



Introduction
Age-related decline in bodily function is associated with increased disease incidence and higher mortality rates [1, 2]. Though the effects of aging are perhaps most immediately recognizable at a physiological level, health decline in older animals is rooted in dysfunction at the level of cells and molecules [3, 4]. Understanding the cellular changes that instigate age-related deterioration might suggest ways to curb their progression and promote longer, healthier lives.
Research over the past few decades has revealed a growing list of cellular hallmarks of aging that characterize an aged state [4]. These cellular aging hallmarks include mitochondrial stress, protein aggregation, telomere shortening, and deregulated signaling [4–8]. Using the nematode Caenorhabditis elegans, we recently identified another cellular change in older animals: loss of peroxisomes via autophagic destruction [9]. Although peroxisomes have received comparably less attention than other organelles in the aging field, peroxisomes are increasingly recognized as essential modulators of cellular-stress responses and metabolic coordination. Indeed, their resident enzyme, catalase, breaks down hydrogen peroxide, and they contain additional enzymes that direct lipid oxidation in concert with mitochondria [10–12]. In our previous study, we developed a tandem-fluorophore reporter to track peroxisome degradation through autophagy (“pexophagy”) during C. elegans aging [9]. This reporter (mCherry-GFP-SKL) is composed of a pH-insensitive red fluorophore fused to a pH-sensitive green fluorophore [13] and contains a peroxisome-targeting Serine-Lysine-Leucine sequence [14] at the C-terminus. As peroxisomes are internalized for degradation at acidic lysosomes, the mCherry-GFP-SKL reporter shows a decrease in GFP fluorescence relative to mCherry fluorescence [9]. Whereas wild-type C. elegans showed a dramatic reduction in peroxisome number and green:red fluorescence ratio by day 5 of adulthood, we found that inhibiting a single peroxisome protein, PEX11 homolog PRX-11, prevented early-age pexophagy [9], most likely by interfering with the normal role of PRX-11/PEX11 in peroxisome fission [15]. Notably, prx-11-defective animals not only retained peroxisomes later in life than control animals, but they lived longer, too [9]. These findings support a model in which early-age autophagic destruction of peroxisomes might ultimately limit animal longevity. However, why pexophagy inhibition increases animal lifespan is poorly understood.
An important consideration in geroscience research is that aging animals are aging systems, and individual aging hallmarks often do not change in isolation. Due to interconnectivities in cellular architecture and signaling, changes to one hallmark can influence others. Among the other parts of the cell, mitochondria show the most obvious connections to peroxisomes. Peroxisomes and mitochondria are metabolically coupled through shared lipid substrates and reactive oxygen species (ROS) signaling, and both organelles rely on common fission machinery [16–19]. Despite these connections, it is unclear whether changes to peroxisomes during aging would have any effect on mitochondria, or vice versa. In many organisms, mitochondrial networks naturally undergo fragmentation with age [20–22]. In C. elegans, this age-associated mitochondrial fragmentation can be easily detected in body wall muscle, where tubular mitochondrial networks present in early adulthood progressively collapse into punctate, fragmented structures by mid-age [22, 23]. These changes to mitochondrial morphology are accompanied by mitochondrial membrane depolarization, mitochondrial calcium (Ca2+) accumulation, energetic dysfunction, and locomotory decline [23–25]. Although mitochondrial quality-control pathways comprising fusion/fission machinery, mitophagy factors, and stress-responsive transcriptional programs can help to maintain the health of this organelle [26–29], the initiating events that destabilize mitochondrial homeostasis during early aging remain unclear. In principle, this might be linked to collapse in functionality of related organelles, including peroxisomes. If so, boosting peroxisomes in older animals might help to maintain more youthful mitochondria. Though an attractive hypothesis, this has yet to be experimentally tested.
In this study, we investigated whether enhanced longevity upon pexophagy inhibition involves coordinated changes to mitochondria. We found that prx-11 downregulation enhanced multiple indications of mitochondrial health with age; namely, inhibiting prx-11 function preserved mitochondrial network architecture, suppressed mitochondrial Ca2+ accumulation, increased energetic resources, and reduced oxidative stress in older animals. Moreover, these changes were associated with not only lifespan extension but also improvements to locomotory function, indicating that inhibition of peroxisome degradation protects mitochondrial integrity and delays associated aspects of physiological aging. We also found that the mitochondrial improvements observed in prx-11-defective animals required the mitochondrial fusion factor FZO-1 [29–31], the UNC-43 protein kinase, which opposes mitochondrial fission [32], and DAF-16, the C. elegans FOXO transcription factor [33], suggesting that mitochondrial-remodeling and biogenesis pathways are engaged downstream of peroxisome retention to promote mitochondrial health in older animals. Intriguingly, not only did peroxisome retention and enhanced mitochondrial health track together, but so did their dysfunction; loss of fzo-1, unc-43, or daf-16 accelerated pexophagy with age, though this was suppressed when prx-11 was additionally knocked down. Consistent with mitochondria being an integral component of the pro-longevity response triggered by pexophagy inhibition, we found that impairing mitochondrial tubulation prevented prx-11 knockdown from extending lifespan. Thus, we posit that the rate of age-related peroxisome loss influences lifespan in part by affecting this separate organelle. This work establishes a new framework for understanding how inter-organelle crosstalk contributes to aging and highlights peroxisome degradation as a potential target for interventions aimed at preserving mitochondrial function and extending lifespan.
Results
Animals lacking prx-11 function retain tubular mitochondria in older ages
We began our investigations by probing whether the enhanced longevity caused by prx-11 RNA interference (RNAi) was associated with any other cellular changes indicative of slower aging. Because peroxisomes and mitochondria share some functional overlap [34, 35], we reasoned that mitochondria might be altered when pexophagy was inhibited. To test this, we visualized mitochondrial morphology using a mitochondrially targeted green fluorescent protein (Mito-GFP) expressed in C. elegans body wall muscles [36]. We confirmed that animals treated with control RNAi showed typical age-dependent mitochondrial fragmentation in mid- and late-adulthood (days 5 and 10 of adulthood, respectively) (Figure 1A), as indicated by decreased mitochondrial lengths and decreased junctions per object (Figure 1B, 1C). Remarkably, this was not the case in prx-11-RNAi animals; at days 5 and 10 of adulthood, prx-11-RNAi animals retained very tubular mitochondria, which resembled those of young, day 2 adult animals (Figure 1A–1C). In contrast, knockdown of HMG-CoA reductase hmgr-1, which accelerates age-dependent pexophagy [9], induced some mitochondrial fragmentation evident from day 2 of adulthood (Figure 1A–1C). We concluded that inhibition of pexophagy, as it occurs by prx-11 knockdown, is associated with retention of tubular, youthful mitochondria in older ages.
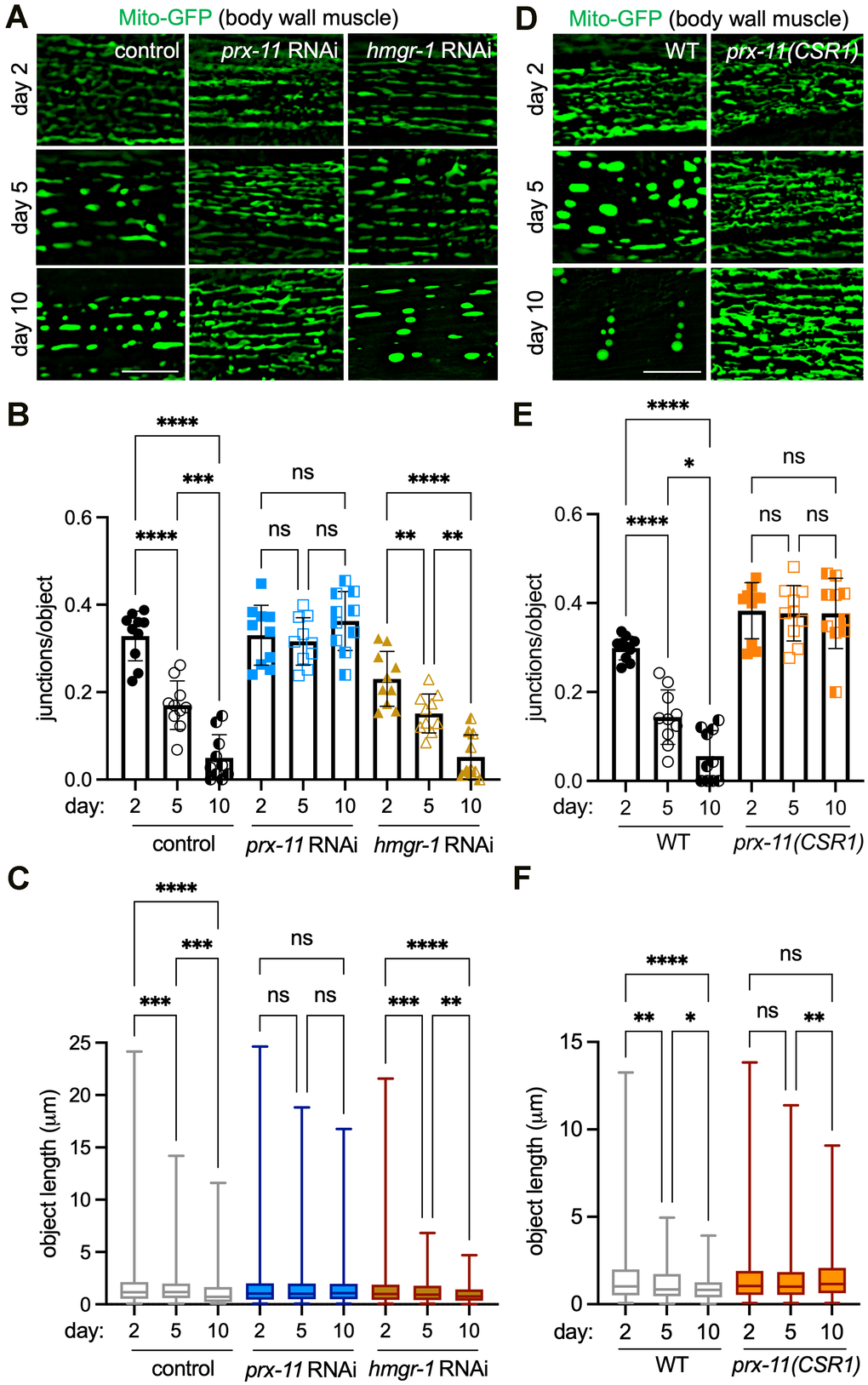
Figure 1. prx-11 depletion maintains mitochondrial junctions and network morphology in aging C. elegans. (A) Representative images of Mito-GFP in body wall muscle of day 2, day 5, and day 10 adult hermaphrodite animals fed either control, prx-11, or hmgr-1 RNAi. (B) Quantification of mitochondrial junctions per object in day 2, day 5, and day 10 adult hermaphrodite animals fed either control, prx-11, or hmgr-1 RNAi (n = 10 worms per condition). Data are presented as mean ± SD. ****, p ≤ 0.0001; ***, p ≤ 0.001; **, p ≤ 0.01; ns, not significant. One-way ANOVA with Tukey’s multiple comparisons. (C) Mito-GFP object lengths in day 2, day 5, and day 10 adult hermaphrodite animals fed either control, prx-11, or hmgr-1 RNAi (n = 10 worms per condition). Data are presented as box-and-whisker plots (minimum, 25th percentile, median, 75th percentile, maximum). ****, p ≤ 0.0001; ***, p ≤ 0.001; **, p ≤ 0.01; ns, not significant. One-way ANOVA with Tukey’s multiple comparisons. (D) Representative images of Mito-GFP in body wall muscle of day 2, day 5, and day 10 adult wild-type and prx-11(CSR1) hermaphrodite animals. (E) Quantification of mitochondrial junctions per object in day 2, day 5, and day 10 adult wild-type and prx-11(CSR1) hermaphrodite animals (n = 10 worms per condition). Data are presented as mean ± SD. ****, p ≤ 0.0001; *, p ≤ 0.05; ns, not significant. One-way ANOVA with Tukey’s multiple comparisons. (F) Mito-GFP object lengths in day 2, day 5, and day 10 adult wild-type and prx-11(CSR1) hermaphrodite animals (n = 10 worms per condition). Data are presented as box-and-whisker plots (minimum, 25th percentile, median, 75th percentile, maximum). ****, p ≤ 0.0001; **, p ≤ 0.01; *, p ≤ 0.05; ns, not significant. One-way ANOVA with Tukey’s multiple comparisons. Bars: 10 μm.
To corroborate this result, we tested whether the same would be true in a previously generated prx-11 mutant, prx-11(CSR1), which contains an internal genetic deletion [37]. First, we verified that prx-11(CSR1) mutants, like prx-11-RNAi animals [9], showed reduced age-dependent pexophagy (Supplementary Figure 1A, 1B), larger peroxisomes (Supplementary Figure 1C, 1D), and increased lifespans (Supplementary Figure 1E). Though loss of prx-11 slowed pexophagy, we noted that it did not appear to impede bulk autophagy, using a tandemly tagged SQST-1/p62 autophagy receptor marker [38] (Supplementary Figure 1F, 1G). Importantly, prx-11(CSR1) mutant animals exhibited robust mitochondrial tubulation in older ages (Figure 1D–1F), confirming that enhanced mitochondrial tubulation is a general result of prx-11 loss of function. Thus, inhibiting prx-11 not only leads to peroxisome retention but also preserves mitochondria with morphological characteristics of a more youthful state.
Aged prx-11-RNAi animals show reductions in a second mitochondrial hallmark of aging, mitochondrial Ca2+
We were curious whether animals having loss of function in prx-11 would display other molecular characteristics of youthful mitochondria in addition to enhanced tubulation. An increase in mitochondrial Ca2+ uptake has been observed with age in both worms and mice and has been associated with age-related diseases such as sarcopenia [24, 39]. Interestingly, blocking mitochondrial Ca2+ uptake via inhibition of the Ca2+ uniporter mcu-1 [40, 41] increases mitochondrial tubulation in older animals [24], though this can perhaps be even further enhanced by prx-11 knockdown (Supplementary Figure 2A, 2B). Given these considerations, we asked whether prx-11-RNAi animals showed reduced mitochondrial Ca2+ uptake in older ages. We used a mitochondrially targeted, low-affinity red fluorescent genetically encoded Ca2+ indicator for optical imaging (Mito-LAR-GECO) [24, 42] to test this hypothesis. Like mcu-1 knockdown (Supplementary Figure 2A–2C), prx-11 knockdown caused diminished Mito-LAR-GECO signal in older animals, which trended together with improvements to mitochondrial morphology (Figure 2A–2C). In contrast, accelerated pexophagy driven by hmgr-1 knockdown was associated with premature Mito-LAR-GECO detection in young adulthood (Figure 2A, 2C). These data support the conclusion that retaining peroxisomes in older age contributes to maintenance of more youthful mitochondria.
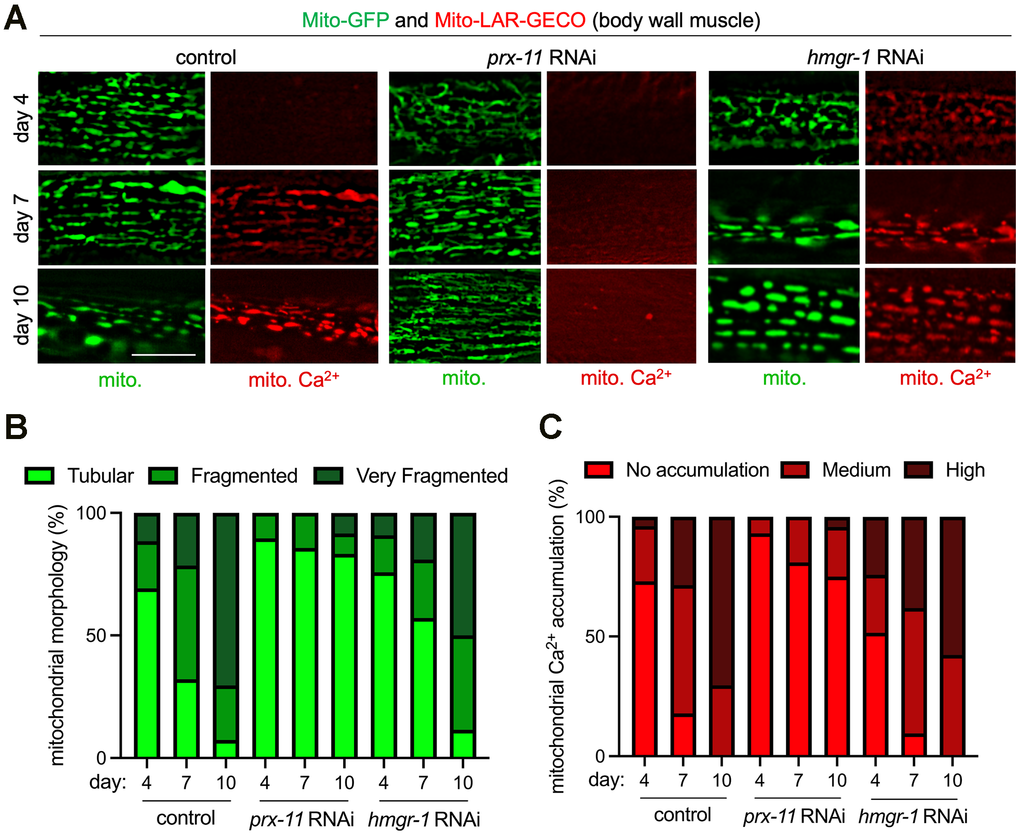
Figure 2. Enhanced mitochondrial tubulation in older prx-11-RNAi animals is associated with reduced mitochondrial Ca2+ accumulation. (A) Representative images of Mito-GFP and the Mito-LAR-GECO Ca2+ reporter in body wall muscle of day 4, day 7, and day 10 adult hermaphrodite animals fed either control, prx-11, or hmgr-1 RNAi. (B, C) Classification of mitochondrial morphology and mitochondrial Ca2+ accumulation in day 4, day 7, and day 10 adult hermaphrodite animals fed either control, prx-11, or hmgr-1 RNAi (n = 20 worms per condition). Bar: 10 μm.
Loss of prx-11 activity alters aspects of physiology related to mitochondrial and peroxisomal health
We aimed to assess whether changes to physiology associated with altered mitochondrial function could be detected upon prx-11 loss of function. We first evaluated whether prx-11 knockdown had any effect on the mitochondrial unfolded protein response (UPRmt). UPRmt is a stress-response pathway that, when activated, can improve the robustness of mitochondrial form and function and support increased longevity [43–45]. Though mitochondria appeared enhanced by prx-11 knockdown, we did not observe a relative induction of UPRmt in prx-11-RNAi animals, as judged using the UPRmt reporters Phsp-6::GFP [45] and Phsp-60::GFP [45] (Supplementary Figure 3A–3D). We also did not observe increased relative expression of the endoplasmic reticulum unfolded protein response (UPRer) reporter Phsp-4::GFP [46] in prx-11-RNAi animals (Supplementary Figure 3E, 3F), suggesting that organelle-based UPR pathways are generally not engaged by prx-11 knockdown.
We next investigated whether prx-11 knockdown was associated with changes in oxidative stress. A previous report linked loss of prx-11 function to decreased oxidative stress [47]. However, this interpretation was made under the assumption that losing prx-11 function would prevent peroxisome formation and lead to an absence of peroxisomes [47]. Our data indicate that losing prx-11 function instead causes an increase in peroxisome area during animal aging (Supplementary Figure 1C, 1D) [9]. We therefore sought to reexamine this relationship. We found that prx-11-RNAi animals indeed exhibited a marked decrease in labeling by the ROS-sensitive dye dihydroethidium (DHE) at days 1 and 5 of adulthood (Figure 3A, 3B). Interestingly, this was associated with reduced expression of a superoxide dismutase transcriptional reporter, Psod-3::GFP [48] (Figure 3C, 3D). These data suggest that the reduction in ROS may not be due to increased superoxide-dismutase activity per se but instead may result from lower intrinsic ROS production, consistent with better mitochondrial function [49, 50].
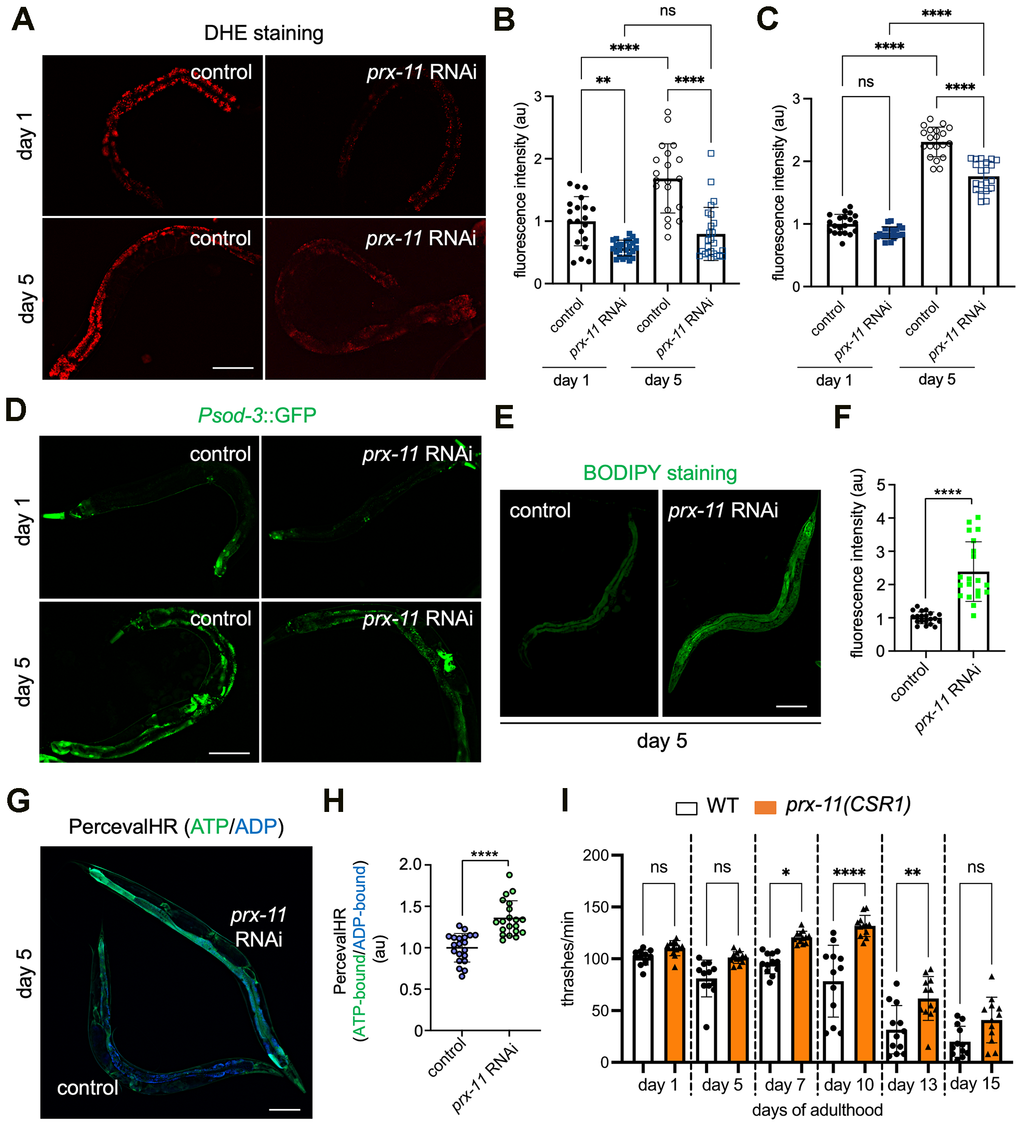
Figure 3. Animals having prx-11 loss of function display attenuated oxidative stress along with enhancements to fat stores, ATP/ADP ratios, and motor function during aging. (A) Representative images of DHE-stained day 1 and day 5 adult hermaphrodite animals fed either control or prx-11 RNAi. (B) Quantification of DHE staining fluorescence intensities in day 1 and day 5 adult hermaphrodite animals fed either control or prx-11 RNAi (n = 20 worms per condition). Data are presented as mean ± SD. ****, p ≤ 0.0001; **, p ≤ 0.01; ns, not significant. One-way ANOVA with Tukey’s multiple comparisons. (C) Quantification of Psod-3::GFP fluorescence intensities in day 1 and day 5 adult hermaphrodite animals fed either control or prx-11 RNAi (n = 20 worms per condition). Data are presented as mean ± SD. ****, p ≤ 0.0001; ns, not significant. One-way ANOVA with Tukey’s multiple comparisons. (D) Representative images of Psod-3::GFP in day 1 and day 5 adult hermaphrodite animals fed either control or prx-11 RNAi. (E) Representative images of BODIPY-stained day 5 adult hermaphrodite animals fed either control or prx-11 RNAi. (F) Quantification of BODIPY staining fluorescence intensities in day 5 adult hermaphrodite animals fed either control or prx-11 RNAi (n = 20 worms per condition). Data are presented as mean ± SD. ****, p ≤ 0.0001. Unpaired t-test. (G) Representative merged image of ATP-bound (colored green) and ADP-bound (colored blue) PercevalHR in day 5 adult hermaphrodite animals fed either control or prx-11 RNAi. (H) Quantification of ATP-bound:ADP-bound PercevalHR in day 5 adult hermaphrodite animals fed either control or prx-11 RNAi (n = 20 worms per condition). Data are presented as mean ± SD. ****, p ≤ 0.0001. Unpaired t-test. (I) Quantification of thrashing rates in adult wild-type and prx-11(CSR1) hermaphrodite animals from day 1 through day 15 of adulthood (n = 12 worms per condition). Data are presented as mean ± SD. ****, p ≤ 0.0001; **, p ≤ 0.01; *, p ≤ 0.05; ns, not significant. One-way ANOVA with Tukey’s multiple comparisons. Bars: 100 μm.
Mitochondria and peroxisomes each show complex interactions with lipids. A previous study found that peroxisomes and lipid droplets are co-regulated in C. elegans and that interventions increasing lipid droplets also increase peroxisome number [51]. Augmented lipid stores can also serve as energy reserves to support enhanced mitochondrial metabolism [52]. We therefore asked whether prx-11-RNAi animals, which retain peroxisomes later in life [9] and show more youthful mitochondria (Figure 1A–1C), also displayed differences in lipid content and storage. We first assessed Pdhs-3::DHS-3::GFP, a marker for C. elegans lipid droplets [53]. Consistent with the previous reported coupling between lipid droplets and peroxisomes in C. elegans [51], we found that the breadth and intensity of Pdhs-3::DHS-3::GFP increased in day 5 prx-11-RNAi animals compared to same-age controls (Supplementary Figure 4A, 4B). Likewise, we found that two fat-soluble dyes that stain neutral lipids in C. elegans, BODIPY and Oil Red O (ORO) [54, 55], showed enhanced labeling following prx-11 knockdown (Figure 3E, 3F and Supplementary Figure 4C, 4D). We concluded that prx-11-RNAi animals have an abundance of fat stores, which may act as a fuel resource when needed. These data fit with emerging evidence [56, 57] indicating that increased fat, though often viewed as deleterious, can be associated with a long lifespan.
We also sought to determine whether enhanced mitochondrial tubulation in prx-11-RNAi animals was associated with changes to energy status. We assessed ATP/ADP ratios using transgenic animals expressing PercevalHR. PercevalHR responds to excitation light differently in ATP-bound and ADP-bound states; emission of the ADP-bound form is maximal when stimulated by violet (~420 nm) light, whereas emission of the ATP-bound form is maximal when stimulated by blue (~500 nm) light [58, 59]. Compared to controls, prx-11-RNAi animals showed increased emission of the ATP-bound form relative to the ADP-bound form at day 5 of adulthood (Figure 3G, 3H). This suggests that prx-11-RNAi animals, which show youthful mitochondria by morphology, also show an improved energy status.
In C. elegans, preservation of energetic resources and mitochondrial function with age has been associated with improved muscle performance [23, 38, 60]. Muscle performance can be assessed by measuring locomotory activity, which in C. elegans is evaluated by counting the number of body thrashes in liquid medium per unit time [38, 61]. We found that loss of prx-11 function in fact enhanced locomotory activity of aging C. elegans (Figure 3I). These data are also consistent with improved muscle mitochondrial function and indicate that prx-11 loss of function not only extends lifespan but boosts some aspects of health in older animals.
Mitochondrial tubulation in aged prx-11-RNAi animals requires FZO-1/Mitofusin, UNC-43 protein kinase, and DAF-16/FOXO
Given the longevity and physiological benefits associated with prx-11 knockdown, we sought to identify genes required for enhanced mitochondrial tubulation in older animals under this condition. A likely candidate was fzo-1, which encodes a Mitofusin GTPase essential for mitochondrial fusion [30, 31, 62]. We found that fzo-1 mutant animals showed hyper-fragmented mitochondria, even when prx-11 was knocked down (Figure 4A–4C). Thus, FZO-1/Mitofusin activity is required for enhanced mitochondrial tubulation upon prx-11 knockdown. Typically, dynamin-related DRP-1, which promotes mitochondrial fission, counteracts the activity of FZO-1/Mitofusin [31, 62, 63]. DRP-1 itself is inhibited by UNC-43, a kinase that blocks DRP-1 activity via phosphorylation [32]. We found that mutation or knockdown of unc-43, like mutation of fzo-1, dampened mitochondrial tubulation in older prx-11-RNAi animals (Figure 4A–4C and Supplementary Figure 5A–5C). Thus, key proteins directly involved in the mitochondrial fusion/fission cycle feed into the regulation of mitochondrial biology in this genetic background.
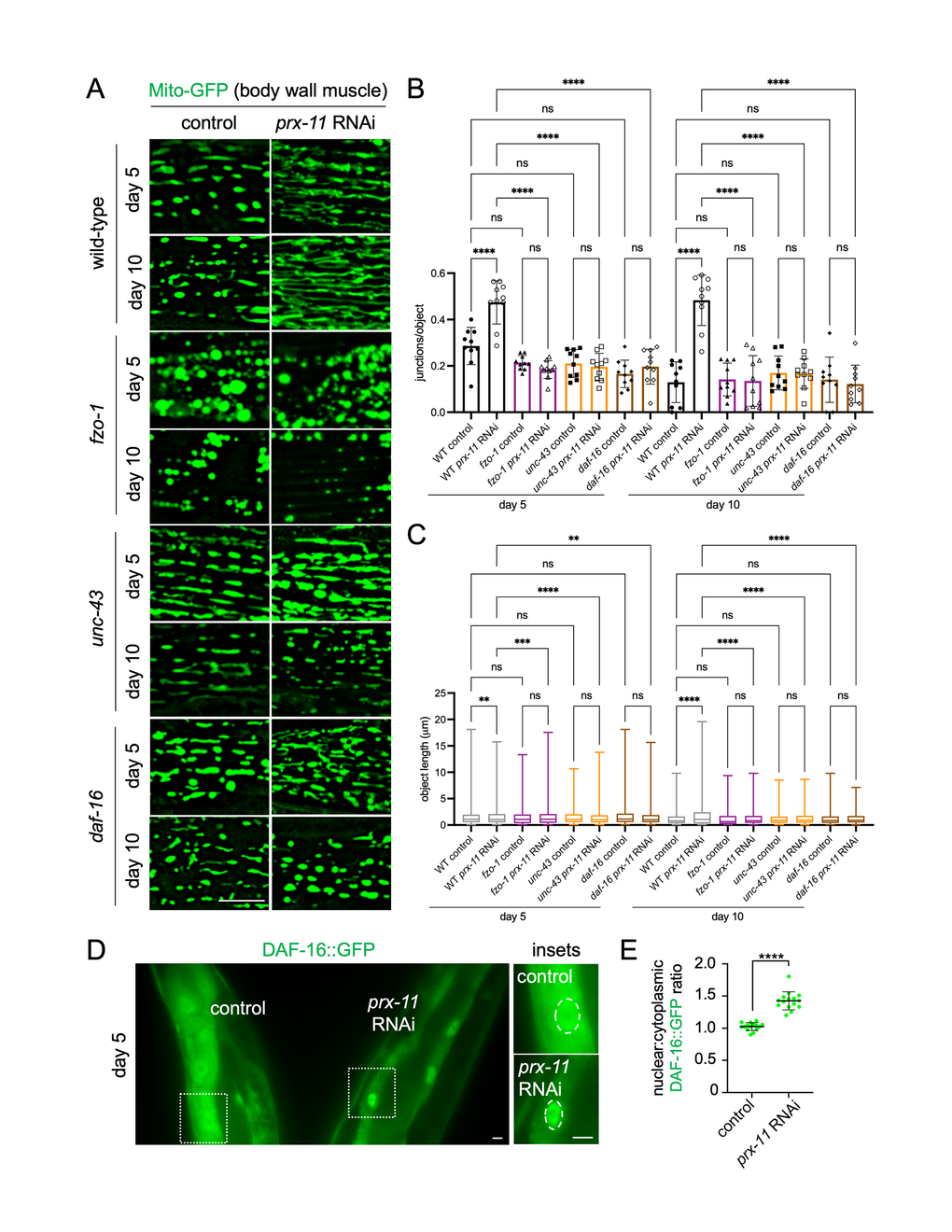
Figure 4. Loss of fzo-1, unc-43, or daf-16 abrogates mitochondrial network preservation in prx-11-RNAi animals. (A) Representative images of Mito-GFP in body wall muscle of day 5 and day 10 adult wild-type, fzo-1, unc-43, and daf-16 hermaphrodite animals fed either control or prx-11 RNAi. (B) Quantification of mitochondrial junctions per object in day 5 and day 10 adult wild-type, fzo-1, unc-43, and daf-16 hermaphrodite animals fed either control or prx-11 RNAi (n = 10 worms per condition). Data are presented as mean ± SD. ****, p ≤ 0.0001; ns, not significant. One-way ANOVA with Tukey’s multiple comparisons. (C) Mito-GFP object lengths in day 5 and day 10 adult wild-type, fzo-1, unc-43, and daf-16 hermaphrodite animals fed either control or prx-11 RNAi (n = 10 worms per condition). Data are presented a box-and-whisker plots (minimum, 25th percentile, median, 75th percentile, maximum). ****, p ≤ 0.0001; ***, p ≤ 0.001; **, p ≤ 0.01; ns, not significant. One-way ANOVA with Tukey’s multiple comparisons. (D) Representative image of DAF-16::GFP in side-by-side day 5 adult hermaphrodite animals fed either control or prx-11 RNAi. Nuclei are encircled by dashed lines in the insets. (E) Quantification of nuclear:cytoplasmic DAF-16::GFP fluorescence ratios in day 5 adult hermaphrodite animals fed either control or prx-11 RNAi (n = 15 worms per condition). Data are presented as mean ± SD. ****, p ≤ 0.0001. Unpaired t-test. Bars: 10 μm.
We also tested whether the improvements to mitochondria seen upon prx-11 knockdown required the function of DAF-16/FOXO, a transcription factor that acts as a master regulator of longevity regulation and mitochondrial homeostasis [29, 33, 64]. We found that prx-11 knockdown stimulated DAF-16 activity. Compared to controls, prx-11-RNAi animals showed increased daf-16 expression by day 2 of adulthood (Supplementary Figure 5D, 5E), and, at day 5 of adulthood, prx-11-RNAi animals displayed enhanced nuclear enrichment of DAF-16::GFP (Figure 4D, 4E). Interestingly, knockdown of prx-11 did not further increase the lifespan of long-lived daf-2 insulin-signaling mutants, which require DAF-16 activation for longevity [65]; if anything, prx-11 RNAi slightly shortened lifespan in the daf-2 mutant background (Supplementary Figure 5F, 5G). The finding that prx-11 RNAi did not additively extend daf-2 mutant lifespan suggests that these two interventions may share some overlapping DAF-16-dependent mechanisms, the effects of which are already maximized by daf-2 mutation alone. Consistent with DAF-16 activation contributing to mitochondrial improvements upon prx-11 loss of function, mutating or knocking down daf-16 reduced mitochondrial tubulation in older prx-11-RNAi animals (Figure 4A–4C and Supplementary Figure 5A–5C). These results identify several factors involved in mitochondrial regulation and biogenesis that are important for retention of tubular mitochondria with age upon loss of prx-11 function.
Mutants with defective mitochondria show faster pexophagy in early adulthood dependent on PRX-11
We were curious whether the mutant strains showing impaired mitochondrial tubulation (i.e., fzo-1, unc-43, and daf-16) also showed a difference in pexophagy with age. Remarkably, each of these mutants showed an acceleration of age-dependent pexophagy, which was apparent even at day 3 of adulthood (Figure 5A, 5B). At this early age, fzo-1, unc-43, and daf-16 mutants expressing the mCherry-GFP-SKL pexophagy reporter showed a noticeable decrease in green:red fluorescence compared to wild-type animals (Figure 5A, 5B). This suggests a degree of bidirectionality in signaling between peroxisomes and mitochondria with aging; while keeping peroxisomes around longer leads to better mitochondrial health in older age, disrupting mitochondrial form and function can hasten peroxisome degradation in early adulthood.
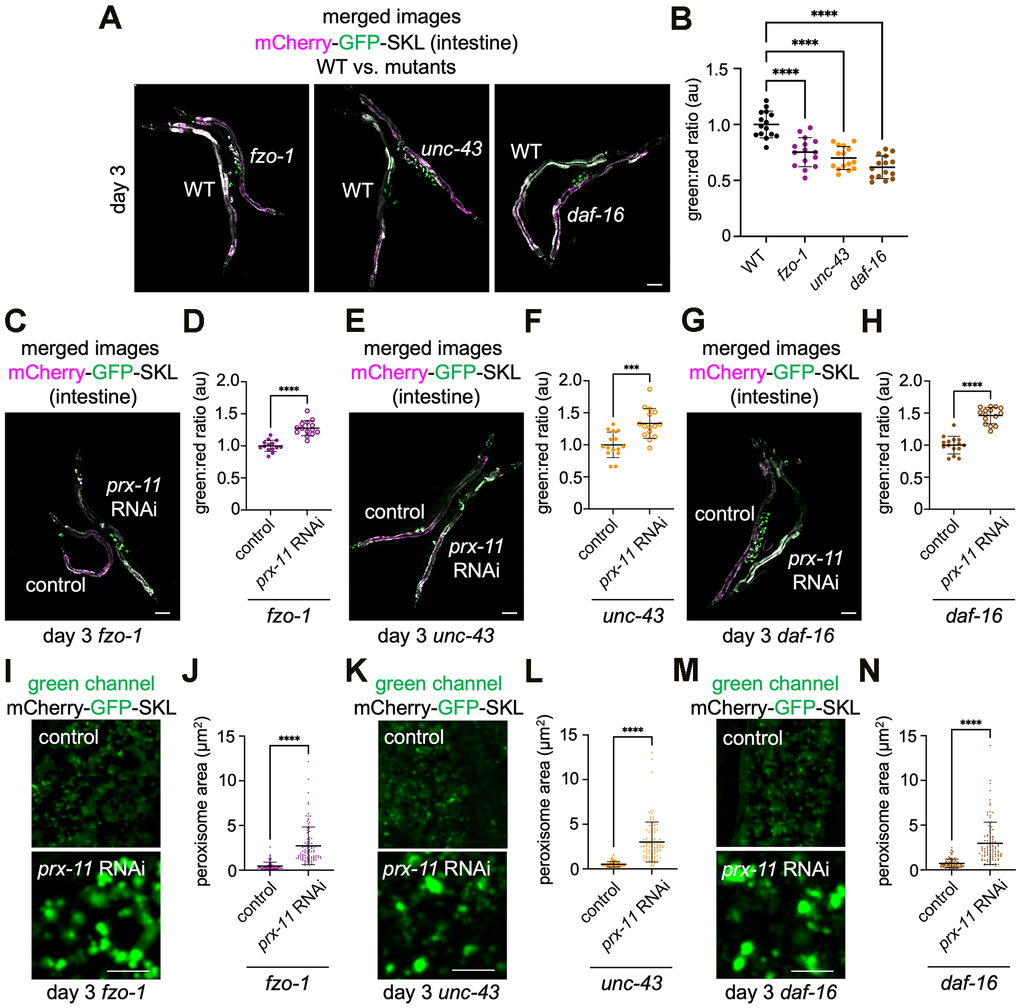
Figure 5. Accelerated pexophagy occurs in fzo-1, unc-43, and daf-16 mutant backgrounds but is suppressed by prx-11 knockdown. (A) Representative merged green and red fluorescence images of the mCherry-GFP-SKL pexophagy reporter in day 3 adult hermaphrodite animals of the indicated genotypes. For comparison, wild-type animals are paired with either fzo-1, unc-43, or daf-16 mutant animals. (B) Quantification of green:red fluorescence ratios for mCherry-GFP-SKL in day 3 adult wild-type, fzo-1, unc-43, and daf-16 hermaphrodite animals (n = 15 worms per condition). Data are presented as mean ± SD. ****, p ≤ 0.0001. One-way ANOVA with Tukey’s multiple comparisons. (C) Representative merged green and red fluorescence image of mCherry-GFP-SKL in day 3 adult hermaphrodite fzo-1 mutant animals fed either control or prx-11 RNAi. (D) Quantification of green:red fluorescence ratios for mCherry-GFP-SKL in day 3 adult hermaphrodite fzo-1 mutant animals fed either control or prx-11 RNAi (n = 15 worms per condition). Data are presented as mean ± SD. ****, p ≤ 0.0001. Unpaired t-test. (E) Representative merged green and red fluorescence image of mCherry-GFP-SKL in day 3 adult hermaphrodite unc-43 mutant animals fed either control or prx-11 RNAi. (F) Quantification of green:red fluorescence ratios for mCherry-GFP-SKL in day 3 adult hermaphrodite unc-43 mutant animals fed either control or prx-11 RNAi (n = 15 worms per condition). Data are presented as mean ± SD. ***, p ≤ 0.001. Unpaired t-test. (G) Representative merged green and red fluorescence image of mCherry-GFP-SKL in day 3 adult hermaphrodite daf-16 mutant animals fed either control or prx-11 RNAi. (H) Quantification of green:red fluorescence ratios for mCherry-GFP-SKL in day 3 adult hermaphrodite daf-16 mutant animals fed either control or prx-11 RNAi (n = 15 worms per condition). Data are presented as mean ± SD. ****, p ≤ 0.0001. Unpaired t-test. (I) Representative green channel images of mCherry-GFP-SKL in day 3 adult hermaphrodite fzo-1 mutant animals fed either control or prx-11 RNAi. (J) Quantification of individual peroxisome areas in day 3 adult hermaphrodite fzo-1 mutant animals fed either control or prx-11 RNAi. Data are presented as mean ± SD. ****, p ≤ 0.0001. Unpaired t-test. (K) Representative green channel images of mCherry-GFP-SKL in day 3 adult hermaphrodite unc-43 mutant animals fed either control or prx-11 RNAi. (L) Quantification of individual peroxisome areas in day 3 adult hermaphrodite unc-43 mutant animals fed either control or prx-11 RNAi. Data are presented as mean ± SD. ****, p ≤ 0.0001. Unpaired t-test. (M) Representative green channel images of mCherry-GFP-SKL in day 3 adult hermaphrodite daf-16 mutant animals fed either control or prx-11 RNAi. (N) Quantification of individual peroxisome areas in day 3 adult hermaphrodite daf-16 mutant animals fed either control or prx-11 RNAi. Data are presented as mean ± SD. ****, p ≤ 0.0001. Unpaired t-test. Bars: 10 μm (I, K, M) or 100 μm (A, C, E, G).
Because prx-11 knockdown [9] or mutation (Supplementary Figure 1A, 1B) slows pexophagy in aging wild-type animals, we next asked whether the accelerated pexophagy observed in fzo-1, unc-43, and daf-16 mutant genetic backgrounds could also be suppressed by prx-11 RNAi. We found, indeed, that knocking down prx-11 enabled retention of more GFP signal in day 3 fzo-1, unc-43, and daf-16 mutant animals (Figure 5C–5H), indicating less peroxisome degradation when PRX-11 was non-functional. As in wild-type animals treated with prx-11 RNAi [9], we observed increased peroxisome size when prx-11 was knocked down in fzo-1, unc-43, and daf-16 mutants (Figure 5I–5N). This suggests that PRX-11 function, perhaps in peroxisome fission, supports peroxisome degradation not only in wild-type animals but also in fzo-1, unc-43, and daf-16 mutants showing even faster pexophagy.
Preventing mitochondrial tubulation negates lifespan extension in prx-11-RNAi animals
Because fzo-1, unc-43, and daf-16 mutants treated with prx-11 RNAi showed dampened pexophagy (Figure 5C–5H) but were unable to support the late-age mitochondrial enhancements typical of prx-11 knockdown (Figure 4A–4C), we asked whether prx-11 knockdown in these genetic backgrounds could still enhance lifespan, as it does in wild-type [9]. If not, this would suggest that mitochondrial enhancements were causal in lifespan extension upon prx-11 RNAi. Though prx-11 RNAi reproducibly extended the lifespan of wild-type animals as previously reported [9, 47], it failed to extend lifespan in any of the three mutant backgrounds where prx-11 RNAi was insufficient to drive mitochondrial tubulation in older age (i.e., fzo-1, unc-43, and daf-16) (Figure 6A–6C). We concluded that prx-11-RNAi animals require improvements to mitochondria in order to live longer. Thus, one mechanism contributing to enhanced lifespan upon pexophagy inhibition is a coordinated improvement to mitochondrial biology.
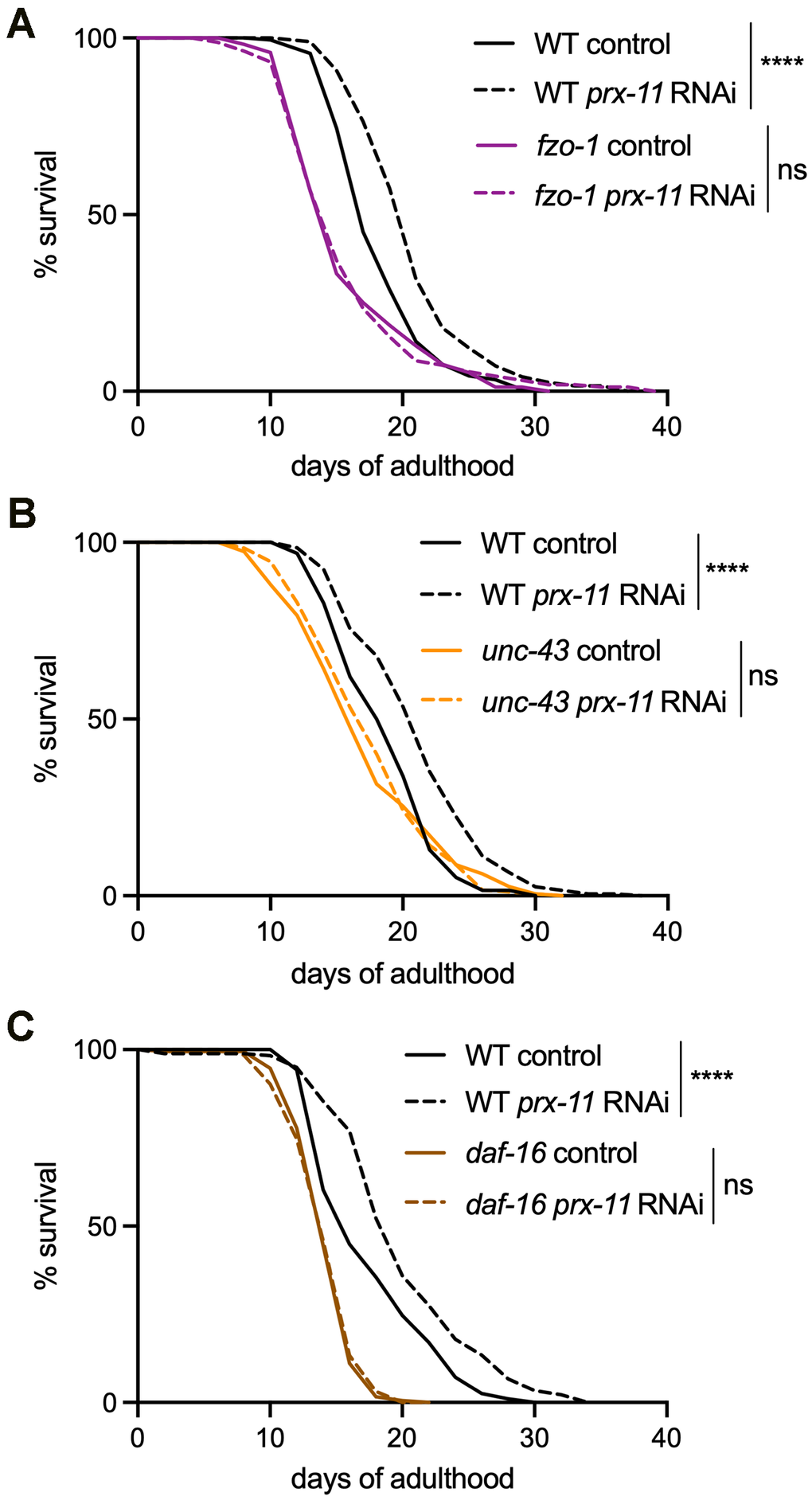
Figure 6. fzo-1, unc-43, and daf-16 mutations block lifespan extension upon prx-11 knockdown. (A) Lifespan analysis of wild-type and fzo-1 hermaphrodite animals fed either control or prx-11 RNAi. Wild-type control: Mean lifespan = 17.47 days, n = 184 worms. Wild-type prx-11 RNAi: Mean lifespan = 19.99 days, n = 195 worms. fzo-1 control: Mean lifespan = 16.08 days, n = 171 worms. fzo-1 prx-11 RNAi: Mean lifespan = 16.07 days, n = 162 worms. Significance was determined using a log-rank test. Wild-type control vs. wild-type prx-11 RNAi: Χ2 = 34.61, p ≤ 0.0001; fzo-1 control vs. fzo-1 prx-11 RNAi: Χ2 = 0.01, ns, not significant. (B) Lifespan analysis of wild-type and unc-43 hermaphrodite animals fed either control or prx-11 RNAi. Wild-type control: Mean lifespan = 18.94 days, n = 192 worms. Wild-type prx-11 RNAi: Mean lifespan = 21.37 days, n = 196 worms. unc-43 control: Mean lifespan = 17.38 days, n = 193 worms. unc-43 prx-11 RNAi: Mean lifespan = 17.77 days, n = 182 worms. Significance was determined using a log-rank test. Wild-type control vs. wild-type prx-11 RNAi: Χ2 = 31.86, p ≤ 0.0001; unc-43 control vs. unc-43 prx-11 RNAi: Χ2 = 0.03, ns, not significant. (C) Lifespan analysis of wild-type and daf-16 hermaphrodite animals fed either control or prx-11 RNAi. Wild-type control: Mean lifespan = 17.75 days, n = 194 worms. Wild-type prx-11 RNAi: Mean lifespan = 20.21 days, n = 178 worms. daf-16 control: Mean lifespan = 14.56 days, n = 189 worms. daf-16 prx-11 RNAi: Mean lifespan = 14.55 days, n = 194 worms. Significance was determined using a log-rank test. Wild-type control vs. wild-type prx-11 RNAi: Χ2 = 24.21, p ≤ 0.0001; daf-16 control vs. daf-16 prx-11 RNAi: Χ2 = 0.13, ns, not significant.
Discussion
Our findings identify peroxisome degradation as a modifiable event that controls mitochondrial aging (Figure 7). This provides a framework for understanding how inter-organelle communication sculpts the aging trajectory and suggests that targeting peroxisome homeostasis may offer a novel approach to preserve cellular function and extend healthy lifespan (Figure 7). These data expand on our previous work, which demonstrated that peroxisome loss begins early in adulthood [9], before overt signs of mitochondrial stress or damage are detectable. While mitochondrial quality control has traditionally been studied in isolation, our data support a model in which early degradation of peroxisomes acts as a gatekeeper event that modulates downstream changes to mitochondria. A recent study similarly suggested that defective peroxisomal import can lead to mitochondrial fragmentation and dysfunction [66]. Together with our study, these findings indicate that the status of mitochondria in aging animals tracks alongside peroxisomes: if peroxisomes are dysfunctional or degraded, mitochondrial health will decline, but, if peroxisomes are better maintained, mitochondrial health will be better maintained as well. Moreover, our data indicate that mitochondrial dysfunction, such as through impaired tubulation, can accelerate pexophagy. The co-regulation of peroxisomes and mitochondria during aging therefore appears to be bidirectional in nature, and mitochondrial dysfunction in response to age-related pexophagy might promote even more peroxisome degradation with time (Figure 7).
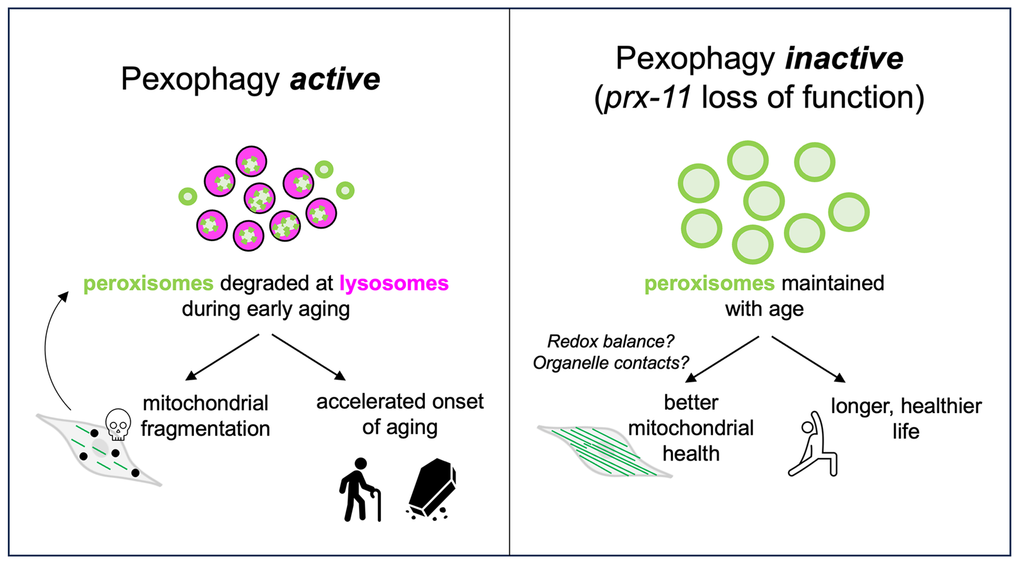
Figure 7. Model for the role of PRX-11 in regulating peroxisome degradation and mitochondrial homeostasis during aging. The schematic in the left panel illustrates that PRX-11 promotes age-associated peroxisome degradation and, in doing so, likely contributes to mitochondrial fragmentation and functional decline in older wild-type animals. Mitochondrial fragmentation and dysfunction can, in turn, drive further pexophagy. The schematic in the right panel illustrates that loss of PRX-11 activity, via gene knockdown or mutation, inhibits pexophagy and preserves peroxisome abundance in older animals, thereby supporting enhanced mitochondrial integrity and lifespan extension. The effects of peroxisome retention on mitochondria quality with age involve crosstalk between the two organelles, potentially mediated by changes to physical interactions or metabolic signaling.
The nature of the peroxisomes retained upon loss of prx-11 function is somewhat mysterious. In both prx-11-RNAi and mutant animals, peroxisomes not only persist with age but also increase in size. It is unclear whether the observed increase in peroxisome size enables them to evade degradation due to impaired fission or altered lysosomal targeting. Peroxisome size is known to influence degradation in other systems; larger or hyper-elongated peroxisomes have been shown to resist autophagic capture [67]. This raises the possibility that prx-11 loss leads to a population of “autophagy-resistant” peroxisomes that remain structurally and potentially functionally intact. Our data showing that SQST-1 turnover is unimpeded in prx-11-RNAi animals suggest that these peroxisomes do not accumulate as autophagy intermediates but may rather bypass the degradative pathway altogether.
An interesting observation is that inhibiting prx-11 prevents mitochondrial fragmentation and delays aging phenotypes without activating canonical stress response pathways such as the UPRmt or UPRer. Instead, our genetic data reveal that mitochondrial maintenance and lifespan extension mediated by prx-11 RNAi require specific regulators of mitochondrial homeostasis, including FZO-1, UNC-43, and DAF-16. How loss of PRX-11, a protein specifically localized to peroxisomes [37], subsequently influences the function and regulation of these mitochondrial regulators is an open question.
One possibility is that peroxisome retention helps to maintain redox balance or buffering capacity, thereby impacting mitochondria (Figure 7). This possibility is consistent with observed reductions in ROS and calcium accumulation in prx-11-RNAi animals. Peroxisomes are known to modulate intracellular redox through catalase activity and fatty-acid oxidation, both of which are tightly linked to mitochondrial function [34, 68]. In mammalian cells, peroxisome-derived NADH and acetyl-CoA feed into mitochondrial metabolism, and catalase plays a key role in buffering hydrogen peroxide produced by both peroxisomal and mitochondrial activity, thereby limiting mitochondrial oxidative damage [69, 70]. The phenotypic suppression of oxidative stress and calcium overload suggests that the retained peroxisomes in prx-11 mutants are not inert. Conversely, animals treated with hmgr-1 RNAi, which accelerates pexophagy [9], exhibited increased mitochondrial calcium accumulation, and animals with mutations in hmgr-1 have previously been reported to show other mitochondrial defects [71]. These data support a model in which early peroxisome loss compromises mitochondrial stability by removing buffering capacity or key metabolic intermediates.
Another possibility is that peroxisomes may engage in direct or indirect signaling with mitochondria to regulate network dynamics and transcriptional adaptation (Figure 7). The requirement for FZO-1 implies that mitochondrial fusion is an essential downstream effector of prx-11 loss. That DAF-16, UNC-43, and mitochondrial biogenesis regulators are also required supports a model in which peroxisome retention activates a broader remodeling program involving transcriptional and signaling components. Although this program has yet to be fully described, plausible components include redox-sensitive transcriptional regulators, lipid-signaling intermediates, or even physical tethering complexes between peroxisomes and mitochondria [29, 33, 70, 72]. Recent work in mammalian cells and yeasts has identified peroxisome-mitochondria contact sites that facilitate lipid exchange and coordinate fission events [73–75]; it is exciting to speculate that similar structures may exist in C. elegans, potentially contributing to peroxisome-mitochondrial crosstalk during aging. Notably, we observed that DAF-16 translocation into the nucleus occurred more robustly in prx-11-RNAi animals, and, once in the nucleus, DAF-16 may alter expression of factors relevant to mitochondrial health and functionality. Of note, DAF-16 activation promotes fat storage [76] and improves energy status [33], suggesting this may be one mechanism contributing to increased lipid staining and ATP/ADP ratios upon prx-11 knockdown. While it is unclear how DAF-16 is initially activated in prx-11-RNAi animals, pro-longevity remodeling of the organelle landscape has been shown to positively influence DAF-16 nuclear translocation [77], and similar principles might apply when peroxisomes are retained longer.
An additional point of note is that our findings challenge the prevailing view that organelle degradation is uniformly beneficial during aging. While mitophagy and other selective autophagy pathways are essential for damage clearance and can promote longevity [78], our findings suggest that premature destruction of functional peroxisomes may act as a destabilizing event that initiates mitochondrial fragmentation and health decline, and that may even amplify with expanding mitochondrial damage (Figure 7). In C. elegans, animals having loss-of-function mutations in the mTORC2 complex show hyperactive autophagy but live short [79]. Interestingly, this short lifespan has been linked to mitochondrial dysfunction [79]. It will be important to investigate whether pexophagy is also accelerated in this genetic background, perhaps explaining scenarios where animals show high autophagic capacity but also have decreased lifespan and impaired mitochondrial functionality.
Materials and Methods
C. elegans strain generation and maintenance
C. elegans strains used in this study are listed in Supplementary Table 1. The prx-11 mutant strain, prx-11(CSR1), which carries a CRISPR-engineered 290 base-pair deletion [37], was graciously provided by the Huanhu Zhu Lab. The site of deletion in the prx-11(CSR1) strain is indicated by a caret symbol in the following sequence: 5’- CCTCACTACAGTACTCTCTT^GACGCTAAGATCCTTGCATT -3’. For genetic crosses, transgenes expressing fluorescent proteins were tracked by stereomicroscopy, and gene deletions and mutations were verified by nested polymerase chain reactions (PCRs) and/or sequencing.
C. elegans were maintained on NGM agar (51.3 mM NaCl [ThermoFisher Scientific, S271-10], 0.25% peptone [ThermoFisher Scientific, BP1420-500], 1.7% agar [ThermoFisher Scientific, BP1423-500], 1 mM CaCl2 [G-Biosciences, R040], 1 mM MgSO4 [ThermoFisher Scientific, M65-500], 25 mM KPO4 [KH2PO4, ThermoFisher Scientific, P285-500; K2HPO4, ThermoFisher Scientific, P288-500], 12.9 μM cholesterol [Alfa Aesar, A11470], pH 6.0) seeded with E. coli OP50 bacteria at 20° C. Synchronous populations of animals were obtained by bleaching adult hermaphrodites; animals were vortexed in 1 mL bleaching solution (0.5 mM NaOH [ThermoFisher Scientific, S318-1], 20% bleach [Clorox, E624362]) for 5 min and then washed three times in M9 buffer (22 mM KH2PO4 [ThermoFisher Scientific, P285-500], 42 mM Na2HPO4 [ThermoFisher Scientific, S374-1], 85.5 mM NaCl [ThermoFisher Scientific, S271-10], 1 mM MgSO4 [ThermoFisher Scientific, M65-500], pH 7.0) before isolated eggs were then plated on OP50-seeded NGM. For aging experiments, adult animals were transferred daily to fresh OP50 and/or RNAi-seeded plates to separate adults from progeny.
For RNAi experiments, L4-stage animals were transferred onto RNAi plates (NGM with 100 ng/μl carbenicillin [ThermoFisher Scientific, BP26481] and 1 mM IPTG [ThermoFisher Scientific, 34060]) seeded with control or RNAi bacteria. The RNAi constructs were obtained from the Julie Ahringer collection [80]. An empty L4440 vector transformed into E. coli HT115 was used as a negative control. Clones were verified by DNA sequencing.
Microscopy and image analysis
Four-percent agarose (ThermoFisher Scientific, BP164) gel pads dried with a Kimwipe (Kimtech) were placed on Gold Seal™ glass microscope slides. A small volume of 10 mM levamisole (Acros Organics, AC18787) was spotted on the agarose gel pad, after which worms were transferred to the levamisole drop and a glass cover slip (ThermoFisher Scientific, 12-540-B) was placed on top to complete the mounting. Live-animal fluorescence microscopy was performed using a Leica DMi8 THUNDER imager, equipped with 10X (NA 0.32), 40X (NA 1.30), and 100X (NA 1.40) objectives, a DFC9000 GTC camera, and LED_405, GFP-T, Texas Red, and quad-band DFT51011 filter cubes.
Mitochondrial networks were analyzed using “Skeleton” plugin in FIJI/ImageJ. Briefly, images were converted to binary 8-bit images and then to skeleton images. Skeleton images were then quantified using the “Analyze Skeleton” function. Numbers of objects, numbers of junctions, and object lengths were scored. An “object” is defined by the “Analyze Skeleton” function as a branch connecting two endpoints, an endpoint and junction, or two junctions. Junctions/object was used as a parameter to quantify network complexity and integrity.
For mitochondrial Ca2+ accumulation analysis, animals were categorized into discrete Ca2+ accumulation states (low, moderate, or high) based on manually defined thresholds.
For fluorescence intensity analysis, regions of interest were outlined using the freehand selection tool in FIJI/ImageJ, and mean intensity values were obtained using the “Analyze>Measure” function. Exposure time and laser intensity were kept the same for compared groups. Fluorescence ratios were calculated, where appropriate, by dividing mean intensity values for the relevant wavelengths and/or regions. For analysis of DAF-16::GFP nuclear translocation, nuclear fluorescence intensity was scored for five nuclei per animal. Each of the five values was normalized to fluorescence intensity in an adjacent, equally sized area in the cytoplasm. The five nuclear:cytoplasmic ratios for each animal were then averaged into a single value per animal for presentation and analysis. To score ATP/ADP ratios using PercevalHR, samples were excited using violet or blue light, and emission was quantified in the 506-532 nm range. Fluorescence from violet-light excitation represents the ADP-bound form, and fluorescence from blue-light excitation represents the ATP-bound form [58].
Individual peroxisome areas were calculated by freehand tracing peroxisomes in FIJI/ImageJ and using the “Analyze>Measure” function. 10 animals were scored per condition for analysis of peroxisome areas.
ROS levels were measured using DHE staining. Synchronized animals were exposed to RNAi from L4 stage and aged until day 5 of adulthood. On days 1 and 5, animals were washed twice in M9 buffer, then incubated in 3 μM DHE (VWR, 76482-322) in M9 buffer for 45 minutes at room temperature in the dark. After incubation, animals were washed twice to remove excess dye and mounted on agarose pads for imaging. Red fluorescence was quantified in individual animals using FIJI/ImageJ, and mean intensity values were compared across conditions.
Lipids were labeled inside animals using ORO (Alfa Aesar, A12989) or BODIPY (ThermoFisher Scientific, D3922). Staining with ORO was performed as previously described [55]. Once animals were fixed and stained with ORO, they were imaged using a Leica S9i digital stereomicroscope with integrated camera. To stain with BODIPY, animals were washed off NGM plates using M9 buffer. Animals were allowed to sediment by gravity, and then they were washed one additional time in M9 buffer before staining with 6.7 μg/mL BODIPY (diluted in M9 buffer). Sample tubes were covered in foil and incubated with nutation for 20 minutes. Following incubation, animals were washed three times in M9 buffer and imaged immediately using the Leica THUNDER DMi8 system.
Lifespan analysis
Synchronous populations of animals were transferred as late L4s to OP50 and/or RNAi seeded plates. The OP50 and/or RNAi plates were spotted with 5 mM FUdR (Acros Organics, 227605000) to prevent progeny production. Animals that exploded, bagged, or crawled off plates were censored during analysis. Lifespans were analyzed using OASIS 2 software [81], and statistical significance was assessed using a log-rank test.
Thrashing assay
Synchronous populations of animals were transferred as late L4s to NGM plates seeded with OP50 bacteria. Worms were transferred to fresh plates every day to separate adults from their progeny. To score thrashing rates, individual worms were transferred into a drop of M9 buffer on an NGM plate, and the number of body thrashes were counted in a 1-minute period.
Statistical analysis
Data were statistically analyzed using GraphPad Prism software. For two-sample comparisons, an unpaired t-test was used to determine significance (α = 0.05). For comparisons of three or more samples, a one-way ANOVA with Tukey's multiple comparisons was used to determine significance (α = 0.05). Statistical significance of lifespan data was determined using a log-rank test.
Abbreviations
Ca2+: calcium; DHE: dihydroethidium; Mito-GFP: mitochondrially targeted green fluorescent protein; Mito-LAR-GECO: mitochondrially targeted low-affinity red fluorescent genetically encoded Ca2+ indicator for optical imaging; ORO: Oil Red O; PCR: polymerase chain reaction; RNAi: RNA interference; ROS: reactive oxygen species; UPRmt: mitochondrial unfolded protein response; UPRer: endoplasmic reticulum unfolded protein response.
Author Contributions
Conceptualization: YF, KAB; Methodology: YF, DS, KRD, KAB; Formal Analysis: YF, DS, KAB; Resources: YF, KRD, KAB; Data Curation: YF, DS, KAB; Visualization: YF, DS, KAB; Funding acquisition: KAB; Project administration: KAB; Supervision: KAB; Writing – original draft: YF, KAB; Writing – review and editing: YF, DS, KRD, KAB.
Acknowledgements
We thank members of the Bohnert lab and the Alyssa Johnson lab at LSU for comments on the study and critical reading of the manuscript. Some strains were provided by the Caenorhabditis Genetics Center (CGC), which is funded by the National Institutes of Health Office of Research Infrastructure Programs grant P40OD010440. We also thank Huanhu Zhu for generously providing the prx-11(CSR1) strain.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This study was supported by National Institutes of Health grant R01AG079970 to K.A.B.
References
- 1. Guo J, Huang X, Dou L, Yan M, Shen T, Tang W, Li J. Aging and aging-related diseases: from molecular mechanisms to interventions and treatments. Signal Transduct Target Ther. 2022; 7:391. https://doi.org/10.1038/s41392-022-01251-0 [PubMed]
- 2. Niccoli T, Partridge L. Ageing as a risk factor for disease. Curr Biol. 2012; 22:R741–52. https://doi.org/10.1016/j.cub.2012.07.024 [PubMed]
- 3. Chimata AV, Deshpande P, Singh A and Singh A. Chapter 2 - Impact of aging at cellular and organ level. In: Singh SK, Lin C-L and Mishra SK, eds. Anti-Aging Drug Discovery on the Basis of Hallmarks of Aging: Academic Press. 2022. pp. 19–39. https://doi.org/10.1016/B978-0-323-90235-9.00009-4
- 4. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: An expanding universe. Cell. 2023; 186:243–78. https://doi.org/10.1016/j.cell.2022.11.001 [PubMed]
- 5. Cuanalo-Contreras K, Schulz J, Mukherjee A, Park KW, Armijo E, Soto C. Extensive accumulation of misfolded protein aggregates during natural aging and senescence. Front Aging Neurosci. 2023; 14:1090109. https://doi.org/10.3389/fnagi.2022.1090109 [PubMed]
- 6. Sun N, Youle RJ, Finkel T. The Mitochondrial Basis of Aging. Mol Cell. 2016; 61:654–66. https://doi.org/10.1016/j.molcel.2016.01.028 [PubMed]
- 7. Rossiello F, Jurk D, Passos JF, d'Adda di Fagagna F. Telomere dysfunction in ageing and age-related diseases. Nat Cell Biol. 2022; 24:135–47. https://doi.org/10.1038/s41556-022-00842-x [PubMed]
- 8. Carlson ME, Silva HS, Conboy IM. Aging of signal transduction pathways, and pathology. Exp Cell Res. 2008; 314:1951–61. https://doi.org/10.1016/j.yexcr.2008.03.017 [PubMed]
- 9. Dolese DA, Junot MP, Ghosh B, Butsch TJ, Johnson AE, Bohnert KA. Degradative tubular lysosomes link pexophagy to starvation and early aging in C. elegans. Autophagy. 2022; 18:1522–33. https://doi.org/10.1080/15548627.2021.1990647 [PubMed]
- 10. Sheikh FG, Pahan K, Khan M, Barbosa E, Singh I. Abnormality in catalase import into peroxisomes leads to severe neurological disorder. Proc Natl Acad Sci USA. 1998; 95:2961–6. https://doi.org/10.1073/pnas.95.6.2961 [PubMed]
- 11. Violante S, Achetib N, van Roermund CW, Hagen J, Dodatko T, Vaz FM, Waterham HR, Chen H, Baes M, Yu C, Argmann CA, Houten SM. Peroxisomes can oxidize medium- and long-chain fatty acids through a pathway involving ABCD3 and HSD17B4. FASEB J. 2019; 33:4355–64. https://doi.org/10.1096/fj.201801498R [PubMed]
- 12. Wanders RJ. Metabolic functions of peroxisomes in health and disease. Biochimie. 2014; 98:36–44. https://doi.org/10.1016/j.biochi.2013.08.022 [PubMed]
- 13. Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007; 3:452–60. https://doi.org/10.4161/auto.4451 [PubMed]
- 14. Gould SJ, Keller GA, Hosken N, Wilkinson J, Subramani S. A conserved tripeptide sorts proteins to peroxisomes. J Cell Biol. 1989; 108:1657–64. https://doi.org/10.1083/jcb.108.5.1657 [PubMed]
- 15. Opaliński Ł, Kiel JA, Williams C, Veenhuis M, van der Klei IJ. Membrane curvature during peroxisome fission requires Pex11. EMBO J. 2011; 30:5–16. https://doi.org/10.1038/emboj.2010.299 [PubMed]
- 16. Park H, He A, Tan M, Johnson JM, Dean JM, Pietka TA, Chen Y, Zhang X, Hsu FF, Razani B, Funai K, Lodhi IJ. Peroxisome-derived lipids regulate adipose thermogenesis by mediating cold-induced mitochondrial fission. J Clin Invest. 2019; 129:694–711. https://doi.org/10.1172/JCI120606 [PubMed]
- 17. Wang B, Van Veldhoven PP, Brees C, Rubio N, Nordgren M, Apanasets O, Kunze M, Baes M, Agostinis P, Fransen M. Mitochondria are targets for peroxisome-derived oxidative stress in cultured mammalian cells. Free Radic Biol Med. 2013; 65:882–94. https://doi.org/10.1016/j.freeradbiomed.2013.08.173 [PubMed]
- 18. Koch A, Yoon Y, Bonekamp NA, McNiven MA, Schrader M. A role for Fis1 in both mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. 2005; 16:5077–86. https://doi.org/10.1091/mbc.e05-02-0159 [PubMed]
- 19. Passmore JB, Carmichael RE, Schrader TA, Godinho LF, Ferdinandusse S, Lismont C, Wang Y, Hacker C, Islinger M, Fransen M, Richards DM, Freisinger P, Schrader M. Mitochondrial fission factor (MFF) is a critical regulator of peroxisome maturation. Biochim Biophys Acta Mol Cell Res. 2020; 1867:118709. https://doi.org/10.1016/j.bbamcr.2020.118709 [PubMed]
- 20. Goulding RP, Charlton BT, Breedveld EA, van der Laan M, Strating AR, Noort W, Kolodyazhna A, Appelman B, van Vugt M, Grootemaat AE, van der Wel NN, de Koning JJ, Bloemers FW, Wüst RC. Skeletal muscle mitochondrial fragmentation predicts age-associated decline in physical capacity. Aging Cell. 2025; 24:e14386. https://doi.org/10.1111/acel.14386 [PubMed]
- 21. Hughes AL, Gottschling DE. An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature. 2012; 492:261–5. https://doi.org/10.1038/nature11654 [PubMed]
- 22. Roux AE, Langhans K, Huynh W, Kenyon C. Reversible Age-Related Phenotypes Induced during Larval Quiescence in C. elegans. Cell Metab. 2016; 23:1113–26. https://doi.org/10.1016/j.cmet.2016.05.024 [PubMed]
- 23. Gaffney CJ, Pollard A, Barratt TF, Constantin-Teodosiu D, Greenhaff PL, Szewczyk NJ. Greater loss of mitochondrial function with ageing is associated with earlier onset of sarcopenia in C. elegans. Aging (Albany NY). 2018; 10:3382–96. https://doi.org/10.18632/aging.101654 [PubMed]
- 24. Higashitani A, Teranishi M, Nakagawa Y, Itoh Y, Sudevan S, Szewczyk NJ, Kubota Y, Abe T, Kobayashi T. Increased mitochondrial Ca2+ contributes to health decline with age and Duchene muscular dystrophy in C. elegans. FASEB J. 2023; 37:e22851. https://doi.org/10.1096/fj.202201489RR [PubMed]
- 25. Berry BJ, Vodičková A, Müller-Eigner A, Meng C, Ludwig C, Kaeberlein M, Peleg S, Wojtovich AP. Optogenetic rejuvenation of mitochondrial membrane potential extends C. elegans lifespan. Nat Aging. 2023; 3:157–61. https://doi.org/10.1038/s43587-022-00340-7 [PubMed]
- 26. Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Cantó C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, Guarente L, Auwerx J. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell. 2013; 154:430–41. https://doi.org/10.1016/j.cell.2013.06.016 [PubMed]
- 27. Leduc-Gaudet JP, Hussain SN, Barreiro E, Gouspillou G. Mitochondrial Dynamics and Mitophagy in Skeletal Muscle Health and Aging. Int J Mol Sci. 2021; 22:8179. https://doi.org/10.3390/ijms22158179 [PubMed]
- 28. Bernhardt D, Müller M, Reichert AS, Osiewacz HD. Simultaneous impairment of mitochondrial fission and fusion reduces mitophagy and shortens replicative lifespan. Sci Rep. 2015; 5:7885. https://doi.org/10.1038/srep07885 [PubMed]
- 29. Chaudhari SN, Kipreos ET. Increased mitochondrial fusion allows the survival of older animals in diverse C. elegans longevity pathways. Nat Commun. 2017; 8:182. https://doi.org/10.1038/s41467-017-00274-4 [PubMed]
- 30. Ichishita R, Tanaka K, Sugiura Y, Sayano T, Mihara K, Oka T. An RNAi screen for mitochondrial proteins required to maintain the morphology of the organelle in Caenorhabditis elegans. J Biochem. 2008; 143:449–54. https://doi.org/10.1093/jb/mvm245 [PubMed]
- 31. Campbell D, Zuryn S. The mechanisms and roles of mitochondrial dynamics in C. elegans. Semin Cell Dev Biol. 2024; 156:266–75. https://doi.org/10.1016/j.semcdb.2023.10.006 [PubMed]
- 32. Jiang HC, Hsu JM, Yen CP, Chao CC, Chen RH, Pan CL. Neural activity and CaMKII protect mitochondria from fragmentation in aging Caenorhabditis elegans neurons. Proc Natl Acad Sci USA. 2015; 112:8768–73. https://doi.org/10.1073/pnas.1501831112 [PubMed]
- 33. Wang H, Webster P, Chen L, Fisher AL. Cell-autonomous and non-autonomous roles of daf-16 in muscle function and mitochondrial capacity in aging C. elegans. Aging (Albany NY). 2019; 11:2295–311. https://doi.org/10.18632/aging.101914 [PubMed]
- 34. Demarquoy J, Le Borgne F. Crosstalk between mitochondria and peroxisomes. World J Biol Chem. 2015; 6:301–9. https://doi.org/10.4331/wjbc.v6.i4.301 [PubMed]
- 35. Schrader M, Costello J, Godinho LF, Islinger M. Peroxisome-mitochondria interplay and disease. J Inherit Metab Dis. 2015; 38:681–702. https://doi.org/10.1007/s10545-015-9819-7 [PubMed]
- 36. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998; 391:806–11. https://doi.org/10.1038/35888 [PubMed]
- 37. Li N, Hua B, Chen Q, Teng F, Ruan M, Zhu M, Zhang L, Huo Y, Liu H, Zhuang M, Shen H, Zhu H. A sphingolipid-mTORC1 nutrient-sensing pathway regulates animal development by an intestinal peroxisome relocation-based gut-brain crosstalk. Cell Rep. 2022; 40:111140. https://doi.org/10.1016/j.celrep.2022.111140 [PubMed]
- 38. Villalobos TV, Ghosh B, DeLeo KR, Alam S, Ricaurte-Perez C, Wang A, Mercola BM, Butsch TJ, Ramos CD, Das S, Eymard ED, Bohnert KA, Johnson AE. Tubular lysosome induction couples animal starvation to healthy aging. Nat Aging. 2023; 3:1091–106. https://doi.org/10.1038/s43587-023-00470-6 [PubMed]
- 39. Del Campo A, Contreras-Hernández I, Castro-Sepúlveda M, Campos CA, Figueroa R, Tevy MF, Eisner V, Casas M, Jaimovich E. Muscle function decline and mitochondria changes in middle age precede sarcopenia in mice. Aging (Albany NY). 2018; 10:34–55. https://doi.org/10.18632/aging.101358 [PubMed]
- 40. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011; 476:341–5. https://doi.org/10.1038/nature10234 [PubMed]
- 41. De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011; 476:336–40. https://doi.org/10.1038/nature10230 [PubMed]
- 42. Wu J, Prole DL, Shen Y, Lin Z, Gnanasekaran A, Liu Y, Chen L, Zhou H, Chen SR, Usachev YM, Taylor CW, Campbell RE. Red fluorescent genetically encoded Ca2+ indicators for use in mitochondria and endoplasmic reticulum. Biochem J. 2014; 464:13–22. https://doi.org/10.1042/BJ20140931 [PubMed]
- 43. Hwang AB, Jeong DE, Lee SJ. Mitochondria and organismal longevity. Curr Genomics. 2012; 13:519–32. https://doi.org/10.2174/138920212803251427 [PubMed]
- 44. Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002; 21:4411–9. https://doi.org/10.1093/emboj/cdf445 [PubMed]
- 45. Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci. 2004; 117:4055–66. https://doi.org/10.1242/jcs.01275 [PubMed]
- 46. Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002; 415:92–6. https://doi.org/10.1038/415092a [PubMed]
- 47. Zhou B, Yang L, Li S, Huang J, Chen H, Hou L, Wang J, Green CD, Yan Z, Huang X, Kaeberlein M, Zhu L, Xiao H, et al. Midlife gene expressions identify modulators of aging through dietary interventions. Proc Natl Acad Sci USA. 2012; 109:E1201–9. https://doi.org/10.1073/pnas.1119304109 [PubMed]
- 48. Libina N, Berman JR, Kenyon C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell. 2003; 115:489–502. https://doi.org/10.1016/s0092-8674(03)00889-4 [PubMed]
- 49. Shi Y, Pulliam DA, Liu Y, Hamilton RT, Jernigan AL, Bhattacharya A, Sloane LB, Qi W, Chaudhuri A, Buffenstein R, Ungvari Z, Austad SN, Van Remmen H. Reduced mitochondrial ROS, enhanced antioxidant defense, and distinct age-related changes in oxidative damage in muscles of long-lived Peromyscus leucopus. Am J Physiol Regul Integr Comp Physiol. 2013; 304:R343–55. https://doi.org/10.1152/ajpregu.00139.2012 [PubMed]
- 50. Teixeira RB, Pfeiffer M, Zhang P, Shafique E, Rayta B, Karbasiafshar C, Ahsan N, Sellke FW, Abid MR. Reduction in mitochondrial ROS improves oxidative phosphorylation and provides resilience to coronary endothelium in non-reperfused myocardial infarction. Basic Res Cardiol. 2023; 118:3. https://doi.org/10.1007/s00395-022-00976-x [PubMed]
- 51. Papsdorf K, Miklas JW, Hosseini A, Cabruja M, Morrow CS, Savini M, Yu Y, Silva-García CG, Haseley NR, Murphy LM, Yao P, de Launoit E, Dixon SJ, et al. Lipid droplets and peroxisomes are co-regulated to drive lifespan extension in response to mono-unsaturated fatty acids. Nat Cell Biol. 2023; 25:672–84. https://doi.org/10.1038/s41556-023-01136-6 [PubMed]
- 52. Enkler L, Spang A. Functional interplay of lipid droplets and mitochondria. FEBS Lett. 2024; 598:1235–51. https://doi.org/10.1002/1873-3468.14809 [PubMed]
- 53. Zhang P, Na H, Liu Z, Zhang S, Xue P, Chen Y, Pu J, Peng G, Huang X, Yang F, Xie Z, Xu T, Xu P, et al. Proteomic study and marker protein identification of Caenorhabditis elegans lipid droplets. Mol Cell Proteomics. 2012; 11:317–28. https://doi.org/10.1074/mcp.M111.016345 [PubMed]
- 54. Klapper M, Ehmke M, Palgunow D, Böhme M, Matthäus C, Bergner G, Dietzek B, Popp J, Döring F. Fluorescence-based fixative and vital staining of lipid droplets in Caenorhabditis elegans reveal fat stores using microscopy and flow cytometry approaches. J Lipid Res. 2011; 52:1281–93. https://doi.org/10.1194/jlr.D011940 [PubMed]
- 55. Escorcia W, Ruter DL, Nhan J, Curran SP. Quantification of Lipid Abundance and Evaluation of Lipid Distribution in Caenorhabditis elegans by Nile Red and Oil Red O Staining. J Vis Exp. 2018: 57352. https://doi.org/10.3791/57352 [PubMed]
- 56. Ghosh B, Guidry HJ, Johnston M, Bohnert KA. A Fat-Promoting Botanical Extract From Artemisia scoparia Exerts Geroprotective Effects on Caenorhabditis elegans Life Span and Stress Resistance. J Gerontol A Biol Sci Med Sci. 2022; 77:1112–20. https://doi.org/10.1093/gerona/glac040 [PubMed]
- 57. Han S, Schroeder EA, Silva-García CG, Hebestreit K, Mair WB, Brunet A. Mono-unsaturated fatty acids link H3K4me3 modifiers to C. elegans lifespan. Nature. 2017; 544:185–90. https://doi.org/10.1038/nature21686 [PubMed]
- 58. Garde A, Sherwood DR. Visualizing cytoplasmic ATP in C. elegans larvae using PercevalHR. STAR Protoc. 2022; 3:101429. https://doi.org/10.1016/j.xpro.2022.101429 [PubMed]
- 59. Tantama M, Martínez-François JR, Mongeon R, Yellen G. Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat Commun. 2013; 4:2550. https://doi.org/10.1038/ncomms3550 [PubMed]
- 60. García-Casas P, Alvarez-Illera P, Gómez-Orte E, Cabello J, Fonteriz RI, Montero M, Alvarez J. The Mitochondrial Na+/Ca2+ Exchanger Inhibitor CGP37157 Preserves Muscle Structure and Function to Increase Lifespan and Healthspan in Caenorhabditis elegans. Front Pharmacol. 2021; 12:695687. https://doi.org/10.3389/fphar.2021.695687 [PubMed]
- 61. Bansal A, Zhu LJ, Yen K, Tissenbaum HA. Uncoupling lifespan and healthspan in Caenorhabditis elegans longevity mutants. Proc Natl Acad Sci USA. 2015; 112:E277–86. https://doi.org/10.1073/pnas.1412192112 [PubMed]
- 62. Rojo M, Legros F, Chateau D, Lombès A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci. 2002; 115:1663–74. https://doi.org/10.1242/jcs.115.8.1663 [PubMed]
- 63. Breckenridge DG, Kang BH, Kokel D, Mitani S, Staehelin LA, Xue D. Caenorhabditis elegans drp-1 and fis-2 regulate distinct cell-death execution pathways downstream of ced-3 and independent of ced-9. Mol Cell. 2008; 31:586–97. https://doi.org/10.1016/j.molcel.2008.07.015 [PubMed]
- 64. Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997; 389:994–9. https://doi.org/10.1038/40194 [PubMed]
- 65. Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993; 366:461–4. https://doi.org/10.1038/366461a0 [PubMed]
- 66. Sharma A, Mistry M, Yao P, Liang Y, Hui S, Mair WB. Peroxisomal Dysfunction Drives Loss of Dynamic Nutrient Responses to Initiate Metabolic Inflexibility During Aging. bioRXiv. 2025. https://doi.org/10.1101/2025.02.14.638340
- 67. Mao K, Liu X, Feng Y, Klionsky DJ. The progression of peroxisomal degradation through autophagy requires peroxisomal division. Autophagy. 2014; 10:652–61. https://doi.org/10.4161/auto.27852 [PubMed]
- 68. Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B, Hacohen N, Chen ZJ, Whelan SP, Fransen M, Nibert ML, Superti-Furga G, Kagan JC. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010; 141:668–81. https://doi.org/10.1016/j.cell.2010.04.018 [PubMed]
- 69. Yoboue ED, Sitia R, Simmen T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018; 9:331. https://doi.org/10.1038/s41419-017-0033-4 [PubMed]
- 70. Tanaka H, Okazaki T, Aoyama S, Yokota M, Koike M, Okada Y, Fujiki Y, Gotoh Y. Peroxisomes control mitochondrial dynamics and the mitochondrion-dependent apoptosis pathway. J Cell Sci. 2019; 132:jcs224766. https://doi.org/10.1242/jcs.224766 [PubMed]
- 71. Ranji P, Rauthan M, Pitot C, Pilon M. Loss of HMG-CoA reductase in C. elegans causes defects in protein prenylation and muscle mitochondria. PLoS One. 2014; 9:e100033. https://doi.org/10.1371/journal.pone.0100033 [PubMed]
- 72. Tao L, Xie Q, Ding YH, Li ST, Peng S, Zhang YP, Tan D, Yuan Z, Dong MQ. CAMKII and calcineurin regulate the lifespan of Caenorhabditis elegans through the FOXO transcription factor DAF-16. Elife. 2013; 2:e00518. https://doi.org/10.7554/eLife.00518 [PubMed]
- 73. Shai N, Yifrach E, van Roermund CW, Cohen N, Bibi C, IJlst L, Cavellini L, Meurisse J, Schuster R, Zada L, Mari MC, Reggiori FM, Hughes AL, et al. Systematic mapping of contact sites reveals tethers and a function for the peroxisome-mitochondria contact. Nat Commun. 2018; 9:1761. https://doi.org/10.1038/s41467-018-03957-8 [PubMed]
- 74. Mattiazzi Ušaj M, Brložnik M, Kaferle P, Žitnik M, Wolinski H, Leitner F, Kohlwein SD, Zupan B, Petrovič U. Genome-Wide Localization Study of Yeast Pex11 Identifies Peroxisome-Mitochondria Interactions through the ERMES Complex. J Mol Biol. 2015; 427:2072–87. https://doi.org/10.1016/j.jmb.2015.03.004 [PubMed]
- 75. Subramani S, Shukla N, Farre JC. Convergent and divergent mechanisms of peroxisomal and mitochondrial division. J Cell Biol. 2023; 222:e202304076. https://doi.org/10.1083/jcb.202304076 [PubMed]
- 76. Ashrafi K, Chang FY, Watts JL, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. Genome-wide RNAi analysis of Caenorhabditis elegans fat regulatory genes. Nature. 2003; 421:268–72. https://doi.org/10.1038/nature01279 [PubMed]
- 77. Ricaurte-Perez C, Gill JP, Wall PK, Dubuisson O, DeLeo KR, Bohnert KA, Johnson AE. DAF-16/FOXO and HLH-30/TFEB comprise a cooperative regulatory axis controlling tubular lysosome induction in C. elegans. Nat Commun. 2025; 16:9878. https://doi.org/10.1038/s41467-025-64832-x [PubMed]
- 78. Butsch TJ, Ghosh B, Bohnert KA. Organelle-Specific Autophagy in Cellular Aging and Rejuvenation. Adv Geriatr Med Res. 2021; 3:e210010. https://doi.org/10.20900/agmr20210010 [PubMed]
- 79. Zhou B, Kreuzer J, Kumsta C, Wu L, Kamer KJ, Cedillo L, Zhang Y, Li S, Kacergis MC, Webster CM, Fejes-Toth G, Naray-Fejes-Toth A, Das S, et al. Mitochondrial Permeability Uncouples Elevated Autophagy and Lifespan Extension. Cell. 2019; 177:299–314.e16. https://doi.org/10.1016/j.cell.2019.02.013 [PubMed]
- 80. Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, Welchman DP, Zipperlen P, Ahringer J. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003; 421:231–7. https://doi.org/10.1038/nature01278 [PubMed]
- 81. Han SK, Lee D, Lee H, Kim D, Son HG, Yang JS, Lee SV, Kim S. OASIS 2: online application for survival analysis 2 with features for the analysis of maximal lifespan and healthspan in aging research. Oncotarget. 2016; 7:56147–52. https://doi.org/10.18632/oncotarget.11269 [PubMed]