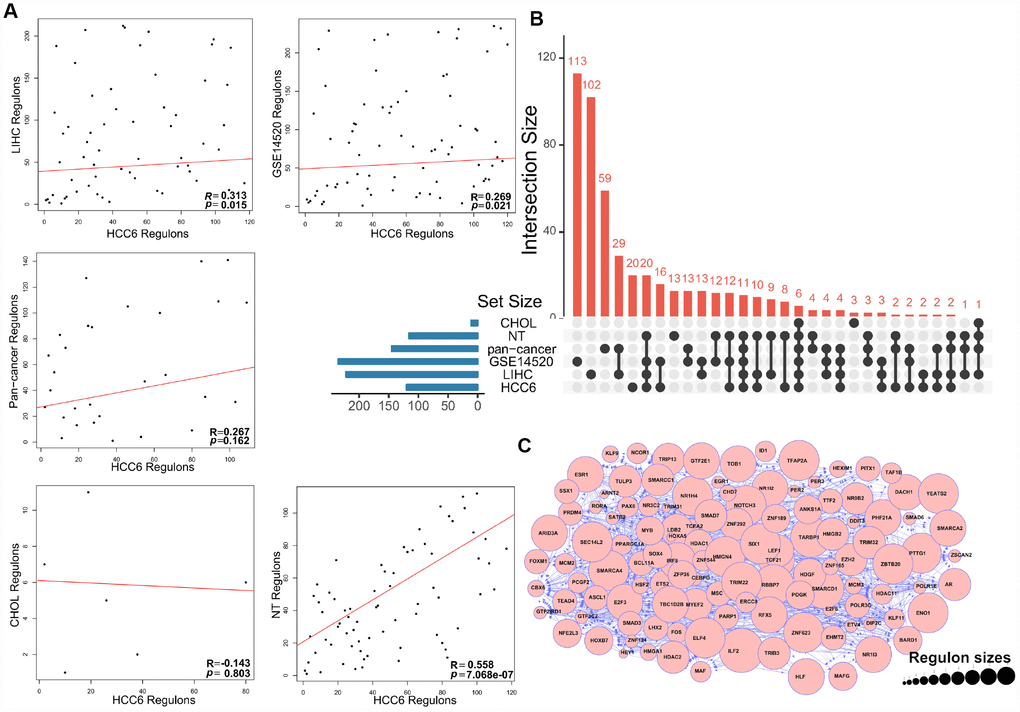
Figure 2. MRA agreement among different cohorts using filtered networks. (A) Networks were filtered by applying a DPI threshold of 0.01 to remove the weakest interactions. The scatter plots show ranking agreement (by enrichment P-value) for all regulons. (B) UpSet plot of the intersection of identified regulons in different cohorts. (C) Network visualization of the 120 MRs. The size of circles represents the size of each regulon.
Validation of master regulators of HCC
We set out to systemically check the quality of predicted MRs. Since both network and signature might affect causal inferences during MR discovery, we performed cross-validation of MRA results using TNs and signatures from different HCC datasets. Compared with the regulons identified in the HCC6 network using different signatures, results showed the HCC6 based network had strong robustness (Figure 3A).
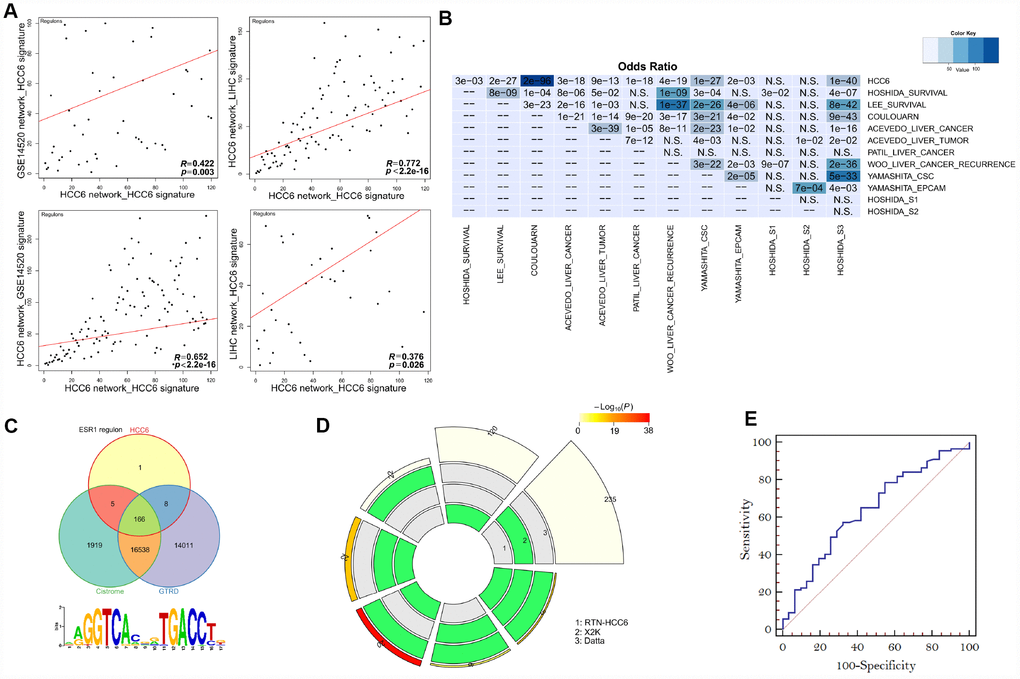
Figure 3. Performance of MR prediction. (A) MRs identified by using LIHC- and GSE14520-derived networks and the HCC6 signatures. The correlation coefficient R is given for each pairwise ranking. (B) Using HCC6 TN and different signatures, enrichment was estimated from the corresponding MRA analyses. The color key represents the odds ratios, and significant P-values are superimposed on the grids. (C) Venn diagrams showing overlap of the ESR1 regulon with corresponding targets from Cistrome and GTRD. The ESR1 motif was enriched in the ESR1 regulon (Pscan analysis). (D) A circular plot visualizing all intersections and the corresponding statistics among three MR discovery methods. The three tracks in the middle represent the three methods, with individual blocks showing “presence” (green) or “absence” (grey) of the gene sets in each intersection. The height of the bars in the outer layer is proportional to the intersection size, as indicated by the numbers on the top of the bars. The color intensity of the bars represents the significance (P value) of the intersections. (E) ROC curves plotted for MR prediction.
Aiming at assessing different features of liver cancer, various studies have reported molecular signatures related to HCC. Thus, we performed signature validation contrasting the TN derived from the HCC6 dataset with reported HCC-related signatures: progenitor tumor cell origin (CSC_Yamashita, EPCAM_Yamashita, CK19_Andersen [15], S2_Hoshida [16] and C2_Cario [17]), cellular proliferation [18], vascular invasion [19], TGF-beta_Coulouarn [20], MET_Kaposi-Novak [21], G3_Boyault [22], S1_Hoshida (TGFβ-WNT) [16]), Recurrence_Woo [23], OS_Kim [24], Interferon_Chiang [18], and G5/6_Boyault (CTTNB1_WNT activation) [22]. Results showed that the HCC6-signature covered almost all of these signatures (Figure 3B).
We also examined enrichment levels for target genes comprising the regulons of individual MRs. To this end, we collected recognized TF targets from the Cistrome Cancer web resource [25] and the Gene Transcription Regulation Database (GTRD) [26], both containing ChIP-seq derived processed data. As an example of this analysis, the ESR1 regulon highly overlapped with the targets from both Cistrome and GTRD. In turn, the motif discovery tool Pscan [27] confirmed that the ESR1 binding motif was enriched among its co-regulated genes (Figure 3C).
Besides ARACNe/RTN, we evaluated other methods designed for finding MRs in gene expression patterns, i.e. the Expression-2-Kinase (X2K) package with default parameters [28], and S. Datta’s approach [5]. Compared to these approaches, RTN identified more risk-MRs (Figure 3D).
Following a prototypic approach to validate cancer-associated MRs, we retrieved TFs annotated as cancer-related in at least one of three cancer gene databases (Bushman Laboratory cancer driver gene list [29], COSMIC somatic mutation catalog [30], and CCGD mouse cancer driver genes [31]). We found that 75% (90/120) of the MRs in our HCC6 gene set were annotated as cancer-associated in the above datasets. Applying information contained in these databases, receiver-operating characteristic curve analysis demonstrated the reliability of MR prediction (Figure 3E).
Identification of MRs associated with HCC subtypes and etiology
To answer the question of which MRs were associated with the different molecular subtypes of HCC, consensus hierarchical clustering [32] was performed on the HCC6 samples. Three tumor subgroups were discriminated from the consensus matrix (Figure 4A). Function annotation analyses (Figure 4B and 4D), MR overlap (Figure 4C), and protein-protein interaction (PPI) network of HCC6 MRs (Figure 4E) were further defined for the three HCC subtypes. For instance, network topology analysis indicated that HDAC1/2 were the top stress genes in the PPI network. As the epigenetic factors, HDACs control gene expression by recruiting multiple transcription factors and other chromatin-related factors. HDAC activate hepatocyte growth factor signaling in HCC [33].
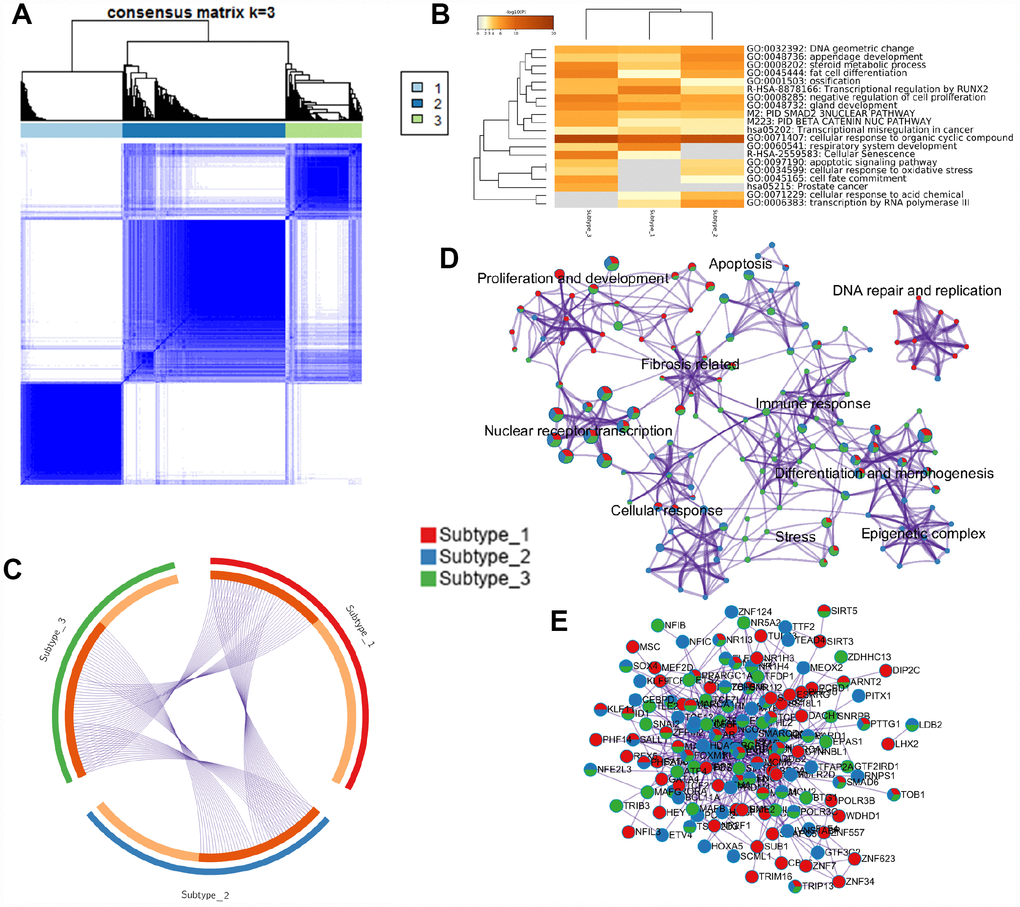
Figure 4. HCC subtyping analysis and functional annotation of MRs. (A) Three subgroups were identified by the non-negative matrix factorization (NMF) method in the HCC6 dataset, using consensus hierarchical clustering. (B) Meta-analysis of function annotation based on three subtype-related MR lists. Heatmap shows the top enrichment clusters (gray color indicates lack of significance). (C) Circos plot of MRs’ overlap among the three HCC subtypes. On the inside arc, dark orange color represents the MRs that appear in multiple subtypes and light orange color represents MRs that are unique to that subtype. (D) Enrichment network visualization for biological function from the three HCC subtypes. Nodes are represented by pie charts indicating their associations with each subtype. (E) Protein-protein interaction (PPI) network of MRs.
We next performed MRA analysis within the HCC subtypes, interpreting subtype-related MRs using Metascape [34]. Associations with ‘cellular response to organic cyclic compound’, ‘nuclear receptor transcription pathway’, ‘chromatin organization’, ‘regulation of cell cycle process’, and ‘hormone- mediated signaling pathway’, among others, were defined for all three HCC subtypes (Supplementary Table 4). Subtype 1 was associated with biological processes such as ‘mitochondrial biogenesis’, ‘gland, mesenchyme, epithelium and tubule development’, ‘circadian clock’, and FOXM1 and NOTCH3 pathways, among others. Subtype 2, containing the BHC (BRAF-HDAC) and the CoREST-HDAC complexes, was linked to ‘cellular response to drug, and antibiotic’, ‘cellular glucose homeostasis’, ‘positive regulation of DNA repair’, ‘telomerase pathway’, and AR and MYC pathways, among others. Subtypes 1 and 2 both showed activated AKT signaling. Lastly, subtype 3 was defined by transcriptional networks related to ‘cellular response to oxygen levels’, ‘oncogene induced senescence’, ‘repression of WNT target genes’, ‘T-helper 17 type immune response’, ‘stem cell proliferation’, and ‘TGF-beta signaling pathway’, among others.
Since risk factors for developing HCC include HBV or HCV infection, alcoholic liver disease, and metabolism disorders like nonalcoholic steatohepatitis, we therefore tested whether the etiology of tumors was linked to candidate MRs. Detailed information on etiology-related MRs is provided in Supplementary Table 5.
Consensus MRs in HCC
To define a smaller set of conserved MRs, we selected the regulons showing significant enrichment across the HCC6, LIHC, and GSE14520 cohorts (Figure 5A). The association map of the resultant 44 MRs is provided in Figure 5B.
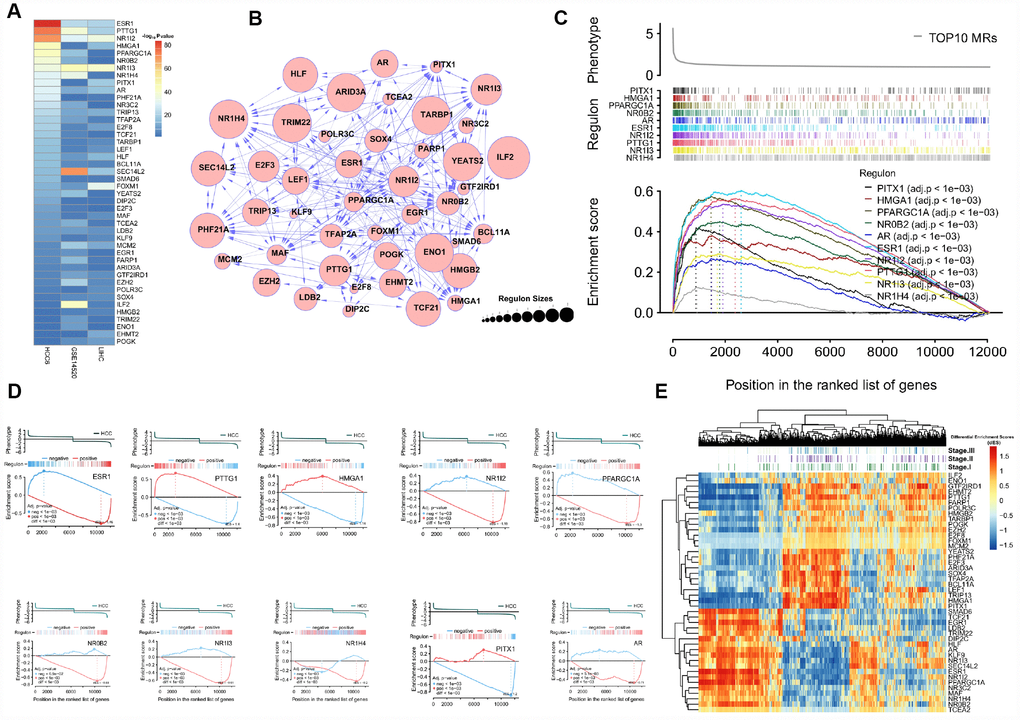
Figure 5. Profiling of conserved MRs. (A) Regulons are ranked by corresponding enrichment P-values, estimated for the HCC6, LIHC, and GSE14520 cohorts. (B) Network view of 44 consistent MRs. (C) GSEA of the top 10 MRs. (D) Examples of the top 10 MRs for which 2-tailed GSEA was carried out. (E) dES heatmap of the 44MRs.
Once identified, we further tested the responsiveness of each regulon to tumor phenotype using gene set enrichment analysis (GSEA) [35]. Figure 5C shows that the top 10 regulons are consistently HCC-responsive. Regulons consist of both MR-induced and MR-repressed genes, and their relative activity influences the phenotype. To evaluate regulon activity, two-tailed GSEA was performed to calculate differential enrichment scores (dES) based on positively and negatively regulated regulon subsets in HCC samples. Detailed target information for each MR is provided in Supplementary Table 6.
Figure 5D presents an example of this analysis, showing GSEA running enrichment scores for the ESR1 regulon after it was split into ESR1-activated and -repressed targets. Details on biological process involvement for the ESR1 regulon, indicating multiple metabolic pathway inhibition mechanisms, are shown in Supplementary Figure 3. Figure 5E shows the dES profile of 44 MRs; two clusters resulted, suggesting opposite biological roles in HCC pathogenesis.
Regulon activity and coordinated MRs as prognostic read-out
Next, dES representing regulon activity were used to investigate the prognostic value of MRs through Kaplan-Meier survival analysis. Almost half (20/44, P < 0.05) of the MRs in our HCC6 cohort were highly correlated with survival phenotype (Supplementary Table 7), compared with 14/44 and 32/44 in the GSE14520 and LIHC cohorts, respectively.
Figure 6 shows dES and survival plots for the top 2 regulons. For the ESR1 regulon, we found a continuous spectrum of dES across the tumors, except near the transition between its active and repressed state, which was characterized by an abrupt change. There was a strong trend for better survival in tumors with high dES. Significant trends were also noted after analyzing the GSE14520 and the LIHC cohorts, evaluated as controls (Figure 6C–6D). Upon stratification for ESR1 expression only, this MR was strongly correlated with prognosis in the LIHC cohort (Figure 6B), but not in the GSE14520 cohort (Supplementary Table 7). Meanwhile, an opposite survival trend was found for the PTTG1 regulon (Figure 6E–6H). These results suggest that regulon activity, expressed as dES, can predict survival outcome in a more context-dependent manner. Detailed MR-based survival analysis results are provided in Supplementary Figure 4.
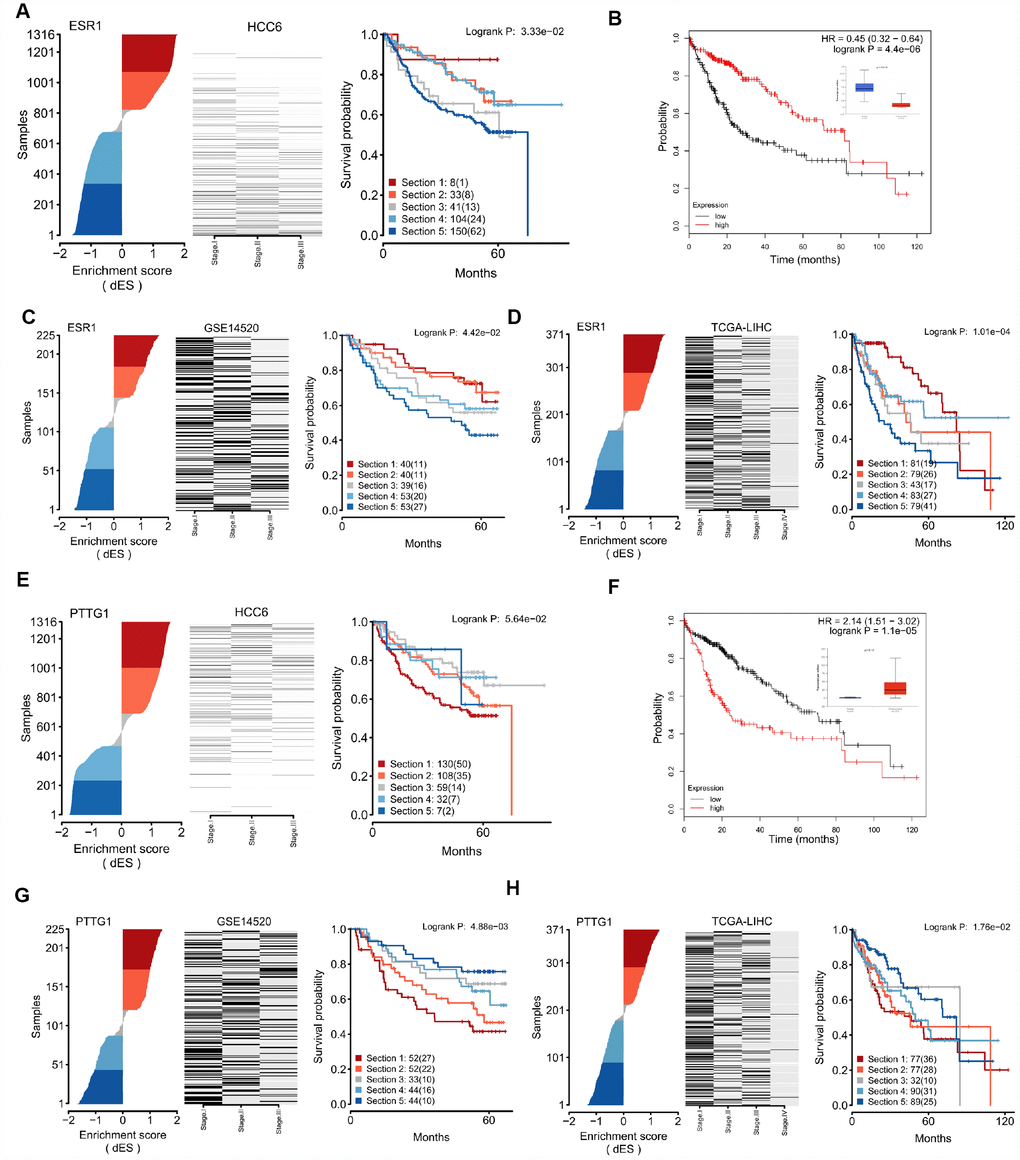
Figure 6. Regulon activity as read-out of survival outcomes. (A) dES of ESR1 calculated for all samples in the HCC6 cohort. Disease-specific survival plots for each tumor subgroup are highlighted; patient numbers are listed for each section. (B) Kaplan-Meier survival curve using ESR1 gene expression data in the LIHC cohort, generated using the KM-plotter tool [56]. (C) dES of ESR1 in the GSE14520 cohort. (D) dES of ESR1 in the LIHC cohort. (E–H) Analysis results for PTTG1.
Since MRs can have divergent effects on the expression of a shared target gene in a real cellular setting, we examined the relationship between regulons. Correlation values were used to assess whether or not MR pairs regulated shared target genes in the same (positive or negative) direction. This analysis was carried out for 44 MRs in our regulatory network, and a correlation heatmap was generated. In addition, a heatmap of Jaccard similarity coefficient (JC) focusing on the overlap between the 44 regulons was also obtained (Figure 7A).
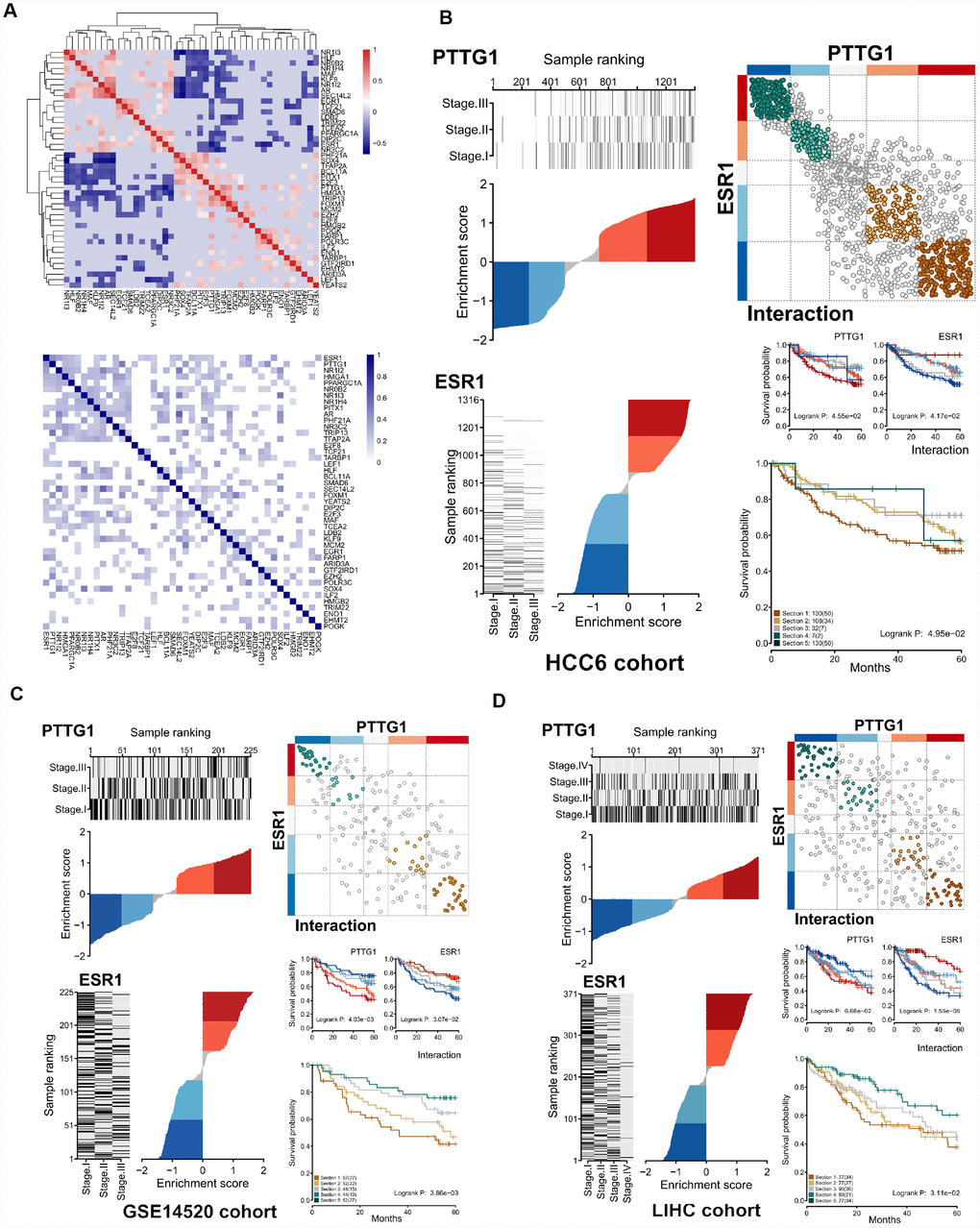
Figure 7. Differential expression effects of MR interactions on shared target genes. (A) Upper panel: Heatmap of gene expression correlation for targets shared by the 44 MRs (HCC6). Lower panel: Hierarchical clustering based on jaccard similarity coefficients (shades of blue) computed among 44 regulons. (B) Interaction of ESR1-PTTG1 pair in the HCC6 cohort. The dES of ESR1 and PTTG1 are shown. On the right upper panel, a cartoon depicts the observed interactions between ESR1 and PTTG1 targets, with brown circles indicating co-activation, and green circles denoting co-repression. Targets are shown in grey if the two MRs have opposing effects. Assuming ESR1 and PTTG1 interaction, survival outcomes for the ESR1 regulon, the PTTG1 regulon, and both interacting regulons are depicted (C, D).
Generally, JC values fell into two distinct groups with high correlation within each group: gene targets shared between two MRs in the same group are regulated in the same direction by both MRs, whereas gene targets shared between a MR in one group and a MR in the other are regulated in opposite directions. This suggests the existence of two distinct regulatory MR groups, each one opposing the effects of the other. Again, using ESR1 and PTTG1 as an example, these two MRs were highly anti-correlated and showed extensive overlap (R = -0.586, JC = 0.431). Considering compounding effect, stratification of activity of interacted-ESR1 and PTTG1 regulon further reveals different survival patterns. This influence was verified in the LIHC and GSE14250 cohorts (Figure 7C–7D).
Functional analysis of a candidate MR
The lipid-binding protein SEC14L2, which possesses putative transcriptional activatory activity, was predicted as a conservative MR in HCC in this study (Figure 8A). Functional enrichment analysis indicated that oxidation-reduction process, metabolic process, PPAR signaling, peroxisome pathway, and fatty acid degradation, etc, were significantly regulated by SEC14L2 regulon in HCC (Figure 8B, 8C).
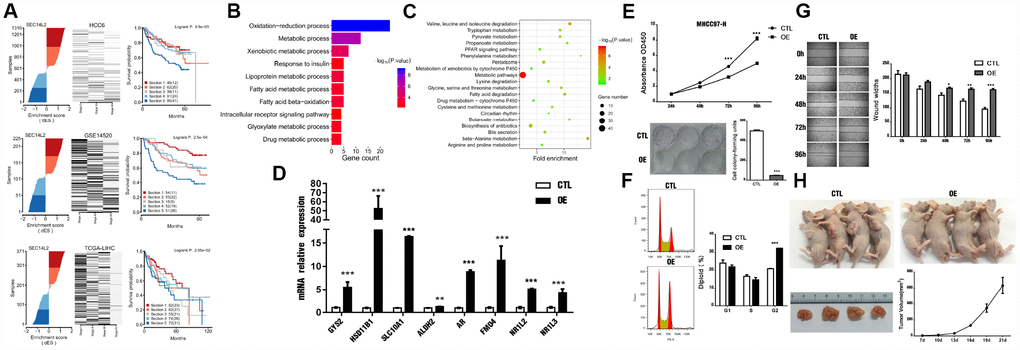
Figure 8. SEC14L2 is a potential HCC suppressor. (A) Calculation of dES and survival responses for SEC14L2 in the HCC6 cohort. (B) GO terms enrichment analysis of SEC14L2 regulon. (C) KEGG pathway enrichment analysis of SEC14L2 regulon. (D) Representative target genes of SEC14L2 regulon were significantly up-regulated after SEC14L2 overexpression. (E) SEC14L2 overexpression significantly inhibited cell proliferation (CCK-8 assay) and colony formation in HCC cell lines. Upper and lower panels show corresponding results in MHCC97-H cells. These assays were repeated in Huh7 cells (Supplementary Figure 6). (F) Cell-cycle distribution analysis of MHCC97-H cells overexpressing SEC14L2. (G) Cell migration (wound-healing) assay results. Data represent the average of three independent experiments in duplicate. (H) Effect of SEC14L2 overexpression on tumor xenografts in vivo. Almost total inhibition of tumor growth was observed after subcutaneous implantation of SEC14L2- overexpressing MHCC97-H cells in nude mice. OE, over-expressed; *P < 0.05, **P < 0.01, ***P < 0.001.
A recent report identified SEC14L2 as a host factor permitting replication of clinical HCV isolates [36], but the relationship between this molecule and HCC was not established. Notably, SEC14L2 expression could not be detected in human hepatoma and non-hepatoma cell lines in vitro [36]. However, primary human hepatocytes, both from fetal and adult sources, expressed readily detectable levels [36]. We thus examined the potential growth-suppressive effects of SEC14L2 re-expression in HCC cells.
Restored expression of SEC14L2 was observed in SEC14L2-transduced MHCC97-H cells, which showed reduced proliferation and decreased colony formation (Figure 8E and Supplementary Figure 5). Also, the number of cells in G2 phase following SEC14L2 ectopic expression was substantially increased (50.96 ± 0.20% vs 43.28 ± 0.95% in control cells; P < 0.05). On wound healing assays, ectopic expression of SEC14L2 distinctly inhibited migration of MHCC97-H cells (P < 0.01) (Figure 8G).
We subsequently investigated the effects of forced SEC14L2 expression on the tumorigenic potential of HCC cells in vivo. To this end, MHCC97-H cells stably transduced with SEC14L2 or empty vector were injected subcutaneously into nude mice. Periodic volume measurements showed that tumor growth was nearly abolished after SEC14L2-transduction, compared with tumors containing control cells (Figure 8H).
As expected, expression of the component genes like GYS2, HSD11B1, SLC10A1, ALDH2, and AR, etc, in SEC14L2 regulon were significantly up-regulated after the ectopic expression of SEC14L2 in MHCC-97H cells (Figure 8D). Briefly, GYS2 was recently found to be responsible for the deregulation of glycogen metabolism in HCC [37]. HSD11B1 was identified as a circulating biomarker candidate for HCC [38]. SLC10A1(NTCP) expression was markedly reduced in most HCC [39]. ALDH2 deficiency promotes liver cancer by activating oncogenic pathways via oxidized DNA-enriched extracellular vesicles [40]. These results suggested that SEC14L2 regulon might have a positive mechanism in metabolic pathways regulated by SEC14L2.
Discussion
Streamlining the analysis of multiple datasets using the same computational processes enhances statistical power and may significantly increase the accuracy of the findings [41, 42]. Our study compiled a large HCC dataset (HCC6) and verified its quality by PVCA analysis. Importantly, we show that the HCC6 signature panel overlaps extensively with a comprehensive 935-gene HCC signature reported by Allain et al. in a meta-analysis of multiple datasets [2]. This lends confidence that the HCC6 dataset captured the main features of HCC.
Several lines of evidence support the relevance of the MRs and associated regulons discovered in the present study. First, two of the most important parameters in MRA, i.e. transcription networks and gene signatures, were widely cross-tested. Upon similar analysis, MRs consistently identified from the HCC6 networks were absent in networks obtained from other malignancies. Second, most MRs identified in this work are cancer-related according to database annotation. Finally, the impact of some MRs on liver cancer predicted by HCC6 is highly consistent with current knowledge.
In this sense, the relation between ESR1 and malignant disease has been asserted in a variety of tissues including breast, colon, bladder, and liver. As a candidate tumor suppressor gene [43], decreased ESR1 expression was significantly correlated to high liver damage score, pathological invasion, and tumor size [44]. PTTG1, a human securin that inhibits sister chromatid separation and is involved in transformation and tumorigenesis, is overexpressed in HCC and has prognostic significance for postoperative survival of patients with HCC [45]. Our prediction also retrieved well-known MRs such as FOXM1, EZH2, and SOX4 [6]. FOXM1, cooperating with YAP, was found to contribute to chromosome instability in liver cancer [46].
Besides reaffirming findings from previous studies (Supplementary Table 8), our analysis also predicted putative MR functions in HCC. For instance, several MRs belonging to the orphan nuclear receptor (NR) family such as NR0B2, NR1I2, NR1H4, and NR1I3 were found.
NR0B2 is a transcriptional corepressor affecting diverse metabolic processes, including bile acid synthesis, cholesterol and lipid metabolism, and glucose and energy homeostasis. Evidence suggests that NR0B2 plays an important suppressing role in the development of liver cancer [47]. Research has revealed broader transcriptional circuits controlled by NR1H4. NR1H4 deficiency led to a 100% incidence of spontaneous liver tumors in male and female mice, indicating that disruption of estrogen-protected pathways promotes hepatic oncogenesis. NR1I2 plays an integral role in xenobiotic and endobiotic metabolism, glucocorticoid and mineralocorticoid homeostasis, vitamin metabolism, and hepatic gluconeogenesis, and was shown to promote tumor growth and chemo-resistance in major cancer types [48]. NR1I3 regulates a set of genes involved in cellular growth, and studies have shown that it may also aid in the promotion of tumor formation. Thus, our prediction suggests that multiple NRs might also be HCC-related MRs. Broader liver transcriptional circuits controlled by multiple orphan NRs warrant further consideration.
Overall, we found that 90 out of 120 MRs analyzed have support from cancer-gene database. As for the rest, little is known about their role in liver cancer according to current literature. For example, no study has so far implicated SEC14L2 in liver cancer, though proteomic analysis identified low levels of SEC14L2 to be prognostic markers for overall breast cancer survival [49]. Saeed et al. identified SEC14L2 as a host factor permitting replication of clinical HCV isolates in cell culture [36]. We found that low SEC14L2 expression is associated with poor patient survival in the HCC6, LIHC, and GSE14520 cohorts. Moreover, SEC14L2 cross-talks with many NRs (Supplementary Figure 5). Further support for the importance of SEC14L2 comes from our functional in vivo experiment, indicating that restoration of SEC14L2 expression in HCC cells significantly inhibited tumor growth.
Earlier, three sub-clusters were defined in HCC by the TCGA research team [50]. Clustering analysis of HCC6 transcriptome profiles also resulted in three clusters, and subtype-related MRs were predicted therein. It would be interesting to investigate the functions of these MRs in future studies.
Importantly, a significant correlation was observed between the regulon activity of various MRs and patient survival. In contrast, simple classification based on molecular expression levels did not unmask prognostic relevance across multiple datasets. For instance, ESR1 and PTTG1 do not have significant prognostic value in the GSE14520 cohort, though these two genes have a clear role in HCC. Therefore, MRs might have increased power to detect prognosis traits that are otherwise concealed by sample heterogeneity and co-regulated genes. The fact that MRs could predict clinical outcome, while MR pairs were differentially linked to opposite prognostic categories, prompted us to deduce that MR interactions have a significant, probably major, contribution to HCC pathogenesis. These findings may reconcile controversies on the prognostic importance of HCC biomarkers and devise an applicable way to subgroup patients based on MR interactions.
In summary, our integrative analysis led to the discovery of many MRs with putative roles in HCC development and progression. Further, the present study offers information to define MRs as biomarkers for early stage diagnosis and to direct targeted therapeutics for HCC.
Materials and Methods
HCC gene expression compendium
The HCC compendium was constructed by collecting 21 HCC-oriented datasets from six main microarray platforms (GPL570, GPL571, GPL3921, GPL96, GPL6244, and GPL10558) in Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) (i.e., GPL570:GSE1898 and GSE4024, GPL571/3921:GSE14520, etc.). All the datasets used fresh frozen liver tissues.
Raw data were uniformly aggregated and normalized using robust multi-array average (RMA) [51] or lumi package [52] according to the different platform. To ensure that the datasets generated from the six types of arrays are comparable, the ComBat batch correction method was performed to remove platform-specific effects [8]. Finally, 2,306 HCC gene expression profiles, including 1,316 tumor tissues (PT) and 990 adjacent non-tumor tissues (NT), were compiled. Genes with multiple probesets were represented by the mean intensity across all samples. RNA-sequencing liver cancer (TCGA-LIHC) Level 3 gene expression data and clinical information were downloaded from the Cancer Genome Atlas data portal.
HCC gene signature
Differentially expressed genes between PT and NT were identified by limma package. FDR adjusted P < 0.05 and an absolute fold change > 1.5 were used as cut-offs for analysis. Enrichment for biological functions or canonical pathways was assessed using DAVID [53] or Metascape [34]. ConsensusClusterPlus (CHC) was used to carry out consensus hierarchical clustering to identify subtypes [32], and consensus indices of each pair of samples were visualized in a consensus matrix.
Network inference and MRA analysis
R package RTN [11, 12] was used to reconstruct and analyze the regulatory network based on mutual information (MI), a measure that evaluates dependencies between two random variables. Briefly, the regulatory structure of the network is derived by mapping significant associations between a known TF and its potential targets. Interactions below a minimum MI threshold are eliminated by a permutation step and unstable interactions are further removed by bootstrap to create a consensus network. In a last step, the data processing inequality (DPI) algorithm is applied with null tolerance to eliminate interactions that are likely to be mediated by another TF. The RedeR package [54] was used to visualize the network.
After network inference, master regulator analysis (MRA) was performed. The algorithm computes the statistical significance of the overlap between the regulon and the signatures (differentially expressed genes obtained from each study), corrected for multiple comparisons.
Regulon activity and survival analysis
Two-tailed gene set enrichment analysis (GSEA) was performed to calculate regulon activity with 1000 permutations, as previously described [11, 12]. Briefly, the resultant regulon was split into two subgroups, positive targets (A) and negative targets (B), using Pearson correlation. Next, independent enrichment scores (ES) for each subgroup were tested by GSEA statistics in the ranked phenotype, with two enrichment distributions. Regulon activity, represented by differential enrichment score (dES=ESA-ESB), was thus computed. A highly positive dES implies that the regulon is induced, while a highly negative dES indicates that the regulatory unit is repressed, in the disease phenotype. The two-tail GSEA P-value cutoff was set to 0.05 and 1000 permutations were used.
Survival analysis was performed using log-rank statistics. For stratified tests, patients were divided into three groups based on dES values: those with an active regulon (dES>0 and ESA>0 and ESB<0), those with a repressed regulon (dES<0 and ESA<0 and ESB>0), and a small group in which the dES values were around zero (inconclusive). The two large groups were further subdivided in half.
Cell culture
The liver cancer cell lines Huh7 and MHCC97-H were obtained from the Cell Bank of Chinese Academy of Sciences (Shanghai, China), and have been authenticated by STR DNA profiling. Cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 100U/ml penicillin, 100μg/ml streptomycin and 10% fetal bovine serum. Cultures were performed at 37°C in a 5% CO2 atmosphere.
Cell transduction
Lentivirus production in HEK293T cells was performed as previously described [55]. MHCC97-H cells were infected with lentivirus expressing SEC14L2 at a multiplicity of infection (M.O.I.) of 5 in the presence of 10 μg/ml polybrene (Sigma, USA) for 16 h, and selected using 2 μg/ml of puromycin (Sigma, USA) for one week. Stable control and SEC14L2-overexpressing MHCC97-H cells were obtained after verification of SEC14L2 expression by qPCR.
Cell proliferation assays
Cell proliferation was detected with CCK8 reagents (Dojindo, Japan). To evaluate colony formation, cells infected with lentivirus were cultured for 3 weeks, stained with gentian violet, and colonies with >50 cells were counted. Cell cycle distribution was analyzed using a cell cycle staining kit (Multisciences Biotech Co., China) and flow cytometry. The experiments were repeated at least three times.
In vivo tumorigenicity
Male athymic nude mice (4-5 weeks old) were purchased from Shanghai Laboratory Animal Co. Ltd (SLAC, China). Mice were injected subcutaneously with 5 × 106 MHCC97-H cells in 0.1 mL of serum-free DMEM. The left flank was implanted with control tumor cells whereas the right side was injected with SEC14L2-transduced tumor cells. Animal procedures were approved by Hangzhou Normal University’s Animal Care and Use Committee (Hangzhou, China).
Statistics
Bioinformatics analysis of microarray data was carried out in R (version 3.4.1) and statistical analysis of experimental results was performed using Prism GraphPad software (version 7). Unpaired Student’s t-test was used to compute statistical significance, set at p < 0.05 unless specified. Data are presented as mean ± SEM.
Supplementary Materials
Author Contributions
JY and JPS conceived, designed, and supervised the study. ZHL, YL, GYT, JYW, and JY carried out data processing and computational analyses. AQL, CJ, BBX, and ZHL performed cellular and animal assays. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare that they have no conflicts of interests.
Funding
This work was supported in part through funding from the National Natural Science Foundation of China (81772520), Zhejiang Provincial Natural Science Foundation (LGF19H030004, LY17H030009), Zhejiang Medical and Health Technology Project (2018PY039), Zhejiang Traditional Chinese Medicine Science and Technology Project (2018ZA098), and Hangzhou Health Technology Project (2017A36).
References
- 1. Llovet JM, Hernandez-Gea V. Hepatocellular carcinoma: reasons for phase III failure and novel perspectives on trial design. Clin Cancer Res. 2014; 20:2072–9. https://doi.org/10.1158/1078-0432.CCR-13-0547 [PubMed]
- 2. Allain C, Angenard G, Clément B, Coulouarn C. Integrative Genomic Analysis Identifies the Core Transcriptional Hallmarks of Human Hepatocellular Carcinoma. Cancer Res. 2016; 76:6374–81. https://doi.org/10.1158/0008-5472.CAN-16-1559 [PubMed]
- 3. Jiang P, Freedman ML, Liu JS, Liu XS. Inference of transcriptional regulation in cancers. Proc Natl Acad Sci USA. 2015; 112:7731–36. https://doi.org/10.1073/pnas.1424272112 [PubMed]
- 4. Moran B, Rahman A, Palonen K, Lanigan FT, Gallagher WM. Master Transcriptional Regulators in Cancer: Discovery via Reverse Engineering Approaches and Subsequent Validation. Cancer Res. 2017; 77:2186–90. https://doi.org/10.1158/0008-5472.CAN-16-1813 [PubMed]
- 5. Sikdar S, Datta S. A novel statistical approach for identification of the master regulator transcription factor. BMC Bioinformatics. 2017; 18:79. https://doi.org/10.1186/s12859-017-1499-x [PubMed]
- 6. Lourenço AR, Coffer PJ. SOX4: Joining the Master Regulators of Epithelial-to-Mesenchymal Transition? Trends Cancer. 2017; 3:571–82. https://doi.org/10.1016/j.trecan.2017.06.002 [PubMed]
- 7. Zhang S, Jing Y, Zhang M, Zhang Z, Ma P, Peng H, Shi K, Gao WQ, Zhuang G. Stroma-associated master regulators of molecular subtypes predict patient prognosis in ovarian cancer. Sci Rep. 2015; 5:16066. https://doi.org/10.1038/srep16066 [PubMed]
- 8. Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007; 8:118–27. https://doi.org/10.1093/biostatistics/kxj037 [PubMed]
- 9. Chen C, Grennan K, Badner J, Zhang D, Gershon E, Jin L, Liu C. Removing batch effects in analysis of expression microarray data: an evaluation of six batch adjustment methods. PLoS One. 2011; 6:e17238. https://doi.org/10.1371/journal.pone.0017238 [PubMed]
- 10. Boedigheimer MJ, Wolfinger RD, Bass MB, Bushel PR, Chou JW, Cooper M, Corton JC, Fostel J, Hester S, Lee JS, Liu F, Liu J, Qian HR, et al. Sources of variation in baseline gene expression levels from toxicogenomics study control animals across multiple laboratories. BMC Genomics. 2008; 9:285. https://doi.org/10.1186/1471-2164-9-285 [PubMed]
- 11. Castro MA, de Santiago I, Campbell TM, Vaughn C, Hickey TE, Ross E, Tilley WD, Markowetz F, Ponder BA, Meyer KB. Regulators of genetic risk of breast cancer identified by integrative network analysis. Nat Genet. 2016; 48:12–21. https://doi.org/10.1038/ng.3458 [PubMed]
- 12. Fletcher MN, Castro MA, Wang X, de Santiago I, O’Reilly M, Chin SF, Rueda OM, Caldas C, Ponder BA, Markowetz F, Meyer KB. Master regulators of FGFR2 signalling and breast cancer risk. Nat Commun. 2013; 4:2464. https://doi.org/10.1038/ncomms3464 [PubMed]
- 13. Nicolle R, Radvanyi F, Elati M. CoRegNet: reconstruction and integrated analysis of co-regulatory networks. Bioinformatics. 2015; 31:3066–68. https://doi.org/10.1093/bioinformatics/btv305 [PubMed]
- 14. Margolin AA, Wang K, Lim WK, Kustagi M, Nemenman I, Califano A. Reverse engineering cellular networks. Nat Protoc. 2006; 1:662–71. https://doi.org/10.1038/nprot.2006.106 [PubMed]
- 15. Andersen JB, Loi R, Perra A, Factor VM, Ledda-Columbano GM, Columbano A, Thorgeirsson SS. Progenitor-derived hepatocellular carcinoma model in the rat. Hepatology. 2010; 51:1401–09. https://doi.org/10.1002/hep.23488 [PubMed]
- 16. Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, Villanueva A, Newell P, Ikeda K, Hashimoto M, Watanabe G, Gabriel S, Friedman SL, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009; 69:7385–92. https://doi.org/10.1158/0008-5472.CAN-09-1089 [PubMed]
- 17. Cairo S, Armengol C, De Reyniès A, Wei Y, Thomas E, Renard CA, Goga A, Balakrishnan A, Semeraro M, Gresh L, Pontoglio M, Strick-Marchand H, Levillayer F, et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell. 2008; 14:471–84. https://doi.org/10.1016/j.ccr.2008.11.002 [PubMed]
- 18. Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, LeBlanc AC, Donovan DJ, Thung SN, Solé M, Tovar V, Alsinet C, Ramos AH, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008; 68:6779–88. https://doi.org/10.1158/0008-5472.CAN-08-0742 [PubMed]
- 19. Mínguez B, Hoshida Y, Villanueva A, Toffanin S, Cabellos L, Thung S, Mandeli J, Sia D, April C, Fan JB, Lachenmayer A, Savic R, Roayaie S, et al. Gene-expression signature of vascular invasion in hepatocellular carcinoma. J Hepatol. 2011; 55:1325–31. https://doi.org/10.1016/j.jhep.2011.02.034 [PubMed]
- 20. Coulouarn C, Factor VM, Thorgeirsson SS. Transforming growth factor-beta gene expression signature in mouse hepatocytes predicts clinical outcome in human cancer. Hepatology. 2008; 47:2059–67. https://doi.org/10.1002/hep.22283 [PubMed]
- 21. Kaposi-Novak P, Lee JS, Gòmez-Quiroz L, Coulouarn C, Factor VM, Thorgeirsson SS. Met-regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J Clin Invest. 2006; 116:1582–95. https://doi.org/10.1172/JCI27236 [PubMed]
- 22. Boyault S, Rickman DS, de Reyniès A, Balabaud C, Rebouissou S, Jeannot E, Hérault A, Saric J, Belghiti J, Franco D, Bioulac-Sage P, Laurent-Puig P, Zucman-Rossi J. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007; 45:42–52. https://doi.org/10.1002/hep.21467 [PubMed]
- 23. Woo HG, Park ES, Cheon JH, Kim JH, Lee JS, Park BJ, Kim W, Park SC, Chung YJ, Kim BG, Yoon JH, Lee HS, Kim CY, Yi NJ, Suh KS, Lee KU, Chu IS, Roskams T, Thorgeirsson SS, Kim YJ. Gene expression-based recurrence prediction of hepatitis B virus-related human hepatocellular carcinoma. Clin Cancer Res. 2008; 14:2056–64. https://doi.org/10.1158/1078-0432.CCR-07-1473 [PubMed]
- 24. Kim SM, Leem SH, Chu IS, Park YY, Kim SC, Kim SB, Park ES, Lim JY, Heo J, Kim YJ, Kim DG, Kaseb A, Park YN, et al. Sixty-five gene-based risk score classifier predicts overall survival in hepatocellular carcinoma. Hepatology. 2012; 55:1443–52. https://doi.org/10.1002/hep.24813 [PubMed]
- 25. Mei S, Meyer CA, Zheng R, Qin Q, Wu Q, Jiang P, Li B, Shi X, Wang B, Fan J, Shih C, Brown M, Zang C, Liu XS. Cistrome Cancer: A Web Resource for Integrative Gene Regulation Modeling in Cancer. Cancer Res. 2017; 77:e19–22. https://doi.org/10.1158/0008-5472.CAN-17-0327 [PubMed]
- 26. Yevshin I, Sharipov R, Valeev T, Kel A, Kolpakov F. GTRD: a database of transcription factor binding sites identified by ChIP-seq experiments. Nucleic Acids Res. 2017; 45:D61–67. https://doi.org/10.1093/nar/gkw951 [PubMed]
- 27. Zambelli F, Pesole G, Pavesi G. Using Weeder, Pscan, and PscanChIP for the Discovery of Enriched Transcription Factor Binding Site Motifs in Nucleotide Sequences. Curr Protoc Bioinformatics. 2014; 47:2.11.1–31. https://doi.org/10.1002/0471250953.bi0211s47 [PubMed]
- 28. Clarke DJ, Kuleshov MV, Schilder BM, Torre D, Duffy ME, Keenan AB, Lachmann A, Feldmann AS, Gundersen GW, Silverstein MC, Wang Z, Ma’ayan A. eXpression2Kinases (X2K) Web: linking expression signatures to upstream cell signaling networks. Nucleic Acids Res. 2018; 46:W171–79. https://doi.org/10.1093/nar/gky458 [PubMed]
- 29. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA
Jr , Kinzler KW. Cancer genome landscapes. Science. 2013; 339:1546–58. https://doi.org/10.1126/science.1235122 [PubMed] - 30. Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat Rev Cancer. 2004; 4:177–83. https://doi.org/10.1038/nrc1299 [PubMed]
- 31. Abbott KL, Nyre ET, Abrahante J, Ho YY, Isaksson Vogel R, Starr TK. The Candidate Cancer Gene Database: a database of cancer driver genes from forward genetic screens in mice. Nucleic Acids Res. 2015; 43:D844–48. https://doi.org/10.1093/nar/gku770 [PubMed]
- 32. Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010; 26:1572–73. https://doi.org/10.1093/bioinformatics/btq170 [PubMed]
- 33. Buurman R, Gürlevik E, Schäffer V, Eilers M, Sandbothe M, Kreipe H, Wilkens L, Schlegelberger B, Kühnel F, Skawran B. Histone deacetylases activate hepatocyte growth factor signaling by repressing microRNA-449 in hepatocellular carcinoma cells. Gastroenterology. 2012; 143:811–820.e15. https://doi.org/10.1053/j.gastro.2012.05.033 [PubMed]
- 34. Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019; 10:1523. https://doi.org/10.1038/s41467-019-09234-6 [PubMed]
- 35. Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics. 2007; 23:3251–53. https://doi.org/10.1093/bioinformatics/btm369 [PubMed]
- 36. Saeed M, Andreo U, Chung HY, Espiritu C, Branch AD, Silva JM, Rice CM. SEC14L2 enables pan-genotype HCV replication in cell culture. Nature. 2015; 524:471–75. https://doi.org/10.1038/nature14899 [PubMed]
- 37. Chen SL, Zhang CZ, Liu LL, Lu SX, Pan YH, Wang CH, He YF, Lin CS, Yang X, Xie D, Yun JP. A GYS2/p53 Negative Feedback Loop Restricts Tumor Growth in HBV-Related Hepatocellular Carcinoma. Cancer Res. 2019; 79:534–45. https://doi.org/10.1158/0008-5472.CAN-18-2357 [PubMed]
- 38. Awan FM, Naz A, Obaid A, Ali A, Ahmad J, Anjum S, Janjua HA. Identification of Circulating Biomarker Candidates for Hepatocellular Carcinoma (HCC): An Integrated Prioritization Approach. PLoS One. 2015; 10:e0138913. https://doi.org/10.1371/journal.pone.0138913 [PubMed]
- 39. Zollner G, Wagner M, Fickert P, Silbert D, Fuchsbichler A, Zatloukal K, Denk H, Trauner M. Hepatobiliary transporter expression in human hepatocellular carcinoma. Liver Int. 2005; 25:367–79. https://doi.org/10.1111/j.1478-3231.2005.01033.x [PubMed]
- 40. Seo W, Gao Y, He Y, Sun J, Xu H, Feng D, Park SH, Cho YE, Guillot A, Ren T, Wu R, Wang J, Kim SJ, et al. ALDH2 deficiency promotes alcohol-associated liver cancer by activating oncogenic pathways via oxidized DNA-enriched extracellular vesicles. J Hepatol. 2019; 71:1000–11. https://doi.org/10.1016/j.jhep.2019.06.018 [PubMed]
- 41. Ballouz S, Verleyen W, Gillis J. Guidance for RNA-seq co-expression network construction and analysis: safety in numbers. Bioinformatics. 2015; 31:2123–30. https://doi.org/10.1093/bioinformatics/btv118 [PubMed]
- 42. Zhu F, Shi L, Engel JD, Guan Y. Regulatory network inferred using expression data of small sample size: application and validation in erythroid system. Bioinformatics. 2015; 31:2537–44. https://doi.org/10.1093/bioinformatics/btv186 [PubMed]
- 43. Li J, Wang Y, Zhu Y, Gong Y, Yang Y, Tian J, Zhang Y, Zou D, Peng X, Ke J, Gong J, Zhong R, Chang J. Breast cancer risk-associated variants at 6q25.1 influence risk of hepatocellular carcinoma in a Chinese population. Carcinogenesis. 2017; 38:447–54. https://doi.org/10.1093/carcin/bgx024 [PubMed]
- 44. Hishida M, Nomoto S, Inokawa Y, Hayashi M, Kanda M, Okamura Y, Nishikawa Y, Tanaka C, Kobayashi D, Yamada S, Nakayama G, Fujii T, Sugimoto H, et al. Estrogen receptor 1 gene as a tumor suppressor gene in hepatocellular carcinoma detected by triple-combination array analysis. Int J Oncol. 2013; 43:88–94. https://doi.org/10.3892/ijo.2013.1951 [PubMed]
- 45. Fujii T, Nomoto S, Koshikawa K, Yatabe Y, Teshigawara O, Mori T, Inoue S, Takeda S, Nakao A. Overexpression of pituitary tumor transforming gene 1 in HCC is associated with angiogenesis and poor prognosis. Hepatology. 2006; 43:1267–75. https://doi.org/10.1002/hep.21181 [PubMed]
- 46. Weiler SME, Pinna F, Wolf T, Lutz T, Geldiyev A, Sticht C, Knaub M, Thomann S, Bissinger M, Wan S, Rössler S, Becker D, Gretz N, Lang H, Bergmann F, Ustiyan V, Kalin TV, Singer S, Lee JS, Marquardt JU, Schirmacher P, Kalinichenko VV, Breuhahn K. Induction of Chromosome Instability by Activation of Yes-Associated Protein and Forkhead Box M1 in Liver Cancer. Gastroenterology. 2017; 152:2037–2051.e22. https://doi.org/10.1053/j.gastro.2017.02.018 [PubMed]
- 47. Zou A, Lehn S, Magee N, Zhang Y. New Insights into Orphan Nuclear Receptor SHP in Liver Cancer. Nucl Receptor Res. 2015; 2:2. https://doi.org/10.11131/2015/101162 [PubMed]
- 48. Braeuning A, Gavrilov A, Brown S, Wolf CR, Henderson CJ, Schwarz M. Phenobarbital-mediated tumor promotion in transgenic mice with humanized CAR and PXR. Toxicol Sci. 2014; 140:259–70. https://doi.org/10.1093/toxsci/kfu099 [PubMed]
- 49. Geiger T, Madden SF, Gallagher WM, Cox J, Mann M. Proteomic portrait of human breast cancer progression identifies novel prognostic markers. Cancer Res. 2012; 72:2428–39. https://doi.org/10.1158/0008-5472.CAN-11-3711 [PubMed]
- 50. Cancer Genome Atlas Research Network. Electronic address: [email protected]; Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell. 2017; 169:1327–1341.e23. https://doi.org/10.1016/j.cell.2017.05.046 [PubMed]
- 51. Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003; 4:249–64. https://doi.org/10.1093/biostatistics/4.2.249 [PubMed]
- 52. Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008; 24:1547–48. https://doi.org/10.1093/bioinformatics/btn224 [PubMed]
- 53. Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4:44–57. https://doi.org/10.1038/nprot.2008.211 [PubMed]
- 54. Castro MA, Wang X, Fletcher MN, Meyer KB, Markowetz F, Rede R. RedeR: R/Bioconductor package for representing modular structures, nested networks and multiple levels of hierarchical associations. Genome Biol. 2012; 13:R29. https://doi.org/10.1186/gb-2012-13-4-r29 [PubMed]
- 55. Benskey MJ, Manfredsson FP. Lentivirus Production and Purification. Methods Mol Biol. 2016; 1382:107–14. https://doi.org/10.1007/978-1-4939-3271-9_8 [PubMed]
- 56. Nagy Á, Lánczky A, Menyhárt O, Győrffy B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep. 2018; 8:9227. https://doi.org/10.1038/s41598-018-27521-y [PubMed]