



Introduction
Tau is a microtubule-associated protein (MAP) that plays an important role in the assembly and stabilization of microtubules. It is predominantly an axonal protein and is involved in the regulation of neuronal morphology, neurite extension and axonal transport of organelles [1–4]. The pathological aggregation of the microtubule associated tau protein into filaments is a histopathological characteristic of tauopathies, including Alzheimer’s disease (AD) [5, 6]. However, recent studies suggest that soluble tau oligomers, the precursors to the higher order paired helical filaments (PHFs) and neurofibrillary tangles (NFTs), are more toxic and have a greater potential in spreading the tau pathology [7–9]. Moreover, it is also documented that the levels of tau oligomerization are elevated prior to NFTs formation in the brain of AD patients supporting a dynamic relationship between tau oligomerization and the progression of tauopathy in AD brains [10, 11].
Aging is one of the greatest risk factors for the development and progression of neurodegenerative diseases, including tauopathies [12, 13]. Moreover, multiple studies have shown an interconnection between tau pathology and mitochondrial dysfunction [14, 15]. Indeed, tau-mediated mitochondrial damage could be due to reduced levels of mitochondrial activity [16–18], its interaction with mitochondrial proteins [11], modulation of mitochondrial dynamics [19] and activation of the mitochondrial apoptotic pathway [20–22]. Mitochondria constantly undergo fusion and fission to regulate their size, shape and numbers in response to nutrient availability, stress conditions and energy demand of the cell [23]. Neurons are particularly vulnerable to impairment of mitochondrial dynamics because they are largely dependent on mitochondria for energy production, especially at the synapses and because distribution of mitochondria to distal parts of the neuron require energy [24–27]. Mitochondrial fusion and fission events are also quality control mechanisms, since fusion complements damaged components of a mitochondrion with those of a healthy mitochondrion, and mitochondrial fission facilitates the selective autophagic removal of defective organelles, known as mitophagy [28]. Previous studies have shown that tau accumulation and mislocalization disturbs microtubule stability and interferes with transport of neuronal organelles, including mitochondria. These disturbances in trafficking partly explain tau mediated synapse and memory deficits in tauopathies [6, 8]. Although the effect of tau lesion on mitochondrial metabolism is frequently reported, it remains unclear whether altered mitochondrial homeostasis is a result of fully developed tau pathology or it is an early event in tauopathies and as such may play a critical role in the disease progression.
Here, we report that excessive mitochondrial damage is an early event in neuronal cells in a nematode model of human tauopathy. Transgenic animals expressing the full length of wild type tau displayed a reduced number of mitochondria in neuronal processes of C. elegans larvae. The morphological characteristics of mitochondria indicated that tau expression promotes deregulation of mitochondrial function during nematode development and adulthood. Despite the fact that tau aggregation was mainly observed in adult animals, locomotion deficits were also present during larval stages underscoring the toxic effects of tau in organelle function and cellular physiology. Finally, we found that chelating calcium by ethylene glycol tetraacetic acid (EGTA) increased the mitochondrial membrane potential and improved motility in the tau-expressing larvae suggesting a positive correlation between mitochondrial function, calcium homeostasis and neuronal performance.
Results
Age-dependent aggregation of wild type tau in C. elegans neurons
To investigate the impact of tau on mitochondrial activity, neuronal function and organismal physiology, we utilized an already characterized nematode strain that expresses the full length of wild type human tau protein under the control of the pan-neuronal promoter of the snb-1 gene (the nematode synaptobrevin homologue) [29]. To avoid possible toxic effects of tau overexpression, we used a transgenic nematode strain expressing wild type tau at low levels (tauwt-low; PIR3, henceforth tau-expressing nematodes), which display mild phenotypic abnormalities [29]. We collected age-synchronized worms and performed total tau protein extraction from the transgenic nematodes to assess the formation of tau oligomers and aggregates over time. We found that high molecular weight tau oligomers started to accumulate from day three of adulthood and gradually increased in C. elegans neurons with age (Figure 1A and Supplementary Figure 1A–1B). Accumulation of tau protein with age was not due to altered transcriptional activity of the tissue specific snb-1 promoter (Gene Expression Omnibus (GEO) dataset GSE832 [30]).
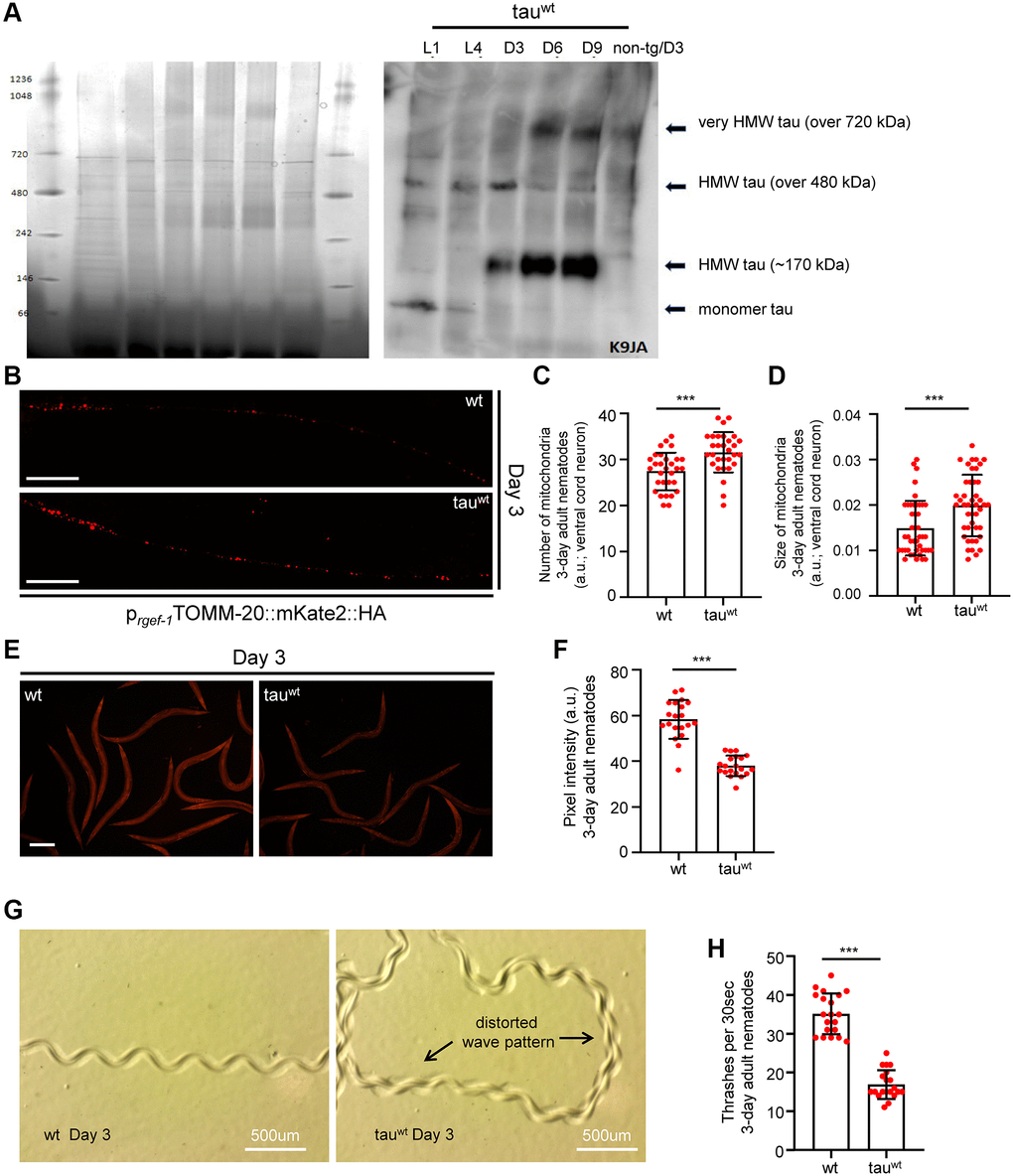
Figure 1. Impaired mitochondrial homeostasis and motility defects in tauwt-expressing 3-day adult animals. (A) Age-dependent accumulation of wild type tau protein in neuronal cells (heigh molecular weight; HMW). (B) Representative fluorescent images of transgenic nematodes expressing pan-neuronally mitochondria-targeted mKate2::HA. Scale bar, 20 μm. Tauwt-expressing 3-day-adult nematodes display (C) increased mitochondrial population and (D) smaller organelle compared to their wild type counterparts (n = 30; ***P < 0.0001; unpaired t-test). (E, F) Tauwt expressing nematodes display decreased mitochondrial membrane potential (n = 40; ***P < 0.0001; unpaired t-test). Scale Bar, 100 μm. (G) Representative images of 3-day adult wild type and tauwt expressing nematodes tracks. (H) Body bends of 3-day adult wild type and tauwt- animals per 30 seconds in M9 buffer (n = 20; ***P < 0.0001; unpaired t-test).
A recent study demonstrated that a low level of tau expression impaired mitochondrial number and distribution in neuronal processes in the tau-expressing C. elegans strain during day 1 and day 3 of adulthood [29]. To further examine the effect of low level of tau expression on neuronal mitochondria integrity, we generated transgenic animals co-expressing mitochondria-targeted mKate2::HA with wild type tau in neuronal cells. Interestingly, we found that 3-day-old tau-expressing animals displayed more and smaller axonal mitochondria compared to the age-matched wild type nematodes (Figure 1B–1D and Supplementary Figure 2A). A growing body of evidence suggests that challenging conditions trigger mitochondrial fragmentation generating smaller organelles to promote the isolation and subsequent removal of damaged mitochondria through mitophagy [31–34]. Notably, several mutant isoforms of tau have been shown to inhibit mitophagy leading to the accumulation of defective mitochondria and subsequently to cellular and tissue deterioration [35, 36]. Thus, our finding suggests that fragmented and damaged mitochondria accumulate in the neurons of tau-expressing worms. Although mitochondrial activity gradually declined with age in wild type animals, the mitochondrial membrane potential was highly reduced even in young tau-expressing nematodes (Figure 1E–1F and Supplementary Figure 2B), underlining the toxic effect of tau expression on energy metabolism.
Proprioception has an essential role in the movement coordination and body balance of an organism. Age-dependent deterioration of biological systems and pathological conditions could cause proprioception impairment leading to uncontrolled and inefficient mobility [37]. In C. elegans, proprioception can be evaluated by monitoring the sinusoidal wave pattern that is generated by the periodic bending of its head and body [38, 39]. Wild type nematodes inscribe a sinusoidal track as they move on an agar plate seeded with OP50 E. coli bacterial strain. The characteristic properties (amplitude and wavelength) of tracks generated by the tau-expression animals were dramatically perturbed indicating severe motility deficits (Figure 1G). Moreover, we assessed the locomotion by measuring bending behavior in both wild type and the tau-expressing nematodes during adulthood. Different age groups of wild type and transgenic nematodes were placed in a 10 μl droplet of M9 buffer and were allowed to swim freely for 1 minute, to become accustomed to their new environment. Body bends were then monitored for 30 seconds. In accord with the abnormal sinusoidal tracks, the tau-expressing nematodes displayed pronounced locomotion defects throughout adulthood compared to the wild type animals (Figure 1H and Supplementary Figure 2C). These results suggest that mitochondrial dysfunction in the tau-expressing nematodes could mediate energy deprivation subsequently leading to neuronal deregulation and eventually to motility defects.
Impaired activity of neuronal mitochondria and motility defects in larval stages
Accumulating evidence underscores the effect of critically high levels of tau and tau lesions on mitochondrial homeostasis [40, 41]. Tau may influence mitochondrial function both directly via its localization on mitochondrial compartments and indirectly through perturbation of cytoskeleton components [40–47]. While the association between tau levels and energy metabolism is established, it is not clear whether mitochondrial dysfunction is an early pathological feature of high levels of tau or a consequence of its excessive formation of protein aggregates. To discriminate between these two scenarios, we focused on the L1 and L4 larval developmental stages. Although the formation of tau oligomers and/or aggregates was not detectable in neuronal cells of L1 and L4 larvae (Figure 1A), tau-expressing animals displayed a reduction in body bends compared to the wild type nematodes (Figure 2A–2B). We next monitored the mitochondrial population in the neuronal processes of the ventral and dorsal cord of both the L1 and L4 larvae, which expressed pan-neuronally mitochondria-targeted mKate2::HA and wild type tau. We found decreased mitochondrial density in ventral (Figure 2C–2D; Figure 3A, 3B) and dorsal cord neurons in larval stages (Supplementary Figure 3A–3B, 3F–3G).
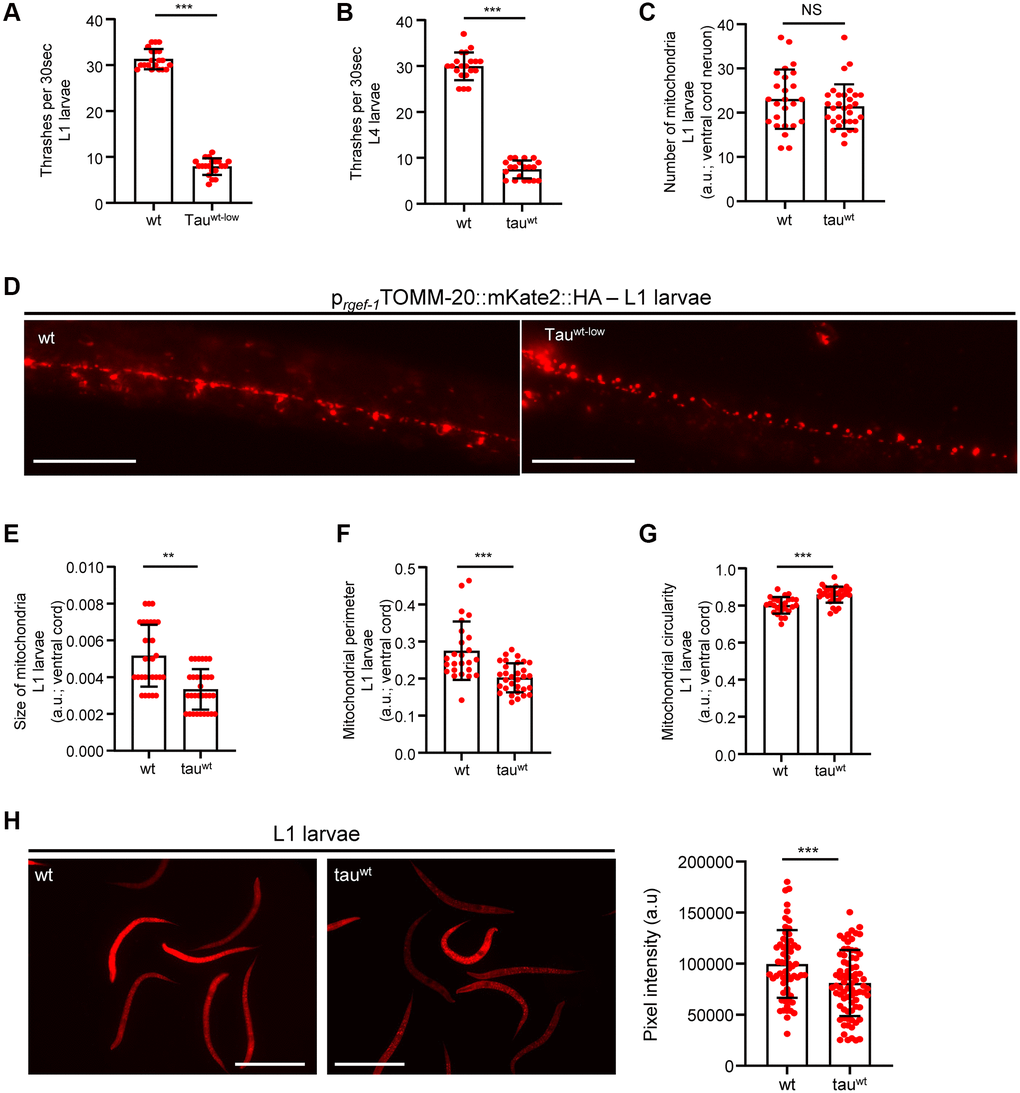
Figure 2. Altered mitochondrial morphology and activity in tauwt-expressing larvae. Wild type and tauwt L1 (A) and L4 (B) larvae body bends per 30 seconds in M9 buffer. (C) Mitochondrial population in the ventral nerve cord of L1 nematodes. (D) Representative fluorescent images of transgenic nematodes expressing pan-neuronally mitochondria-targeted mKate2::HA. Scale bar, 20 μm. Tauwt-expressing L1 larvae display (E) smaller, (F, G) more circular and (H) Fewer active mitochondria compared to wild type animals. Scale bar, 100 μm. (n = 50; NS P > 0.05, **P < 0.001, ***P < 0.0001; unpaired t-test).
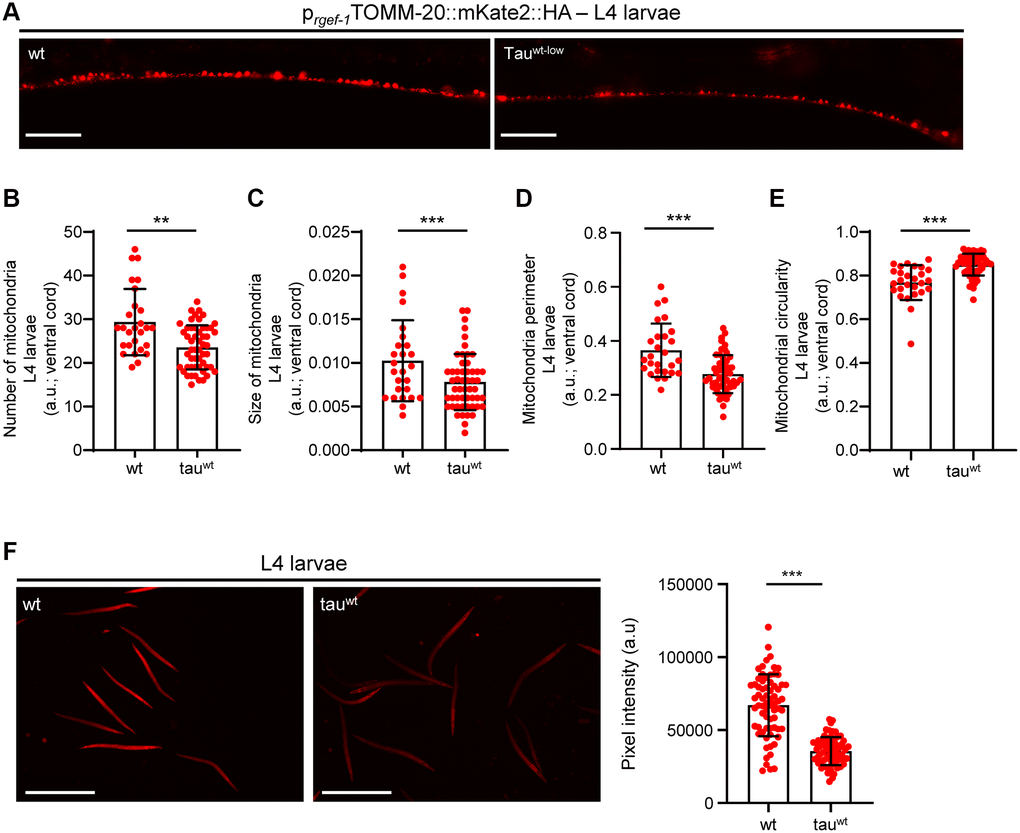
Figure 3. Tauwt-expressing L4 nematodes display impaired mitochondrial morphology and function. (A) Representative fluorescent images of L4 transgenic nematodes expressing panneuronally mitochondria-targeted mKate2::HA. Scale Bar, 20 μm. (B) Mitochondrial population in the ventral nerve cord of L4 nematodes. Neuronal processes of tauwt-expressing L4 larvae display (C) smaller, (D, E) more circular and (F) fewer active mitochondria compared to wild type animals (n = 30–50; **P < 0.001, ***P < 0.0001; unpaired t-test).
To investigate whether the locomotion deficits were generated because of excessive mitochondrial damage, we evaluated several aspects of mitochondrial morphology, including size, perimeter and circularity, which have been used to evaluate mitochondrial dysfunction [48–51]. Tau-expressing larvae presented smaller and more globular mitochondria compared to the wild type counterparts (Figure 2E–2G, Figure 3C–3E and Supplementary Figure 3C–3E, 3H). Moreover, mitochondrial membrane potential assessment of the tau-expressing nematodes displayed fewer active organelles indicating that tau expression is sufficient to alter not only the shape of mitochondrial networks but also their activity during early development (Figure 2H and Figure 3F). These results indicate that increased mitochondrial damage and impaired energy generation might drive neuronal dysfunction and the abnormal locomotion of the tau-expressing nematodes and highlight mitochondrial defects as an early pathological feature of tauopathy.
Calcium homeostasis modulates mitochondrial activity and neuronal fitness in tau-expressing nematodes
Ca2+ is an important second messenger that controls multiple cellular processes. Local Ca2+ signals are widely recognized as broad regulators of neuronal function and survival [52, 53]. Neurons are highly dependent on balanced Ca2+ homeostasis, since they have developed intricate regulatory mechanisms coupling Ca2+ signaling with their molecular and biochemical machineries. Indeed, altered cytosolic Ca2+ fluctuations lead to impaired neurotransmission, axon guidance, spine formation and subsequently to neuronal loss and cognitive dysfunction [54, 55].
A recent study in C. elegans demonstrated that overexpression of a mutant isoform of tau (tauA152T) promoted necrotic cell death of glutamatergic neurons through dysregulation of the cytosolic calcium levels [56]. Therefore, we examined whether calcium deregulation is implicated in the impairment of mitochondrial activity in the tau-expressing nematodes. Interestingly, supplementation with 10 mM EGTA, a calcium chelating agent, restored mitochondrial membrane potential in tau-expressing animals without any detectable effect on wild type worms (Figure 4A). Moreover, the bending behavior, the sinusoidal wave pattern and the velocity of tau-expressing L4 larvae were improved following calcium chelation without any substantial effect on the behavior in wild type (Figure 4B–4E and Supplementary Figure 4A). However, EGTA treatment could not rescue the severe motility defect in the tau-expressing nematodes during adulthood (Supplementary Figure 4B). Moreover, EGTA supplementation did not affect tau oligomer and aggregate formation, suggesting that calcium chelation solely impacts mitochondrial homeostasis (Supplementary Figure 5A–5B). These findings suggest that tau expression results in altered calcium homeostasis enhancing neuronal vulnerability to degeneration during aging, and demonstrate tight interplay between tau overexpression, calcium homeostasis, and mitochondrial function, in neuronal health.
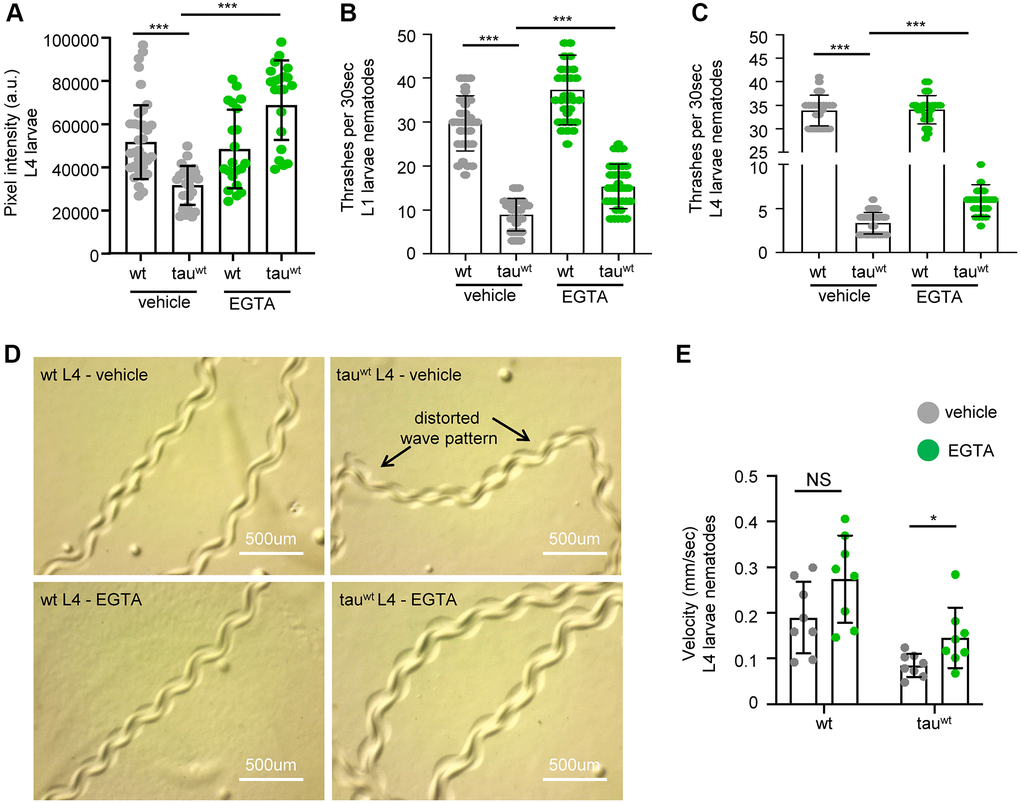
Figure 4. Cytoplasmic calcium chelation rescues motility defects in tauwt-expressing larvae. (A) Mitochondrial activity is increased in tauwt-expressing L4 nematodes upon 10 mM EGTA supplementation. Locomotion defects are ameliorated in both (B) L1 and (C) L4 tauwt-expressing nematodes in response to 10 mM EGTA treatment (n = 50; ***P < 0.0001; unpaired t-test). (D) Representative images of wild type and tauwt-expressing nematodes tracks with or without 10 mM EGTA treatment. (E) Velocity assessment of wild type and tauwt-expressing nematodes with or without 10 mM EGTA treatment (n = 8; NS P > 0.05, *P < 0.03; unpaired t-test).
Discussion
Aging is universally associated with a marked decrease in brain function and increased susceptibility to neurodegeneration. In human populations, this is manifested as an ever-increasing prevalence of devastating neurodegenerative conditions, including AD and other types of dementia. Therefore, the development of novel therapeutic interventions against human aging and age-related pathologies is a top research priority.
Mitochondrial dysfunction is a recognized hallmark of aging and age-associated diseases [12, 57]. Recent studies in AD mouse models suggest that Aβ plaques and tau tangles formation are accompanied with mitochondrial dysfunction [58–60]. However, it remains elusive whether impaired mitochondrial metabolism is an early pathological feature of disease development. It is shown that Aβ and tau are localized to mitochondria, where they perturb energy metabolism through either their interference with mitochondrial import machinery or inhibition of mitochondrial enzymes and electron transport chain function [61–63]. Moreover, tau aggregates promote mitochondrial dysfunction and exacerbate Aβ-related mitochondrial damage [18, 64]. Although these results implicate Aβ and tau lesions in energy homeostasis, the role of mitochondrial impairment on the initiation and progression of AD is not well understood.
Our model recapitulates several key features of the human neurodegenerative disorders, including adult onset, progressive neurodegeneration, accumulation of abnormal tau, and shorter lifespan. The cumulated results of our study indicate that, at least in this tauopathy model, neurotoxicity depends on protein alterations and mitochondrial dysfunction, which takes place before the formation of detectable levels of aggregates during larval stages, underscoring the detrimental neurotoxic effect of high level of tau (Supplementary Figure 6). Tau-expressing nematodes presented an increased number of defective mitochondria in neuronal processes, which may be a result of impaired removal of damaged organelles. Indeed, recent studies in nematodes and mammalian cells have demonstrated that mutant isoforms of tau block neuronal mitophagy and result in pronounced mitochondrial damage and neuronal loss [35, 36]. In sharp contrast, mitochondrial density was markedly reduced in the L1 and L4 tau-expressing larvae suggesting that mitophagy is still efficient in eliminating dysfunctional organelles at least in the larvae. Thus, aging and tau aggregate formation act synergistically to deregulate mitochondrial activity and quality surveillance mechanisms, leading to a gradual accumulation of defective organelles and subsequently to neurodegeneration.
A growing body of evidence demonstrates that calcium homeostasis collapse is a critical modulator of tau-mediated neurotoxicity. Transgenic nematodes expressing the mutant tau isoform A152T (tauA152T) displayed progressive degeneration of glutamatergic neurons through impairment of cellular calcium levels that eventually lead to necrotic cell death induction. Interestingly, depletion of the Ca2+ binding chaperones calreticulin (CRT-1) and calnexin (CNX-1) and the Ca2+ -dependent phosphatase calcineurin (CNB-1) delayed neuronal cell death in L4 larvae [56]. Due to the subcellular localization of CRT-1 and CNX-1 in endoplasmic reticulum (ER), these findings support that tauA152T expression impairs ER homeostasis resulting in enhanced release of Ca2+ into the cytoplasm. A very recent study in rat primary cortical neurons and human iPSC-derived neurons documented that tau mutations inhibit mitochondrial Na+/Ca2+ exchanger (NCLX) function and deregulate mitochondrial calcium efflux, leading to altered cytosolic Ca2+ levels and subsequently to increased vulnerability of neuronal cells to cell death. [65]. Therefore, tau mutations could modulate calcium homeostasis by influencing the main cellular storage sites ER and mitochondria.
Although we cannot exclude possible differences in mechanisms of tau toxicity between C. elegans and human disease, the enhanced biological toxicity of mutant tau as reported previously, the degenerative nature of the pathology, and the selective accumulation of abnormal tau in areas of neuronal degeneration, all argue that the mechanisms of tau neurotoxicity are conserved between C. elegans and humans. Collectively, our findings underline the essential impact of early tau oligomer formation on mitochondrial dysfunction and disease development. Investigating the tight interplay between tau oligomers and energy metabolism will enlighten new avenues for therapeutic strategies to slow or halt the progression of dementia-related diseases such as AD. The C. elegans tauopathy model can be used as a screening platform to identify novel genes and chemical compounds that protect against early tau-mediated mitochondrial damage and neurotoxicity.
Materials and Methods
C. elegans strains and culture methods
We followed standard procedures for C. elegans strain maintenance (Brenner, 1974 Genetics 77). Nematode rearing temperature was kept at 20°C, unless noted otherwise. The following strains were used in this study: N2: wild-type Bristol isolate, PIR3: pirIs3[psnb-1 htau40WT-low; pmyo-2GFP]. To monitor mitochondrial morphology in neuronal cells, we used the following transgenic animals: SJZ216: foxSi44[prgef-1TOMM-20::mKate2::HA]I and foxSi44[prgef-1TOMM-20::mKate2::HA]I; pirIs3[psnb-1htau40WT-low; pmyo-2GFP]. We used Ethylene glycol tetraacetic acid (EGTA) as a chemical to reduce specifically cytosolic calcium. EGTA was administered at a final concentration of 10 mM. EGTA concentration was prepared by dilutions in 150 ml of sterilized water, from a concentrated stock solution (0.5 M), and applied to the top of the agar medium. Plates were then gently swirled to allow the drug to spread to the entire OP50-seeded NGM surface. Identical drug-free water solutions were used for the control plates. Animals were treated with EGTA for two generations.
Thrashing assay
Wild type and tauwt-expressing L1, L4 and 3-day-adult nematodes were transferred in 10 μl M9 buffer using an eyelash pick. Animals were allowed to swim freely for 1 minute to be accustomed to the new environment. Then, body bends were assessed for 30 seconds. For each experiment, at least 50 animals were examined for each strain/condition. Each assay was repeated at least two times. The Prism software package (GraphPad Software) was used for statistical analyses.
Mitochondrial imaging
TMRE (tetramethylrhodamine, ethyl ester, perchlorate) is a dye that accumulates in intact, respiring mitochondria. Embryos/eggs were placed and grown at 20°C in the presence of 150 nM TMRE. Stained and washed L1, L4 and 3-day-adult nematodes were immobilized with levamisole before mounting for microscopic examination with a Zeiss AxioImager Z2 epifluorescence microscope. Images were acquired under the same exposure. Average pixel intensity values were calculated by sampling images of different animals. The mean and maximum pixel intensity were calculated for each animal in these images using the ImageJ software (http://rsb.info.nih.gov/ij/). For each experiment, at least 30–50 animals were examined for each strain/condition. The assessment of the mitochondrial morphology was performed by using ImageJ software, as it is previously described [66]. For quantitative characterization of mitochondrial morphology fluorescent images were acquired and used for the analysis. Background was subtracted and the resulting images were thresholded at the default setting. The resulting particles were analyzed and the following parameters were collected for each mito-mKate2-labeled particle: area, perimeter and circularity. Mitochondrial number was evaluated by counting the average number of mito-mKate2-labeled puncta per 100 μm of axonal length. Each assay was repeated at least three times. The Prism software package (GraphPad Software) was used for statistical analyses.
Worm tracking software
To obtain synchronized populations of worm, 5–7 L4 worms were transferred onto NGM plates (with or without EGTA) and allowed to lay eggs for 24 h. After removing the adult worms, each synchronized progeny was cultured to L4 larvae. For worm imaging, tracking, and describing the worm crawling, synchronized L4 larvae were recorded using the Wormlab software (MBF Bioscience) and captured at rates 15 frames per second. Each worm was manually detected, and their locomotion were analyzed for each frame. The movements were exported to Excel files and worm tracks were generated by GraphPad Prism software. The velocity was evaluated by measuring the distance of each worm that moved for 30 seconds and was calculated by utilizing ImageJ based on the worm track graph. Statistical analysis was performed using GraphPad Prism software.
Western blot analysis
For total tau protein extraction, age-synchronized worms were washed off NGM plates using M9 buffer. To completely remove the bacteria, the washing steps were repeated thrice. The resulting worm pellets were resuspended in 1X protein sample buffer containing (20 mM Hepes, pH 7.9, 25% glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT) and lysed by sonication (3 × 10 s, 10 s break) on ice. After a brief centrifugation at 40,000 g for 15 min, the supernatants were analyzed on 3–12% native PAGE. The entire extraction procedures were carried out on ice and centrifugation steps were performed at 4°C. All buffers contained Complete Protease and Phosphatase Inhibitor cocktail (Sigma-Aldrich). The proteins were transferred to nitrocellulose membranes and immunoblotted. The following antibodies were used: K9JA (1:5,000; no. A0024; Dako), Anti-Tau (T22), oligomeric antibody (1:1000; ABN454; Sigma-Aldrich). Coomassie staining serves as loading control. For SDS-PAGE, 100 synchronized worms of the indicated ages were washed off NGM plates with M9 buffer and the bacteria were removed in the subsequent washing steps. The resulting worms were allowed to settle under gravity to form a pellet and were resuspended in 100 μl of 2X protein sample buffer containing 355 mM 2- mercaptoethanol and boiled at 96°C for 10 min. The supernatant was collected by brief centrifugation. Proteins were separated in Tris-glycine SDS gel and transferred onto PVDF membrane. Each experiment was done in two biological replicates. The following antibodies were used: Anti-Human Tau (#A0024; Dako), Tau monoclonal antibody (TAU-5) (#AHB0042, Invitrogen) Pan actin (ACTN05 (C4) #MA5-11869; Life Technologies), GAPDH (#sc25778; Santa Cruz Biotechnology).
Supplementary Materials
Author Contributions
K.P., A.K. and M.A. wrote the paper. K.P. and A.K. conceptualized, performed and analyzed the experiments. M.A and V.A.B. conceived the project. All authors edited the paper. K.P., M.A. and V.A.B. supervised the project.
Acknowledgments
Some nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the National Center for Research Resources of the National Institutes of Health. We thank E.M. Mandelkow for PIR3 transgenic nematodes.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Funding
Supported by the Olav Thon Foundation, CESA and the intramural Program at the National Institute on Aging, NIH, USA.
References
- 1. Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985; 101:1371–78. https://doi.org/10.1083/jcb.101.4.1371 [PubMed]
- 2. Drubin D, Kobayashi S, Kirschner M. Association of tau protein with microtubules in living cells. Ann N Y Acad Sci. 1986; 466:257–68. https://doi.org/10.1111/j.1749-6632.1986.tb38398.x [PubMed]
- 3. Fellous A, Francon J, Lennon AM, Nunez J. Microtubule assembly in vitro. Purification of assembly-promoting factors. Eur J Biochem. 1977; 78:167–74. https://doi.org/10.1111/j.1432-1033.1977.tb11726.x [PubMed]
- 4. Medina M, Avila J. The role of extracellular Tau in the spreading of neurofibrillary pathology. Front Cell Neurosci. 2014; 8:113. https://doi.org/10.3389/fncel.2014.00113 [PubMed]
- 5. Arendt T, Stieler JT, Holzer M. Tau and tauopathies. Brain Res Bull. 2016; 126:238–92. https://doi.org/10.1016/j.brainresbull.2016.08.018 [PubMed]
- 6. Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016; 17:5–21. https://doi.org/10.1038/nrn.2015.1 [PubMed]
- 7. Castillo-Carranza DL, Gerson JE, Sengupta U, Guerrero-Muñoz MJ, Lasagna-Reeves CA, Kayed R. Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J Alzheimers Dis. 2014 (Suppl 1); 40:S97–111. https://doi.org/10.3233/JAD-132477 [PubMed]
- 8. Kopeikina KJ, Hyman BT, Spires-Jones TL. Soluble forms of tau are toxic in Alzheimer's disease. Transl Neurosci. 2012; 3:223–33. https://doi.org/10.2478/s13380-012-0032-y [PubMed]
- 9. Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001; 293:711–14. https://doi.org/10.1126/science.1062382 [PubMed]
- 10. Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener. 2011; 6:39. https://doi.org/10.1186/1750-1326-6-39 [PubMed]
- 11. Maeda S, Sahara N, Saito Y, Murayama S, Ikai A, Takashima A. Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer's disease. Neurosci Res. 2006; 54:197–201. https://doi.org/10.1016/j.neures.2005.11.009 [PubMed]
- 12. Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, Bohr VA. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019; 15:565–81. https://doi.org/10.1038/s41582-019-0244-7 [PubMed]
- 13. Wang Y, Xu E, Musich PR, Lin F. Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci Ther. 2019; 25:816–24. https://doi.org/10.1111/cns.13116 [PubMed]
- 14. Cheng Y, Bai F. The Association of Tau With Mitochondrial Dysfunction in Alzheimer's Disease. Front Neurosci. 2018; 12:163. https://doi.org/10.3389/fnins.2018.00163 [PubMed]
- 15. Schulz KL, Eckert A, Rhein V, Mai S, Haase W, Reichert AS, Jendrach M, Müller WE, Leuner K. A new link to mitochondrial impairment in tauopathies. Mol Neurobiol. 2012; 46:205–16. https://doi.org/10.1007/s12035-012-8308-3 [PubMed]
- 16. David DC, Hauptmann S, Scherping I, Schuessel K, Keil U, Rizzu P, Ravid R, Dröse S, Brandt U, Müller WE, Eckert A, Götz J. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J Biol Chem. 2005; 280:23802–14. https://doi.org/10.1074/jbc.m500356200 [PubMed]
- 17. Kim SH, Vlkolinsky R, Cairns N, Fountoulakis M, Lubec G. The reduction of NADH ubiquinone oxidoreductase 24- and 75-kDa subunits in brains of patients with Down syndrome and Alzheimer's disease. Life Sci. 2001; 68:2741–50. https://doi.org/10.1016/s0024-3205(01)01074-8 [PubMed]
- 18. Quintanilla RA, Matthews-Roberson TA, Dolan PJ, Johnson GV. Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer disease. J Biol Chem. 2009; 284:18754–66. https://doi.org/10.1074/jbc.M808908200 [PubMed]
- 19. DuBoff B, Götz J, Feany MB. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron. 2012; 75:618–32. https://doi.org/10.1016/j.neuron.2012.06.026 [PubMed]
- 20. Wang ZF, Yin J, Zhang Y, Zhu LQ, Tian Q, Wang XC, Li HL, Wang JZ. Overexpression of tau proteins antagonizes amyloid-beta-potentiated apoptosis through mitochondria-caspase-3 pathway in N2a cells. J Alzheimers Dis. 2010; 20:145–57. https://doi.org/10.3233/jad-2010-1351 [PubMed]
- 21. Wang JZ, Wang ZH, Tian Q. Tau hyperphosphorylation induces apoptotic escape and triggers neurodegeneration in Alzheimer's disease. Neurosci Bull. 2014; 30:359–66. https://doi.org/10.1007/s12264-013-1415-y [PubMed]
- 22. Li HL, Wang HH, Liu SJ, Deng YQ, Zhang YJ, Tian Q, Wang XC, Chen XQ, Yang Y, Zhang JY, Wang Q, Xu H, Liao FF, Wang JZ. Phosphorylation of tau antagonizes apoptosis by stabilizing beta-catenin, a mechanism involved in Alzheimer's neurodegeneration. Proc Natl Acad Sci U S A. 2007; 104:3591–96. https://doi.org/10.1073/pnas.0609303104 [PubMed]
- 23. Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013; 17:491–506. https://doi.org/10.1016/j.cmet.2013.03.002 [PubMed]
- 24. Knott AB, Bossy-Wetzel E. Impairing the mitochondrial fission and fusion balance: a new mechanism of neurodegeneration. Ann N Y Acad Sci. 2008; 1147:283–92. https://doi.org/10.1196/annals.1427.030 [PubMed]
- 25. Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, Taguchi N, Morinaga H, Maeda M, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009; 11:958–66. https://doi.org/10.1038/ncb1907 [PubMed]
- 26. Flippo KH, Strack S. Mitochondrial dynamics in neuronal injury, development and plasticity. J Cell Sci. 2017; 130:671–81. https://doi.org/10.1242/jcs.171017 [PubMed]
- 27. Cagalinec M, Safiulina D, Liiv M, Liiv J, Choubey V, Wareski P, Veksler V, Kaasik A. Principles of the mitochondrial fusion and fission cycle in neurons. J Cell Sci. 2013; 126:2187–97. https://doi.org/10.1242/jcs.118844 [PubMed]
- 28. Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008; 27:433–46. https://doi.org/10.1038/sj.emboj.7601963 [PubMed]
- 29. Pir GJ, Choudhary B, Mandelkow E, Mandelkow EM. Tau mutant A152T, a risk factor for FTD/PSP, induces neuronal dysfunction and reduced lifespan independently of aggregation in a C. elegans Tauopathy model. Mol Neurodegener. 2016; 11:33. https://doi.org/10.1186/s13024-016-0096-1 [PubMed]
- 30. McCarroll SA, Murphy CT, Zou S, Pletcher SD, Chin CS, Jan YN, Kenyon C, Bargmann CI, Li H. Comparing genomic expression patterns across species identifies shared transcriptional profile in aging. Nat Genet. 2004; 36:197–204. https://doi.org/10.1038/ng1291 [PubMed]
- 31. Evans CS, Holzbaur ELF. Autophagy and mitophagy in ALS. Neurobiol Dis. 2019; 122:35–40. https://doi.org/10.1016/j.nbd.2018.07.005 [PubMed]
- 32. Montava-Garriga L, Ganley IG. Outstanding Questions in Mitophagy: What We Do and Do Not Know. J Mol Biol. 2020; 432:206–30. https://doi.org/10.1016/j.jmb.2019.06.032 [PubMed]
- 33. Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. 2018; 20:1013–22. https://doi.org/10.1038/s41556-018-0176-2 [PubMed]
- 34. Lou G, Palikaras K, Lautrup S, Scheibye-Knudsen M, Tavernarakis N, Fang EF. Mitophagy and Neuroprotection. Trends Mol Med. 2020; 26:8–20. https://doi.org/10.1016/j.molmed.2019.07.002 [PubMed]
- 35. Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan-Olive MM, Caponio D, Dan X, Rocktäschel P, Croteau DL, Akbari M, et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nat Neurosci. 2019; 22:401–12. https://doi.org/10.1038/s41593-018-0332-9 [PubMed]
- 36. Cummins N, Tweedie A, Zuryn S, Bertran-Gonzalez J, Götz J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019; 38:e99360. https://doi.org/10.15252/embj.201899360 [PubMed]
- 37. Goble DJ, Coxon JP, Wenderoth N, Van Impe A, Swinnen SP. Proprioceptive sensibility in the elderly: degeneration, functional consequences and plastic-adaptive processes. Neurosci Biobehav Rev. 2009; 33:271–78. https://doi.org/10.1016/j.neubiorev.2008.08.012 [PubMed]
- 38. Wen Q, Po MD, Hulme E, Chen S, Liu X, Kwok SW, Gershow M, Leifer AM, Butler V, Fang-Yen C, Kawano T, Schafer WR, Whitesides G, et al. Proprioceptive coupling within motor neurons drives C. elegans forward locomotion. Neuron. 2012; 76:750–61. https://doi.org/10.1016/j.neuron.2012.08.039 [PubMed]
- 39. Fieseler C, Kunert-Graf J, Kutz JN. The control structure of the nematode Caenorhabditis elegans: Neuro-sensory integration and proprioceptive feedback. J Biomech. 2018; 74:1–8. https://doi.org/10.1016/j.jbiomech.2018.03.046 [PubMed]
- 40. Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J Cell Biol. 1998; 143:777–94. https://doi.org/10.1083/jcb.143.3.777 [PubMed]
- 41. Szabo L, Eckert A, Grimm A. Insights into Disease-Associated Tau Impact on Mitochondria. Int J Mol Sci. 2020; 21:6344. https://doi.org/10.3390/ijms21176344 [PubMed]
- 42. Torres AK, Jara C, Olesen MA, Tapia-Rojas C. Pathologically phosphorylated tau at S396/404 (PHF-1) is accumulated inside of hippocampal synaptic mitochondria of aged Wild-type mice. Sci Rep. 2021; 11:4448. https://doi.org/10.1038/s41598-021-83910-w [PubMed]
- 43. Cieri D, Vicario M, Vallese F, D'Orsi B, Berto P, Grinzato A, Catoni C, De Stefani D, Rizzuto R, Brini M, Calì T. Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca2+ handling. Biochim Biophys Acta Mol Basis Dis. 2018; 1864:3247–56. https://doi.org/10.1016/j.bbadis.2018.07.011 [PubMed]
- 44. Pérez MJ, Jara C, Quintanilla RA. Contribution of Tau Pathology to Mitochondrial Impairment in Neurodegeneration. Front Neurosci. 2018; 12:441. https://doi.org/10.3389/fnins.2018.00441 [PubMed]
- 45. Matenia D, Mandelkow EM. The tau of MARK: a polarized view of the cytoskeleton. Trends Biochem Sci. 2009; 34:332–42. https://doi.org/10.1016/j.tibs.2009.03.008 [PubMed]
- 46. Alonso AD, Cohen LS, Corbo C, Morozova V, ElIdrissi A, Phillips G, Kleiman FE. Hyperphosphorylation of Tau Associates With Changes in Its Function Beyond Microtubule Stability. Front Cell Neurosci. 2018; 12:338. https://doi.org/10.3389/fncel.2018.00338 [PubMed]
- 47. Sohn PD, Tracy TE, Son HI, Zhou Y, Leite RE, Miller BL, Seeley WW, Grinberg LT, Gan L. Acetylated tau destabilizes the cytoskeleton in the axon initial segment and is mislocalized to the somatodendritic compartment. Mol Neurodegener. 2016; 11:47. https://doi.org/10.1186/s13024-016-0109-0 [PubMed]
- 48. Faitg J, Davey T, Turnbull DM, White K, Vincent AE. Mitochondrial morphology and function: two for the price of one!. J Microsc. 2020; 278:89–106. https://doi.org/10.1111/jmi.12891 [PubMed]
- 49. Karbowski M, Youle RJ. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003; 10:870–80. https://doi.org/10.1038/sj.cdd.4401260 [PubMed]
- 50. Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. 2008; 9:505–18. https://doi.org/10.1038/nrn2417 [PubMed]
- 51. Leonard AP, Cameron RB, Speiser JL, Wolf BJ, Peterson YK, Schnellmann RG, Beeson CC, Rohrer B. Quantitative analysis of mitochondrial morphology and membrane potential in living cells using high-content imaging, machine learning, and morphological binning. Biochim Biophys Acta. 2015; 1853:348–60. https://doi.org/10.1016/j.bbamcr.2014.11.002 [PubMed]
- 52. Augustine GJ, Santamaria F, Tanaka K. Local calcium signaling in neurons. Neuron. 2003; 40:331–46. https://doi.org/10.1016/s0896-6273(03)00639-1 [PubMed]
- 53. Heine M, Heck J, Ciuraszkiewicz A, Bikbaev A. Dynamic compartmentalization of calcium channel signalling in neurons. Neuropharmacology. 2020; 169:107556. https://doi.org/10.1016/j.neuropharm.2019.02.038 [PubMed]
- 54. Brini M, Calì T, Ottolini D, Carafoli E. Neuronal calcium signaling: function and dysfunction. Cell Mol Life Sci. 2014; 71:2787–814. https://doi.org/10.1007/s00018-013-1550-7 [PubMed]
- 55. Burgoyne RD, Helassa N, McCue HV, Haynes LP. Calcium Sensors in Neuronal Function and Dysfunction. Cold Spring Harb Perspect Biol. 2019; 11:a035154. https://doi.org/10.1101/cshperspect.a035154 [PubMed]
- 56. Choudhary B, Mandelkow E, Mandelkow EM, Pir GJ. Glutamatergic nervous system degeneration in a C. elegans TauA152T tauopathy model involves pathways of excitotoxicity and Ca2+ dysregulation. Neurobiol Dis. 2018; 117:189–202. https://doi.org/10.1016/j.nbd.2018.06.005 [PubMed]
- 57. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013; 153:1194–217. https://doi.org/10.1016/j.cell.2013.05.039 [PubMed]
- 58. Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2009; 106:14670–75. https://doi.org/10.1073/pnas.0903563106 [PubMed]
- 59. Blanchard V, Moussaoui S, Czech C, Touchet N, Bonici B, Planche M, Canton T, Jedidi I, Gohin M, Wirths O, Bayer TA, Langui D, Duyckaerts C, et al. Time sequence of maturation of dystrophic neurites associated with Abeta deposits in APP/PS1 transgenic mice. Exp Neurol. 2003; 184:247–63. https://doi.org/10.1016/s0014-4886(03)00252-8 [PubMed]
- 60. Zheng J, Akbari M, Schirmer C, Reynaert ML, Loyens A, Lefebvre B, Buée L, Croteau DL, Galas MC, Bohr VA. Hippocampal tau oligomerization early in tau pathology coincides with a transient alteration of mitochondrial homeostasis and DNA repair in a mouse model of tauopathy. Acta Neuropathol Commun. 2020; 8:25. https://doi.org/10.1186/s40478-020-00896-8 [PubMed]
- 61. Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006; 26:9057–68. https://doi.org/10.1523/JNEUROSCI.1469-06.2006 [PubMed]
- 62. Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004; 304:448–52. https://doi.org/10.1126/science.1091230 [PubMed]
- 63. Swerdlow RH, Burns JM, Khan SM. The Alzheimer's disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta. 2014; 1842:1219–31. https://doi.org/10.1016/j.bbadis.2013.09.010 [PubMed]
- 64. Quintanilla RA, von Bernhardi R, Godoy JA, Inestrosa NC, Johnson GV. Phosphorylated tau potentiates Aβ-induced mitochondrial damage in mature neurons. Neurobiol Dis. 2014; 71:260–69. https://doi.org/10.1016/j.nbd.2014.08.016 [PubMed]
- 65. Britti E, Ros J, Esteras N, Abramov AY. Tau inhibits mitochondrial calcium efflux and makes neurons vulnerable to calcium-induced cell death. Cell Calcium. 2020; 86:102150. https://doi.org/10.1016/j.ceca.2019.102150 [PubMed]
- 66. Morsci NS, Hall DH, Driscoll M, Sheng ZH. Age-Related Phasic Patterns of Mitochondrial Maintenance in Adult Caenorhabditis elegans Neurons. J Neurosci. 2016; 36:1373–85. https://doi.org/10.1523/JNEUROSCI.2799-15.2016 [PubMed]