



Introduction
The prevalence of chronic kidney disease (CKD) is high and increasing continuously [1, 2]. Renal fibrosis is a common outcome of a wide variety of CKD and is associated with compromised kidney functions, leading to eventual end-stage renal disease, for which the renal function needs to be ameliorated and/or mitigated by undergoing dialysis or kidney transplantation [3]. However, no effective therapy is available to inhibit or reverse renal fibrosis.
Renal fibrosis is characterized by tubular atrophy, inflammatory cell infiltration, activated myofibroblasts accumulation, and excessive extracellular matrix (ECM) deposition [4, 5]. Myofibroblasts, characterized by being α-smooth muscle actin (α-SMA) positive in renal interstitium, play an important role in the process of fibrosis and are proportionally correlated with severity of renal fibrosis [6]. In addition, many cytokines contribute to renal fibrosis, including TGF-β1, and directly promote ECM production in renal interstitium by epithelial cells and fibroblasts.
Tumor necrosis factor superfamily protein 14 (TNFSF14), also called as LIGHT or CD258, is a 29-kD type II transmembrane protein expressed primarily on activated T lymphocytes and other immunocytes [7]. TNFSF14 plays an important role in immune and inflammatory responses and can exist in a soluble form by proteolytic cleavage [8–10]. Recently, several studies have reported that TNFSF14 contributes to tissue remodeling and fibrosis, which are initiated by inflammatory conditions such as skin fibrosis, pulmonary fibrosis, and asthmatic airway remodeling, and rheumatoid arthritis [11–14]. In addition, TNFSF14 expression is induced by epithelial damage and directly increases the level of primary human bronchial epithelial cells (hBECs) undergoing EMT and expressing matrix metallopeptidase-9 [15]. However, the role of TNFSF14 in renal fibrosis pathogenesis remains unknown.
Sphingosine kinase 1 (Sphk1), an enzyme that produces sphingosine-1-phosphate (S1P), has gained considerable attention owing to its potential involvement in renal inflammation and fibrosis progression [16]. Sphk1 is up-regulated in inflammatory related kidney diseases, such as diabetic nephropathy and polycystic kidney disease [17, 18]. Moreover, it is well-established that Sphk1/S1P signaling promotes renal fibrosis by up-regulating miR-21, and Sphk1 silencing by siRNA treatment results in a reduction in fibronectin [19, 20]. However, how TNFSF14 pathway correlates Sphk1 expression during renal fibrosis progression is unclear.
In the present study, we have provided direct evidence, for the first time, that TNFSF14 is a novel pro-fibrotic factor in renal fibrosis progression, for which TNFSF14 up-regulates Sphk1 expression. Inhibition of the TNFSF14 pathway is plausible for the clinical treatment of patients with renal fibrosis.
Results
Increased TNFSF14 expression in the fibrotic kidney
To address TNFSF14 relevance in determining kidney fibrosis, we used the unilateral ureteral obstruction (UUO)-induced renal fibrosis mouse model. It was found that TNFSF14 levels both in kidney tissues (Figure 1A and 1B) and in serum (Figure 1C) increased rapidly within 3 days and peaked at day 7 after UUO surgery (Figure 1A and 1C). TNFSF14 receptors, HVEM and LTβR, also increased remarkably in kidney tissues at day 7 after UUO surgery (Supplementary Figure 2A and 2B). Moreover, both TNFSF14 and HVEM/LTβR were primarily expressed in the tubular epithelia rather than in the interstitium (Figure 1B and Supplementary Figure 2).
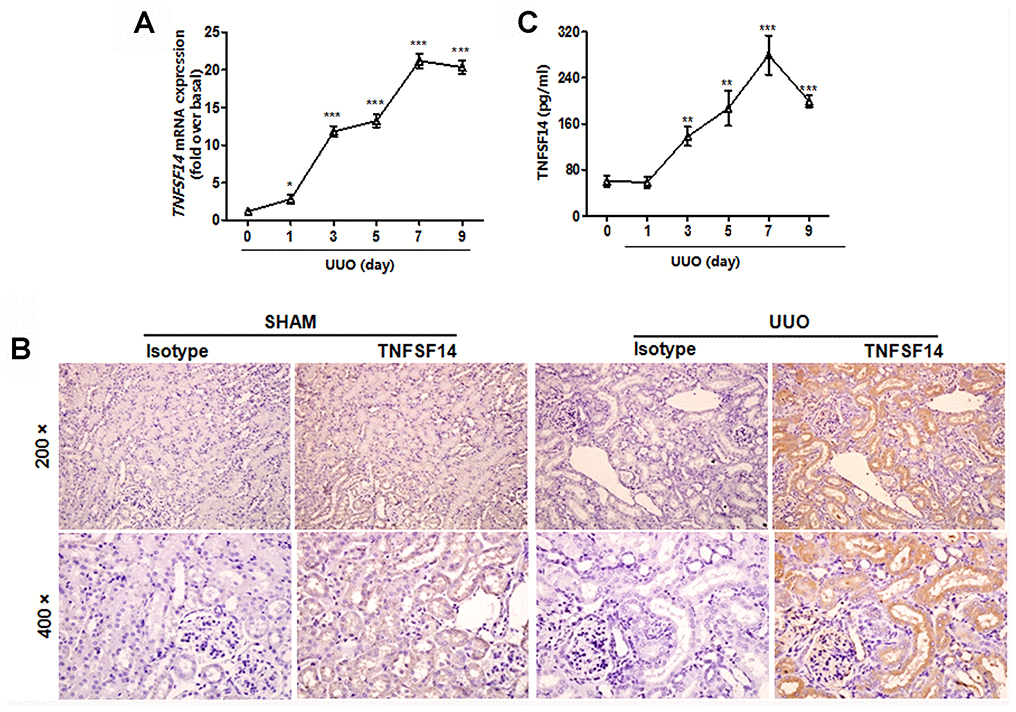
Figure 1. Increased TNFSF14 expression in mice after UUO surgery. (A) TNFSF14 expression in kidney tissues at the indicated time points in Tnfsf14+/+ mice after UUO surgery was assessed by qRT-PCR. GAPDH was used as the internal control. (B) The expression of TNFSF14 in the kidney tissues of Tnfsf14+/+ mice after UUO for 7 days was detected by immunohistochemistry (upper lane, original magnification ×200; lower lane, original magnification ×400). (C) TNFSF14 levels in serum in Tnfsf14+/+ mice at the indicated time points after UUO surgery were measured by ELISA. The data were representative of the results of three independent experiments. All values are represented as mean ± SEM. Sham group was used as the UUO control. n = 5 per group. *P < 0.01, **P < 0.01 and ***P < 0.001.
Through specimen examination of patients with CKD (including membranous nephritis, focal segmental glomerulosclerosis and thrombotic microangiopathy), which showed fibrotic lesions in kidney tissues (Supplementary Figure 3), we also detected increased expression of TNFSF14 and its receptors in biopsy kidney tissues (Figure 2A and 2B) and in serum (Figure 2C).
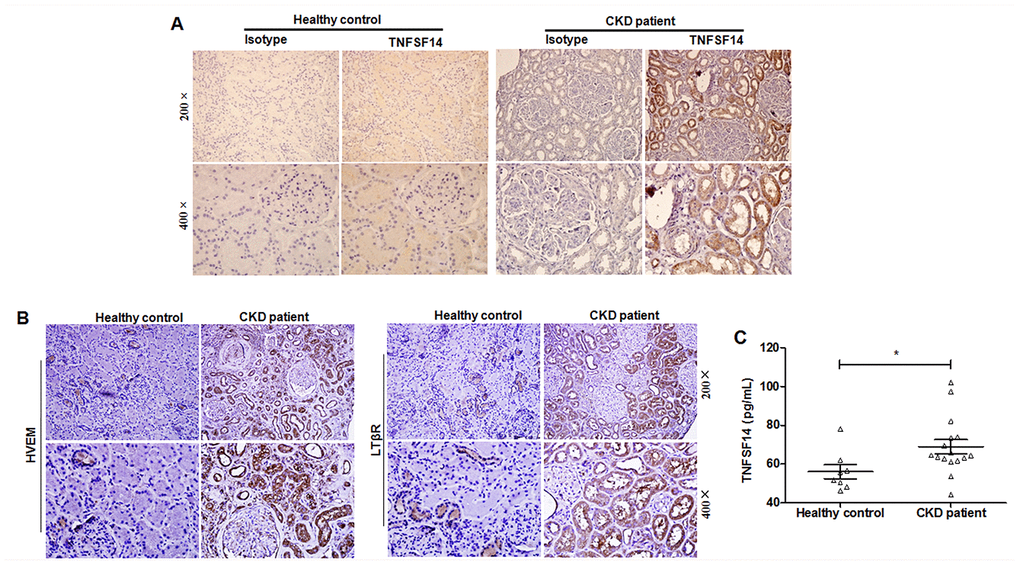
Figure 2. Increased TNFSF14 expression in patients with fibrotic nephropathy. (A) The expression of TNFSF14 in human kidney sections from healthy control, CKD patients were detected by immunohistochemistry (upper lane, original magnification×200; lower lane, original magnification×400). (B) The expression of TNFSF14 receptors (HVEM and LTβR) in biopsied human kidney specimens from patients with CKD were detected by immunohistochemistry. (upper lane, original magnification×200; lower lane, original magnification×400). Nontumoral kidney tissue from patients with renal cell carcinoma was used as healthy control. (C) TNFSF14 levels in serum from healthy controls (n = 8) and patients with CKD (n = 16) were measured by ELISA. The data were representative of the results of three independent experiments. Values are represented as mean ± SEM. *P < 0.05.
These results suggest that TNFSF14 plays a critical role during renal fibrosis development.
Tnfsf14 deficiency reduces renal fibrosis
To further examine the functional importance of TNFSF14 in kidney fibrosis, we used Tnfsf14-deficient mice. Tnfsf14 absence apparently alleviated UUO-induced tubular injury and renal fibrosis, as demonstrated by significant reductions in collagen deposition by staining with Sirius Red and Masson’s trichrome (Figure 3A), in the expression of α-SMA and fibronectin by immunohistochemical staining and western blot (Figure 3B and 3C), in the mRNA levels of pro-fibrotic markers Cola1,Vim, and TGF-β1 by qRT-PCR (Figure 3D), and in tubular dilation, brush border disruption and tubular atrophy (Supplementary Figure 4A and 4B).
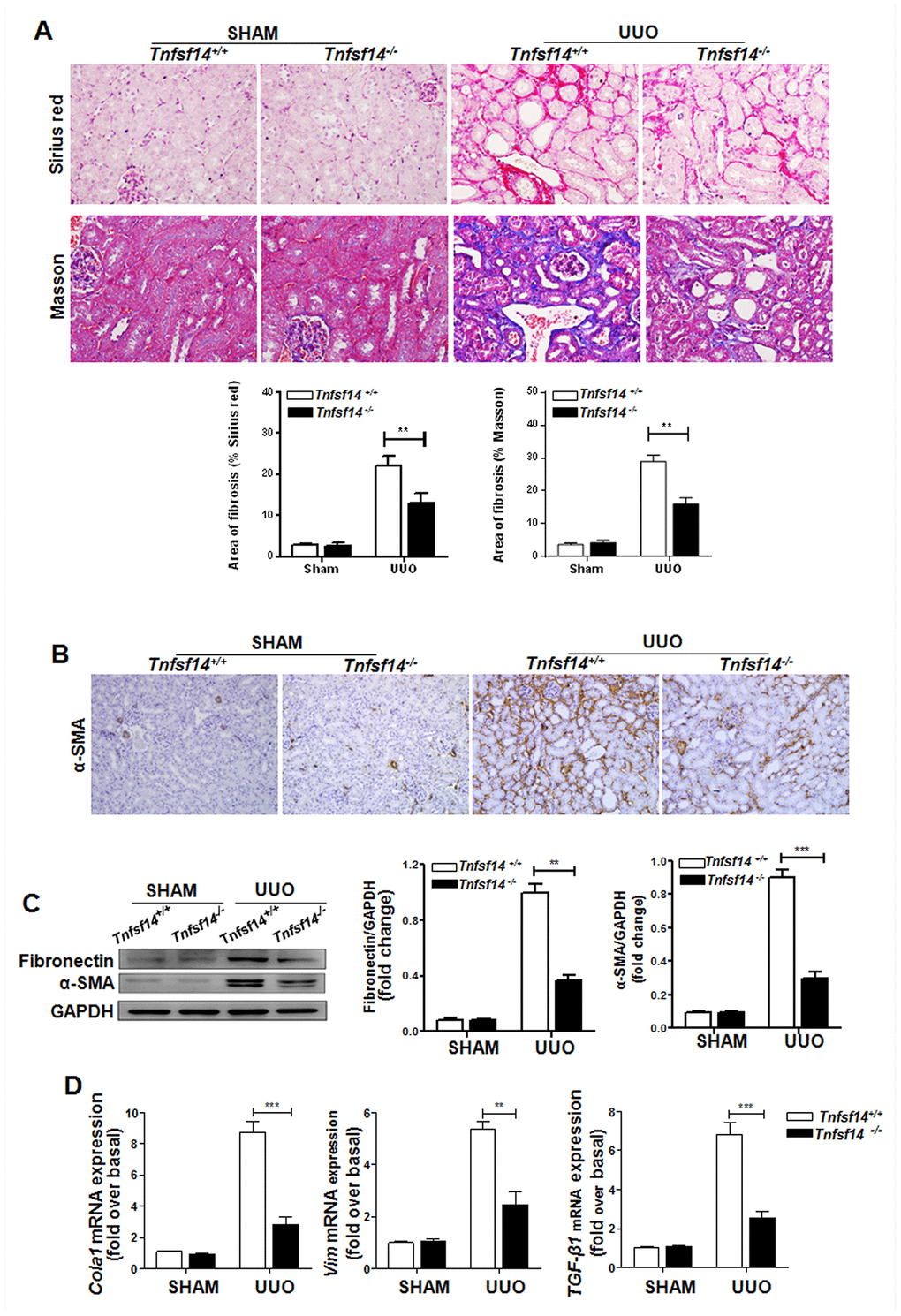
Figure 3. Tnfsf14 deficiency ameliorates UUO-induced renal fibrosis. Kidney tissues from Tnfsf14+/+ and Tnfsf14−/− mice were collected at 7 days after UUO surgery. Sham group was used as the control of UUO. (A) Sirius Red and Masson staining of kidney tissues sections. Original magnification ×400. (B) α-SMA expression in kidney tissues was measured by immunohistochemistry. Original magnification ×200. (C) Western blot analyses of renal fibronectin and α-SMA expression in kidney tissues. Representative western blot (Left) and quantitative data (Right) are presented. (D) The mRNA levels of pro-fibrotic mediators Cola1, Vim, and TGF-β1 were measured by qRT-PCR. The data were representative of the results of three independent experiments. All values are represented as mean ± SEM. n = 5 per group. **P < 0.01 and ***P < 0.001.
E-cadherin expression is an important marker of epithelial integrity and its absence is closely related to tissue fibrosis [21]. Therefore, we detected the expression of E-cadherin in the obstructed kidney, and found that it was significantly higher in Tnfsf14 KO mice than that of WT mice after UUO surgery (Figure 4A and 4B).
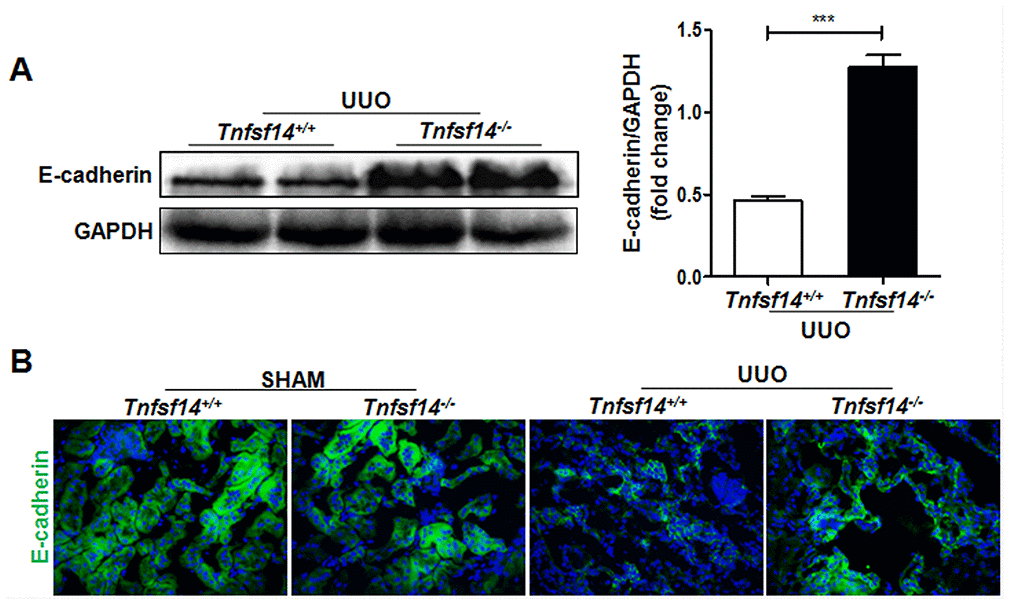
Figure 4. Increased renal tubular cell integrity in Tnfsf14-deficient mice after UUO. Kidney tissues of Tnfsf14+/+ and Tnfsf14−/− mice were collected after UUO surgery for 7 days. (A) E-cadherin expression in kidney tissues was measured by western blot. Representative western blot (Left) and quantitative data (Right) are presented. (B) E-cadherin expression in kidney tissues was measured by immunofluorescence. Sham group was used as the control of UUO. The data were representative of the results of three independent experiments. Values are represented as mean ± SEM. Original magnification ×400. n = 5 per group. ***P < 0.001.
Tnfsf14 deficiency attenuates inflammatory cytokines expression in psoriatic skin lesions
Persistent inflammatory cell infiltration in the tissue and inflammatory cytokines secretion play an important role in renal fibrosis progression [22]. It was demonstrated that Tnfsf14 deficiency led to a remarkable reduction in infiltration of macrophages (F4/80+), neutrophils (Ly-6G+), and T lymphocytes (CD3+) (Figure 5A) and in the mRNA levels of pro-inflammatory cytokines TNF-α,IL-6, and IL-1β (Figure 5B); However, an increase was observed in anti-inflammatory factor IL-10 (Figure 5B) level in UUO-induced obstructed damaged kidney.
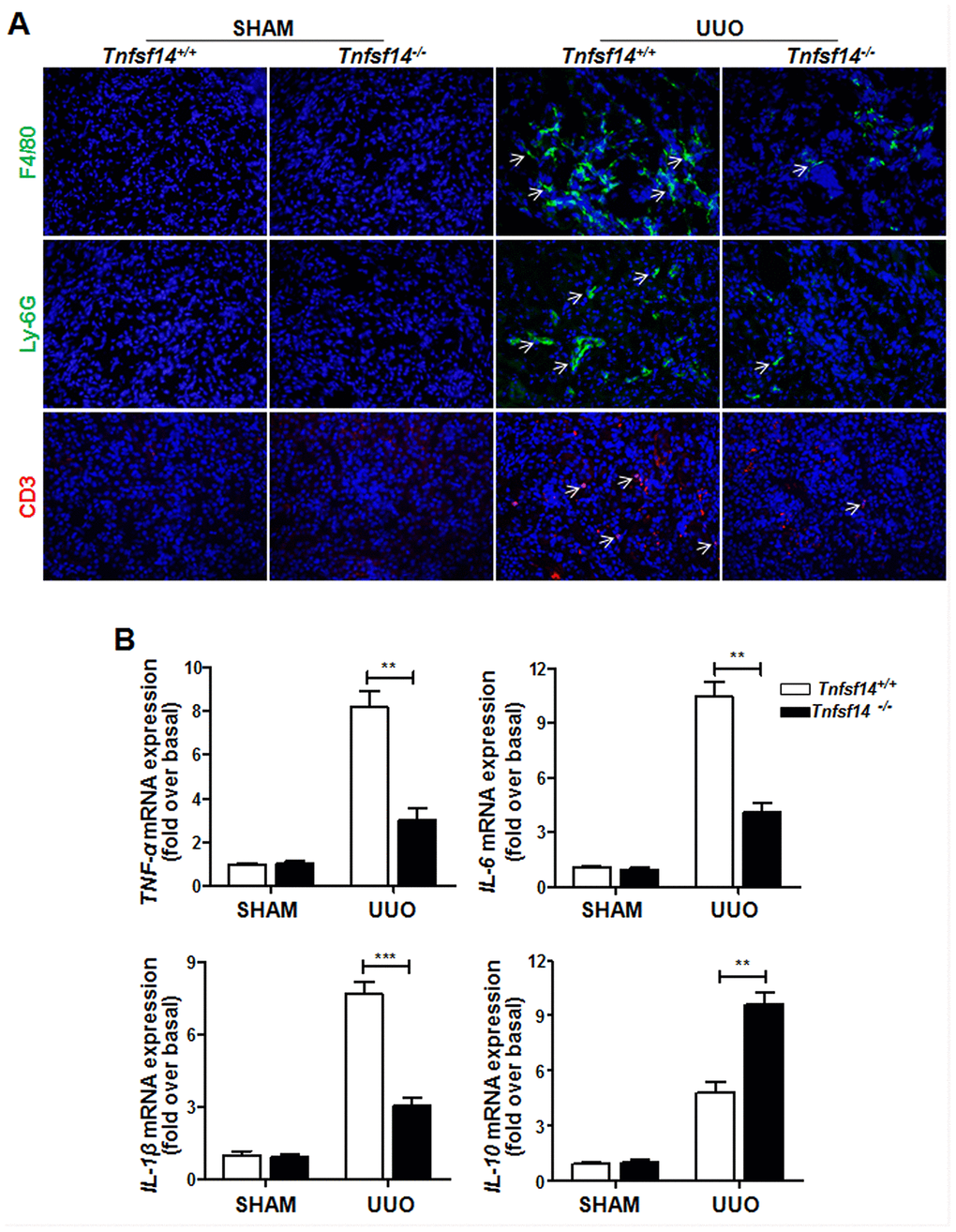
Figure 5. Reduced inflammatory responses in Tnfsf14-deficient mice after UUO. Kidney tissues of Tnfsf14+/+ and Tnfsf14−/− mice were collected after UUO surgery for 7 days. (A) Infiltration of neutrophils (Ly-6G+, white arrows), macrophages (F4/80+, white arrows), and T lymphocytes (CD3+, white arrows) in kidney tissues was assessed by immunofluorescence. Original magnification ×400. (B) The mRNA levels of inflammatory cytokines TNF-α, IL-6, IL-1β, and IL-10 in kidney tissues were measured by qRT-PCR. Sham group was used as the control of UUO. The data were representative of the results of three independent experiments. Values are represented as means ± SEM. n = 5 per group. **P < 0.01 and ***P < 0.001.
These data clearly demonstrate that Tnfsf14 deficiency can alleviate disease severity during UUO-induced renal fibrosis in mice.
Sphk1 is critical for the development of renal fibrosis
Sphk1 participates in some types of tissue fibrosis, including pulmonary, liver, and cardiac fibrosis [23–26]. Moreover, Sphk1 is closely associated with CKD [27, 28] Consistent with these results and similar to increased Sphk1 expression in renal biopsy of patients with CKD (Supplementary Figure 5A), we found that Sphk1 was significantly up-regulated in the UUO-induced obstructed kidney in mice (Figure 6A and Supplementary Figure 5B). Furthermore, Sphk1 expression exhibited a close association with fibrosis-related factors expression, including Cola1 (Figure 6B, r2 = 0.736) and Acta2 (Figure 6B, r2 = 0.745).
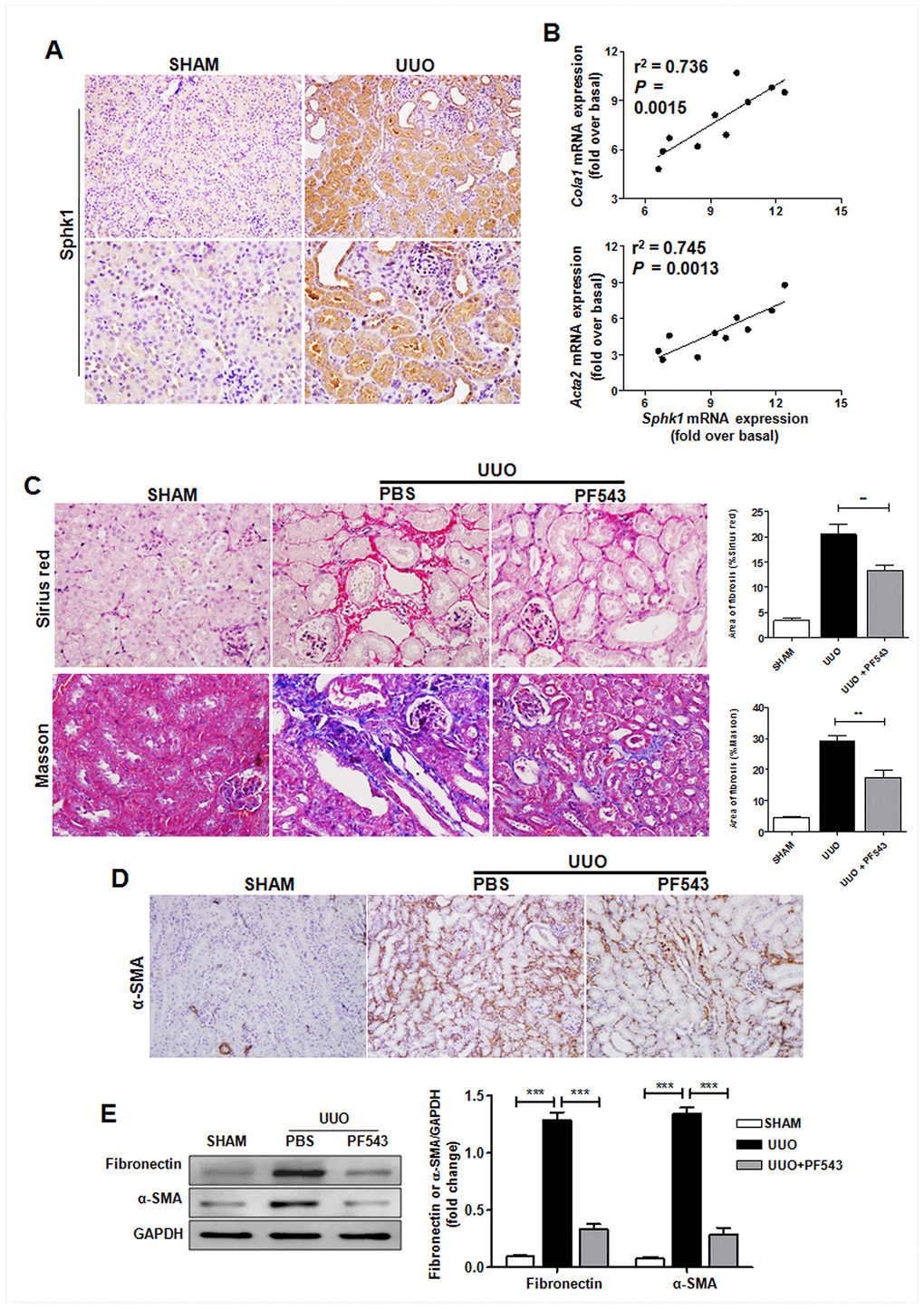
Figure 6. Sphk1 is critical for UUO-induced kidney fibrosis in mice. (A) Sphk1 expression in kidney tissues of Tnfsf14+/+ mice after UUO surgery for 7 days was measured by immunohistochemistry (upper lane, original magnification ×200; lower lane, original magnification ×400). (B) Linear regression showed a close correlation between Sphk1 mRNA expression and Cola1 and Acta2 mRNA expression in kidney tissues of Tnfsf14+/+ mice after UUO surgery for 7 days. Spearman’s correlation coefficient and P value are shown (n = 10). (C–E) After UUO surgery, PF543 (1 mg/kg/day) was injected intraperitoneally for consecutive 7 days, and then kidney tissues were collected from each group. (C) Sirius Red and Masson staining of kidney tissues sections. Original magnification ×400. (D) α-SMA expression in kidney tissues was measured by immunohistochemistry. Original magnification ×200. (E) Western blot analyses of renal fibronectin and α-SMA protein in kidney tissues. Representative western blot (Left and quantitative data (Right) are presented. Sham group was used as the control of UUO. The data were representative of the results of three independent experiments. All values are represented as means ± SEM. n = 5 per group. ***P < 0.001.
To further assess the role of Sphk1 during renal fibrosis development, we used PF543, a specific inhibitor of Sphk1 activation, to block Sphk1 endogenous activity in vivo, and found that PF543 treatment led to an evident down-regulation in UUO-induced collagen deposition (Figure 6C) and the expression of α-SMA (Figure 6D) and fibronectin (Figure 6E) in kidney tissues in mice. All these results suggest that Sphk1 is an important factor during UUO-induced renal fibrosis.
TNFSF14 signaling is essential for pro-fibrotic factor Sphk1 production during the development of renal fibrosis
To explore the association of pro-fibrotic factor Sphk1 with TNFSF14 pathway during renal fibrosis pathogenesis, we measured Sphk1 expression in Tnfsf14-deficient mice. Compared with Tnfsf14+/+ controls, Tnfsf14−/− mice displayed significantly reduced Sphk1 expression in UUO-induced obstructed damaged kidney (Figure 7A–7C). To further investigate the effect of TNFSF14 signaling on Sphk1 expression, we isolated primary mTECs, which exhibited the typical characteristic cobblestone morphology of epithelial cells under light microscopy (Supplementary Figure 6A) and were CK-18, HVEM and LTβR positive (Supplementary Figure 6B). The Sphk1 production of primary cultured mTECs was remarkably increased after stimulating with rmTNFSF14 (100 ng/mL) for 24 h (Figure 7D and Supplementary Figure 7). Moreover, TNFSF14 showed a stronger ability for the induction of Sphk1 expression when compared with TNF-α, IFN-γ, TGFβ-1, IL-1β and IL-6 (Supplementary Figure 7). These data indicate that the TNFSF14 pathway is critical for pro-fibrotic factor Sphk1 expression during the development of renal fibrosis, which may be the underlying mechanism of TNFSF14-mediated renal fibrosis.
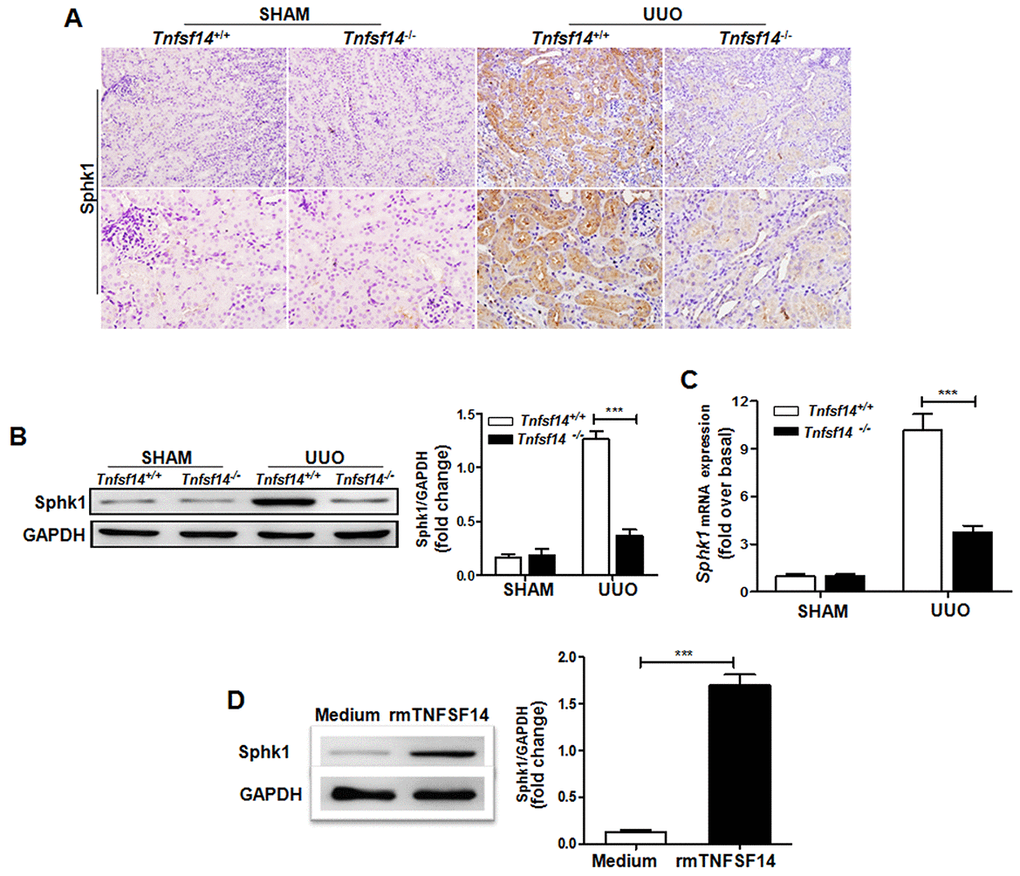
Figure 7. Tnfsf14 deficiency leads to a remarkable reduction in UUO-induced Sphk1 expression in kidney tissues of mice. After UUO surgery for 7 days, kidney tissues from Tnfsf14+/+ and Tnfsf14−/− mice were collected. (A) The expression of Sphk1 in kidney tissues was measured by immunohistochemistry (upper lane, original magnification ×200; lower lane, original magnification ×400). (B) The expression of Sphk1 in kidney tissues was measured by western blot. Representative western blot (Left) and quantitative data (Right) are presented. (C) The expression of Sphk1 mRNA in kidney tissues was measured by qRT-PCR. Sham group was used as the control of UUO. (D) Primary cultured mTECs were stimulated with rmTNFSF14 (100 ng/mL) for 24 h. Medium was used as the negative control. Western blot analyses of Sphk1 protein of mTECs. Representative western blot (Left) and quantitative data (Right) are presented. The data were representative of the results of three independent experiments. All values are represented as means ± SEM. n = 5 per group. **P < 0.01 and ***P < 0.001.
Discussion
TNFSF14 has been reported to be involved in various tissues fibrosis, including pulmonary and skin fibrosis [11, 12]. However, no studies had evaluated the role of TNFSF14 in renal fibrosis development. In the present study, we found that the expression of TNFSF14 and its receptors HVEM and LTβR were rapidly up-regulated in UUO-induced mouse renal fibrosis model and in patients with fibrosis nephropathy. Notably, although TNFSF14 is a transmembrane protein [7], it can be proteolyzed releasing a soluble, bioactive form since a metalloprotease cleavage site is present in TNFSF14 [29]. In agreement with previous results from the concanavalin A-induced hepatitis model [30], the morbidly obese subjects [31] and the diabetic patients [32], circulating TNFSF14 concentrations were significantly increased in fibrotic nephropathy patients. Furthermore, the important role of soluble form TNFSF14 is confirmed in the pathogenesis of liver inflammation [30] and DSS-induced colitis [8]. However, the cellular source of plasma levels of TNFSF14 is complex and not mentioned in the most of literatures since TNFSF14 is expressed and can also be shed from different types of immune-inflammatory cells, including T and B lymphocytes, monocytes/macrophages, granulocytes, spleen cells, and dendritic cells [33–35]. Although it was confirmed that platelets in the diabetic patients [32] or activated T cells in acute DSS-induced colitis [8] were the important cellular source of plasma levels of TNFSF14, the possibility could not be dismissed that other cell types might be capable of shedding TNFSF14. Herein, the source and the functions of increased circulating TNFSF14 in fibrotic nephropathy patients was not evaluated because of the limited human samples, and further studies are needed in the future.
“No inflammation, no fibrosis”, and in agreement with previous studies [2, 36], UUO induced a significant increase in the expression of several pro-inflammatory cytokines as well as pro-fibrotic factors. However, Tnfsf14 deficiency markedly down-regulated UUO-induced inflammatory responses and renal fibrosis, which indicating that the protection observed in Tnfsf14 deficient mice, at least partially, is associated to the reduced immune-inflammatory response. Consistent with these results, anti-inflammation drugs showed a potentially effectiveness for inflammatory renal diseases [37, 38].
There are several cell types that can generate proliferating and activated myofibroblast, such as resident fibroblasts, pericytes, epithelial-to-mesenchymal transition (EMT), endothelial-to-mesenchymal transition, and circulating bone-marrow-derived cells [39]. Here, our results showed a remarkable reduced α-SMA level in renal tissues of UUO-induced Tnfsf14 KO mice, indicating a lower level of fibrosis and fewer myofibroblasts in kidney tissues. By using lineage tracing techniques, it was demonstrated that approximately 36% of fibroblasts in UUO-induced fibrosis were derived from tubular EMT [40]. TNFSF14 has been proved to induce EMT effectively [15]. That may be the reason why fewer myofibroblasts and then the decreased renal fibrosis were observed in UUO-induced Tnfsf14 KO mice. On the other hand, several studies showed that epithelia contributed little to the myofibroblast population after injury [41–43]. The reasons for the discrepancy may be due to the different experimental models and animal strains [44]. In addition, because it is difficult to demonstrate EMT in vivo, and a concept of “partial EMT” is proposed in UUO model; partial EMT means that tubular epithelia acquire mesenchymal features but do not fully transform into myofibroblasts [45], which explains the conflicting views previously.
E-cadherin is essential for the maintenance of epithelial cell integrity and its absence is closely related to renal fibrosis [46, 47]. TNFSF14 directly induces E-cadherin degradation [15, 48]. Consistent with these results, our data showed that Tnfsf14 deficiency substantially preserved E-cadherin expression. Taken together, these results indicate a profound involvement of endogenous TNFSF14 in kidney fibrosis pathogenesis.
Sphk1 plays a pivotal role in renal inflammatory response and fibrosis. Sphk1/S1P was shown to induce pro-fibrotic factors production by cross-activating the TGF-β/Smad signaling pathway [49], and over-expression of Shpk1 promoted the release of inflammatory factors leading to glomerulosclerosis and interstitial fibrosis [28]. Sphk1 deficiency induced renal fibrosis to a lesser extent in diabetic mice [50]. In the present study, the pro-fibrotic properties of Sphk1 were further confirmed in the UUO-induced kidney fibrosis mouse model. In contrast to the pro-fibrotic role of Sphk1, it was found that Sphk2-deficient rather than Sphk1-deficient mice were protected from folic acid or ischemia-reperfusion injury-induced renal fibrosis [51]. The discrepancy between these results may be related to the different renal fibrosis models, and the pro-fibrotic effects of Sphk1 were not obvious in acute kidney injury model. In addition, we found that UUO-induced Sphk1 expression was remarkably reduced in Tnfsf14 KO mice, and in vitro recombinant TNFSF14 administration markedly up-regulated Sphk1 expression of mTECs, indicating TNFSF14 was required for Sphk1 production during renal fibrosis and TNFSF14 may mediate renal fibrosis by way of potentiating pro-fibrotic factor Sphk1 expression.
Collectively, our results provide direct evidence of the role of TNFSF14 pathway in kidney fibrosis development, indicating that disturbing the TNFSF14 signaling pathway can be a useful immunotherapeutic strategy for kidney fibrosis in humans. Moreover, this is the first report showing that the TNFSF14 pathway potentiates pro-fibrotic factor Sphk1 expression, which may be the underlying mechanism of TNFSF14-mediated renal fibrosis.
Materials and Methods
Human serum and kidney biopsy samples
Human serum samples and kidney specimens were obtained from 16 patients with CKD stage 3 or 4 at the Southwest Hospital, Third Military Medical University, China. All patients were diagnosed with CKD for the first time. Serum samples were also obtained from healthy volunteers, considered as controls. After centrifugation at 3000 rpm for 10 min to remove debris, serum samples were aliquot and stored at −80° C for further study. Optimal cutting temperature compound (O.C.T, Sakura Finetek, USA)-embedded human kidney biopsy sections (4-μm) were prepared as described previously [52]. Peritumoral renal tissues from patients with renal cell carcinoma who underwent nephrectomy were used as normal controls. Informed consent was obtained from all patients included in this study, and all experiments were conducted according to the principles of the Declaration of Helsinki. The study was approved by the ethical committee of the First Affiliated Hospital (Southwest Hospital) of Third Military Medical University.
Animal models
Male C57BL/6 mice were purchased from the Peking University Animal Center (Beijing, China). Tnfsf14−/− mice with a C57BL/6 genetic background were provided by Prof. Pfeffer (Institute of Medical Microbiology and Hospital Hygiene, University of Duesseldorf, Germany). Genotyping for Tnfsf14 in mice was performed by PCR with the following primers, provided by Dr. Preffer [53]. For Tnfsf14, wild type: 5’-CGACAGACATGCCAGGAATGG-3’; common: 5’-ACG CATGTGTCCTGCGTGTGG-3’; mutant: 5’-GACGTAAACTCCTCTTCAGAC-3. Eight to twelve-week-old male mice were used for the present study. UUO-induced renal fibrosis was performed as described previously [54]. Briefly, mice were anesthetized with 1 % pentobarbital (10 μL/g), and the left ureter was exposed by a midline incision. The ureter was obstructed by two point ligations with 6–0 silk sutures. Sham-operated mice had their ureters exposed and manipulated and underwent the same procedure but were not ligated. The incision was sutured, and mice were allowed to recover and were provided ad libitum access to food and water. Mice were euthanized at day 1, 3, 5, 7, or 9 after the surgery, and kidney tissues and serum were collected for further study. For assessing PF543 therapeutic efficacy (Sphk1 activation specific inhibitor) on UUO-induced kidney fibrosis, three groups of mice were used: (1) Sham control, (2) UUO injected with vehicle, and (3) UUO injected with PF543. There were 5 mice in each group except for special instructions. Mice were treated with vehicle (PBS) or PF543 (1 mg/kg, Medchemexpress, USA) by intraperitoneal injection daily and were euthanized at 7 days after UUO. This dosage of PF543 is widely used in studies including in UUO-induced mouse model of renal fibrosis [55, 56], and the inhibitory effect of PF-543 was also determined by western blot (Supplementary Figure 1). All animal studies were approved by the Institutional Animal Care and Use Committee of the Third Military Medical University.
Primary mouse renal tubular epithelial cells (mTECs) culture and treatment
mTECs were isolated as previous described [57, 58]. Briefly, the kidney was removed and washed with cold PBS. After the capsule was removed, kidney cortices from WT mice were cut into pieces and digested with collagenase (2 mg/mL) at 37° C for 30 min, followed by PBS washing. Next, the suspension was passed through a series of cell sieves (mesh diameters of 100 and 70 μm). Cortical tubular cells were centrifuged at 1300 rpm for 5 min, followed by PBS washing. Cells were cultured in DMEM/F12 medium supplemented with 10% fetal bovine serum (ScienCell, Carlsbad, USA) and 100 U/mL penicillin/streptomycin (Life Technologies, Grand Island, USA). For in vitro studies, serum-starved mTECs were stimulated with recombinant murine TNFSF14 (100 ng/mL), TNF-α (10 ng/mL), IFN-γ (100 ng/mL), TGFβ-1 (10 ng/mL), IL1-β (10 ng/mL), or IL-6 (10 ng/ml) (All from Peprotech, Rocky Hill, NJ, USA) for 24 h.
Immunofluorescence staining
Kidney cryosections were fixed with 4 % paraformalin for 15 min at room temperature. mTECs cultured on coverslips were fixed with cold acetone for 10 min at −20° C. After blocking with 5 % BSA for 1 h, the slides were immunostained with primary antibodies against fibronectin, E-cadherin, CD3, CK-18, LTβR (All diluted by 1:100, Abcam, Cambridge, MA, USA), and TNFSF14, F4/80, Ly-6G, HVEM (All diluted by 1:100; Santa Cruz, Dallas, TX, USA). These slides were then stained with DyLight- or Cy3-conjugated secondary antibody (1:300; Biolegend, San Diego, CA, USA), respectively. Nuclei were stained using Hoechst33258 (Enzo, Lausen, Switzerland). The slides were visualized by fluorescent microscopy (Olympus BX51, Japan).
Histology and immunohistochemistry
Kidney tissues were fixed in 4 % formalin and embedded in paraffin. Paraffin sections (4-μm) were stained with Masson’s trichrome and Sirius Red. Renal fibrosis was assessed and quantified by imaging analysis (ImageJ software; Bethesda, MD, USA). Briefly, 6–8 corticomedullary junction viewing fields were selected from appropriate areas for each kidney examined. Fibrosis was expressed as the percentage of the total area. A total of 4–5 images were evaluated per kidney, and mean values were calculated. Immunohistochemical staining was performed on kidney by using routine protocols [59]. Briefly, sections were blocked with 5 % BSA for 1 h at room temperature and incubated at 4° C overnight with primary antibodies against α-SMA, LTβR, Sphk1 (All diluted by 1:150; Abcam, Cambridge, MA, USA), and TNFSF14, HVEM (All diluted by 1:150; Santa Cruz, Dallas, TX, USA). These slides were then stained with a horseradish peroxidase-conjugated secondary antibody (1:800; Beyotime, Shanghai, China). The results were analyzed using the DAB assay kit (ZSGB-BIO, Beijing, China). The slides were visualized by microscopy (Olympus BX51, Japan).
Quantitative real-time PCR
Total RNA was extracted from the tissues using the TRIzol reagent (Takara, Tokyo, Japan) according to the manufacturer’s protocol. First-strand cDNA was synthesized using a reverse transcription system (Takara, Tokyo, Japan) according to the manufacturer’s instruction, and the cDNA was used for quantitative real-time PCR analysis using SYBR Premix Ex Taq (Takara, Tokyo, Japan). mRNA levels were normalized to those of GAPDH. Primer sequences used for amplifications are presented in Table 1. All samples were measured in triplicates. Differences in gene expression were calculated using 2−ΔΔct method.
Table 1. Sequences of primers used for qRT-PCR.
| Gene | Forward primer | Reverse primer |
| Tnfsf14 | ATCTTACAGGAGCCAACGCC | ACGTCAAGCCCCTCAAGAAG |
| Acta2 | CCCAGACATCAGGGAGTAATGG | TCTATCGGATACTTCAGCGTCA |
| Cola1 | TTCTCCTGGCAAAGACGGAC | CTCAAGGTCACGGTCACGAA |
| Vim | CAAACGAGTACCGGAGACAG | TAGCAGCTTCAAGGGCAAAA |
| TGF-β1 | CTCCCGTGGCTTCTAGTGC | GCCTTAGTTTGGACAGGATCTG |
| TNF-α | CCTGTAGCCCACGTCGTAG | GGGAGTAGACAAGGTACAACCC |
| IL-1β | GCAACTGTTCCTGAACTCAACT | ATCTTTTGGGGTCCGTCAACT |
| IL-6 | TTCCTCTGGTCTTCTGGAGT | GTGACTCCAGCTTATCTCTTGG |
| IL-10 | GCTGGACAACATACTGCTAACC | ATTTCCGATAAGGCTTGGCAA |
| Sphk1 | TTTGGAGGTTGCTGACGAG | GGGGCGGCCAGATTTTTAG |
| GAPDH | GGTTGTCTCCTGCGACTTCA | TAGGGCCTCTCTTGCTCAGT |
ELISA
TNFSF14 levels in serum were measured using commercially available ELISA kit (Cloud-Clone, Houston, USA) according to the manufacturer’s instructions. OD values were detected using a microplate absorbance reader (BIO-RAD, California, USA) at a wavelength of 450 nm and calculated in the linear part of the curve.
Western blot
Total proteins were isolated from the renal tissues using the RIPA buffer, and protein concentrations were quantified using the BCA protein assay kit (Beyotime, Shanghai, China). Protein samples (35 μg/lane) were resolved via sodium dodecyl sulfate-polyacrylamide gel electrophoresis and were transferred onto polyvinylidene difluoride membranes (Beyotime, Shanghai, China). The membranes were incubated overnight at 4° C with rabbit anti-mouse fibronectin (1:1000), mouse anti-mouse α-SMA (1:200), mouse anti-mouse E-cadherin (1:500), rabbit anti-mouse Sphk1 (1:1000) (All from Abcam, Cambridge, MA, USA) followed by incubation with horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit IgG secondary antibodies (1:3000; ZSGB-BIO, Beijing, China). Immunoblots were visualized using the ECL Western blot Detection System (Millipore, Billerica, MA, USA). GAPDH was used as the loading control.
Statistics
All data are represented as mean ± SEM. Statistical significances between experimental and control groups were assessed by the Student’s t-test or one-way ANOVA. Spearman (nonparametric) correlation analysis was used to assess the relationship between Sphk1 mRNA expression in kidney and other variables. P < 0.05 was considered significant.
Supplementary Materials
Author Contributions
Y.L. and M.T. for study design, acquisition of data, statistical analysis and drafting of the manuscript; X.C., B. H., F. X., K.Z., Q.Z., Q.H., S.L., G.L., S.W., J.C. and D.Y. for technical and material support; G.X. and K.Q.Z. for study design, data analysis, and writing and revision of the article. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflicts of interests.
Funding
This work was supported by National Natural Science Foundation of China (NO. 81670684 and 81570703), General Logistics Department Health Research Project (15BJZ28) and New Clinical Technology Program for Military Medicine and War Trauma Treatment of Southwest Hospital (SWH2016JSTSYB-38).
References
- 1. Zhang L, Wang F, Wang L, Wang W, Liu B, Liu J, Chen M, He Q, Liao Y, Yu X, Chen N, Zhang JE, Hu Z, et al. Prevalence of chronic kidney disease in China: a cross-sectional survey. Lancet. 2012; 379:815–22. https://doi.org/10.1016/S0140-6736(12)60033-6 [PubMed]
- 2. Zhou D, Tian Y, Sun L, Zhou L, Xiao L, Tan RJ, Tian J, Fu H, Hou FF, Liu Y. Matrix metalloproteinase-7 is a urinary biomarker and pathogenic mediator of kidney fibrosis. J Am Soc Nephrol. 2017; 28:598–611. https://doi.org/10.1681/ASN.2016030354 [PubMed]
- 3. Grande MT, Sánchez-Laorden B, López-Blau C, De Frutos CA, Boutet A, Arévalo M, Rowe RG, Weiss SJ, López-Novoa JM, Nieto MA. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med. 2015; 21:989–97. https://doi.org/10.1038/nm.3901 [PubMed]
- 4. Wang W, Zhou PH, Xu CG, Zhou XJ, Hu W, Zhang J. Baicalein ameliorates renal interstitial fibrosis by inducing myofibroblast apoptosis in vivo and in vitro. BJU Int. 2016; 118:145–52. https://doi.org/10.1111/bju.13219 [PubMed]
- 5. Song CJ, Zimmerman KA, Henke SJ, Yoder BK. Inflammation and fibrosis in polycystic kidney disease. Results Probl Cell Differ. 2017; 60:323–44. https://doi.org/10.1007/978-3-319-51436-9_12 [PubMed]
- 6. Phan SH. Biology of fibroblasts and myofibroblasts. Proc Am Thorac Soc. 2008; 5:334–37. https://doi.org/10.1513/pats.200708-146DR [PubMed]
- 7. Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, Ruben S, Murphy M, Eisenberg RJ, Cohen GH, Spear PG, Ware CF. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity. 1998; 8:21–30. https://doi.org/10.1016/s1074-7613(00)80455-0 [PubMed]
- 8. Jungbeck M, Daller B, Federhofer J, Wege AK, Wimmer N, Männel DN, Hehlgans T. Neutralization of LIGHT ameliorates acute dextran sodium sulphate-induced intestinal inflammation. Immunology. 2009; 128:451–58. https://doi.org/10.1111/j.1365-2567.2009.03131.x [PubMed]
- 9. Choi EK, Kim WK, Sul OJ, Park YK, Kim ES, Suh JH, Yu R, Choi HS. TNFRSF14 deficiency protects against ovariectomy-induced adipose tissue inflammation. J Endocrinol. 2013; 220:25–33. https://doi.org/10.1530/JOE-13-0341 [PubMed]
- 10. Ware CF, Sedý JR. TNF superfamily networks: bidirectional and interference pathways of the herpesvirus entry mediator (TNFSF14). Curr Opin Immunol. 2011; 23:627–31. https://doi.org/10.1016/j.coi.2011.08.008 [PubMed]
- 11. Herro R, Antunes RD, Aguilera AR, Tamada K, Croft M. The tumor necrosis factor superfamily molecule LIGHT promotes keratinocyte activity and skin fibrosis. J Invest Dermatol. 2015; 135:2109–18. https://doi.org/10.1038/jid.2015.110 [PubMed]
- 12. Herro R, Da Silva Antunes R, Aguilera AR, Tamada K, Croft M. Tumor necrosis factor superfamily 14 (LIGHT) controls thymic stromal lymphopoietin to drive pulmonary fibrosis. J Allergy Clin Immunol. 2015; 136:757–68. https://doi.org/10.1016/j.jaci.2014.12.1936 [PubMed]
- 13. Doherty TA, Soroosh P, Khorram N, Fukuyama S, Rosenthal P, Cho JY, Norris PS, Choi H, Scheu S, Pfeffer K, Zuraw BL, Ware CF, Broide DH, Croft M. The tumor necrosis factor family member LIGHT is a target for asthmatic airway remodeling. Nat Med. 2011; 17:596–603. https://doi.org/10.1038/nm.2356 [PubMed]
- 14. Pierer M, Brentano F, Rethage J, Wagner U, Hantzschel H, Gay RE, Gay S, Kyburz D. The TNF superfamily member LIGHT contributes to survival and activation of synovial fibroblasts in rheumatoid arthritis. Rheumatology (Oxford). 2007; 46:1063–70. https://doi.org/10.1093/rheumatology/kem063 [PubMed]
- 15. Hung JY, Chiang SR, Tsai MJ, Tsai YM, Chong IW, Shieh JM, Hsu YL. LIGHT is a crucial mediator of airway remodeling. J Cell Physiol. 2015; 230:1042–53. https://doi.org/10.1002/jcp.24832 [PubMed]
- 16. Huang J, Huang K, Lan T, Xie X, Shen X, Liu P, Huang H. Curcumin ameliorates diabetic nephropathy by inhibiting the activation of the SphK1-S1P signaling pathway. Mol Cell Endocrinol. 2013; 365:231–40. https://doi.org/10.1016/j.mce.2012.10.024 [PubMed]
- 17. Ren S, Babelova A, Moreth K, Xin C, Eberhardt W, Doller A, Pavenstädt H, Schaefer L, Pfeilschifter J, Huwiler A. Transforming growth factor-beta2 upregulates sphingosine kinase-1 activity, which in turn attenuates the fibrotic response to TGF-beta2 by impeding CTGF expression. Kidney Int. 2009; 76:857–67. https://doi.org/10.1038/ki.2009.297 [PubMed]
- 18. Natoli TA, Husson H, Rogers KA, Smith LA, Wang B, Budman Y, Bukanov NO, Ledbetter SR, Klinger KW, Leonard JP, Ibraghimov-Beskrovnaya O. Loss of GM3 synthase gene, but not sphingosine kinase 1, is protective against murine nephronophthisis-related polycystic kidney disease. Hum Mol Genet. 2012; 21:3397–407. https://doi.org/10.1093/hmg/dds172 [PubMed]
- 19. Lan T, Liu W, Xie X, Xu S, Huang K, Peng J, Shen X, Liu P, Wang L, Xia P, Huang H. Sphingosine kinase-1 pathway mediates high glucose-induced fibronectin expression in glomerular mesangial cells. Mol Endocrinol. 2011; 25:2094–105. https://doi.org/10.1210/me.2011-0095 [PubMed]
- 20. Liu X, Hong Q, Wang Z, Yu Y, Zou X, Xu L. Transforming growth factor-β-sphingosine kinase 1/S1P signaling upregulates microRNA-21 to promote fibrosis in renal tubular epithelial cells. Exp Biol Med (Maywood). 2016; 241:265–72. https://doi.org/10.1177/1535370215605586 [PubMed]
- 21. Zhang Y, Wu X, Li Y, Zhang H, Li Z, Zhang Y, Zhang L, Ju J, Liu X, Chen X, Glybochko PV, Nikolenko V, Kopylov P, et al. Endothelial to mesenchymal transition contributes to arsenic-trioxide-induced cardiac fibrosis. Sci Rep. 2016; 6:33787. https://doi.org/10.1038/srep33787 [PubMed]
- 22. Liu S, Jiang L, Li H, Shi H, Luo H, Zhang Y, Yu C, Jin Y. Mesenchymal stem cells prevent hypertrophic scar formation via inflammatory regulation when undergoing apoptosis. J Invest Dermatol. 2014; 134:2648–57. https://doi.org/10.1038/jid.2014.169 [PubMed]
- 23. Milara J, Navarro R, Juan G, Peiró T, Serrano A, Ramón M, Morcillo E, Cortijo J. Sphingosine-1-phosphate is increased in patients with idiopathic pulmonary fibrosis and mediates epithelial to mesenchymal transition. Thorax. 2012; 67:147–56. https://doi.org/10.1136/thoraxjnl-2011-200026 [PubMed]
- 24. Gorshkova I, Zhou T, Mathew B, Jacobson JR, Takekoshi D, Bhattacharya P, Smith B, Aydogan B, Weichselbaum RR, Natarajan V, Garcia JG, Berdyshev EV. Inhibition of serine palmitoyltransferase delays the onset of radiation-induced pulmonary fibrosis through the negative regulation of sphingosine kinase-1 expression. J Lipid Res. 2012; 53:1553–68. https://doi.org/10.1194/jlr.M026039 [PubMed]
- 25. Li C, Zheng S, You H, Liu X, Lin M, Yang L, Li L. Sphingosine 1-phosphate (S1P)/S1P receptors are involved in human liver fibrosis by action on hepatic myofibroblasts motility. J Hepatol. 2011; 54:1205–13. https://doi.org/10.1016/j.jhep.2010.08.028 [PubMed]
- 26. Pchejetski D, Foussal C, Alfarano C, Lairez O, Calise D, Guilbeau-Frugier C, Schaak S, Seguelas MH, Wanecq E, Valet P, Parini A, Kunduzova O. Apelin prevents cardiac fibroblast activation and collagen production through inhibition of sphingosine kinase 1. Eur Heart J. 2012; 33:2360–69. https://doi.org/10.1093/eurheartj/ehr389 [PubMed]
- 27. Deng Y, Lan T, Huang J, Huang H. Sphingosine Kinase-1/sphingosine 1-phosphate pathway in diabetic nephropathy. Chin Med J (Engl). 2014; 127:3004–10. [PubMed]
- 28. Schwalm S, Pfeilschifter J, Huwiler A. Targeting the sphingosine kinase/sphingosine 1-phosphate pathway to treat chronic inflammatory kidney diseases. Basic Clin Pharmacol Toxicol. 2014; 114:44–9. https://doi.org/10.1111/bcpt.12103 [PubMed]
- 29. Granger SW, Butrovich KD, Houshmand P, Edwards WR, Ware CF. Genomic characterization of LIGHT reveals linkage to an immune response locus on chromosome 19p13.3 and distinct isoforms generated by alternate splicing or proteolysis. J Immunol. 2001; 167:5122–28. https://doi.org/10.4049/jimmunol.167.9.5122 [PubMed]
- 30. Anand S, Wang P, Yoshimura K, Choi IH, Hilliard A, Chen YH, Wang CR, Schulick R, Flies AS, Flies DB, Zhu G, Xu Y, Pardoll DM, et al. Essential role of TNF family molecule LIGHT as a cytokine in the pathogenesis of hepatitis. J Clin Invest. 2006; 116:1045–51. https://doi.org/10.1172/JCI27083 [PubMed]
- 31. Bassols J, Moreno-Navarrete JM, Ortega F, Ricart W, Fernandez-Real JM. LIGHT is associated with hypertriglyceridemia in obese subjects and increased cytokine secretion from cultured human adipocytes. Int J Obes (Lond). 2010; 34:146–56. https://doi.org/10.1038/ijo.2009.199 [PubMed]
- 32. Halvorsen B, Santilli F, Scholz H, Sahraoui A, Gulseth HL, Wium C, Lattanzio S, Formoso G, Di Fulvio P, Otterdal K, Retterstøl K, Holven KB, Gregersen I, et al. LIGHT/TNFSF14 is increased in patients with type 2 diabetes mellitus and promotes islet cell dysfunction and endothelial cell inflammation in vitro. Diabetologia. 2016; 59:2134–44. https://doi.org/10.1007/s00125-016-4036-y [PubMed]
- 33. Wang J, Lo JC, Foster A, Yu P, Chen HM, Wang Y, Tamada K, Chen L, Fu YX. The regulation of T cell homeostasis and autoimmunity by T cell-derived LIGHT. J Clin Invest. 2001; 108:1771–80. https://doi.org/10.1172/JCI13827 [PubMed]
- 34. Wang J, Fu YX. The role of LIGHT in T cell-mediated immunity. Immunol Res. 2004; 30:201–14. https://doi.org/10.1385/IR:30:2:201 [PubMed]
- 35. Lim SG, Suk K, Lee WH. Reverse signaling from LIGHT promotes pro-inflammatory responses in the human monocytic leukemia cell line, THP-1. Cell Immunol. 2013; 285:10–17. https://doi.org/10.1016/j.cellimm.2013.08.002 [PubMed]
- 36. Lecru L, Desterke C, Grassin-Delyle S, Chatziantoniou C, Vandermeersch S, Devocelle A, Vernochet A, Ivanovski N, Ledent C, Ferlicot S, Dalia M, Saïd M, Beaudreuil S, et al. Cannabinoid receptor 1 is a major mediator of renal fibrosis. Kidney Int. 2015; 88:72–84. https://doi.org/10.1038/ki.2015.63 [PubMed]
- 37. Kou Y, Koag MC, Cheun Y, Shin A, Lee S. Application of hypoiodite-mediated aminyl radical cyclization to synthesis of solasodine acetate. Steroids. 2012; 77:1069–74. https://doi.org/10.1016/j.steroids.2012.05.002 [PubMed]
- 38. Kou Y, Cheun Y, Koag MC, Lee S. Synthesis of 14',15'-dehydro-ritterazine Y via reductive and oxidative functionalizations of hecogenin acetate. Steroids. 2013; 78:304–11. https://doi.org/10.1016/j.steroids.2012.10.021 [PubMed]
- 39. Sun YB, Qu X, Caruana G, Li J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation. 2016; 92:102–07. https://doi.org/10.1016/j.diff.2016.05.008 [PubMed]
- 40. Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002; 110:341–50. https://doi.org/10.1172/JCI15518 [PubMed]
- 41. LeBleu VS, Taduri G, O’Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013; 19:1047–53. https://doi.org/10.1038/nm.3218 [PubMed]
- 42. Koesters R, Kaissling B, Lehir M, Picard N, Theilig F, Gebhardt R, Glick AB, Hähnel B, Hosser H, Gröne HJ, Kriz W. Tubular overexpression of transforming growth factor-beta1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol. 2010; 177:632–43. https://doi.org/10.2353/ajpath.2010.091012 [PubMed]
- 43. Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010; 176:85–97. https://doi.org/10.2353/ajpath.2010.090517 [PubMed]
- 44. Inoue T, Umezawa A, Takenaka T, Suzuki H, Okada H. The contribution of epithelial-mesenchymal transition to renal fibrosis differs among kidney disease models. Kidney Int. 2015; 87:233–38. https://doi.org/10.1038/ki.2014.235 [PubMed]
- 45. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012; 18:1028–40. https://doi.org/10.1038/nm.2807 [PubMed]
- 46. Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009; 119:1429–37. https://doi.org/10.1172/JCI36183 [PubMed]
- 47. Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, Wu CC, Hagos Y, Burckhardt BC, Pentcheva-Hoang T, Nischal H, Allison JP, Zeisberg M, Kalluri R. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med. 2015; 21:998–1009. https://doi.org/10.1038/nm.3902 [PubMed]
- 48. Mikami Y, Yamauchi Y, Horie M, Kase M, Jo T, Takizawa H, Kohyama T, Nagase T. Tumor necrosis factor superfamily member LIGHT induces epithelial-mesenchymal transition in A549 human alveolar epithelial cells. Biochem Biophys Res Commun. 2012; 428:451–57. https://doi.org/10.1016/j.bbrc.2012.10.097 [PubMed]
- 49. Xin C, Ren S, Kleuser B, Shabahang S, Eberhardt W, Radeke H, Schäfer-Korting M, Pfeilschifter J, Huwiler A. Sphingosine 1-phosphate cross-activates the smad signaling cascade and mimics transforming growth factor-beta-induced cell responses. J Biol Chem. 2004; 279:35255–62. https://doi.org/10.1074/jbc.M312091200 [PubMed]
- 50. Yaghobian D, Don AS, Yaghobian S, Chen X, Pollock CA, Saad S. Increased sphingosine 1-phosphate mediates inflammation and fibrosis in tubular injury in diabetic nephropathy. Clin Exp Pharmacol Physiol. 2016; 43:56–66. https://doi.org/10.1111/1440-1681.12494 [PubMed]
- 51. Bajwa A, Huang L, Kurmaeva E, Ye H, Dondeti KR, Chroscicki P, Foley LS, Balogun ZA, Alexander KJ, Park H, Lynch KR, Rosin DL, Okusa MD. Sphingosine kinase 2 deficiency attenuates kidney fibrosis via IFN- γ. J Am Soc Nephrol. 2017; 28:1145–61. https://doi.org/10.1681/ASN.2016030306 [PubMed]
- 52. Li SS, Wang LL, Liu M, Jiang SK, Zhang M, Tian ZL, Wang M, Li JY, Zhao R, Guan DW. Cannabinoid CB₂ receptors are involved in the regulation of fibrogenesis during skin wound repair in mice. Mol Med Rep. 2016; 13:3441–50. https://doi.org/10.3892/mmr.2016.4961 [PubMed]
- 53. Scheu S, Alferink J, Pötzel T, Barchet W, Kalinke U, Pfeffer K. Targeted disruption of LIGHT causes defects in costimulatory T cell activation and reveals cooperation with lymphotoxin beta in mesenteric lymph node genesis. J Exp Med. 2002; 195:1613–24. https://doi.org/10.1084/jem.20020215 [PubMed]
- 54. Choi HY, Lee HG, Kim BS, Ahn SH, Jung A, Lee M, Lee JE, Kim HJ, Ha SK, Park HC. Mesenchymal stem cell-derived microparticles ameliorate peritubular capillary rarefaction via inhibition of endothelial-mesenchymal transition and decrease tubulointerstitial fibrosis in unilateral ureteral obstruction. Stem Cell Res Ther. 2015; 6:18. https://doi.org/10.1186/s13287-015-0012-6 [PubMed]
- 55. Zhang F, Xia Y, Yan W, Zhang H, Zhou F, Zhao S, Wang W, Zhu D, Xin C, Lee Y, Zhang L, He Y, Gao E, Tao L. Sphingosine 1-phosphate signaling contributes to cardiac inflammation, dysfunction, and remodeling following myocardial infarction. Am J Physiol Heart Circ Physiol. 2016; 310:H250–61. https://doi.org/10.1152/ajpheart.00372.2015 [PubMed]
- 56. Du C, Ren Y, Yao F, Duan J, Zhao H, Du Y, Xiao X, Duan H, Shi Y. Sphingosine kinase 1 protects renal tubular epithelial cells from renal fibrosis via induction of autophagy. Int J Biochem Cell Biol. 2017; 90:17–28. https://doi.org/10.1016/j.biocel.2017.07.011 [PubMed]
- 57. Wang JL, Cheng HF, Zhang MZ, McKanna JA, Harris RC. Selective increase of cyclooxygenase-2 expression in a model of renal ablation. Am J Physiol. 1998; 275:F613–22. https://doi.org/10.1152/ajprenal.1998.275.4.F613 [PubMed]
- 58. Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, Alexander SI, Sharland AF, Chadban SJ. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007; 117:2847–59. https://doi.org/10.1172/JCI31008 [PubMed]
- 59. Pan JS, Huang L, Belousova T, Lu L, Yang Y, Reddel R, Chang A, Ju H, DiMattia G, Tong Q, Sheikh-Hamad D. Stanniocalcin-1 inhibits renal ischemia/reperfusion injury via an AMP-activated protein kinase-dependent pathway. J Am Soc Nephrol. 2015; 26:364–78. https://doi.org/10.1681/ASN.2013070703 [PubMed]