



Introduction
Colorectal cancer (CRC) is one of the most common malignancies in the world [1]. Despite improvement in medical technology over the past years, including surgical resection, chemotherapy, radiotherapy and molecular targeted therapy, the five-year survival rate of CRC still remains unsatisfied [1,2]. Therefore, exploring the molecular mechanisms underlying CRC initiation and development is urgently needed.
Long non-coding RNAs(lncRNAs) are a class of more than 200 necleotides in length with limited protein-coding potential [3]. Accumulating studies reported that many lncRNAs were dysregulated and involved in various biological process, such as cell proliferation, apoptosis, migration, invasion and metastasis [4,5].
TINCR, a lncRNA about 3.7kb, controls human epidermal differentiation and is frequently deleted and downregulated in human squamous cell carcinoma (SCC) [6,7]. Moreover, multiple evidences have shown that aberrant expression of TINCR is closely associated with various human cancers, for example, T-p Xu, et al. [8] reported that TINCR was overexpressed and regulated cell cycle progression and apoptosis via modulating CDKN1A/P21, KLF2 and CDKN2B/P15 expression in gastric cancer. Xiaochun Liu, et al. [9] reported that TINCR was downregulated and suppressed cell proliferation and invasion via regulating miR-544a/FBXW7 axis in lung cancer. These prompted us to investigate the role of TINCR in human CRC.
In this study, we found that TINCR was upregulated in CRC cells and tissue samples, and the upregulation of TINCR was induced by SP1. We also revealed that TINCR, acting as a miR-7-5p sponge, may promote CRC progression via miR-7-5p-mediated PI3K/ Akt/mTOR signaling pathway. Our findings uncover the role of TINCR as a regulator of CRC progression, and shed new light on our understanding of TINCR-mediated malignancy progression.
Results
TINCR is obviously upregulated in CRC and increased TINCR expression predicts poor prognosis
Firstly, we investigated the expression level of TINCR in CRC cell lines and tissues. TINCR is significantly overexpressed in CRC cell lines (HCT116, HCT8, HT29, SW480 and SW620) in comparison with normal colon mucosal epithelial cell (FHC) (Fig. 1A). We also measured TINCR expression levels in CRC tissue samples and matched adjacent normal tissues (ANTs), and the results showed that TINCR expression was markedly higher in CRC tissue samples than that in the ANTs (Fig. 1B).
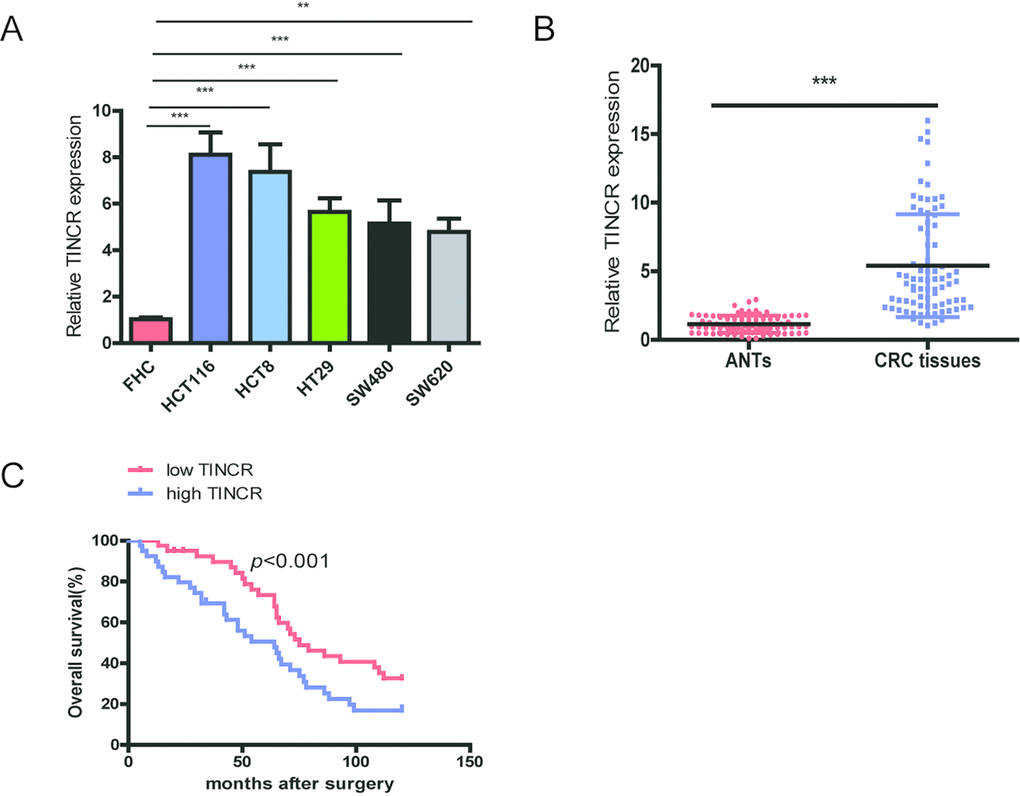
Figure 1. TINCR is frequently upregulated in CRC tissues and cell lines and predicts poor prognosis. (A) qRT-PCR analysis of TINCR expression in HCT116, HCT8, HT29, SW620, SW480 and FHC cells.GAPDH was used as an internal control. (B) qRT-PCR analysis of TINCR expression in 80 paired CRC tissues and and corresponding adjacent normal tissues. (C) Association of TINCR expression with OS (Kaplan-Meier plot). Data were shown as mean±S.D. of three independent experiments. **P<0.01, *** P<0.001.
Next, we analyze the relationship between TINCR expression and clinicopathological characteristics of CRC patients, and total 80 CRC cases were divided into low-expressing group (n=40) and high-expressing group (n=40) based on median value of TINCR expression. As shown in Table 1, high-expressing of TINCR was obviously correlated with lymph node metastasis (p=0.012), differentiation (p=0.033) and TNM stage (p<0.001).
Table 1. Correlation between TINCR expression and different clinical characteristics.
| Patient characteristics | No. of Patients (%) (n=80) | TINCR expression | p value | |
| High (%) (n=40) | Low (%) (n=40) | |||
| Age (years) | 0.637 | |||
| <60 | 27 (33.75%) | 12 (30.0%) | 15 (37.5%) | |
| ≥60 | 53 (66.25%) | 28 (70.0%) | 25 (62.5%) | |
| Gender | 0.367 | |||
| Female | 35 (43.75%) | 15 (37.5%) | 20 (50.0%) | |
| Male | 45 (56.25%) | 25 (62.5%) | 20 (50.0%) | |
| Tumor location | 0.073 | |||
| Colon | 41 (51.25%) | 25 (62.5%) | 16 (40.0%) | |
| Rectum | 39 (48.75%) | 15 (37.5%) | 24 (60.0%) | |
| Tumor size (cm) | 0.822 | |||
| <5 | 36 (45.0%) | 17 (42.5%) | 19 (47.5%) | |
| ≥5 | 44(55.0%) | 23 (57.5%) | 21 (52.5%) | |
| Differentiation | 0.033 | |||
| Well | 15 (18.75%) | 9 (22.5%) | 6 (15.0%) | |
| Moderate | 44 (55.0%) | 13 (32.5%) | 31 (77.5%) | |
| Poor | 21 (26.25%) | 18 (45.0%) | 3(7.5%%) | |
| Serum CEA level (ng/mL) | 0.261 | |||
| <10 | 36 (45.0%) | 21 (52.5%) | 15 (37.5%) | |
| ≥10 | 44 (55.0%) | 19 (47.5%) | 25 (62.5%) | |
| Local invasion | 0.647 | |||
| T1-T2 | 31 (38.75%) | 17 (42.5%) | 14 (35.0%) | |
| T3-T4 | 49 (61.25%) | 23 (57.5%) | 26 (65.0%) | |
| Lymph node metastasis | 0.012 | |||
| N0-N1 | 46 (57.5%) | 17 (42.5%) | 29 (72.5%) | |
| N2 | 34 (42.5%) | 23 (57.5%) | 11 (27.5%) | |
| TNM stage | <0.001 | |||
| I-II | 39(48.75%) | 11(27.5%) | 28(70.0%) | |
| III-IV | 41(51.25%) | 29(72.5%) | 12(30.0%) | |
Furthermore, CRC patients with high expression of TINCR predicted a poor overall survival (OS) using the analysis of Kaplan-Meier survival curve (p<0.001).
Univariate analysis revealed that differentiation, Lymph node metastasis, TNM stage and TINCR were predictors for poor OS. Multivariate analysis showed that upregulation of TINCR was obviously correlated with unfavorable OS (Table 2). These results suggest that TINCR upregulation as an early event in CRC development and have a pivotal role in CRC progression.
Table 2. Univariate and multivariate analysis for OS in patients with CRC.
| Characteristics | Multivariate analysis for OS | Univariate analysis for OS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HR (95% CI) | HR (95% CI) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age (<60/≥60) | - | - | 1.023(0.681-1.232) | 0.332 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Gender (Female/Male) | - | - | 0.909(0.616-1.331) | 0.215 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Tumor location (colon/rectum) | - | - | 0.918(0.661-1.421) | 0.571 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Tumor size (<5/≥5cm) | - | - | 0.911(0.751-1.434) | 0.314 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Differentiation (well/moderate/poor) | 1.878 (1.212-3.132) | 0.019 | 3.125(2.019-5.111) | 0.007 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CEA (<10/≥10ng/mL) | - | - | 0.799(0.516-1.114) | 0.261 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Location invasion (T1+T2/T3+T4) | - | - | 1.091(0.781-1.298) | 0.315 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Lymph node metastasis (N0-N1/N2) | 1.516(1.093-2.978) | 0.012 | 1.902(1.311-3.391) | 0.006 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TNM stage (I-II/III-IV) | 1.742 (1.065-3.145) | 0.002 | 3.289 (2.141-4.995) | <0.001 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TINCR (low/high) | 0.459 (0.265-0.912) | 0.027 | 0.414(0.226-0.624) | 0.005 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviations: CI, confidence interval; HR, hazard ratio. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
SP1 induces TINCR expression by functioning as a transcription factor
The promoter region of TINCR was identified by JASPAR database (http://jaspardev.genereg.net/), and found that three putative SP1-binding sites at the sites E1(-163bp to-153bp, ACTCCGCCTCT), E2 (-88to -78bp, GCCCCGCCCCG) and E3 (-16 to -6bp, GGGGGCGGGCG) in the TINCR promoter (Fig. 2A). ChIP assay was performed to determine which region in the TINCR promoter mediated SP1-binding to the TINCR promoter. The ChIP data indicated that SP1 could bind to E1 sites (Fig. 2B). To further verify this result, we cloned the full promoter region of TINCR and E1 deleted promoter region into pGL3-basic reporter. Our results showed that the deletion of E1 sites significantly impaired the effect of SP1 on TINCR transcription activation (Fig. 2C), implying that SP1 could bind to the promoter of TINCR to regulate TINCR transcription.
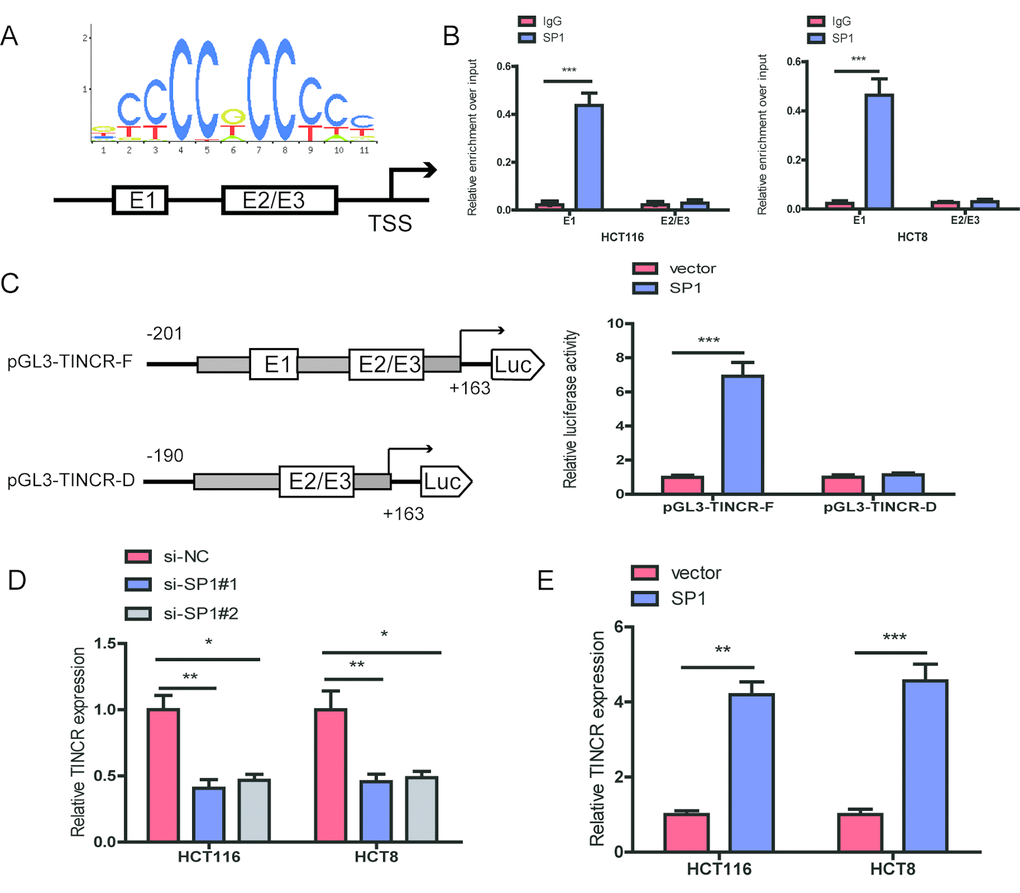
Figure 2. SP1 is involved in TINCR upregulation. (A)The predicted positions of putative SP1 binding motif in human TINCR promoter. (B) ChIP assays were employed to show direct binding of SP1 to endogenous TINCR promoter regions. (C) A luciferase reporter assay was performed by cotransfecting the full TINCR promoter(pGL3-TINCR-F) or deleted TINCR promoter fragment E1 (pGL3-TINCR-D) with SP1 or blank vector in 293T cells. (D, E) qRT-PCR analysis of TINCR expression levels following SP1 upregulation and knockdown. Data were shown as mean ±S.D. of three independent experiments. * P<0.05, **P<0.01, ***P<0.001.
Moreover, we upregulated SP1 by transfecting pcDNA3.1-TINCR and knocked down SP1 using an siRNA targeting SP1. Upon SP1 downregulation, TINCR expression was significantly decreased in HCT116 and HCT8 cells, while upon SP1 overexpression, TINCR expression was markedly increased in HCT116 and HCT8 cells (Fig. 2D, 2E).
Silencing of TINCR inhibits CRC cells proliferation, migration and invasion in vitro
To further explore the biological role of TINCR in CRC cells, we designed two siRNAs targeting TINCR to silence TINCR expression in HCT116 and HCT8 cells. The two siRNAs obviously decreased TINCR expression levels, and we chose si-TINCR#1 for the further study due to the higher inhibitory efficiency (Fig. 3A). CCK-8 assays showed that silencing of TINCR significantly inhibited CRC cell proliferation in HCT116 and HCT8 cells (Fig. 3B). The colony formation assay revealed that TINCR knockdown markedly suppressed colony forming ability of these two cells (Fig. 3C). Moreover, wound healing and transwell invasion assay demonstrated that TINCR knockdown obviously impeded HCT116 and HCT8 cells migration and invasion (Fig. 3D, 3E), respectively.
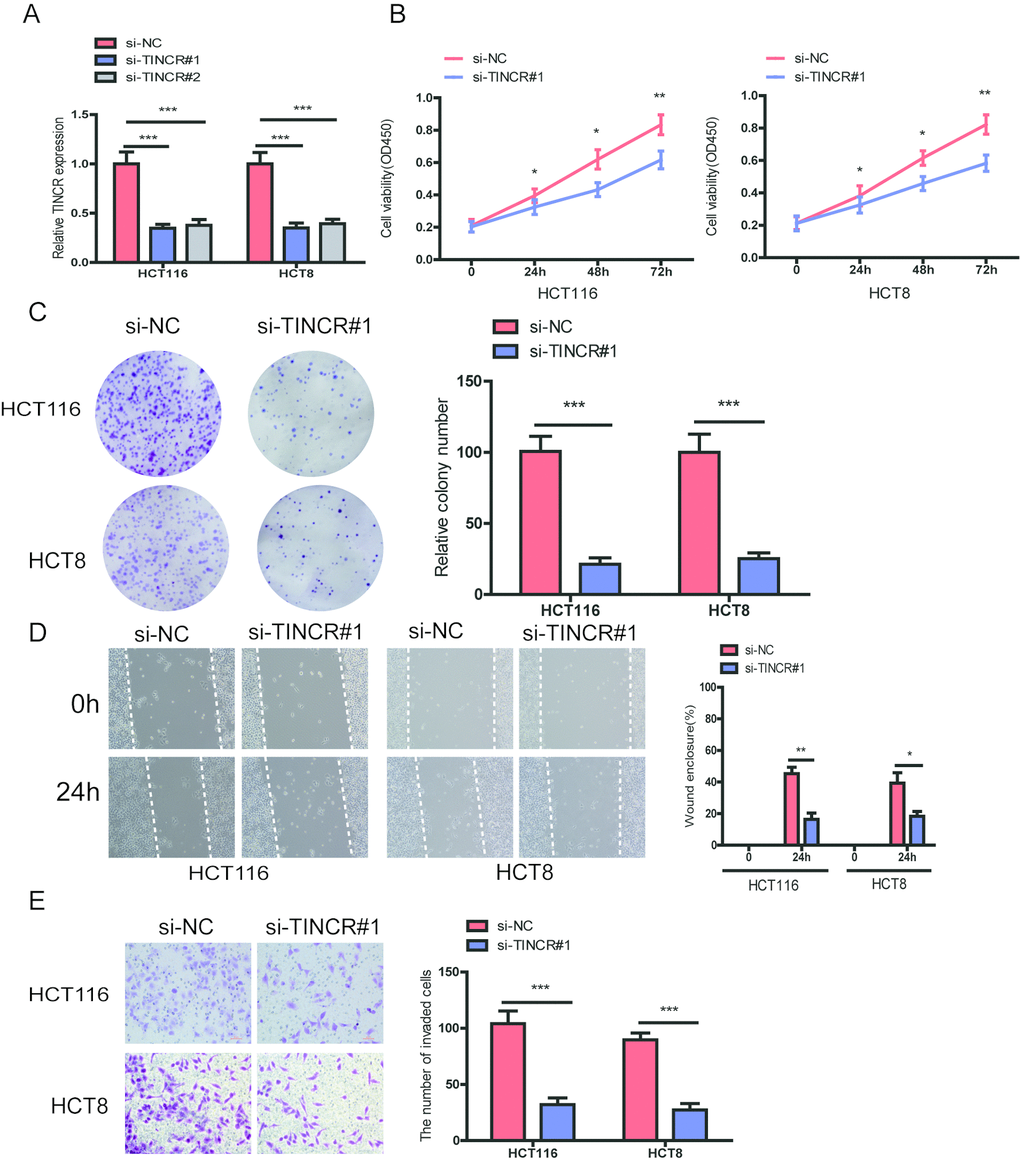
Figure 3. TINCR knockdown suppressed CRC cells proliferation, migration and invasion. (A) The relative expression of TINCR were detected after transfecting with siTINCR#1, siTINCR#2 or negative control (si-NC) in HCT116 and HCT8 cells. (B, C) Cells proliferation were evaluated in TINCR knockdown CRC cells using CCK-8 (B) and colony formation (C). (D, E) Wound healing (D) and transwell invasion (E) assays were performed to assess the ability of TINCR knockdown CRC cells. Data were shown as mean±S.D.. *P<0.05, **P<0.01. ***P<0.001.
Based on above results, we preliminarily conclude that TINCR could retard the progression of CRC cells.
Silencing of TINCR inhibits CRC cells growth and metastasis in vivo
To further explore the biological function of TINCR on CRC cells in vivo, we established the xenograft mice models by injecting TINCR stable knockdown HCT116 cells. As illustrated in Fig. 4A, TINCR depletion could inhibit tumor growth. We also established the lung metastasis model via tail vein injecting cells into nude mice (n=4 for each group), as shown in Fig 4B, an average of 24 lung metastatic nodules per mouse was detected in Lv-ShNC group, while only 8 were observed in Lv-shTINCR group. Overall, these results indicate that silencing of TINCR suppresses CRC cells growth and metastasis in vivo.
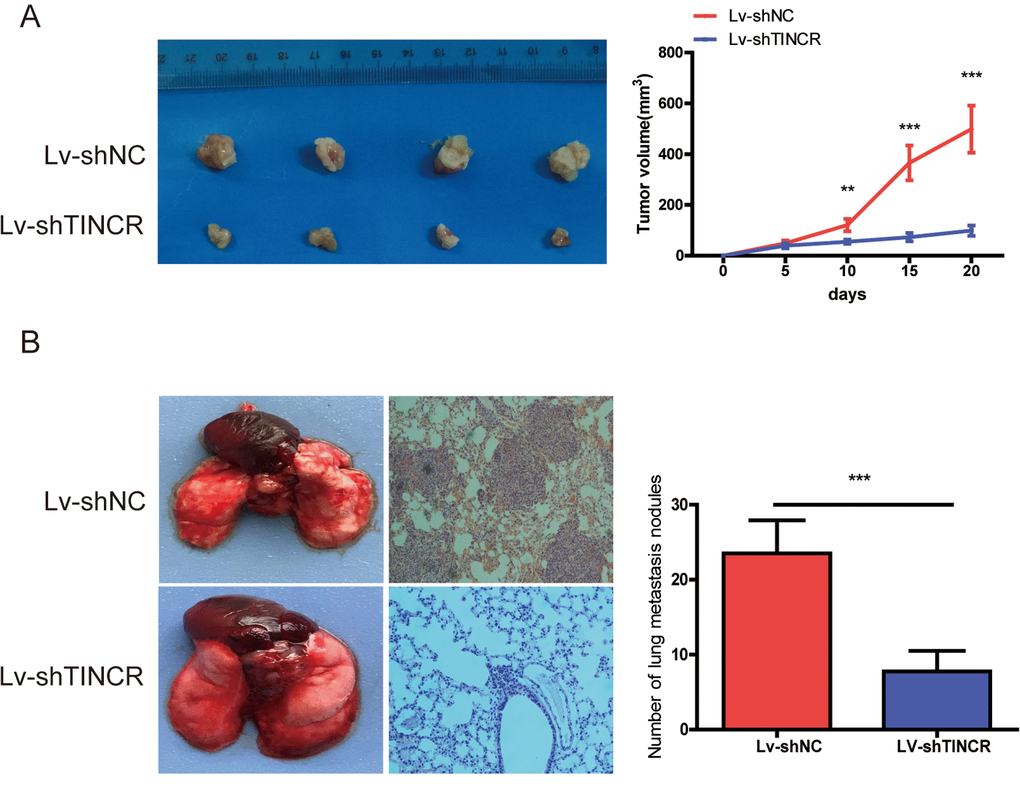
Figure 4. TINCR knockdown inhibited tumor growth and metastasis. (A) Subcutaneous implant model was established using TINCR stable knockdown HCT116 cells. The volume of xenograft tumors in two groups (n=4). (B) The number of metastatic nodules in the lungs of mice (three sections evaluated per lung) from two groups(n=4). Data are presented as the mean±S.D. **P<0.01. ***P<0.001.
TINCR can sponge miR-7-5p in CRC cells
To detect the subcellular location of TINCR, the result of fluorescence in situ hybridization (FISH) indicated that TINCR mainly located in the cytoplasm (Fig. 5A).
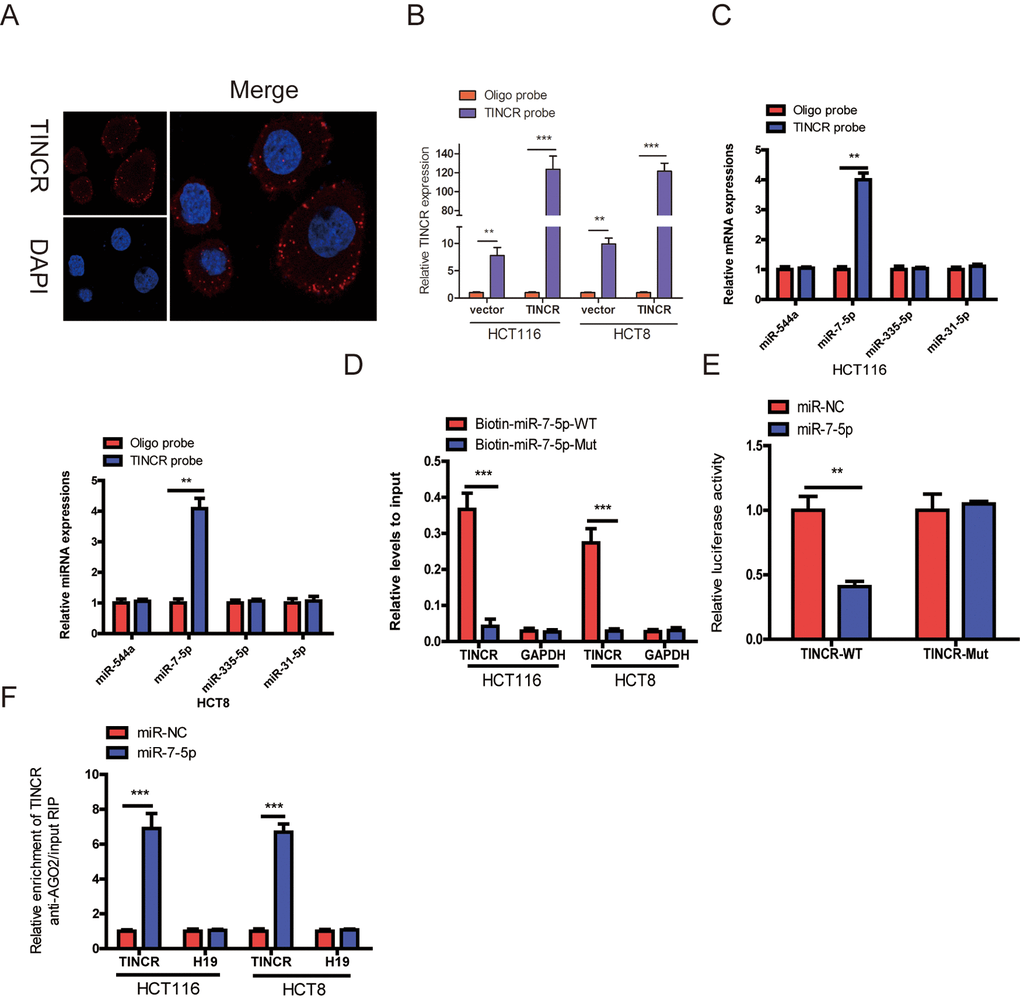
Figure 5. TINCR functioned as a ceRNA by sponging miR-7-5p. (A) RNA-FISH were employed to verify that TINCR was located mainly in the cytoplasm. (B) Lysates from HCT116 and HCT8 cells with TINCR overexpression were subject to biotinylated-TINCR pull-down assay and the expression of TINCR were measured by qRT-PCR. (C) The expression of four candidate miRNAs predicted by starbase database were quantified by qRT-PCR after the biotinylated-TINCR pull-down assay in HCT116 and HCT8 cells. (D) The biotinylated wild-type/mutant miR-7-5p was, respectively, transfected into HCT116 and HCT8 cells with TINCR overexpression. The expression of TINCR were measured by qRT-PCR after streptavidin capture. (E) Luciferase activity in HCT116 and HCT8 cells cotransfected with luciferase reporter containing TINCR sequences with wild-type or mutated miR-7-5p binding sites and miR-7-5p or its control. (F) Anti-AGO2 RIP was used in HCT116 and HCT8 cells overexpressing miR-7-5p, followed by qRT-PCR to assess the expression of TINCR or H19 (control) associated with AGO2. The data are presented as the mean±S.D. of three independent experiments. **P<0.01. ***P<0.001.
Mounting evidences have proved that lncRNAs could function as competing endogenous RNA (ceRNA) to indirectly regulate miRNAs [11,12]. To explore whether TINCR has a similar mechanism in CRC cells, we selected four (miR-544a, miR-7-5p, miR-335-5p and miR-31-5p) candidate miRNAs through Starbase database (http://starbase.sysu.edu.cn/). A 3’terminal-biotinylated-TINCR probe was designed to investigate which miRNAs could interact with TINCR. The probe was proved to pull down TINCR in CRC cells and TINCR upregulation increased the pull-down efficiency (Fig. 5B), the results showed that miR-7-5p was the only miRNA that was abundantly pulled down by TINCR probe (Fig. 5C).
To further verify the interaction between them, TINCR- overexpressing cells were transfected with wild-type or mutant biotinylated miR-7-5p mimics, and the data suggested that wild-type miR-7-5p mimics captured more TINCR than mutant miR-7-5p mimics (Fig. 5D).
To further prove that TINCR could bind to miR-7-5p, the wild type sequence of TINCR (TINCR-WT) or its mutant sequence (TINCR-Mut) was cloned into the pmirGLO luciferase reporter, miR-7-5p significantly decreased luciferase activity of the pmirGLO-TINCR-WT vector, but failed to reduce luciferase activity of pmirGLO-TINCR-Mut vector. We preliminary revealed that TINCR was a target of miR-7-5p (Fig. 5E).
miRNAs bind their targets and exert the ability of translational repression or RNA degradation mainly in an AGO2-dependent manner [13,14]. To further determine whether TINCR binds to miR-7-5p in this manner, RIP was performed in CRC cells and confirmed the interaction between TINCR and miR-7-5p (Fig. 5F). These results strongly supported the idea that TINCR acted as a miR-7-5p sponge in CRC.
miR-7-5p inhibition could reverse the inhibitory effect of TINCR downregulation
To further demonstrated that TINCR promoted CRC progression via sponging miR-7-5p. Firstly, we downregulated miR-7-5p expression in TINCR-silencing cells (Fig. 6A), and found that downregulation of miR-7-5p could partially counteract the decrease in proliferation (Fig. 6B), migration (Fig. 6C) and invasion (Fig. 6D) by the TINCR downregulation.
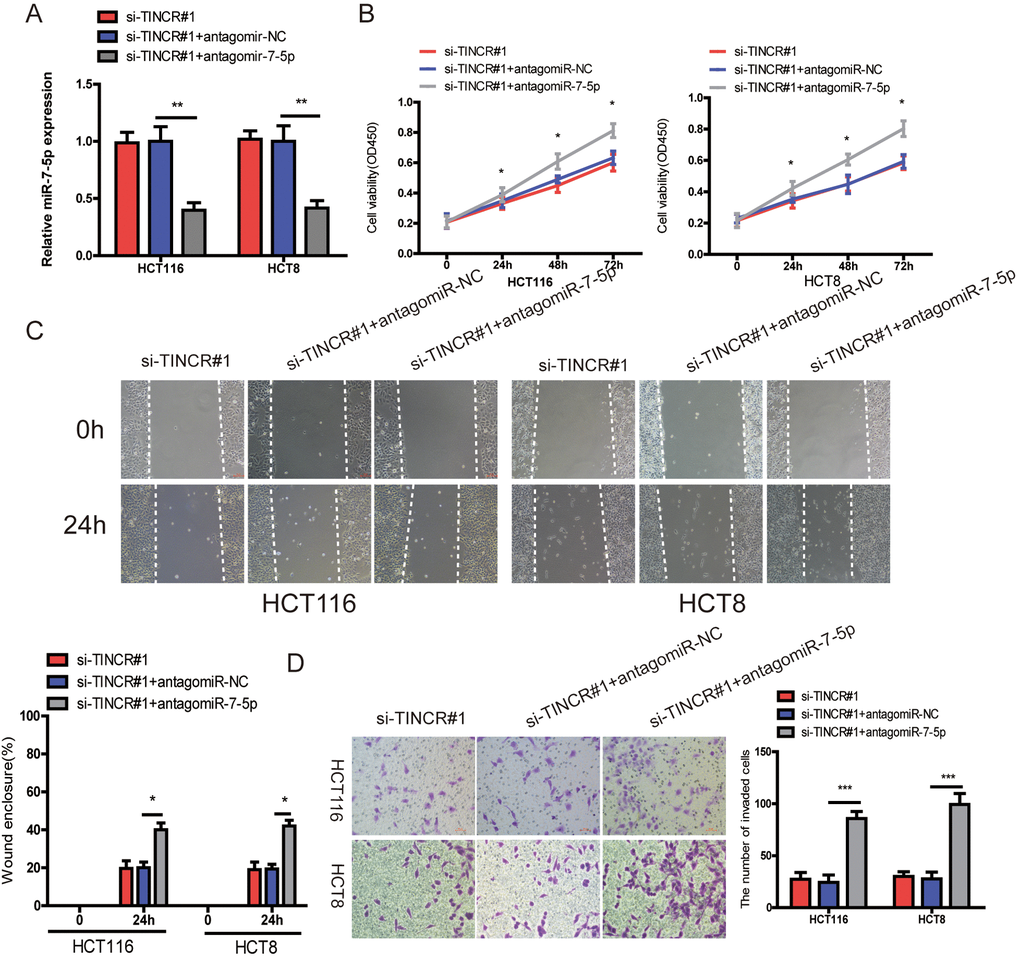
Figure 6. miR-7-5p inhibition could reverse the inhibitory effect of TINCR downregulation. (A) miR-7-5p was downregulated in TINCR knockdown CRC cells. (B, C, D) The cell proliferation, migration and invasion were assessed using CCK-8 (B), Wound-healing (C) and transwell invasion (D) assays in cells cotransfected with siTINCR#1 and antagomiR-7-5p or antagomiR-NC. The data are shown as the mean±S.D. of three independent experiments. *P<0.05, **P<0.01. ***P<0.001.
TINCR may promote CRC progression via PI3K/Akt/mTOR signaling pathway.
Previous studies have revealed that miR-7 regulated cell proliferation and metastasis via PI3K/Akt/mTOR signaling pathway [15–17]. Here, we found that miR-7-5p overexpression could suppress the expression of Akt and mTOR as well as phosphorylation of Akt and mTOR(p-Akt, p-mTOR) (Fig. 7A), while TINCR upregulation could restore the expression of Akt, mTOR, p-Akt and p-mTOR (Fig. 7B). Therefore, we could speculate that TINCR may promote CRC progression via miR-7-5p-mediated PI3K/Akt/mTOR signaling pathway.
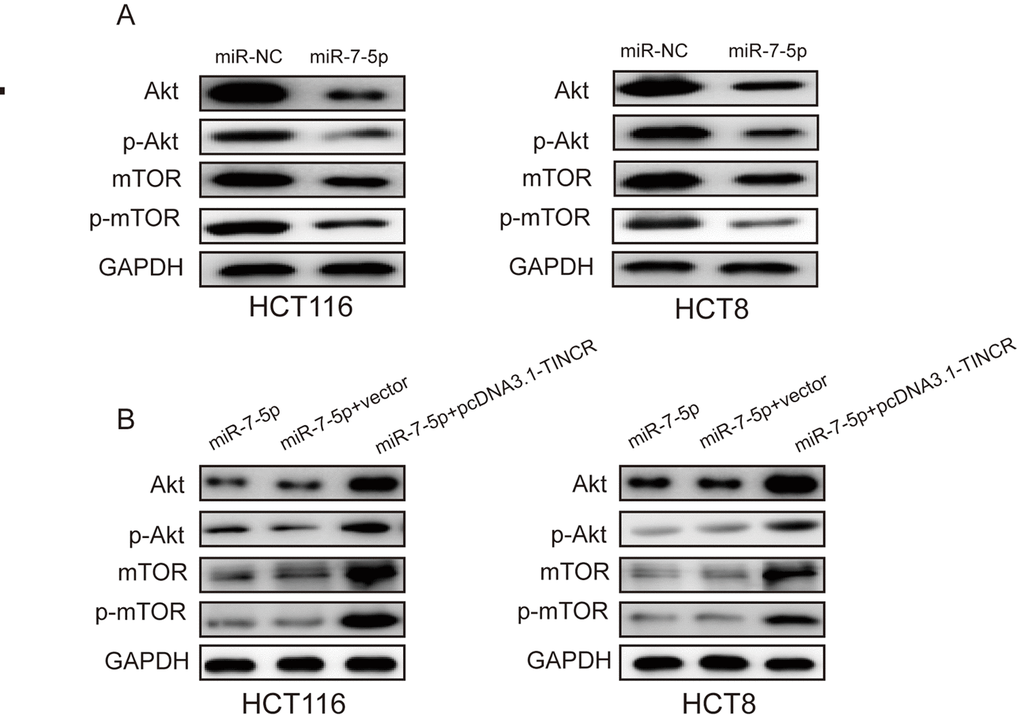
Figure 7. TINCR may promote CRC progression via PI3K/Akt/mTOR pathway. (A) miR-7-5p overexpression markedly inhibited the expression of Akt, p-Akt, mTOR and p-mTOR. (B) TINCR upregulation could restore the expression of Akt, p-Akt, mTOR and p-mTOR.
TINCR might be a potential diagnostic biomarker of CRC
To determine whether plasma TINCR could be a potential biomarker for CRC, we compared plasma TINCR levels between healthy volunteers and CRC patients. The data showed that plasma TINCR level in CRC patients group was significantly higher than that in the healthy controls group (Fig. 8A). An ROC curve curve was performed for distinguishing healthy controls from patients with CRC. The results showed that the AUC was up to 0.922 (95% CI, 0.878-0.966; p<0.001). On the cutoff values from ROC curves, when the cutoff value of plasma TINCR was 1.815, sensitivity was 97.5%, and specificity was 80.0% (Fig. 8B).
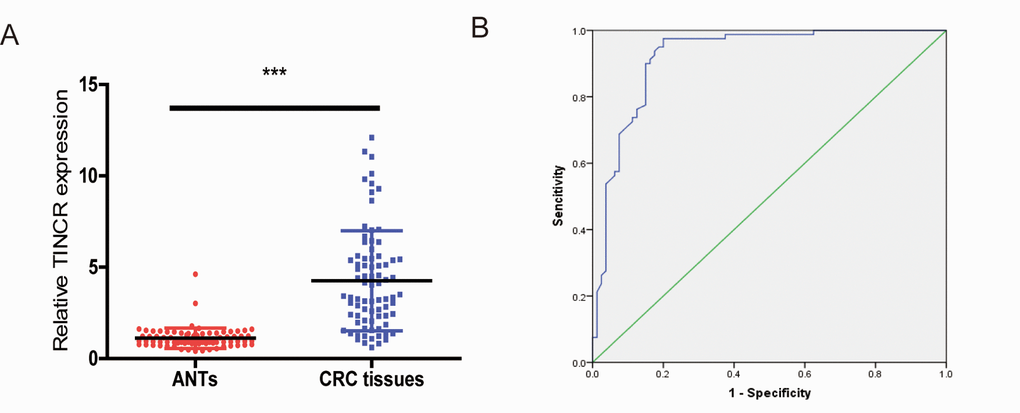
Figure 8. Plasma levels of TINCR were upregulated in CRC patients. (A) The relative levels of TINCR in CRC patients and healthy controls. All data were normalized to GAPDH. (B) ROC curves for TINCR in 80 patients with CRC and 80 healthy controls. ***P<0.001.
Discussion
Increasing researches have shown that lncRNAs play pivotal roles in cancer initiation and progression, and dysregulated lncRNAs have also been identified in CRC [18,19]. Previous studies have reported that TINCR exerts discrepant function in various cancers. For example, Tongpeng Xu, et al. [20] reported that TINCR was aberrantly expressed in gastric cancer, and contributed gastric cancer progression through activating TINCR/STAU1/CDKN2B signaling axis. Zhijun Zhu, et al. [21] reported that TINCR was upregulated in non-small cell lung cancer and promoted NSCLC tumorigenesis and progression via BARF-activated MAPK pathway. Here, we showed that TINCR was upregulated in CRC tissues and cells, and TINCR overexpression was closely associated with differentiation, TNM stage, Lymph node metastasis and poor OS. Moreover, we found that silencing of TINCR significantly inhibited CRC cells proliferation, migration, invasion and metastasis.
SP1, as a transcription factor, could promote cancer progression through altering the expression of other genes, for example, Guanghua Liu, et al. [22] reported that SP1 could bind to the lncRNA-SNHG14 promoter region and promote its transcription. In this work, ChIP and luciferase reporter assays were performed and determine that SP1 could bind to TINCR promoter region, and then induce its transcription.
Increasing studies have verified that lncRNAs could regulate mRNA levels via competing for miRNAs, by acting as ceRNAs [23,24]. For example, Dong Liu, et al. [25] reported that lncRNA SPRY4-IT4 functioned as miR-101-3p sponge to promote bladder cancer cells growth and metastasis. Xiangsong Wu, et al. [26] reported that lncRNA-PAGBC competitively binds to miR-133b and miR-511 and promote tumor growth and metastasis in gallbladder cancer. In this study, we showed TINCR mainly located in the cytoplasm, bioinformatics analysis indicated that TINCR could interact with miR-7-5p, RNA pull down, RIP and luciferase reporter assay were performed to further verify that TINCR is able to bind to miR-7-5p. Furthermore, we found that downregulation of miR-7-5p could partially abrogate the decrease in proliferation, migration and invasion by the TINCR downregulation.
Previous studies have reported that Akt/mTOR signaling pathway played a critical role in various biological processes of cancers, such as proliferation, metastasis and survival [27–29]. Here, we revealed that TINCR may promote CRC progression via miR-7-5p-mediated PI3K/Akt/mTOR signaling pathway.
Recently, lncRNAs have been identified as novel biomarkers in various cancers [30,31]. Here, we tested the potential diagnostic value of plasma TINCR in differentiating people without CRC from CRC patients. We detected that plasma TINCR levels from patients with CRC were significantly higher than the levels from healthy controls. Our data suggested that plasma TINCR had a potential diagnostic value in CRC.
In summary, our study found that TINCR was upregulated in CRC tissues and cells, and was activated by transcription factor SP1. We also observed that TINCR, acting as a miR-7-5p sponge, promoted tumor growth, migration, invasion and metastasis in CRC. Our data will provide new insights into the underlying mechanism of CRC progression and suggested that TINCR might serve as a promising prognostic biomarker and a potential therapeutic target for CRC.
Materials and Methods
Patients and tissue samples
A total of 80 CRC tissues and their matched adjacent normal tissues (ANTs) were obtained from CRC patients during operation in the Second Affiliated Hospital, Zhejiang University School of Medicine between January 2006 and July 2009. None patients achieve preoperative chemotherapy and radiotherapy before surgery. All these samples were placed immediately in liquid nitrogen and stored at -80°C until use. Informed consent was obtained from each patient and this study was approved by the Institutional Review Board of The Second Affiliated Hospital, Zhejiang University School of Medicine. Detailed patient information is listed in Table 1.
Cell culture
Human normal colon epithelial cell (FHC) and CRC cell lines (HCT116, HCT8, HT29, SW620 and SW480) were obtained from ATCC. These cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, USA) with 100U/ml penicillin, 0.1mg/ml streptomycin and 10% fetal bovine serum (FBS, Gibco, USA) at 37°C supplied with 5% CO2 atmosphere.
Cell transfection and stable cell line construction
Si-TINCR (siTINCR#1, siTINCR#2), si-SP1(si-SP1#1, siSP1#2), miR-7-5p agomir, miR-7-5p antagomir and their corresponding negative control (NC) were obtained from Genepharma (Shanghai, China) and the sequences of siRNAs were listed in Table S1. To overexpress TINCR, human TINCR was synthesized and cloned into pcDNA3.1 vector. Transfections were performed using Lipofectamine 2000(Invitrogen) in accordance with the manufacture’s protocol.
To prove the function of TINCR in vivo, we constructed stable TINCR knockdown cells using lentivirus-mediated short hairpin RNA (Lv-shTINCR or LV-shNC) against the more effective siRNA target site (si-TINCR#1) or its negative control (si-NC). HCT116 cells were infected with concentrated virus, then the cells were selected by treatment of puromycin (2µg/ml).
Luciferase reporter assay
For the promoter of TINCR luciferase reporter assay, the wild-type (WT) and mutant (Mut) promoters of TINCR were synthesized and cloned into PGL3-basic vectors (GeneCreat, China), and cotransfected with SP1 plasmid into 293T cells.
For TINCR and miR-7-5p luciferase reporter assay, the TINCR sequences containing WT or Mut miR-7-5p binding sites (BS) were respectively synthesized and cloned into pmirGLO luciferase vector (GeneCreat, China), then cotransfected with miR-7-5p mimics or its negative control(miR-NC) into 293T cells. Cells were harvested at 48 h after transfection, and the luciferase activity was detected using the dual-luciferase reporter assay system (Promega).
Fluorescent in situ hybridization (FISH)
FISH assay was performed using a Ribo™ lncRNA FISH Probe Mix and Ribo™ Fluorescent In Situ Hybridization Kit and (Ribo, China) following the manufacturer’s protocols.
Chromatin immunoprecipitation (ChIP) assays
The ChIP assay kit (Beyotime, China) was used to conduct ChIP assays. In brief, the cells were fixed with 1% formaldehyde solution for 20 min and quenched with 0.125M glycine for 10 min. DNA fragments ranging from 200 bp to 300 bp were generated using sonication. Antibodies including anti-SP1(ab227383, Abcam) and IgG were employed for each immunoprecipitation. The precipitated DNA was analyzed using qPCR. The primer sequences were listed in Table S2.
RNA immunoprecipitation
An EZMagna RNA immunoprecipitation (RIP) Kit (Millipore, Billerica, USA) was used according to the manufacturer’s protocol. Antibody (anti-AGO2) for RIP assay was from Abcam.
Biotinylated RNA pull-down assay
The pull-down assay with biotinylated RNA was performed as described previously [10]. In brief, for TINCR pulled down miRNAs, the biotinylated-TINCR probe was incubated with C-1 magnetic beads (Life Technology) to generate probe-coated beads, incubated with sonicated cells at 4 °C overnight,followed by eluted and qRT-PCR. For miR-7-5p pulled down TINCR, cells with TINCR overexpression were transfected with biotinylated miR-7-5p mimics or mutant using Lipofectamine 2000. Then the cells were harvested, lysed, sonicated, and incubated with C-1 magnetic beads (Life Technologies), followed by washed and qRT-PCR.
RNA isolation and qRT-PCR
Total RNA from tissue samples, cells and plasma were isolated using TRIzol reagent (Invitrogen). The miR-7-5p levels were quantified using Hairpin-itTM MicroRNAs Quantitation PCR (GenePharma, China), TINCR levels were measured by SYBR Green PCR kit (Takara, Japan). U6 was selected as reference gene for miR-7-5p and GAPDH was chosen as an internal control for TINCR. The relative expression of TINCR or miR-7-5p using the 2-ΔΔCt method. The primer sequences were shown in Table S3.
Western blot
Proteins were extracted from cells using RIPA lysis buffer. Equal amounts of protein (30µg) were separated by SDS-PAGE and transferred onto polyvinylidene fluoride membranes (PVDF), and incubated with primary antibody at 4 °C overnight. Afterwards, the membranes were incubated with secondary antibody for 1 h. The blots were detected by using enhanced chemiluminescence reagents (ECL, KeyGEN BioTECH, China).
Cell counting kit-8 assay
CCK-8 assay was employed to evaluate the proliferative ability of CRC cells following manufacturer’s protocol. In brief, 1×103 transfected cells were seeded into 96-well plates and treated with 10 µl CCK-8 solution. Absorbance was detected at 450 nm by using a microplate reader (Bio-Tek Instruments Inc., Winooski, VT, USA).
Colony formation assay
1000 transfected cells were plated into 6-well plates and cultured for two weeks. The colonies were fixed with 100% methanol for 15 min and stained using 0.1% crystal violet for 20 min at room temperature.
Wound-healing assay
Wound-healing assay was used to assess cell migration. In brief, transfected cells were plated into six well plates (3×105 cells/well) and grown to about 90% confluence. Cells were scratched by using 200 µL pipette tips, and then washed with PBS. Cells were further cultured with a medium containing 1% FBS for 24 h. Images were detected using an inverted microscope (Nikon Corporation, Tokyo, Japan) at 100×magnifcation.
Transwell invasion assay
The cell invasion was performed using a 24-well transwell chamber (Corning, NY, USA. Cells were plated into upper chamber precoated with 2% Matrigel (BD Biosciences, USA). The lower chambers were loaded with 500µl DMEM containing 20% FBS. The invaded cells were fixed with 100% methanol for 15 min and stained using 0.1% crystal violet for 20 min at room temperature. The number of invaded cells were counted by a microscope (Nikon Corporation, Tokyo, Japan) at 200×magnification.
In vivo experiments
All animal experiments were approved by the animal care Committee of The Second Affiliated Hospital, Zhejiang University School of Medicine.
For xenograft tumor model, 1×107 TINCR stable knockdown HCT116 cells were subcutaneously into the the armpit region of 8 six-week old male BABL/c nude mice which were randomly divided into two groups (n=4 each group) into the tumors. The tumors were measured every 5 days. The volume of tumors was calculated using the following equation: V=(L×W2)/2, L is the length and W is the width of tumor.
For metastasis assay, 2 × 106 stable knockdown HCT116 cells were injected into 8 six-week-old male BALB/c nude mice (n=4 for each group) which were randomly divided into two groups (n = 4 for each group). Six weeks later, the mice were sacrificed, and their lungs were removed and stained by Hematoxylin and Eosin (HE) Staining.
Statistical analysis
All experiments were performed in triplicate. The data are presented as mean ± S.D., and statistical analysis were conducted using SPSS version 17.0. One-way analysis of variance (ANOVA) and the Student’s t-test were used to estimate the differences between groups. χ2 test or Pearson’s Mann-Whitney U test was employed to analyze the relationship between TINCR expression and clinicopathological factors. Kaplan-Meier method was performed to evaluate overall survival (OS). The survival curves were compared with log-rank test. Cox proportional hazards model was applied to perform multivariate analysis and calculate the 95% confidence interval (95% CI). p<0.05 was considered to be statistically significant.
Supplementary Materials
Abbreviations
CRC: colorectal cancer; ceRNA: competing endogenous RNA; OS: overall survival; RIP: RNA immunoprecipitation; FISH: fluorescence in situ hybridization; p-Akt: phosphorylation of Akt; p-mTOR: phosphorylation of mTOR.
Conflicts of Interest
The authors have declared that no competing interest exists.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66:7–30. https://doi.org/10.3322/caac.21332 [PubMed]
- 2. Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017; 66:683–91. https://doi.org/10.1136/gutjnl-2015-310912 [PubMed]
- 3. Necsulea A, Soumillon M, Warnefors M, Liechti A, Daish T, Zeller U, Baker JC, Grützner F, Kaessmann H. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature. 2014; 505:635–40. https://doi.org/10.1038/nature12943 [PubMed]
- 4. Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009; 10:155–59. https://doi.org/10.1038/nrg2521 [PubMed]
- 5. Wang P, Ning S, Zhang Y, Li R, Ye J, Zhao Z, Zhi H, Wang T, Guo Z, Li X. Identification of lncRNA-associated competing triplets reveals global patterns and prognostic markers for cancer. Nucleic Acids Res. 2015; 43:3478–89. https://doi.org/10.1093/nar/gkv233 [PubMed]
- 6. Wan D, Gong Y, Qin W, Zhang P, Li J, Wei L, Zhou X, Li H, Qiu X, Zhong F, He L, Yu J, Yao G, et al. Large-scale cDNA transfection screening for genes related to cancer development and progression. Proc Natl Acad Sci USA. 2004; 101:15724–29. https://doi.org/10.1073/pnas.0404089101 [PubMed]
- 7. Kretz M, Siprashvili Z, Chu C, Webster DE, Zehnder A, Qu K, Lee CS, Flockhart RJ, Groff AF, Chow J, Johnston D, Kim GE, Spitale RC, et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature. 2013; 493:231–35. https://doi.org/10.1038/nature11661 [PubMed]
- 8. Xu TP, Liu XX, Xia R, Yin L, Kong R, Chen WM, Huang MD, Shu YQ. SP1-induced upregulation of the long noncoding RNA TINCR regulates cell proliferation and apoptosis by affecting KLF2 mRNA stability in gastric cancer. Oncogene. 2015; 34:5648–61. https://doi.org/10.1038/onc.2015.18 [PubMed]
- 9. Liu X, Ma J, Xu F, Li L. TINCR suppresses proliferation and invasion through regulating miR-544a/FBXW7 axis in lung cancer. Biomed Pharmacother. 2018; 99:9–17. https://doi.org/10.1016/j.biopha.2018.01.049 [PubMed]
- 10. Xu TP, Wang YF, Xiong WL, Ma P, Wang WY, Chen WM, Huang MD, Xia R, Wang R, Zhang EB, Liu YW, De W, Shu YQ. E2F1 induces TINCR transcriptional activity and accelerates gastric cancer progression via activation of TINCR/STAU1/CDKN2B signaling axis. Cell Death Dis. 2017; 8:e2837. https://doi.org/10.1038/cddis.2017.205 [PubMed]
- 11. Chen X, Zeng K, Xu M, Hu X, Liu X, Xu T, He B, Pan Y, Sun H, Wang S. SP1-induced lncRNA-ZFAS1 contributes to colorectal cancer progression via the miR-150-5p/VEGFA axis. Cell Death Dis. 2018; 9:982. https://doi.org/10.1038/s41419-018-0962-6 [PubMed]
- 12. Lü MH, Tang B, Zeng S, Hu CJ, Xie R, Wu YY, Wang SM, He FT, Yang SM. Long noncoding RNA BC032469, a novel competing endogenous RNA, upregulates hTERT expression by sponging miR-1207-5p and promotes proliferation in gastric cancer. Oncogene. 2016; 35:3524–34. https://doi.org/10.1038/onc.2015.413 [PubMed]
- 13. Bagga S, Bracht J, Hunter S, Massirer K, Holtz J, Eachus R, Pasquinelli AE. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell. 2005; 122:553–63. https://doi.org/10.1016/j.cell.2005.07.031 [PubMed]
- 14. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009; 136:215–33. https://doi.org/10.1016/j.cell.2009.01.002 [PubMed]
- 15. Fang Y, Xue JL, Shen Q, Chen J, Tian L. MicroRNA-7 inhibits tumor growth and metastasis by targeting the phosphoinositide 3-kinase/Akt pathway in hepatocellular carcinoma. Hepatology. 2012; 55:1852–62. https://doi.org/10.1002/hep.25576 [PubMed]
- 16. Kefas B, Godlewski J, Comeau L, Li Y, Abounader R, Hawkinson M, Lee J, Fine H, Chiocca EA, Lawler S, Purow B. microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res. 2008; 68:3566–72. https://doi.org/10.1158/0008-5472.CAN-07-6639 [PubMed]
- 17. Guo J, Xu L, Teng X, Sun M. MicroRNA-7-5p regulates the proliferation and migration of intestinal epithelial cells by targeting trefoil factor 3 via inhibiting the phosphoinositide 3-kinase/Akt signalling pathway. Int J Mol Med. 2017; 40:1435–43. https://doi.org/10.3892/ijmm.2017.3120 [PubMed]
- 18. Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011; 1:391–407. https://doi.org/10.1158/2159-8290.CD-11-0209 [PubMed]
- 19. Gutschner T, Diederichs S. The hallmarks of cancer: a long non-coding RNA point of view. RNA Biol. 2012; 9:703–19. https://doi.org/10.4161/rna.20481 [PubMed]
- 20. Xu TP, Wang YF, Xiong WL, Ma P, Wang WY, Chen WM, Huang MD, Xia R, Wang R, Zhang EB, Liu YW, De W, Shu YQ. E2F1 induces TINCR transcriptional activity and accelerates gastric cancer progression via activation of TINCR/STAU1/CDKN2B signaling axis. Cell Death Dis. 2017; 8:e2837. https://doi.org/10.1038/cddis.2017.205 [PubMed]
- 21. Zhu ZJ, He JK. TINCR facilitates non-small cell lung cancer progression through BRAF-activated MAPK pathway. Biochem Biophys Res Commun. 2018; 497:971–77. https://doi.org/10.1016/j.bbrc.2018.02.059 [PubMed]
- 22. Liu G, Ye Z, Zhao X, Ji Z. SP1-induced up-regulation of lncRNA SNHG14 as a ceRNA promotes migration and invasion of clear cell renal cell carcinoma by regulating N-WASP. Am J Cancer Res. 2017; 7:2515–25. [PubMed]
- 23. Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, Tramontano A, Bozzoni I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011; 147:358–69. https://doi.org/10.1016/j.cell.2011.09.028 [PubMed]
- 24. Lü MH, Tang B, Zeng S, Hu CJ, Xie R, Wu YY, Wang SM, He FT, Yang SM. Long noncoding RNA BC032469, a novel competing endogenous RNA, upregulates hTERT expression by sponging miR-1207-5p and promotes proliferation in gastric cancer. Oncogene. 2016; 35:3524–34. https://doi.org/10.1038/onc.2015.413 [PubMed]
- 25. Liu D, Li Y, Luo G, Xiao X, Tao D, Wu X, Wang M, Huang C, Wang L, Zeng F, Jiang G. LncRNA SPRY4-IT1 sponges miR-101-3p to promote proliferation and metastasis of bladder cancer cells through up-regulating EZH2. Cancer Lett. 2017; 388:281–91. https://doi.org/10.1016/j.canlet.2016.12.005 [PubMed]
- 26. Wu XS, Wang F, Li HF, Hu YP, Jiang L, Zhang F, Li ML, Wang XA, Jin YP, Zhang YJ, Lu W, Wu WG, Shu YJ, et al. LncRNA-PAGBC acts as a microRNA sponge and promotes gallbladder tumorigenesis. EMBO Rep. 2017; 18:1837–53. https://doi.org/10.15252/embr.201744147 [PubMed]
- 27. Trinh XB, Tjalma WA, Vermeulen PB, Van den Eynden G, Van der Auwera I, Van Laere SJ, Helleman J, Berns EM, Dirix LY, van Dam PA. The VEGF pathway and the AKT/mTOR/p70S6K1 signalling pathway in human epithelial ovarian cancer. Br J Cancer. 2009; 100:971–78. https://doi.org/10.1038/sj.bjc.6604921 [PubMed]
- 28. Bauer TM, Patel MR, Infante JR. Targeting PI3 kinase in cancer. Pharmacol Ther. 2015; 146:53–60. https://doi.org/10.1016/j.pharmthera.2014.09.006 [PubMed]
- 29. Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009; 10:307–18. https://doi.org/10.1038/nrm2672 [PubMed]
- 30. Xie Z, Zhou F, Yang Y, Li L, Lei Y, Lin X, Li H, Pan X, Chen J, Wang G, Liu H, Jiang J, Wu B. Lnc-PCDH9-13:1 Is a Hypersensitive and Specific Biomarker for Early Hepatocellular Carcinoma. EBioMedicine. 2018; 33:57–67. https://doi.org/10.1016/j.ebiom.2018.06.026 [PubMed]
- 31. Xu H, Chen Y, Dong X, Wang X. Serum Exosomal Long Noncoding RNAs ENSG00000258332.1 and LINC00635 for the Diagnosis and Prognosis of Hepatocellular Carcinoma. Cancer Epidemiol Biomarkers Prev. 2018; 27:710–16. https://doi.org/10.1158/1055-9965.EPI-17-0770 [PubMed]