




Introduction
Non-homologous end-joining (NHEJ) is a DNA repair pathway that recognizes, processes and ligates DNA double-stranded breaks (DSB) throughout the cell cycle. NHEJ is required for lymphocyte development; in particular, to repair DSBs induced by the recombination activating genes (RAG) 1 and 2 in developing B and T lymphocytes, and by activation-induced cytidine deaminase (AID) in mature B cells [1]. NHEJ is initiated when Ku70 and Ku80 (Ku) are recruited to the DSB sites. Ku, together with DNA-dependent protein kinase, catalytic subunit (DNA-PKcs), forms the DNA-PK holoenzyme [2]. Subsequently, the nuclease Artemis is recruited to the DSB sites to process DNA hairpins and overhangs [3]. Finally, DNA ligase IV (LIG4), X-ray repair cross-complementing protein 4 (XRCC4) and XRCC4-like factor (XLF) mediate DNA end ligation. The NHEJ complex is stabilized by a paralogue of XRCC4 and XLF (PAXX) and a modulator of retroviral infection (MRI/CYREN) [4, 5].
Inactivation of Ku70, Ku80, Dna-pkcs or Artemis results in severe combined immunodeficiency (SCID) characterized by lack of mature B and T lymphocytes [2, 3, 6–8]. Deletion of both alleles of Xrcc4 [9] or Lig4 [10] results in late embryonic lethality in mice, which correlates with increased apoptosis in the central nervous system (CNS). Inactivation of Xlf (Cernunnos) only results in modest immunodeficiency in mice [11–13], while mice lacking Paxx [14–17] or Mri [5, 18] display no overt phenotype.
The mild phenotype observed in mice lacking XLF could be explained by functional redundancy between XLF and multiple DNA repair factors, including Ataxia telangiectasia mutated (ATM), histone H2AX [19], Mediator of DNA Damage Checkpoint 1 (MDC1) [20, 21], p53-binding protein 1 (53BP1) [17, 22], RAG2 [23], DNA-PKcs [20, 24, 25], PAXX [4, 14, 15, 20, 26–28] and MRI [5]. However, combined inactivation of Xlf and Paxx [4, 14, 15, 20], as well as Xlf and Mri [5], results in late embryonic lethality in mice, presenting a challenge to the study of B and T lymphocyte development in vivo. It has also been shown that both embryonic lethality and increased levels of CNS neuronal apoptosis in mice with deficiency in Lig4 [9, 10, 29, 30], Xrcc4 [9, 31], Xlf and Paxx [20], or Xlf and Dna-pkcs [24, 25] is p53-dependent.
In this study, we rescue synthetic lethality from Xlf and Mri by inactivating one or two alleles of Trp53. We also show that both Xlf-/-Mri-/-Trp53+/- and Xlf-/-Paxx-/-Trp53+/- mice possess a leaky SCID phenotype with severely reduced mature B and T lymphocyte counts in the spleen, low mature T cell counts in the thymus, and accumulated progenitor B cells in the bone marrow. Finally, we demonstrate that MRI is functionally redundant with DNA-PKcs and PAXX.
Results
Inactivation of Trp53 gene rescued embryonic lethality in mice lacking XLF and MRI
Combined inactivation of Xlf and Mri has previously been shown to result in synthetic lethality in mice [5]. To generate XLF/MRI deficient mice with altered expression of Trp53, we intercrossed an Mri-/- strain [18] with an Xlf-/-Trp53+/- [20] strain. Next, we selected and intercrossed triple heterozygous (Xlf+/-Mri+/-Trp53+/-), and later, Xlf-/-Mri+/-Trp53+/- mice. With PCR screening, we identified Xlf-/-Mri-/-Trp53+/- (n=11), Xlf-/-Mri-/-Trp53-/- (n=2), and Xlf-/-Mri-/-Trp53+/+ (n=1) (Figure 1A) among the resulting offspring. Mice lacking both XLF and MRI possessed reduced weight (12 g on average, p<0.0001) when compared with gender- and age-matched WT (19 g), Xlf-/- (19 g) and Mri-/- (20 g) controls (Figure 1B and 1C). In addition, Xlf-/-Mri-/-Trp53+/- and Xlf-/-Mri-/-Trp53-/- mice were viable up to 63 days and died for unknown reasons. We used Xlf-/-Mri-/-Trp53+/- mice to further characterize the development of B and T lymphocytes in vivo.
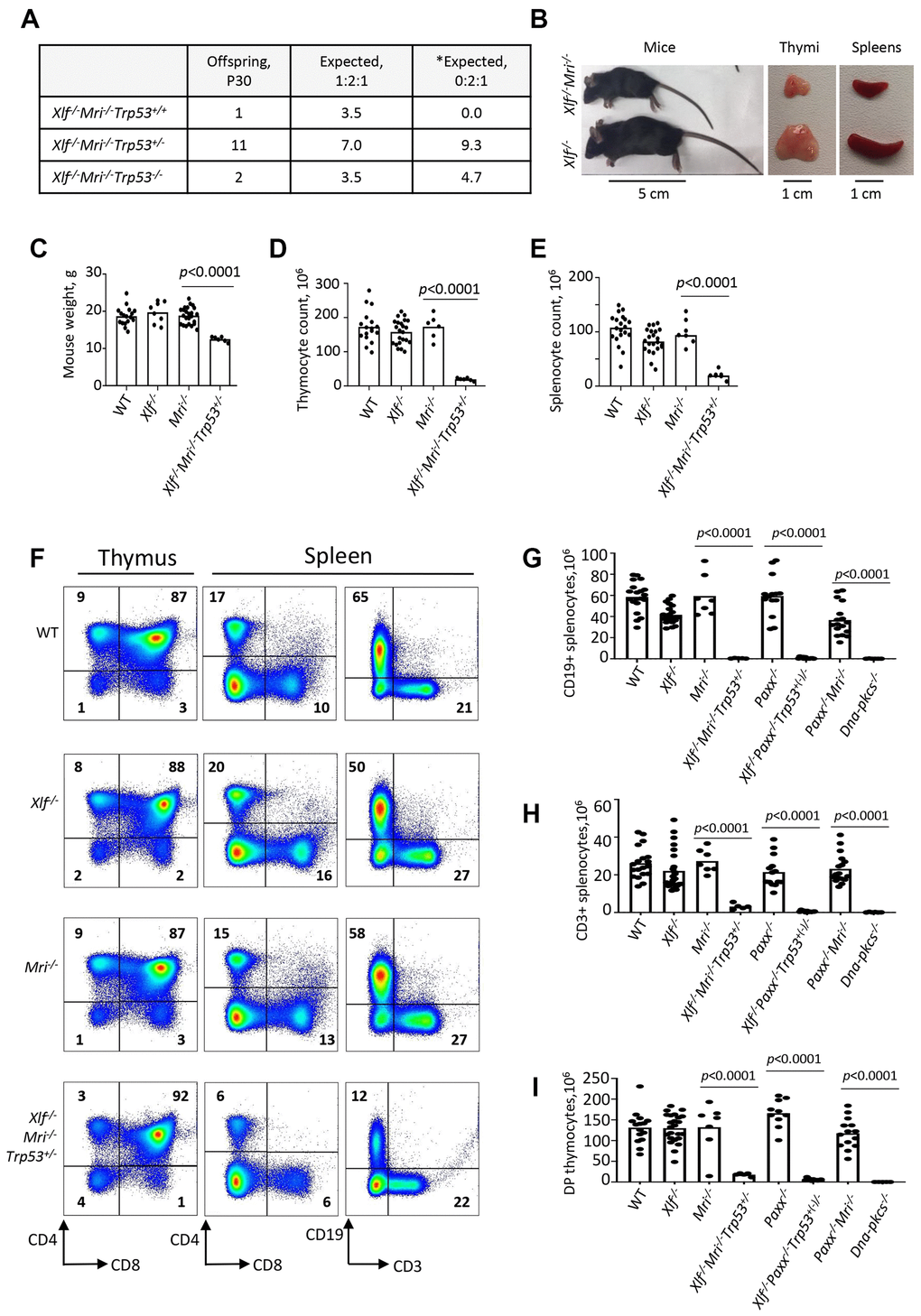
Figure 1. Development of B and T lymphocytes in Xlf-/-Mri-/-Trp53+/- mice. (A) Number of thirty-day-old mice (P30) of indicated genotypes. *Expected distribution assuming lethality. (B) Comparison of body size, thymi and spleens of XLF/MRI-deficient and XLF-deficient mice of the same age. (C) Weights of WT, Xlf-/-, Mri-/-, Xlf-/-Mri-/-Trp53+/- mice. (D, E) Number (×106) of thymocytes (D) and splenocytes (E) in WT, Xlf-/-, Mri-/-, Xlf-/-Mri-/-Trp53+/- mice. (F) Flow cytometric analysis of thymic and splenic T cell subsets and splenic B cells. (G, H, I) Number (×106) of splenic CD19+ B cells (G), splenic CD3+ T cells (H) and thymic CD4+CD8+ double positive (DP) T cells (I) in WT, Xlf-/-, Mri-/-, Xlf-/-Mri-/-Trp53+/-, Paxx-/-, Xlf-/-Paxx-/-Trp53+(-)/- and Paxx-/-Mri-/- mice. Dna-pkcs-/- mice were used as an immunodeficient control. Comparisons between every two groups were made using one-way ANOVA, GraphPad Prism 8.0.1. Xlf-/-Paxx-/-Trp53+(-)/- is a combination of Xlf-/-Paxx-/-Trp53+/- and Xlf-/-Paxx-/-Trp53-/-. Not shown in the graph for (G): WT vs Paxx-/-Mri-/-, p<0.0001 (****), Paxx-/- vs Paxx-/-Mri-/-, p<0.0001 (****), Mri-/- vs Paxx-/-Mri-/-, p<0.0025 (**), Xlf-/- vs Paxx-/-Mri-/-, p=0.9270 (n.s), Xlf-/-Mri-/-Trp53+/- vs Paxx-/-Mri-/-, p<0.0001 (****), and Xlf-/-Paxx-/-Trp53+(-)/- vs Paxx-/-Mri-/-, p<0.0001 (****).
Leaky SCID in Xlf-/-Mri-/-Trp53+/- mice
To determine the roles of XLF and MRI in lymphocyte development in vivo, we isolated the thymus, spleen, and femur from Xlf-/-Mri-/-Trp53+/- mice, as well as from Xlf-/-, Mri-/-, Trp53+/- and WT controls. Combined deficiency for XLF and MRI resulted in a 3-fold reduction in thymus size (32 mg on average, p<0.0001) and a 9-fold reduction in thymocyte count (1.9x107, p<0.0001) when compared to single deficient or WT controls (Figure 1D). Similarly, both average spleen weight (22 mg, p<0.0001) and splenocyte count (2.0x107, p<0.0001) in Xlf-/-Mri-/-Trp53+/- mice decreased approximately 4-5 fold when compared with WT and single deficient controls (Figure 1E). The reduced number of splenocytes in XLF/MRI double-deficient mice could be explained by decreased populations of B and T lymphocytes observed in the Xlf-/-Mri-/-Trp53+/- mice (Figure 1F–1H and Supplementary Tables 1–4). Specifically, CD3+ T cells were reduced 6-fold (p<0.0001), while CD19+ B cells were reduced 50-fold (p<0.0001) when compared with single deficient and WT controls (Figure 1F–1H). Likewise, counts of CD4+ and CD8+ T cells in the spleen (Supplementary Tables 3 and 4), were all dramatically reduced when compared with single deficient and WT controls (about 4-fold, p<0.0001; Figure 1F, 1H) as well as counts of CD4+, CD8+ and CD4+CD8+ T cells in the thymus (Figure 1F, 1I and Supplementary Tables 5–7). From these observations, we conclude that XLF and MRI are functionally redundant during B and T lymphocytes development in mice.
Leaky SCID in mice lacking XLF and PAXX
Combined inactivation of XLF and PAXX has been shown to result in embryonic lethality in mice [4, 14, 15, 20]. To determine the impact of XLF and PAXX on B and T cell development in vivo, we rescued the synthetic lethality by inactivating one allele of Trp53, as described previously [20]. We did not detect any direct influence of altered Trp53 genotype on lymphocyte development (Supplementary Tables 1–9). The resulting Xlf-/-Paxx-/-Trp53+/- and Xlf-/-Paxx-/-Trp53-/- mice possess 30- to 40-fold reduced thymocyte count (4.0x106, p<0.0001) when compared to WT (1.3x108), Xlf-/- (1.4x108) and Paxx-/- (1.7x108) mice. This is reflected in decreased levels of double-positive CD4+CD8+ cells, as well as decreased levels of single-positive CD4+ and CD8+ T cells (Figure 1, Supplementary Figure 1, and Supplementary Tables 5–7). Spleen development was dramatically affected in mice lacking XLF and PAXX compared to WT and single-deficient controls, due to the lack of B cells and decreased T cell count (Figure 1, Supplementary Figure 1, and Supplementary Tables 1–4). When compared with the WT and single knockout controls, Xlf-/-Paxx-/-Trp53+/- and Xlf-/-Paxx-/-Trp53-/- mice had a 100- to 600-fold reduction in CD19+ B splenocyte count (0.7x106, p<0.0001) and a 50- to 90-fold reduction in CD3+ splenocyte count (to 0.5x106) (Figure 1F–1H and Supplementary Figure 1). From these results, we concluded that XLF and PAXX are functionally redundant during the B and T lymphocyte development in vivo.
Early B cell development is abrogated in mice lacking XLF and MRI, or XLF and PAXX
Reduced counts and proportions of mature B lymphocytes in Xlf-/-Mri-/-Trp53+/- mice suggest a blockage in B cell development in the bone marrow. To investigate this further, we isolated the bone marrow cells from femora of mice lacking XLF, MRI or both XLF/MRI, and analyzed the proportions of B220+CD43+IgM- progenitor B cells and B220+CD43-IgM+ immature and mature B cells. We detected only background levels of B220+CD43-IgM+ B cells in bone marrows isolated from Xlf-/-Mri-/-Trp53+/- mice (Figure 2A, 2B and Supplementary Table 8). However, these mice exhibited a 2- to 3-fold higher proportion of pro-B cells when compared with WT, Xlf-/- and Mri-/- controls (Figure 2A, 2C and Supplementary Table 9). Similarly, Xlf-/-Paxx-/-Trp53+/- and Xlf-/-Paxx-/-Trp53-/- mice also possess background levels of IgM+ B cells (p<0.0001; Figure 2A, 2B and Supplementary Table 8) while having 3- to 4-fold higher proportion of pro-B cells when compared with WT, Xlf-/- and Paxx-/- controls (p<0.0001; Figure 2A, 2C and Supplementary Table 9). Therefore, we conclude that B cell development is blocked at the pro-B cell stage of Xlf-/-Mri-/-Trp53+/- and Xlf-/-Paxx-/-Trp53+/- mice.
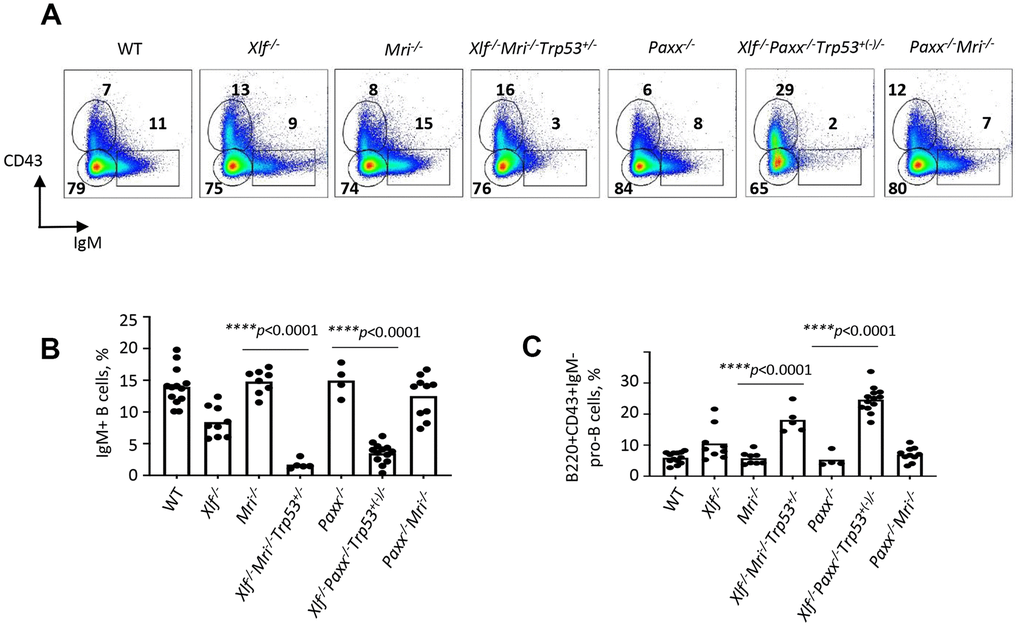
Figure 2. Development of B cells is abrogated in bone marrow of Xlf-/-Mri-/-Trp53+/- and Xlf-/-Paxx-/-Trp53+/- mice. (A) Flow cytometric analysis of developing B cells. Upper left boxes mark B220+CD43+IgM- progenitor B cell populations, and lower right boxes mark the B220+CD43-IgM+ B cells. (B, C) Frequencies (%) of B220+CD43-IgM+ B cells (B) and B220+CD43+IgM- progenitor B cells (C) in WT, Xlf-/-, Mri-/-, Xlf-/-Mri-/-Trp53+/-, Paxx-/-, Xlf-/-Paxx-/-Trp53+(-)/- and Paxx-/-Mri-/- mice. Comparisons between groups were made using one-way ANOVA, GraphPad Prism 8.0.1. Xlf-/-Paxx-/-Trp53+(-)/- is a combination of Xlf-/-Paxx-/-Trp53+/- and Xlf-/-Paxx-/-Trp53-/-.
Paxx-/-Mri-/- mice possess a modest phenotype
Both PAXX and MRI are NHEJ factors that are functionally redundant with XLF in mice. Combined inactivation of Paxx and Xlf [4, 14, 15, 20], or Mri and Xlf ([5]; this study) results in synthetic lethality in mice, as well as in abrogated V(D)J recombination in vAbl pre-B cells [4, 5, 14, 15, 27]. To determine if Paxx genetically interacts with Mri, we intercrossed mice that are heterozygous or null for both genes (such as Paxx-/-Mri+/- and Paxx+/-Mri+/-). We found that resulting Paxx-/-Mri-/- mice are live-born, fertile, and are similar in size to WT littermates (17 g, p>0.9999) (Figure 3A and 3B). Specifically, we observe that Paxx-/-Mri-/- mice have normal thymocyte and splenocyte counts. Furthermore, Paxx-/-Mri-/- mice underwent normal T cell development that was indistinguishable from the WT, Paxx-/-, and Mri-/- controls (Figures 1H, 1I and 3C). However, Paxx-/-Mri-/- mice had reduced CD19+ B cell counts (Figure 1G) when were compared to WT, Paxx-/- and Mri-/- controls (p<0.0025). Moreover, CD19+ B cell counts were similar in Paxx-/-Mri-/- and Xlf-/- mice (p>0.9270), suggesting that combined depletion of PAXX and MRI has modest phenotype similar to the one in XLF-deficient mice. CSR to IgG1 was performed in order to determine if DNA repair-dependent immunoglobulin production is affected in mature B cells lacking PAXX and MRI [16, 18]. Paxx inactivation did not affect Ig switch to IgG1 in MRI-deficient B cells (Figure 3D and 3E). The quantity of IgG1+ cells after CSR stimulation was similar between Paxx-/-Mri-/- and Mri-/- naïve B cells (p>0.73). From this, we can conclude that there is a genetic interaction between Paxx and Mriin vivo, and it is only detected in B cells.
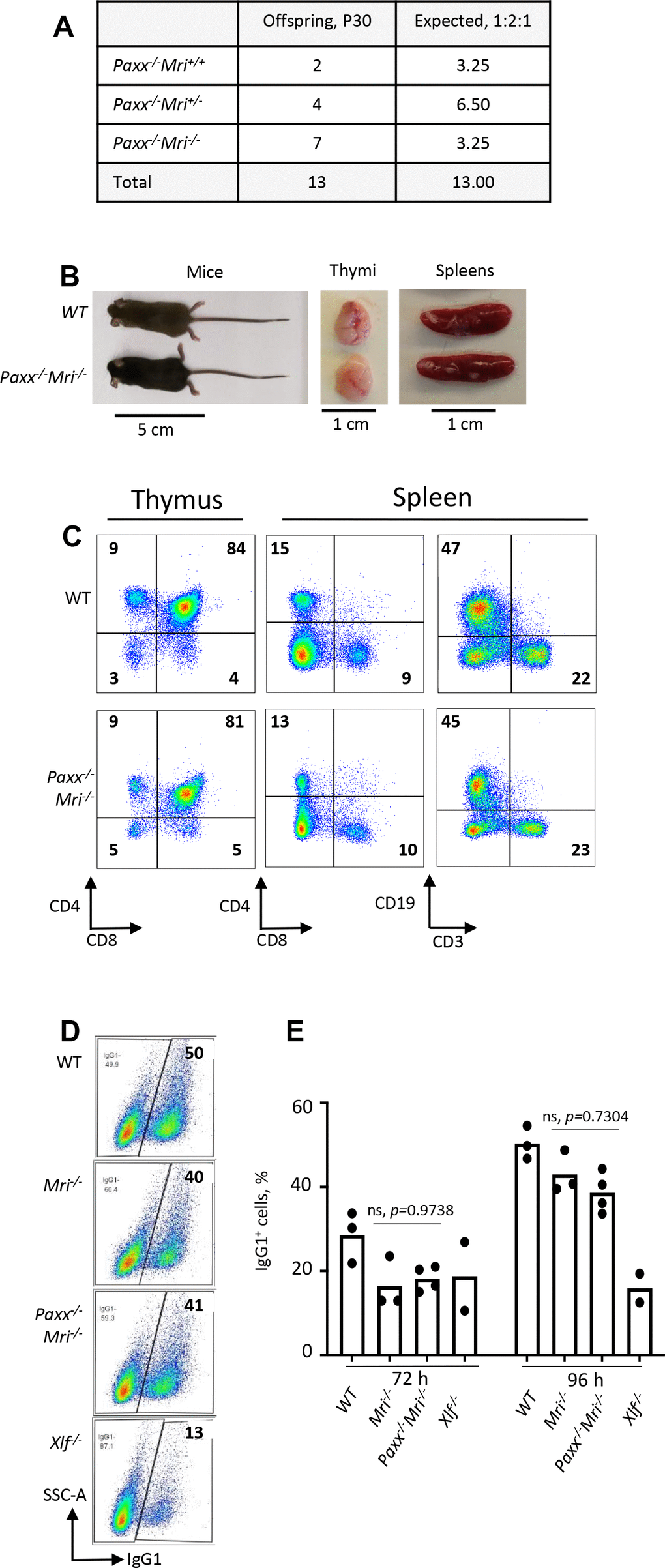
Figure 3. Development of B and T cells in Paxx-/-Mri-/- mice. (A) Number of thirty-day-old mice (P30) of indicated genotypes. Parents were Paxx+/-Mri+/- and Paxx-/-Mri+/-. (B) Example of thirty-day-old Paxx-/-Mri-/- and WT male littermates with their respective thymi and spleens. (C) Example of flow cytometry analyzes of B and T cells in Paxx-/-Mri-/- and WT mice. (D, E) Class switching analyzes of in vitro activated naïve B cells of indicated genotypes.
Synthetic lethality between Mri and Dna-pkcs in mice
Both MRI and DNA-PKcs are functionally redundant with XLF in mouse development [5, 24]. Combined inactivation of Paxx and Mri (this study), or Paxx and Dna-pkcs [20] genes results in live-born mice that are indistinguishable from single deficient controls. To determine if Mri genetically interacts with Dna-pkcs, we crossed Mri+/- and Dna-pkcs+/- mouse strains, then intercrossed the double-heterozygous Mri+/-Dna-pkcs+/-, and then Mri-/-Dna-pkcs+/- mice (Figure 4A). We identified 12 Mri-/-Dna-pkcs+/+ and 12 Mri-/-Dna-pkcs+/, but no Mri-/-Dna-pkcs-/- mice (out of 6 expected). To determine if double-deficient Mri-/-Dna-pkcs-/- embryos are present at day E14.5, we intercrossed Mri-/-Dna-pkcs+/- mice, extracted and genotyped the embryos (Figure 4B). We identified two Mri-/-Dna-pkcs-/- mice at E14.5 (63mg), which were about 40% lighter than Mri-/- littermates (108mg) (Figure 4C and 4D). A Chi-Square test (χ2) was performed to determine if the embryonic distribution data fits the mendelian ratio of 1:2:1 that is expected from Mri-/-Dna-pkcs+/- parents. With DF=2 and χ2=1.8, the corresponding p-value lies within the range 0.25<p<0.5. This affirms that our data fit the expected 1:2:1 distribution and suggests that Mri-/-Dna-pkcs-/- is synthetic lethal. Therefore, we can conclude that there is genetic interaction between Mri and Dna-pkcsin vivo.
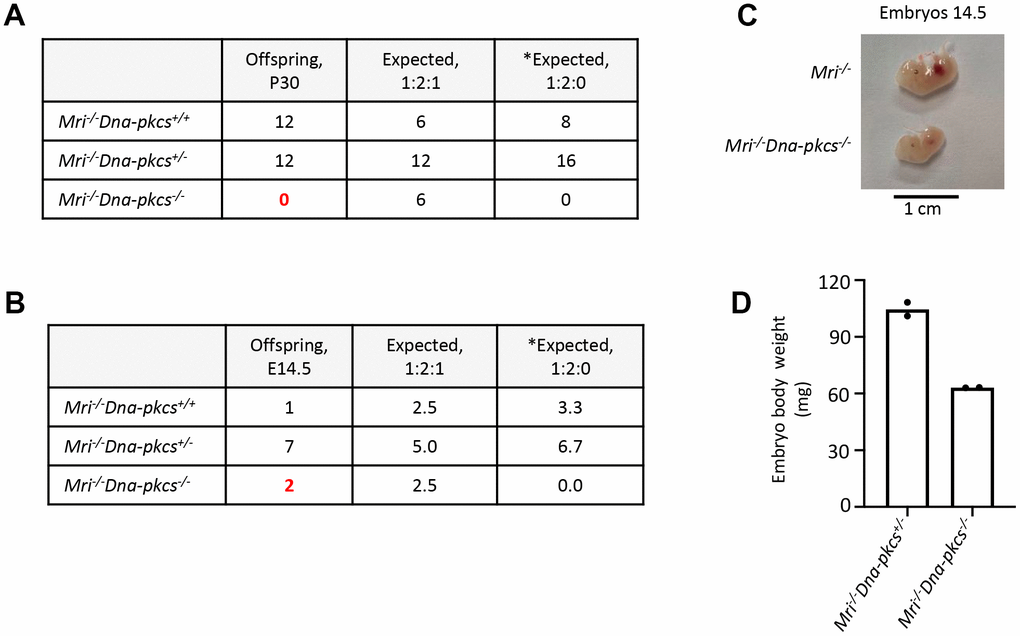
Figure 4. Genetic interaction between Mri and Dna-pkcsin vivo. (A) No live-born Mri-/-Dna-pkcs-/- mice were detected. (B, C) Mri-/-Dna-pkcs-/- embryos were detected at day E14.5. (D) Body weight in milligrams (mg) from two E14.5 Mri-/-Dna-pkcs-/- and Mri-/-Dna-pkcs+/- embryos from the same litter. The mendelian ratio 1:2:1 in embryos was verified by the Chi-Square test (χ2). The χ2 was 1.8 and its corresponding probability was between 25 and 50%. *Expected distribution assuming lethality.
Discussion
Recent findings by our and other research groups suggest that MRI forms heterogeneous complexes involving PAXX or XLF, which function during DNA DSB repair by NHEJ [5]. Furthermore, genetic inactivation of Xlf [11], Paxx [4, 14–16], or Mri [5, 18] in mice leads to development of modest or no detectable phenotype. However, combined inactivation of Xlf and Mri [5] or Xlf and Paxx [4, 14, 15] results in embryonic lethality, which correlates with increased levels of neuronal apoptosis in the CNS (Figure 5). Here, we show that synthetic lethality produced by combined inactivation of Xlf and Mri can be rescued by altered Trp53 expression, similar to our previous Xlf-/-Paxx-/-Trp53+(-)/- [20] mouse model. Furthermore, we have developed and presented here Paxx-/-Mri-/- and Mri-/-Dna-pkcs-/- double deficient models.
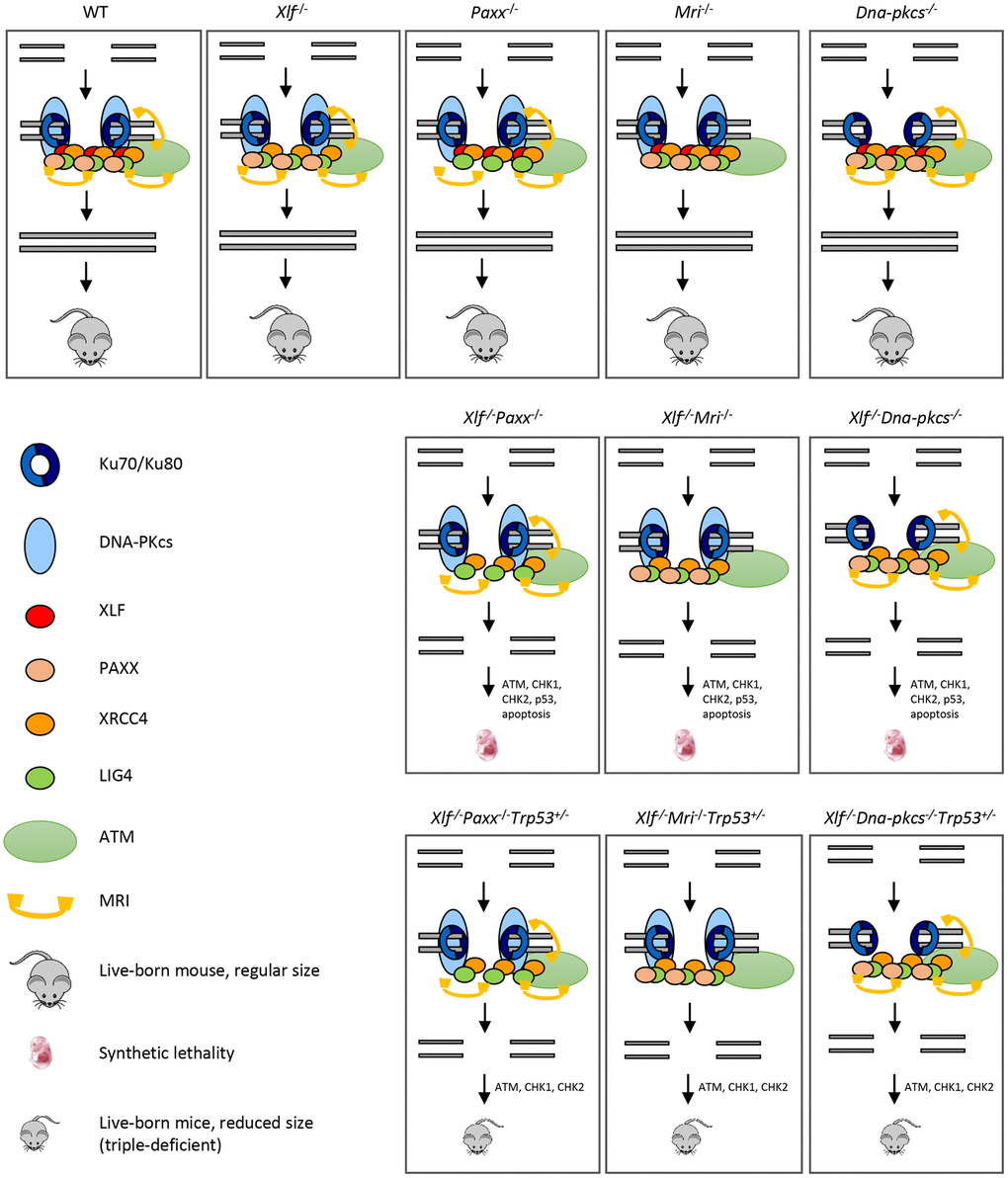
Figure 5. Mutations in NHEJ genes result in distinct phenotypes. Suggested models. Inactivation of Paxx or Mri results in live-born mice with nearly no DNA repair defects. Inactivation of Xlf or Dna-pkcs results in live-born mice with increased levels of genomic instability due to reduced NHEJ activity. Combined inactivation of Xlf/Paxx, Xlf/Mri or Xlf/Dna-pkcs leads to embryonic lethality in mice that correlate with high levels of genomic instability and nearly no NHEJ. Accumulated DSBs activate the ATM-dependent DNA damage response (DDR) pathway; ATM phosphorylates CHK checkpoint proteins that further trigger cell cycle arrest and apoptosis. Alternative end-joining is blocked by presence of Ku70/Ku80. Inactivation of one or two alleles of Trp53 rescues embryonic lethality of Xlf/Paxx, Xlf/Mri and Xlf/Dna-pkcs mice. While in these mice the levels of DSBs are increased and ATM-dependent DDR response is activated, lack of p53 prevents massive apoptosis and thus results in alive mice. Sizes of the triple-deficient mice are reduced, as one option, due to DNA damage-dependent cell cycle arrest in multiple cells of the body. The embryonic lethality in mice lacking Xlf/Paxx and Xlf/Mri is likely to be rescued by inactivation of Ku70 or Ku80.
Our findings have demonstrated that mice lacking XLF, MRI and p53, although live-born, possess a leaky SCID phenotype. Xlf-/-Mri-/-Trp53+/- mice have a clear fraction of mature B cells in the spleens (CD19+) and bone marrow (B220+CD43-IgM+) (Figures 1 and 2), as well as clear fractions of double- and single-positive T cells in the thymus (CD4+CD8+, CD4+, CD8+) and single-positive T cells in the spleen (CD4+ and CD8+) (Figure 1). However, the cell fractions from these mice are noticeably smaller than those of WT or single-deficient mice. Strikingly, we were able to identify one Xlf-/-Mri-/-Trp53+/+ mouse at day P30 post-birth. This mouse resembled Xlf-/-Mri-/-Trp53+/- mice of similar age with respect of B and T cell development (Supplementary Table 10), although this mouse was generally sicker than its littermates and had to be euthanized. Similarly, one live-born Xlf-/-Paxx-/- mouse was reported by Balmus et al. 2016 [15], indicating that, exceptionally, embryonic lethality in NHEJ ligation-deficient mice can be overcome, likely due to activity of alternative end-joining. Previously, in 2018, Hung et al. [5] reported that combined inactivation of Xlf and Mri in vAbl pre-B cells results in a severe block in V(D)J recombination and accumulation of unrepaired DSBs in vitro, although it was unclear whether this combined inactivation would lead to a deficiency in B lymphocytes when translated to a mouse model [5]. Similarly, double deficient vAbl pre-B cells lacking Xlf and Paxx are also unable to sustain V(D)J recombination. Importantly, the lack of a progenitor T cell model system left the question of T cell development in Xlf-/-Mri-/- and Xlf-/-Paxx-/- mice completely unexplored.
Previously, we showed that mice lacking XLF, PAXX and p53 were live-born and had nearly no B and T cells, reduced size of spleen and hardly detectable thymus [20] (Figure 5). Consistent with this model, a conditional knockout mouse model, which results in double-deficiency of XLF/PAXX in early hematopoietic progenitor cells, was also able to overcome the embryonic lethality of Xlf-/-Paxx-/- mice [33]. With this model, impairment of V(D)J recombination in Xlf-/-Paxx-/- cells, as well as the resulting depletion of mature B cells and lack of a visible thymus could also be observed in vivo [33]. Our new data provide evidence that Xlf-/-Paxx-/-Trp53+/- and Xlf-/-Paxx-/-Trp53-/- mice possess a very small number of mature B cells in the spleen and bone marrow, as well as very minor fractions of single positive T cells in thymus and spleen (Figures 2, 5 and Supplementary Figure 1). Therefore, both mature B and T cells are present in mice lacking XLF/PAXX and XLF/MRI. This can be explained by incomplete blockage in NHEJ and V(D)J recombination, in which the process is dramatically reduced but still possible. We also detected more mature T cells than B cells in these double-deficient mice. Potential explanations include longer lifespan of T cells, which accumulate over time following low efficiency of V(D)J recombination, while B cells are eliminated faster from the pool due to the different physiology [34, 35]. It is also possible that the T cells we detected are a resultant subpopulation that is descendent from the few cells that were able to bypass V(D)J recombination [12]. In this case, the repertoire of T cells based on T cell receptor in mice lacking XLF/PAXX and XLF/MRI would be significantly lower than in control mice, even if normalized to the total cell count. Due to the small presence of mature B and T cells in Xlf-/-Mri-/-Trp53+/-, Xlf-/-Paxx-/-Trp53+/- and Xlf-/-Paxx-/-Trp53-/- mice, we categorize the observed immunodeficient phenotypes as “leaky SCID”. Previously, leaky SCID has been described in mice lacking other NHEJ factors, such as Ku70-/- [6], Artemis-/- [3], Lig4-/-Trp53-/- [10, 30], Xrcc4-/-Trp53-/- [9, 31], Xlf-/-Atm-/- [19] and Xlf-/-Rag2c/c [23].
In addition to XLF/MRI and XLF/PAXX deficient mice, inactivation of one or two alleles of Trp53 also rescues the embryonic lethality of Xrcc4-/- [9, 31], Lig4-/- [10, 30] and Xlf-/-Dna-pkcs-/- [20] mice. We propose a model (Figure 5), when single deficiency for DNA-PKcs, PAXX or MRI results in no or modest phenotypes, and DSBs are efficiently repaired. Combined inactivation of Xlf/Dna-pkcs, Xlf/Paxx and Xlf/Mri results in inefficient DSB ligation, accumulation of DNA breaks, activation of ATM-dependent DDR, checkpoint protein CHK2, stabilization of p53 and massive apoptosis. This results in embryonic lethality in mice. Furthermore, inactivation of Trp53 results in Xlf/Dna-pkcs/Trp53, Xlf/Paxx/Trp53 and Xlf/Mri/Trp53 triple-deficient mice. While DNA breaks in these mice are not repaired, ATM-dependent DDR response and activation of CHK proteins takes place. However, without p53, apoptosis is not activated, allowing survival of mice (Figure 5). Moreover, we propose that inactivation of Atm will also rescue embryonic lethality of Xlf/Paxx and Xlf/Mri mice due to the mechanisms proposed above. However, inactivation of Atm will not rescue embryonic lethality of Xlf/Dna-pkcs mice, due to synthetic lethality between Atm and Dna-pkcs.
It is important to note that altered Trp53 expression is not always sufficient to rescue embryonic lethality in mice; for example, PLK1-interacting checkpoint helicase (PICH)-deficient mice possess developmental defects in the presence or absence of p53 [36], and ATR mutants (Seckel syndrome) are not completely rescued from embryonic lethality with the inactivation of Trp53 [37]. Embryonic lethality of XLF/PAXX and XLF/MRI double-deficient mice can be explained by the presence of Ku70/Ku80 heterodimer at the DSBs sites, which blocks DNA repair by alternative end-joining pathway(s), leading to massive apoptosis and cell cycle arrest [38]. Previously, it was shown that embryonic lethality of LIG4-deficient [39] and XLF/DNA-PKcs double-deficient mice [25] could be rescued by inactivating Ku70 or Ku80 genes. Similarly, we propose that inactivation of either Ku70 or Ku80 gene will rescue the embryonic lethality of XLF/PAXX and XLF/MRI double-deficient mice and will result in mice indistinguishable from Ku70- or Ku80-deficient controls (Figure 5).
Recent studies have shown that Xlf genetically interacts with Rag2 [23] and DDR factors, such as Atm, 53bp1, H2ax, and Mdc1 [17, 19–22, 38]. Xlf-/-Rag2c/c mice almost completely lack mature B cells and have significantly fewer mature T cells than single deficient controls [23]. Xlf-/-Atm-/- and Xlf-/-53bp1-/- mice are live-born and exhibit reduced body weight, increased genomic instability, and severe lymphocytopenia as a result of V(D)J recombination impairment in developing B and T cells [1, 17, 19, 22]. Xlf-/-H2ax-/- and Xlf-/-Mdc1-/-, on the other hand, are embryonic lethal [19–21]. There are several possible explanations for the functional redundancy observed between DNA repair genes. For instance, the two factors could have identical (e.g., if both proteins are involved in ligation or DNA end tethering) or complementary (e.g., if one protein stimulates ligation while the other is required for DNA end tethering) functions. To date, XLF has been shown to genetically interact with multiple DNA repair factors [1, 4, 5, 14, 15, 19, 20, 24, 25], and this list is likely to grow [38, 40]. However, no clear genetic interaction has been shown between Xlf and Artemis or Xrcc4 in the context of mouse development and V(D)J recombination [24], meaning that it remains difficult to predict genetic interactions without developing and characterizing genetic models.
We found that mice with combined inactivation of Paxx and Mri (Paxx-/-Mri-/-) are live-born, fertile, and undergo almost normal B and T cell development (Figure 3), where only the number of splenic B cells is affected, giving rise to a modest phenotype. Moreover, inactivation of Paxx did not affect the CSR efficiency in in vitro stimulated MRI-deficient B cells (Figure 3), thereby confirming our observations in vitro. It has been also shown that combined inactivation of Paxx and Mri genes in vAbl pre-B cells lead to similar V(D)J recombination efficiency to single-deficient Mri-/-, Paxx-/- and WT controls [5]. Thus, we conclude that there is a genetic interaction between Paxx and Mri, which results in a modest phenotype.
Lastly, we found that combined inactivation of Mri and Dna-pkcs (Mri-/-Dna-pkcs-/-) leads to embryonic lethality, and that E14.5 Mri-/-Dna-pkcs-/- murine embryos were about 40% smaller than single-deficient siblings (Figure 4). DNA-PKcs is associated with the N-terminus of the MRI and Ku heterodimer in the process of recognizing DSBs [5], which may account for genetic interaction between Mri and Dna-pkcs. Thus, inactivation of Trp53, Ku70 or Ku80 may be a viable method to rescue synthetic lethality from Mri-/-Dna-pkcs-/- mice.
In conclusion, we have developed and described several complex genetic mouse models (Figure 5). Xlf-/-Mri-/-Trp53+/- and Xlf-/-Paxx-/-Trp53+(-)/- mice possessed severely impaired B and T lymphocyte development, leaky SCID; Paxx-/-Mri-/- mice develop a modest B cell phenotype; and Mri-/-Dna-pkcs-/- mice are embryonic lethal.
Materials and Methods
Mice
All experiments involving mice were performed according to the protocols approved by the Comparative Medicine Core Facility (CoMed) at the Norwegian University of Science and Technology (NTNU, Trondheim, Norway). Xlf+/- [11] and Dna-pkcs+/- [2] mice were imported from the laboratory of Professor Frederick W. Alt at Harvard Medical School. Trp53+/- mice [32] were imported from Jackson Laboratories. Paxx+/- [16] and Mri+/- [18] mice were generated by the Oksenych group and described previously.
Lymphocyte development
Lymphocyte populations were analyzed by flow cytometry [16, 18, 19, 22]. In summary, cells were isolated from the spleen, thymus, and femur of 5-7-week-old mice and treated with red blood cell lysis buffer Hybri-MaxTM (Sigma Aldrich, St. Louis, MO, USA; #R7757). The cells were resuspended in PBS (Thermo Scientific, Basingstoke, UK; #BR0014G) containing 5% Fetal bovine serum, FCS (Sigma Life Science, St. Louis, Missouri, United States; #F7524), and counted using a Countess™ II Automated Cell Counter (Invitrogen, Carlsbad, CA, United States; #A27977). Then, the cell suspension was diluted with PBS to get a final cell concentration of 2.5 x 107 cells/mL. Finally, surface markers were labeled with fluorochrome-conjugated antibodies and the cell populations were analyzed using flow cytometry.
Class switch recombination (CSR)
Spleens were isolated from 5-7-week-old mice and stored in cold PBS. Splenocytes were obtained by mincing the spleens, and naïve B cells were negatively selected using an EasySep Isolation kit (Stemcell™, Cambridge, UK; #19854). Lipopolysaccharide (LPS; 40 μg/mL; Sigma Aldrich, St. Louis, MO, USA; #437627-5MG) and interleukin 4 (IL-4; 20 ng/mL; PeproTech, Stockholm, Sweden; #214-14) were used to induce CSR to IgG1. Expression of IgG1 was analyzed by flow cytometry.
Antibodies
The following antibodies were used for flow cytometric analysis: rat anti-CD4-PE-Cy7 (BD PharmingenTM, Allschwil, Switzerland, #552775; 1:100); rat anti-CD8-PE-Cy5 (BD PharmingenTM, Allschwil, Switzerland, #553034; 1:100); anti-CD19-PE-Cy7 (Biolegend, San Diego, CA, USA, #115520; 1:100); hamster anti-mouse anti-CD3-FITC (BD PharmingenTM, Allschwil, Switzerland, #561827; 1:100); rat anti-mouse anti-CD43-FITC (BD PharmingenTM, Allschwil, Switzerland, #561856; 1:100); rat anti-mouse anti-CD45R/B220-APC (BD PharmingenTM, Allschwil, Switzerland; #553092; 1:100); rat anti-mouse anti-IgM-PE-Cy7 (BD PharmingenTM, Allschwil, Switzerland, #552867; 1:100); rat anti-mouse IgG1-APC (BD PharmingenTM, Allschwil, Switzerland; #550874; 1:100). A LIVE/DEAD™ fixable violet dead cell stain kit (ThermoFisher Scientific, Waltham, MA, USA; #L34955; 1:1000) was used to identify dead cells.
Statistics
Statistical analyses were performed using one-way ANOVA, GraphPad Prism 8.0.1.244 (San Diego, CA, USA). In all statistical tests, p<0.05 were taken to be significant (*p<0.05; **p<0.01; ***p<0.001; ****p<0.0001).
Author Contributions
VO, SCZ, QZ, AL and MFB designed the study, analyzed and interpreted the results. SCZ, QZ, AL and MFB performed most of the experiments. VO wrote the paper with the help of SCZ and RY. All the authors contributed to writing of the final manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding
This work was supported by the Research Council of Norway Young Talent Investigator grant (#249774) to V.O. In addition, VO group was supported by the Liaison Committee for Education, Research, and Innovation in Central Norway (#13477; #38811); the Norwegian Cancer Society (#182355); the Research Council of Norway FRIMEDBIO grants (#270491 and #291217), and The Outstanding Academic Fellow Program at NTNU (2017–2021). Karolinska Institutet Stiftelser och Fonder (#2020-02155).
References
- 1. Kumar V, Alt FW, Oksenych V. Functional overlaps between XLF and the ATM-dependent DNA double strand break response. DNA Repair (Amst). 2014; 16:11–22. https://doi.org/10.1016/j.dnarep.2014.01.010 [PubMed]
- 2. Gao Y, Chaudhuri J, Zhu C, Davidson L, Weaver DT, Alt FW. A targeted DNA-PKcs-null mutation reveals DNA-PK-independent functions for KU in V(D)J recombination. Immunity. 1998; 9:367–76. https://doi.org/10.1016/s1074-7613(00)80619-6 [PubMed]
- 3. Rooney S, Sekiguchi J, Zhu C, Cheng HL, Manis J, Whitlow S, DeVido J, Foy D, Chaudhuri J, Lombard D, Alt FW. Leaky scid phenotype associated with defective V(D)J coding end processing in artemis-deficient mice. Mol Cell. 2002; 10:1379–90. https://doi.org/10.1016/s1097-2765(02)00755-4 [PubMed]
- 4. Liu X, Shao Z, Jiang W, Lee BJ, Zha S. PAXX promotes KU accumulation at DNA breaks and is essential for end-joining in XLF-deficient mice. Nat Commun. 2017; 8:13816. https://doi.org/10.1038/ncomms13816 [PubMed]
- 5. Hung PJ, Johnson B, Chen BR, Byrum AK, Bredemeyer AL, Yewdell WT, Johnson TE, Lee BJ, Deivasigamani S, Hindi I, Amatya P, Gross ML, Paull TT, et al. MRI is a DNA damage response adaptor during classical non-homologous end joining. Mol Cell. 2018; 71:332–42.e8. https://doi.org/10.1016/j.molcel.2018.06.018 [PubMed]
- 6. Gu Y, Seidl KJ, Rathbun GA, Zhu C, Manis JP, van der Stoep N, Davidson L, Cheng HL, Sekiguchi JM, Frank K, Stanhope-Baker P, Schlissel MS, Roth DB, Alt FW. Growth retardation and leaky SCID phenotype of Ku70-deficient mice. Immunity. 1997; 7:653–65. https://doi.org/10.1016/s1074-7613(00)80386-6 [PubMed]
- 7. Nussenzweig A, Chen C, da Costa Soares V, Sanchez M, Sokol K, Nussenzweig MC, Li GC. Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature. 1996; 382:551–55. https://doi.org/10.1038/382551a0 [PubMed]
- 8. Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002; 108:781–94. https://doi.org/10.1016/s0092-8674(02)00671-2 [PubMed]
- 9. Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, Seidl KJ, Sekiguchi JM, Rathbun GA, Swat W, Wang J, Bronson RT, Malynn BA, Bryans M, et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998; 95:891–902. https://doi.org/10.1016/s0092-8674(00)81714-6 [PubMed]
- 10. Frank KM, Sekiguchi JM, Seidl KJ, Swat W, Rathbun GA, Cheng HL, Davidson L, Kangaloo L, Alt FW. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature. 1998; 396:173–77. https://doi.org/10.1038/24172 [PubMed]
- 11. Li G, Alt FW, Cheng HL, Brush JW, Goff PH, Murphy MM, Franco S, Zhang Y, Zha S. Lymphocyte-specific compensation for XLF/cernunnos end-joining functions in V(D)J recombination. Mol Cell. 2008; 31:631–40. https://doi.org/10.1016/j.molcel.2008.07.017 [PubMed]
- 12. Vera G, Rivera-Munoz P, Abramowski V, Malivert L, Lim A, Bole-Feysot C, Martin C, Florkin B, Latour S, Revy P, de Villartay JP. Cernunnos deficiency reduces thymocyte life span and alters the T cell repertoire in mice and humans. Mol Cell Biol. 2013; 33:701–11. https://doi.org/10.1128/MCB.01057-12 [PubMed]
- 13. Roch B, Abramowski V, Chaumeil J, de Villartay JP. Cernunnos/Xlf deficiency results in suboptimal V(D)J recombination and impaired lymphoid development in mice. Front Immunol. 2019; 10:443. https://doi.org/10.3389/fimmu.2019.00443 [PubMed]
- 14. Abramowski V, Etienne O, Elsaid R, Yang J, Berland A, Kermasson L, Roch B, Musilli S, Moussu JP, Lipson-Ruffert K, Revy P, Cumano A, Boussin FD, de Villartay JP. PAXX and Xlf interplay revealed by impaired CNS development and immunodeficiency of double KO mice. Cell Death Differ. 2018; 25:444–52. https://doi.org/10.1038/cdd.2017.184 [PubMed]
- 15. Balmus G, Barros AC, Wijnhoven PW, Lescale C, Hasse HL, Boroviak K, le Sage C, Doe B, Speak AO, Galli A, Jacobsen M, Deriano L, Adams DJ, et al. Synthetic lethality between PAXX and XLF in mammalian development. Genes Dev. 2016; 30:2152–57. https://doi.org/10.1101/gad.290510.116 [PubMed]
- 16. Gago-Fuentes R, Xing M, Sæterstad S, Sarno A, Dewan A, Beck C, Bradamante S, Bjørås M, Oksenych V. Normal development of mice lacking PAXX, the paralogue of XRCC4 and XLF. FEBS Open Bio. 2018; 8:426–34. https://doi.org/10.1002/2211-5463.12381 [PubMed]
- 17. Liu X, Jiang W, Dubois RL, Yamamoto K, Wolner Z, Zha S. Overlapping functions between XLF repair protein and 53BP1 DNA damage response factor in end joining and lymphocyte development. Proc Natl Acad Sci USA. 2012; 109:3903–08. https://doi.org/10.1073/pnas.1120160109 [PubMed]
- 18. Castañeda-Zegarra S, Huse C, Røsand Ø, Sarno A, Xing M, Gago-Fuentes R, Zhang Q, Alirezaylavasani A, Werner J, Ji P, Liabakk NB, Wang W, Bjørås M, Oksenych V. Generation of a mouse model lacking the non-homologous end-joining factor mri/cyren. Biomolecules. 2019; 9:798. https://doi.org/10.3390/biom9120798 [PubMed]
- 19. Zha S, Guo C, Boboila C, Oksenych V, Cheng HL, Zhang Y, Wesemann DR, Yuen G, Patel H, Goff PH, Dubois RL, Alt FW. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature. 2011; 469:250–54. https://doi.org/10.1038/nature09604 [PubMed]
- 20. Castañeda-Zegarra S, Xing M, Gago-Fuentes R, Sæterstad S, Oksenych V. Synthetic lethality between DNA repair factors Xlf and Paxx is rescued by inactivation of Trp53. DNA Repair (Amst). 2019; 73:164–69. https://doi.org/10.1016/j.dnarep.2018.12.002 [PubMed]
- 21. Beck C, Castañeda-Zegarra S, Huse C, Xing M, Oksenych V. Mediator of DNA damage checkpoint protein 1 facilitates V(D)J recombination in cells lacking DNA repair factor XLF. Biomolecules. 2019; 10:60. https://doi.org/10.3390/biom10010060 [PubMed]
- 22. Oksenych V, Alt FW, Kumar V, Schwer B, Wesemann DR, Hansen E, Patel H, Su A, Guo C. Functional redundancy between repair factor XLF and damage response mediator 53BP1 in V(D)J recombination and DNA repair. Proc Natl Acad Sci U S A. 2012; 109:2455–60. https://doi.org/10.1073/pnas.1121458109 [PubMed]
- 23. Lescale C, Abramowski V, Bedora-Faure M, Murigneux V, Vera G, Roth DB, Revy P, de Villartay JP, Deriano L. RAG2 and XLF/cernunnos interplay reveals a novel role for the RAG complex in DNA repair. Nat Commun. 2016; 7:10529. https://doi.org/10.1038/ncomms10529 [PubMed]
- 24. Oksenych V, Kumar V, Liu X, Guo C, Schwer B, Zha S, Alt FW. Functional redundancy between the XLF and DNA-PKcs DNA repair factors in V(D)J recombination and nonhomologous DNA end joining. Proc Natl Acad Sci U S A. 2013; 110:2234–9. https://doi.org/10.1073/pnas.1222573110 [PubMed]
- 25. Xing M, Bjørås M, Daniel JA, Alt FW, Oksenych V. Synthetic lethality between murine DNA repair factors XLF and DNA-PKcs is rescued by inactivation of Ku70. DNA Repair (Amst). 2017; 57:133–38. https://doi.org/10.1016/j.dnarep.2017.07.008 [PubMed]
- 26. Lescale C, Lenden Hasse H, Blackford AN, Balmus G, Bianchi JJ, Yu W, Bacoccina L, Jarade A, Clouin C, Sivapalan R, Reina-San-Martin B, Jackson SP, Deriano L. Specific roles of XRCC4 paralogs PAXX and XLF during V(D)J recombination. Cell Rep. 2016; 16:2967–79. https://doi.org/10.1016/j.celrep.2016.08.069 [PubMed]
- 27. Kumar V, Alt FW, Frock RL. PAXX and XLF DNA repair factors are functionally redundant in joining DNA breaks in a G1-arrested progenitor b-cell line. Proc Natl Acad Sci USA. 2016; 113:10619–24. https://doi.org/10.1073/pnas.1611882113 [PubMed]
- 28. Hung PJ, Chen BR, George R, Liberman C, Morales AJ, Colon-Ortiz P, Tyler JK, Sleckman BP, Bredemeyer AL. Deficiency of XLF and PAXX prevents DNA double-strand break repair by non-homologous end joining in lymphocytes. Cell Cycle. 2017; 16:286–95. https://doi.org/10.1080/15384101.2016.1253640 [PubMed]
- 29. Barnes DE, Stamp G, Rosewell I, Denzel A, Lindahl T. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr Biol. 1998; 8:1395–98. https://doi.org/10.1016/s0960-9822(98)00021-9 [PubMed]
- 30. Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C, Manis JP, Horner J, DePinho RA, Alt FW. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell. 2000; 5:993–1002. https://doi.org/10.1016/s1097-2765(00)80264-6 [PubMed]
- 31. Gao Y, Ferguson DO, Xie W, Manis JP, Sekiguchi J, Frank KM, Chaudhuri J, Horner J, DePinho RA, Alt FW. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature. 2000; 404:897–900. https://doi.org/10.1038/35009138 [PubMed]
- 32. Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994; 4:1–7. https://doi.org/10.1016/s0960-9822(00)00002-6 [PubMed]
- 33. Musilli S, Abramowski V, Roch B, de Villartay JP. An in vivo study of the impact of deficiency in the DNA repair proteins PAXX and XLF on development and maturation of the hemolymphoid system. J Biol Chem. 2020; 295:2398–406. https://doi.org/10.1074/jbc.AC119.010924 [PubMed]
- 34. Di Rosa F, Ramaswamy S, Ridge JP, Matzinger P. On the lifespan of virgin T lymphocytes. J Immunol. 1999; 163:1253–57. [PubMed]
- 35. Fulcher DA, Basten A. B cell life span: a review. Immunol Cell Biol. 1997; 75:446–55. https://doi.org/10.1038/icb.1997.69 [PubMed]
- 36. Albers E, Sbroggiò M, Pladevall-Morera D, Bizard AH, Avram A, Gonzalez P, Martin-Gonzalez J, Hickson ID, Lopez-Contreras AJ. Loss of PICH results in chromosomal instability, p53 activation, and embryonic lethality. Cell Rep. 2018; 24:3274–84. https://doi.org/10.1016/j.celrep.2018.08.071 [PubMed]
- 37. Murga M, Bunting S, Montaña MF, Soria R, Mulero F, Cañamero M, Lee Y, McKinnon PJ, Nussenzweig A, Fernandez-Capetillo O. A mouse model of ATR-seckel shows embryonic replicative stress and accelerated aging. Nat Genet. 2009; 41:891–98. https://doi.org/10.1038/ng.420 [PubMed]
- 38. Castañeda-Zegarra S, Fernandez-Berrocal M, Tkachov M, Yao R, Upfold NL, Oksenych V. Genetic interaction between the non-homologous end-joining factors during B and T lymphocyte development: in vivo mouse models. Scand J Immunol. 2020; 92:e12936. https://doi.org/10.1111/sji.12936 [PubMed]
- 39. Karanjawala ZE, Adachi N, Irvine RA, Oh EK, Shibata D, Schwarz K, Hsieh CL, Lieber MR. The embryonic lethality in DNA ligase IV-deficient mice is rescued by deletion of ku: implications for unifying the heterogeneous phenotypes of NHEJ mutants. DNA Repair (Amst). 2002; 1:1017–26. https://doi.org/10.1016/s1568-7864(02)00151-9 [PubMed]
- 40. Wang XS, Lee BJ, Zha S. The recent advances in non-homologous end-joining through the lens of lymphocyte development. DNA Repair (Amst). 2020; 94:102874. https://doi.org/10.1016/j.dnarep.2020.102874 [PubMed]